

Abstract
The cardiac ryanodine receptor (RyR2), a Ca2+ release channel on the membrane of the sarcoplasmic reticulum (SR), plays a key role in determining the strength of the heartbeat by supplying Ca2+ required for contractile activation. Abnormal RyR2 function is recognized as an important part of the pathophysiology of heart failure (HF). While in the normal heart, the balance between the cytosolic and intra-SR Ca2+ regulation of RyR2 function maintains the contraction–relaxation cycle, in HF, this behaviour is compromised by excessive post-translational modifications of the RyR2. Such modification of the Ca2+ release channel impairs the ability of the RyR2 to properly deactivate leading to a spectrum of Ca2+-dependent pathologies that include cardiac systolic and diastolic dysfunction, arrhythmias, and structural remodelling. In this article, we present an overview of recent advances in our understanding of the underlying causes and pathological consequences of abnormal RyR2 function in the failing heart. We also discuss the implications of these findings for HF therapy.
Keywords: Ryanodine receptor, Heart failure, Arrhythmia, Ca2+ release, Sarcoplasmic reticulum
1. Introduction
The cardiac ryanodine receptor (RyR2) plays an important role in the physiology of the heart by regulating Ca2+ release from the sarcoplasmic reticulum (SR). Such Ca2+ release is required for the activation of systolic contraction. Over the past decade, evidence has accumulated which suggests that altered RyR2 function contributes to the pathological manifestation of heart failure (HF). A consistent finding observed both in human as well as in various animal models of HF is that RyR2s become abnormally active, or ‘leaky’, thereby unable to remain closed during diastole. This dysregulated Ca2+ handling has the potential to decrease systolic contraction while introducing unwanted irregular contractile and electrical activity, and predisposing the myocardium to cellular death and other Ca2+-dependent detrimental processes as part of aetiology of HF. A considerable effort has been directed at identifying the causes as well as the consequences of RyR2 dysfunction in the failing heart. Moreover, ongoing research focuses on strategies to mitigate the phenomenon of the ‘leaky’ RyR2 channels as a potential therapeutic strategy for the management of HF symptoms. Despite substantial progress, however, many uncertainties and controversies remain. In this review, we will summarize the current state of this important and dynamic area of cardiovascular research, focusing on existing controversies and highlighting emerging consensuses. In particular, we will consider, based on available evidence, the mechanisms underlying altered RyR2s activity in the failing heart. We will also present an overview of the functional consequences of altered RyR2 behaviour ultimately leading to a spectrum of pathological outcomes encompassing contractile dysfunction, arrhythmogenesis, and pathological structural remodelling. Finally, we will discuss the prospects and present the state of HF therapies that are based on our current understanding of Ca2+ handling.
2. Control and modulation of RyR2s in normal heart
2.1. RyR2 activation and deactivation during excitation–contraction coupling
The chain of events leading to myocyte contraction in the normal beating heart proceeds as follows: opening of the L-type Ca2+ channels during the systolic action potential initiates Ca2+ influx into the cell that in turn activates RyR2s on the SR membrane, thereby triggering Ca2+-induced Ca2+ release (CICR) from the SR.1,2 Importantly, the aforementioned Ca2+ release is controlled locally, such that the activity of RyR2s arranged in clusters is determined by Ca2+ influx through adjacent L-type Ca2+ channels, as well as by rapid accumulation and dissipation of Ca2+ in a sub-membrane domain (reviewed in Cheng and Lederer3). The RyR2 channel opens when several Ca2+ ions bind to activation sites on the RyR2 homotetramer.4 The ensuing elevation of Ca2+ in the cytoplasm (Ca2+ transient) facilitates Ca2+ binding to contractile proteins and activates myocyte systolic contraction. After its rapid activation, SR Ca2+ release abruptly terminates and stays refractory through most of the diastolic period.5,6 A significant body of evidence indicates that this refractory process involves changes in luminal Ca2+ causing deactivation of RyR2.7–9 The precise molecular steps and players responsible for luminal regulation of RyR2 remain to be elucidated. However, the SR Ca2+-binding protein calsequestrin (CASQ2) may play a role as a luminal sensor by inhibiting RyR2 function at low luminal Ca2+ following SR Ca2+ release.10 During the diastolic relaxation phase, cytosolic Ca2+ is re-sequestered back into the SR by SR Ca2+ ATPase (SERCA2a) to be liberated again during next cardiac cycle. In HF, this orderly Ca2+ cycling is severely altered and is discussed in the subsequent sections.
2.2. Physiological modulation of RyR2s
Apart from its dependence on cytosolic and luminal Ca2+, RyR2 is subject to modulation by multiple cytosolic factors, including Mg2+, ATP, and a number of auxiliary proteins, such as calmodulin (CaM), FKBP12.6, sorcin, triadin, junctin, and CASQ2, that are tethered to the RyR2 from both the cytosolic and luminal sides (reviewed elsewhere10–12). Additionally, RyR2 undergoes post-translational modifications through phosphorylation of at least three distinct sites: by multiple protein kinases including protein kinase A (PKA) and Ca2+/calmodulin-dependent protein kinase II (CaMKII) at Ser-2808,12–14 by CaMKII at Ser-2814,12 and by PKA at Ser-2030,13 respectively. The RyR2 is accordingly equipped with the necessary molecular machinery including kinases (PKA, CaMKII) and phosphatases (PP1 and PP2A) as part of the Ca2+-release complex.12,15,16 Furthermore, the RyR2 contains more than 90 free thiols available for redox modifications via oxidation, nitrosylation, and glutathionylation (reviewed elsewhere11,17,18).
The physiological role of this complex system of signalling and the manner by which it operates in cardiac myocytes is not yet fully understood at the present time. Indeed as emphasized by Eisner et al.,19 modulatory influences on RyR2 alone are only expected to affect several heartbeats due to compensatory readjustments of the SR Ca2+ content. For example, enhancing RyR2 functional activity while increasing Ca2+ release initially will fail to increase release long-term because the extrusion of the additional Ca2+ from the cell via NCX will decrease the SR Ca2+ content. Thus modulatory changes in RyR2 alone appear to be of little consequence to cardiac contractile function (unless the changes are profound such as with high caffeine concentrations which deplete the SR of Ca2+ 20). It is perhaps for this reason alterations in RyR2 typically occur as part of an integral physiological response that also includes changes in Ca2+ resequestration into the SR by SERCA2a and in the L-type Ca2+ current amplitude. For example, the positive inotropic effects of β-adrenergic stimulation involve co-ordinated increases in L-type Ca2+ current, SR Ca2+ uptake, and RyR2 functional activity that results in more synchronous, faster and abbreviated Ca2+ release as required for maintaining faster heart rates. However, as discussed below, this integrated Ca2+ signalling becomes compromised in the failing heart.
3. Functional outcomes and underlying causes of increased RyR2 activity in HF
3.1. RyR2s in HF: an overview
Reduction in the amplitude and prolongation of Ca2+ transient duration are classic hallmark features of HF (reviewed in 21–23). These aberrations in Ca2+ handling are believed to contribute to compromised systolic and diastolic cardiac functions in the failing heart by decreasing systolic contractility and impairing diastolic relaxation. HF is also characterized by an increased propensity of the cardiomyocytes towards spontaneous Ca2+ release and arrhythmogenic membrane potential oscillations, delayed after-depolarizations (DADs).24,25 This is consistent with the increased arrhythmia vulnerability in patients with cardiomyopathy. Increased functional activity of RyR2 has been linked to the abnormal Ca2+ handling in various forms of HF.26–29 More recently, abnormal Ca2+ release via RyR2s has also been associated with cardiomyocyte death and pathological hypertrophy in HF.30–33
About a decade ago Marks et al.15 first brought RyR2 as an important player in HF to the attention of the scientific community. They reported that the functional activity of RyR2s from failing human hearts becomes abnormally high, i.e. ‘leaky’, and attributed this abnormality to hyperphosphorylation of RyR2s (at Ser-2808) which in turn lead to dissociation of the auxiliary protein FKBP12.6 from the RyR2. They further postulated that diastolic Ca2+ leak via hyperactive RyR2s causes depletion of the SR Ca2+ store that leads to reduced systolic Ca2+ transient and contraction. Subsequent studies by multiple groups provided more evidence for RyR2 hyperactivity in HF and its role in the pathophysiology of HF.20,27,34–36 However, the specific mechanism for increased RyR2 leak proposed by Marks et al., involving PKA phopshorylation and FKBP12.6 has been disputed in a number of studies and is not supported by most available evidence.14,37 It is important to note here that intensive investigation into the causes and consequences of RyR2 dysfunction in HF is ongoing and highlights of this work are summarized below.
3.2. Forms of diastolic Ca2+ release: persistent Ca2+ leak, Ca2+ waves, and runaway Ca2+ oscillations
RyR2 hyperactivity in HF was first directly demonstrated in single RyR2 studies as an increase in open probability of RyR2s from failing hearts.15 Subsequently, increased RyR2 functional activity in HF was corroborated in cardiomyocyte experiments both as an increase in Ca2+ spark-mediated SR Ca2+ leak,36,38 as well as a RyR2-mediated loss of SR Ca2+ content measured over time either by application of caffeine or directly by tracking intra-SR Ca2+.20,38 It has been also shown that, in addition to persistent diastolic SR Ca2+ leak, increased RyR2 activity can lead to several different patterns of abnormal Ca2+ handling. These include self-propagating diastolic Ca2+ waves39 and runaway rapid Ca2+ oscillations decoupled from electrical excitation in paced failing myocytes.27 Besides the different degrees of RyR2 leakiness, these different patterns may vary depending on other factors such as adrenergic stimulation and pacing frequency (Figure 1) (see below).
Figure 1.
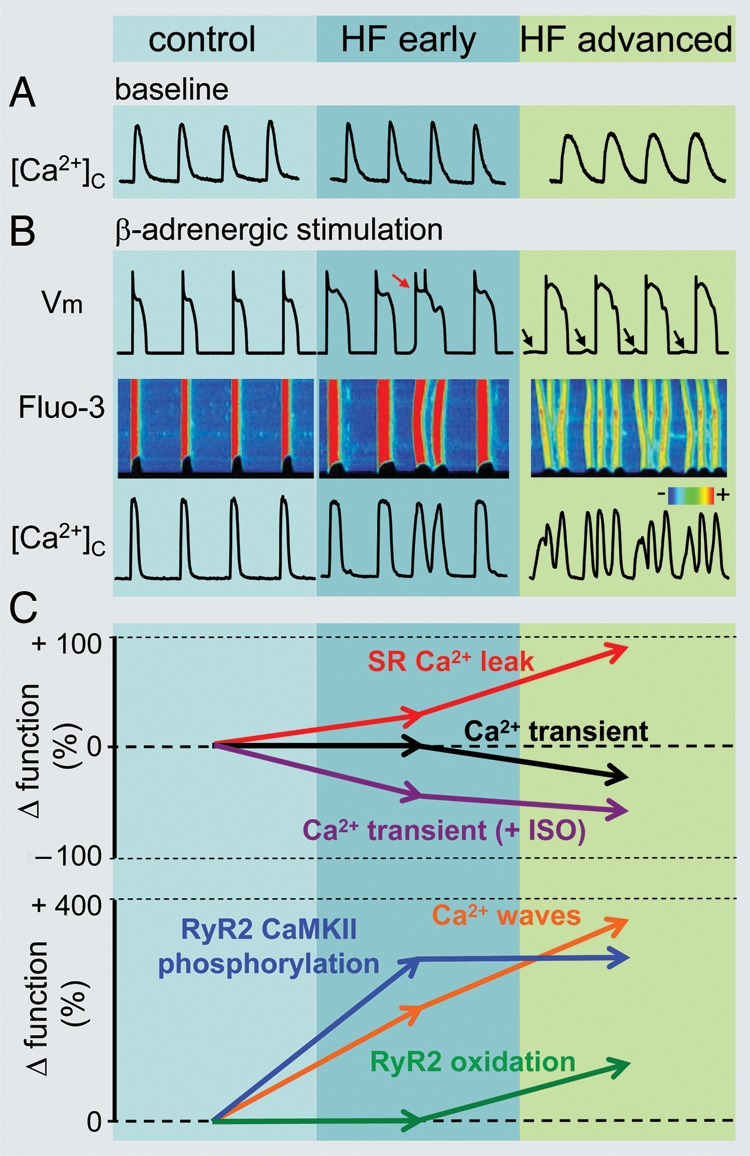
Remodelling of intracellular Ca2+cycling during heart failure (HF) progression. (A) Representative Ca2+ transients recorded in control as well as in ventricular myocytes isolated from hearts with early and advanced stages of HF. (B) Representative action potential (AP) traces (top) along with corresponding line scan images of Fluo-3 fluorescence (middle) and Ca2+ transients ([Ca2+]c; bottom) recorded during β-adrenergic receptor stimulation in the three different types of cardiomyocytes. Arrows indicate delayed after-depolarizations. Red arrow points to extrasystolic AP. (C) Summary graphs illustrate that progression of HF is associated with early and progressive increase in the rate of SR Ca2+ leak (red line). The amplitude of Ca2+ transients recorded under baseline conditions (black line) decreases with significant time-delay in respect to elevation of leak during the onset of HF. Such relationships are attributable to the capacity of myocyte Ca2+ handling for autoregulation until SR is depleted by a drastic increase in the SR Ca2+ leak (see text for detailed explanation). In contrast, the frequency of diastolic Ca2+ waves recorded in the presence of β-adrenergic agonist isoproterenol (ISO) (brown line) parallels increases in the SR Ca2+ leak. Ca2+ waves facilitate diastolic SR Ca2+ loss resulting in decrease in Ca2+ transient amplitude (+ISO; purple line). Note that in advanced stages of HF persistent cytosolic Ca2+ oscillations effectively uncouple electrical excitation from mechanical response. Progressive alterations in Ca2+ cycling in HF are coupled to sequential modification of RyR2 by CaMKII-dependent phosphorylation (blue line) and oxidation (green line). Adapted from Belevych et al.27
Several studies have examined the contribution of elevated SR Ca2+ leak to HF-related alterations in the cytosolic Ca2+ transient. Such studies are challenging due to the presence of competing Ca2+ fluxes, including those mediated by SERCA2a and NCX. HF is commonly associated with reduced SERCA2a and increased NCX expression/activity,21,40 which would tend to slow SR Ca2+ uptake and drive Ca2+ out of the cell thus contributing to the reduced amplitude and slowed decay of Ca2+ transients in HF myocytes. It is important to note, however, that a number of reports have indicated no substantial decrease in SERCA2a abundance/function in animal models and human HF.20,22,40,41 Shannon et al.,34 using a rabbit model of HF, found a significantly elevated SR Ca2+ leak and increased NCX activity without a substantial decrease in SERCA2a function. Using computational analysis, these authors estimated that the significance of leak is secondary to enhanced NCX-mediated Ca2+ extrusion in this model. Direct measurements of SR Ca2+ leak and competing Ca2+ fluxes via NCX and SERCA2a led Belevych et al.20 to conclude that increased RyR2-mediated Ca2+ leak is a major determinant of the reduced SR Ca2+ content and of the decreased amplitude and slowed time course of cytosolic Ca2+ transients in cardiac myocytes from canine hearts in advanced stages of HF. Of note, early stages of HF are characterized by an unchanged and even increased Ca2+ transient amplitude despite evidence of altered RyR2 function.27,42 For example, a longitudinal study27 using a model of rapid-pacing induced HF showed that the decrease in Ca2+ transient amplitude lags measurably behind elevation of leak during the onset of HF (see Figure 1). This delay is consistent with a capacity of Ca2+ handling for autoregulation19 and can be attributed to increased fractional release, which compensates the effects of decreased SR Ca2+ content until the SR Ca2+ content falls below a critical level at more advanced stages of HF.27 Thus persistent SR Ca2+ leak in some models as well as at certain stages of HF can indeed decrease the amplitude of Ca2+ transients and weaken contractility by decreasing the SR Ca2+ content. The precise contributions of various Ca2+ fluxes including SR Ca2+ leak, Ca2+ resequestration into SR, or Ca2+ extrusion via NCX to pathology of HF are likely to vary and remain to be clarified in different models of HF as well as in human cardiomyopathy.
HF is characterized by increased levels of circulating catecholamines.43 To account for the enhanced adrenergic tonus in HF, several studies examined HF-related alterations in myocyte Ca2+ handling under the conditions of β-adrenergic receptor (β-AR) stimulation. When exposed to β-AR agonists, myocytes from failing hearts display an increased predisposition for diastolic Ca2+ waves as well as elevated persistent Ca2+ leak.27,28,44–46 In addition to exacerbating leak-induced SR Ca2+ depletion and weakened contractility,27,47 diastolic Ca2+ waves are considered arrhythmogenic for their potential to induce DADs, which in turn can lead to triggered activity and arrhythmias.24,25 Notably, diastolic Ca2+ waves in myocytes from the hearts at advanced stages of HF have been shown to lead to runaway Ca2+ oscillations that occur independently of electrical pacing.27 This aberrant Ca2+ cycling rather than being arrhythmogenic is expected to severely compromise contractility by uncoupling myocyte Ca2+ cycling from electrical excitation (Figure 1B). Thus the forms and role of increased diastolic SR Ca2+ release leak appear to vary among different models and stages of HF. In general, moderate alterations in RyR2 function at the onset of HF combined with increased adrenergic tonus are associated with Ca2+-dependent arrhythmogenesis; more severe RyR2 dysfunction at later stages of HF in addition to the pro-arrhythmogenic effects could compromise contractile performance by either decreasing the SR Ca2+ content or decoupling Ca2+ release from electrical activity. In addition to arrhythmias and compromised inotropy, diastolic release has been shown to contribute to the progression of HF by activating Ca2+-dependent hypertrophic pathways,31,32 as well as by causing cell death30,33 and impairing myocyte energetics.23 The specific relationships between parameters of leak (persistent vs. oscillating) and its effect on various signalling pathways that potentially may modulate progression of HF (e.g. calcineurin/NFAT48 and CaMKII/HDAC49) warrants further investigation.
3.3. Role of altered RyR2 regulation by luminal Ca2+
Increased diastolic Ca2+ release observed in the form of both Ca2+ sparks and Ca2+ waves is commonly associated with elevation in the SR Ca2+ content.2 Moreover, Ca2+ waves arise only when the SR Ca2+ load rises above a certain critical, ‘threshold’ level.50,51 However, myocytes from failing hearts are characterized by lowered rather than elevated SR Ca2+ content.21–23,41,52 These seemingly contradictory findings raise a question of how increased diastolic SR Ca2+ release can emerge in failing myocytes whose SR Ca2+ content is decreased? To address this paradox, Kubalova et al.36 investigated the mechanism of enhanced SR Ca2+ leak and its dependence upon luminal Ca2+ ([Ca2+]SR) in myocytes from normal and failing hearts using a canine model of chronic HF. This study found that [Ca2+]SR (measured with the low affinity Ca2+ indicator Fluo5N) was markedly reduced in HF myocytes and the increased leak (measured as Ca2+ sparks) was associated with abnormally high RyR2 open probability at low luminal Ca2+ measured in planar lipid bilayer experiments. Subsequent studies using a rabbit model of HF showed that, in HF myocytes, Ca2+ waves indeed occur at reduced (or unchanged) rather than increased SR Ca2+ content when compared with control cells.28,53 These studies led to the conclusion that a decrease in threshold SR Ca2+ content accounts for increased propensity for spontaneous Ca2+ release in HF myocytes. It was also concluded that adrenergic stimulation exacerbates this condition by further lowering the threshold (via RyR2 phosphorylation) and/or enhancing SR Ca2+ load, bringing the content closer to the threshold (via stimulation of SERCA2a).
While gaining insights into the mechanisms of increased diastolic release during steady-state conditions these studies left unanswered the question of how diastolic Ca2+ release, and Ca2+ waves, in particular, arise during dynamic changes in luminal Ca2+ that occur in beating cardiomyocytes. To address this question, Belevych et al.27,54 performed simultaneous measurements of cytosolic and luminal Ca2+ in myocytes isolated from normal and diseased dog hearts (both chronic non-ischaemic HF and post-infarction cardiac disease) paced in the presence of β-AR agonist isoproterenol. These studies revealed that although spontaneous Ca2+ release was influenced by the levels of intra-SR Ca2+, it did not arise immediately when a final [Ca2+]SR level was reached, but rather occurred with a distinct time delay, or latency (see Figure 2). This latency was significantly shorter in cells from diseased hearts compared with control myocytes, thus accounting for the increased arrhythmogenic potential in HF myocytes. Notably, the refractory period was significantly shortened in post-infarction myocytes, thereby accounting for the shortened latency to spontaneous release in these cells.54 The reduced refractoriness in myocytes from diseased hearts was attributable to the modification of RyR2s by CaMKII-phosphorylation and oxidation (see below). These results demonstrated that the attainment of a certain threshold intra-SR [Ca2+] is insufficient for the generation of spontaneous Ca2+ release. Furthermore, this study also demonstrated that shortened Ca2+-signalling refractoriness due to abnormal post-translational modifications of RyR2 contributes to the increased rate of diastolic Ca2+ waves in HF myocytes.
Figure 2.

Shortened Ca2+ signalling refractoriness in heart disease. (A) Schematic illustration of time-dependent processes occurring between systolic SR Ca2+ depletion and onset of diastolic spontaneous Ca2+ wave (DCW). This latency is composed of a refractory period, reflecting recovery from store-dependent deactivation, and an idle period required for the transition of stochastic release events through functionally recovered RyR2s to regenerating Ca2+ waves. A shorter time delay between systolic Ca2+ release and DCW in diseased myocytes is attributed to reduced refractoriness of the SR Ca2+ release determined by recording the restitution of the Ca2+ transient amplitude measured with a two-pulse protocol. (B) Shortened refractoriness in post-myocardial infarction (MI) myocytes is associated with altered regulation of Ca2+ release by SR luminal Ca2+ resulting in a diminished capability of reduced SR Ca2+ to inhibit RyR2 activity during diastole. Fast restitution of Ca2+ transient (C) and abnormally high activity of RyR2 (D) observed in diseased hearts can be normalized by treatment with reducing agents monopropyonylglycine (MPG; C) and dithiothreitol (DTT; D and E), respectively. Adapted from Belevych et al.27,54 and Terentyev et al.62
3.4. Molecular mechanisms of altered RyR2 function in HF
As mentioned above, early work in the failing myocardium suggested a role for PKA phosphorylation of RyR2 at Ser-2808 and FKBP12.6 dissociation in the pathogenesis of HF.12,15 However, this particular mechanism has been challenged and is not supported by most available evidence.14,37,55 Unlike that of PKA, the role of CaMKII in modulation of RyR2 in health and disease is less controversial and at this time well established.49 Initial studies using transgenic mouse lines of cardiac up-regulation of CaMKII evidenced increased RyR2 phosphorylation, altered Ca2+ cycling, dilated cardiomyopathy, and catecholamine-induced arrhythmias.56–58 These studies were then followed by investigations conducted in various models of HF.27,35,59,60 For instance, Ai et al.,35 using a rabbit model of HF, demonstrated that inhibition of CaMKII resulted in a reduction in diastolic Ca2+ leak from the SR, which in turn was unaffected by blockade of PKA. These observations in an animal model of hypertrophic cardiomyopathy were later confirmed by Sossalla et al.26 in diseased human hearts. As stated earlier, RyR2 phosphorylation by CaMKII has also been implicated in arrhythmogenesis in HF. Pharmacological inhibition of CaMKII resulted in a reduction in diastolic spontaneous Ca2+ release events and DADs in both murine and large animal models of HF.27,35,60 Besides Ser-2808 and Ser-2814, the RyR2 is phosphorylated at Ser-203013 and potentially at several other sites. 14,61 The role of these sites and of possible interactions between the various sites in both normal physiology and HF remains to be investigated.
In addition to phosphorylation, redox and nitroso modifications are known to affect RyR2 function11,17,18 and have been shown to contribute to the altered Ca2+ signalling in HF. Work by Terentyev et al., in non-ischaemic cardiomyopathy, revealed that the number of oxidized thiols on RyR2s increased in HF when compared with control.62 Oxidation of RyR2 resulted in cardiac dysfunction marked by reduced Ca2+ transient amplitude as well as an increased incidence of diastolic spontaneous Ca2+ waves. Conversely, treatment of normal myocytes with oxidizing agents produced alterations in SR Ca2+ leak and intra-SR Ca2+ concentration similar to those observed in HF.62 In addition to increased oxidation, reductions in S-nitrosylation of RyR2s has also been observed in the failing cardiomyocytes.63 Physiological S-nitrosylation, whose baseline level is high,64 may play a protective role by preventing oxidation of reactive thiols on RyR2. This protective mechanism seems to be compromised in HF, thus leaving the exposed thiols vulnerable to hyperoxidation. Taken together, these studies highlight that nitroso-redox imbalance can directly lead to RyR2 oxidation, hyponitrosylation, and elevated SR Ca2+ leak in myocytes from failing hearts. Of note, increased oxidative burden in the failing myocytes in addition to oxidizing RyR2s could alter RyR2 activity through methionine oxidation-dependent activation of CaMKII65 with subsequent phosphorylation of RyR2. Thus CaMKII provides yet another site for cross-talk between Ca2+ and redox signalling in HF.
With advancement of HF, RyR2 function increasingly deteriorates as modifications of RyR2 structure due to phosphorylation and oxidation progressively accumulate. Interestingly, in the course of HF development, CaMKII-mediated phosphorylation occurs first and is followed at a later stage by an increase in thiol oxidation27 (see Figure 1). It appears that in terms of functional effects on RyR2 gating, both CaMKII phosphorylation and oxidation act in a similar manner. Both types of modifications appear to increase RyR2 activity by shifting RyR2 responsiveness to luminal Ca2+ to lower intra-SR Ca2+ concentrations,56,62 thereby shortening RyR2 refractoriness (Figure 2). Indeed, antioxidant treatment restored Ca2+ sensitivity of RyR2 from failing hearts,62 while both antioxidant treatment and CaMKII inhibition prolonged Ca2+ release refractoriness in diseased myocytes towards normal values (Figure 2).54 As discussed above, RyR2 deactivation occurs in response to the drop of intra-SR Ca2+ in the wake of systolic Ca2+ release6 and excessive covalent modifications of the channel protein through phosphorylation and oxidation impairs the ability of RyR2 to appropriately deactivate and become refractory during the diastolic period.54
3.5. Is altered RyR2 function a cause or a consequence of HF?
Although dysregulated RyR2 activity and diastolic SR Ca2+ release are key features of advanced HF, whether and under what conditions abnormal RyR2-mediated Ca2+ signalling plays a causal role in HF development remains unclear. One way to address this question is to determine whether abnormal RyR2 function precedes or follows the development of HF. In a longitudinal study using a chronic model of HF Belevych et al.27 found that enhanced diastolic SR Ca2+ leak and increased CaMKII phosphorylation of RyR2 emerged and progressed in parallel with deterioration of in vivo contractile function during HF development. In this study, both the increased leak and cardiac contractile impairment preceded the reduction in the amplitude of the Ca2+ transient that occurred only at later stages of HF development (Figure 1). These results are consistent with (although do not prove) the notion that altered RyR2 function plays a causal role in HF. They also suggest that increased RyR2 leak may contribute to HF through mechanisms other than reducing Ca2+ transient amplitude at least at early stages of disease (for further discussion, see below).
Another approach to addressing the causal role of altered RyR2 function is to test whether experimental manipulations that increase RyR2-mediated leak will lead to HF. Notably, although more than 100 mutations in RyR2 and 12 in CASQ2 (http://www.fsm.it/cardmoc) are implicated in Ca2+-dependent arrhythmias (CPVT), none of these mutations have been reliably linked to the chronic contractile weakening or anatomical remodelling associated with HF. It is possible that RyR2 dysfunction caused by these naturally occurring mutations is too mild or that other factors besides RyR2 dysfunction are needed to induce HF. Interestingly, combining dysregulated RyR2 function with up-regulated SR Ca2+ uptake by cross-breeding mice with SERCA1a overexpression and CASQ2 knock-out mice led to dilated cardiomyopathy and early mortality.30 The pathophysiology of HF in this model appears to involve sustained diastolic Ca2+ release leading to mitochondria-dependent cell death.
Thus based on available evidence, excessive diastolic SR Ca2+ release through RyR2s can indeed causally contribute to HF. However, the manner in which SR Ca2+ leak relates to manifestations of HF seems to differ with disease progression. At early stages, elevated diastolic release rather than weakening myocyte contractility directly (by depleting SR Ca2+) seems to impair cardiac function through more complex mechanisms that involve pathological remodelling, myocyte death, and abnormal energy utilization.23,30–33 Leak-dependent SR Ca2+ depletion becomes a dominant factor in limiting myocyte contractility only at later stages of HF when alterations in RyR2 function grow more severe.27 Additionally, abnormal RyR2 function does not seem to be the sole cause of HF. Thus, facilitation/augmentation of SR Ca2+ uptake in addition to RyR2 leakiness seems essential for the maintenance of an adequate level of diastolic release that is sufficient to result in HF.30 This is understandable, considering that RyR2-mediated leak is a self-limiting process that is continuously countered by a reciprocal decrease in the SR Ca2+ content.66 As referred to earlier, RyR2-mediated leak is an early alteration in myocyte Ca2+ handling. Similarly, increased β-AR stimulation of the myocardium expected to facilitate the SR Ca2+ uptake (via phosphorylation of phospholamban) occurs early, during the compensatory phase of HF development.43 Thus, altered RyR2 function would be expected to causally contribute to HF when coinciding with adrenergically mediated increase in the SR Ca2+ uptake. Moreover, as part of the natural course HF, this feed-forward cyclic cross-talk between Ca2+ signalling and neurohormonal modulation that are coupled to contractile performance could contribute to progressive deterioration of both RyR2 function and cardiac contractility (see Figure 3). In this context, abnormal RyR2 function can be viewed as both a cause and a consequence of HF. Therefore, braking this vicious cycle by therapeutically targeting RyR2s is a logical strategy for the management of HF.
Figure 3.

Flow diagram summarizing the events involved in time and Ca2+-dependent progression to heart failure (HF). Initial myocardial injury/stress via post-translational modifications of RyR2 and stimulation of SERCA2a activity leads to abnormal regulation of RyR2 by cytosolic and luminal Ca2+. Dysregulated intracellular Ca2+ handling results in myocardial contractile dysfunction, increased arrhythmogenesis, and altered intracellular metabolic and cell survival pathways.
4. Potential therapeutic interventions in HF targeting RyR2
Despite the advances in medical management, HF remains a critical healthcare problem.67 Abnormalities in Ca2+ handling that prevail in failing myocardium have prompted an interest in the development of therapies for HF. For instance, the ‘leaky’ RyR2 has been targeted by RyR2 stabilizing therapy with JTV519. Such RyR2 stabilization resulted in ventricular arrhythmia suppression as well as improvement of the contractile performance in human HF.68 Interestingly, one of the commonly used β-blockers in management of HF, carvedilol, also proved to reduce RyR2 open probability in addition to its β-blocking properties.69,70 Another possible therapeutic target for mitigation of ‘leaky’ RyR2 is modulation of RyR2 hyperphosphorylation by CaMKII. Either pharmacological or genetic inhibition of CaMKII in a murine model of cardiomyopathy secondary to chronic β-AR stimulation resulted in a prevention of cardiomyopathy.71 Also the protective action of these agents with a broad range of action may include targets other than RyR2. Taken together these studies support the concept that mitigating the RyR2 leak may have a therapeutic potential.
Redox modification in the failing myocardium is yet another possible target in management and/or prevention of HF. Recent advances in the clinical realm revealed that inhibition with allopurinol of xanthine oxidase, one possible source of reactive oxygen species in cardiomyopathy, resulted in an improvement of myocardial function.72 A subsequent large clinical study in patients with advanced HF did not confirm these findings.73 However, a post hoc analysis revealed a reduction in morbidity and mortality in a subset of patients with elevated levels of uric acid, a product of xanthine oxidase.73 It is important to note that late stages of HF development are marked by redox modification that contributes to abnormal function of RyR2. At this stage, antioxidant therapy is less effective than at the early one, which is marked by hyperphosphorylation of RyR2 by CaMKII.27 This in part may serve as an explanation for a lack of overall efficacy of an antioxidant therapy in advanced HF. Future studies are needed in order to determine whether therapy with antioxidants in earliest stages of HF will prevent the progression of the disease.
As discussed above, diminished intra-SR Ca2+ stores in the failing heart are likely due to increased diastolic RyR2-mediated Ca2+ release20,36 and possibly also due to a decreased SERCA2a function.21,40 Outside of the aforementioned approaches of stabilizing of RyR2 either directly or by reducing its hyperphosphorylation state, another approach for mitigation of reduced ionotropy in HF might be increasing Ca2+ resequestration into the SR. The beneficial effects of up-regulation of the SR Ca2+ re-uptake through overexpression of SERCA2a or ablation of PLB have been demonstrated in a number of animal studies and more recently in human patients with HF.74–77 Although decreased SR Ca2+ content is an important feature of HF and a viable target for HF therapy, facilitation of SR Ca2+ uptake does not always necessarily prevent or reverse HF development.33,78–80 Moreover, up-regulation of SR Ca2+ uptake could exacerbate disease by further increasing SR Ca2+ leak especially in settings of dysregulated RyR2 function. Indeed, overexpression of SERCA molecules or ablation of PLB have been shown to increase diastolic Ca2+ release and lead to cardiac hypertrophy, contractile dysfunction, and early mortality in mice overexpressing CaMKIIδC 33 or deficient in CASQ2.30 These mice are characterized by leaky RyR2s via either RyR2 hyperphosphorylation 56,57 or lack of regulation by CASQ2.10 Taken together, these studies show a ‘proof of concept’ that up-regulation of resequestration of Ca2+ into SR might prove a promising option in the management of certain types of HF although these therapies must be applied with caution in the context of abnormal RyR2 function.
5. Conclusions
During the past decade RyR2 has emerged as an important player in the pathophysiology of HF. Defective RyR2 behaviour marked by excessive activity has been linked to key pathological manifestations of HF, which encompasses malignant arrhythmias, contractile dysfunction, and adverse structural remodelling. This set of conditions associated with RyR2 dysfunction can be dubbed as ‘ryanopathy’. Such aberrancy in RyR2 function hinges on an inability of RyR2s to appropriately deactivate and become/remain refractory during the diastolic relaxation period. This process of RyR2 deactivation is normally mediated by a decrease in the SR Ca2+ concentration that in turn is precipitated by the preceding Ca2+ release. The abnormal RyR2 behaviour involves cumulative post-translational modifications of the channel by CaMKII phosphorylation and oxidation. Rather than a sole cause of HF, RyR2 dysfunction, in combination with adrenergically mediated enhanced SR Ca re-uptake, is jointly responsible for pathological diastolic SR Ca2+ release. Progressive deterioration of RyR2 function is a result of a vicious cycle of Ca2+ leak-induced Ca2+ leak that underlies a gradual deterioration of the cardiac function and as such represents a target for therapeutic interventions with the aim to halt or potentially even reverse the progression of HF.
Conflict of interest: none declared.
Funding
This work was supported by National Institutes of Health grants HL074045 and HL063043 (to S.G.), HL089836 (to C.A.C.).
References
- 1.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245:C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 2.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht, Boston: Kluwer Academic Publishers; 2001. [Google Scholar]
- 3.Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- 4.Li P, Chen SR. Molecular basis of Ca2+ activation of the mouse cardiac Ca2+ release channel (ryanodine receptor) J Gen Physiol. 2001;118:33–44. doi: 10.1085/jgp.118.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng H, Lederer MR, Lederer WJ, Cannell MB. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol. 1996;270:C148–C159. doi: 10.1152/ajpcell.1996.270.1.C148. [DOI] [PubMed] [Google Scholar]
- 6.Radwanski PB, Belevych AE, Brunello L, Carnes CA, Gyorke S. Store-dependent deactivation: cooling the chain-reaction of myocardial calcium signaling. J Mol Cell Cardiol. doi: 10.1016/j.yjmcc.2012.10.008. Advance Access published October 27, 2012, doi: 10.1016/j.yjmcc.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL, Gyorke S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res. 2002;91:414–420. doi: 10.1161/01.res.0000032490.04207.bd. [DOI] [PubMed] [Google Scholar]
- 8.Szentesi P, Pignier C, Egger M, Kranias EG, Niggli E. Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+-induced Ca2+ release refractoriness in heart muscle. Circ Res. 2004;95:807–813. doi: 10.1161/01.RES.0000146029.80463.7d. [DOI] [PubMed] [Google Scholar]
- 9.Sobie EA, Dilly KW, dos Santos Cruz J, Lederer WJ, Jafri MS. Termination of cardiac Ca2+ sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J. 2002;83:59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gyorke S, Terentyev D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res. 2008;77:245–255. doi: 10.1093/cvr/cvm038. [DOI] [PubMed] [Google Scholar]
- 11.Meissner G. Regulation of mammalian ryanodine receptors. Front Biosci. 2002;7:d2072–d2080. doi: 10.2741/A899. [DOI] [PubMed] [Google Scholar]
- 12.Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 13.Xiao B, Zhong G, Obayashi M, Yang D, Chen K, Walsh MP, et al. Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem J. 2006;396:7–16. doi: 10.1042/BJ20060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valdivia HH. Ryanodine receptor phosphorylation and heart failure: phasing out S2808 and “criminalizing” S2814. Circ Res. 2012;110:1398–1402. doi: 10.1161/CIRCRESAHA.112.270876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 16.Currie S, Loughrey CM, Craig MA, Smith GL. Calcium/calmodulin-dependent protein kinase IIδ associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biochem J. 2004;377:357–366. doi: 10.1042/BJ20031043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donoso P, Sanchez G, Bull R, Hidalgo C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front Biosci. 2011;16:553–567. doi: 10.2741/3705. [DOI] [PubMed] [Google Scholar]
- 18.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 19.Eisner DA, Trafford AW, Diaz ME, Overend CL, O'Neill SC. The control of Ca release from the cardiac sarcoplasmic reticulum: regulation versus autoregulation. Cardiovasc Res. 1998;38:589–604. doi: 10.1016/s0008-6363(98)00062-5. [DOI] [PubMed] [Google Scholar]
- 20.Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Gyorke S. Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J. 2007;93:4083–4092. doi: 10.1529/biophysj.107.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol. 2002;34:951–969. doi: 10.1006/jmcc.2002.2037. [DOI] [PubMed] [Google Scholar]
- 22.Lou Q, Janardhan A, Efimov IR. Remodeling of calcium handling in human heart failure. Adv Exp Med Biol. 2012;740:1145–1174. doi: 10.1007/978-94-007-2888-2_52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006;21:380–387. doi: 10.1152/physiol.00019.2006. [DOI] [PubMed] [Google Scholar]
- 24.Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med. 2004;14:61–66. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 25.Janse MJ. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc Res. 2004;61:208–217. doi: 10.1016/j.cardiores.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 26.Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, et al. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res. 2010;107:1150–1161. doi: 10.1161/CIRCRESAHA.110.220418. [DOI] [PubMed] [Google Scholar]
- 27.Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, Hamlin RL, et al. The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc Res. 2011;90:493–502. doi: 10.1093/cvr/cvr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curran J, Brown KH, Santiago DJ, Pogwizd S, Bers DM, Shannon TR. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca2+-calmodulin-dependent protein kinase II. J Mol Cell Cardiol. 2010;49:25–32. doi: 10.1016/j.yjmcc.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dincer UD. Cardiac ryanodine receptor in metabolic syndrome: is JTV519 (K201) future therapy? Diabetes Metab Syndr Obes. 2012;5:89–99. doi: 10.2147/DMSO.S30005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalyanasundaram A, Lacombe VA, Belevych AE, Brunello L, Carnes CA, Janssen PM, et al. Up-regulation of sarcoplasmic reticulum Ca2+ uptake leads to cardiac hypertrophy, contractile dysfunction and early mortality in mice deficient in CASQ2. Cardiovasc Res. 2013;98:297–306. doi: 10.1093/cvr/cvs334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Oort RJ, Respress JL, Li N, Reynolds C, De Almeida AC, Skapura DG, et al. Accelerated development of pressure overload-induced cardiac hypertrophy and dysfunction in an RyR2-R176Q knockin mouse model. Hypertension. 2010;55:932–938. doi: 10.1161/HYPERTENSIONAHA.109.146449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamaguchi N, Takahashi N, Xu L, Smithies O, Meissner G. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca release channel. J Clin Invest. 2007;117:1344–1353. doi: 10.1172/JCI29515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, et al. Phospholamban ablation rescues sarcoplasmic reticulum Ca2+ handling but exacerbates cardiac dysfunction in CaMKIIδC transgenic mice. Circ Res. 2010;106:354–362. doi: 10.1161/CIRCRESAHA.109.207423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- 35.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 36.Kubalova Z, Terentyev D, Viatchenko-Karpinski S, Nishijima Y, Gyorke I, Terentyeva R, et al. Abnormal intrastore calcium signaling in chronic heart failure. Proc Natl Acad Sci USA. 2005;102:14104–14109. doi: 10.1073/pnas.0504298102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bers DM. Ryanodine receptor S2808 phosphorylation in heart failure: smoking gun or red herring. Circ Res. 2012;110:796–799. doi: 10.1161/CIRCRESAHA.112.265579. [DOI] [PubMed] [Google Scholar]
- 38.Domeier TL, Blatter LA, Zima AV. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J Physiol. 2009;587:5197–5209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Venetucci LA, Trafford AW, O'Neill SC, Eisner DA. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res. 2008;77:285–292. doi: 10.1093/cvr/cvm009. [DOI] [PubMed] [Google Scholar]
- 40.Frank KF, Bolck B, Brixius K, Kranias EG, Schwinger RH. Modulation of SERCA: implications for the failing human heart. Basic Res Cardiol. 2002;97(Suppl 1):I72–I78. doi: 10.1007/s003950200033. [DOI] [PubMed] [Google Scholar]
- 41.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 42.Mork HK, Sjaastad I, Sande JB, Periasamy M, Sejersted OM, Louch WE. Increased cardiomyocyte function and Ca2+ transients in mice during early congestive heart failure. J Mol Cell Cardiol. 2007;43:177–186. doi: 10.1016/j.yjmcc.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. 2009;54:1747–1762. doi: 10.1016/j.jacc.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 44.Baartscheer A, Schumacher CA, Belterman CN, Coronel R, Fiolet JW. SR calcium handling and calcium after-transients in a rabbit model of heart failure. Cardiovasc Res. 2003;58:99–108. doi: 10.1016/s0008-6363(02)00854-4. [DOI] [PubMed] [Google Scholar]
- 45.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 46.Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 47.Lakatta EG. Functional implications of spontaneous sarcoplasmic reticulum Ca2+ release in the heart. Cardiovasc Res. 1992;26:193–214. doi: 10.1093/cvr/26.3.193. [DOI] [PubMed] [Google Scholar]
- 48.Colella M, Grisan F, Robert V, Turner JD, Thomas AP, Pozzan T. Ca2+ oscillation frequency decoding in cardiac cell hypertrophy: role of calcineurin/NFAT as Ca2+ signal integrators. Proc Natl Acad Sci USA. 2008;105:2859–2864. doi: 10.1073/pnas.0712316105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–473. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stern MD, Capogrossi MC, Lakatta EG. Spontaneous calcium release from the sarcoplasmic reticulum in myocardial cells: mechanisms and consequences. Cell Calcium. 1988;9:247–256. doi: 10.1016/0143-4160(88)90005-x. [DOI] [PubMed] [Google Scholar]
- 51.Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ Res. 2007;100:105–111. doi: 10.1161/01.RES.0000252828.17939.00. [DOI] [PubMed] [Google Scholar]
- 52.Hobai IA, O'Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103:1577–1584. doi: 10.1161/01.cir.103.11.1577. [DOI] [PubMed] [Google Scholar]
- 53.Desantiago J, Ai X, Islam M, Acuna G, Ziolo MT, Bers DM, et al. Arrhythmogenic effects of β2-adrenergic stimulation in the failing heart are attributable to enhanced sarcoplasmic reticulum Ca load. Circ Res. 2008;102:1389–1397. doi: 10.1161/CIRCRESAHA.107.169011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, et al. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res. 2012;110:569–577. doi: 10.1161/CIRCRESAHA.111.260455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, et al. Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–829. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- 56.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIδC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 57.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, et al. The δC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 58.Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, et al. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 59.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sag CM, Wadsack DP, Khabbazzadeh S, Abesser M, Grefe C, Neumann K, et al. Calcium/calmodulin-dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ Heart Fail. 2009;2:664–675. doi: 10.1161/CIRCHEARTFAILURE.109.865279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yuchi Z, Lau K, Van Petegem F. Disease mutations in the ryanodine receptor central region: crystal structures of a phosphorylation hot spot domain. Structure. 2012;20:1201–1211. doi: 10.1016/j.str.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 62.Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gonzalez DR, Treuer AV, Castellanos J, Dulce RA, Hare JM. Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J Biol Chem. 2010;285:28938–28945. doi: 10.1074/jbc.M110.154948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 65.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lukyanenko V, Viatchenko-Karpinski S, Smirnov A, Wiesner TF, Gyorke S. Dynamic regulation of sarcoplasmic reticulum Ca2+ content and release by luminal Ca2+-sensitive leak in rat ventricular myocytes. Biophys J. 2001;81:785–798. doi: 10.1016/S0006-3495(01)75741-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kong MH, Fonarow GC, Peterson ED, Curtis AB, Hernandez AF, Sanders GD, et al. Systematic review of the incidence of sudden cardiac death in the United States. J Am Coll Cardiol. 2011;57:794–801. doi: 10.1016/j.jacc.2010.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Toischer K, Lehnart SE, Tenderich G, Milting H, Korfer R, Schmitto JD, et al. K201 improves aspects of the contractile performance of human failing myocardium via reduction in Ca2+ leak from the sarcoplasmic reticulum. Basic Res Cardiol. 2010;105:279–287. doi: 10.1007/s00395-009-0057-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mochizuki M, Yano M, Oda T, Tateishi H, Kobayashi S, Yamamoto T, et al. Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J Am Coll Cardiol. 2007;49:1722–1732. doi: 10.1016/j.jacc.2007.01.064. [DOI] [PubMed] [Google Scholar]
- 70.Zhou Q, Xiao J, Jiang D, Wang R, Vembaiyan K, Wang A, et al. Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat Med. 2011;17:1003–1009. doi: 10.1038/nm.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 72.Cappola TP, Kass DA, Nelson GS, Berger RD, Rosas GO, Kobeissi ZA, et al. Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation. 2001;104:2407–2411. doi: 10.1161/hc4501.098928. [DOI] [PubMed] [Google Scholar]
- 73.Hare JM, Mangal B, Brown J, Fisher C, Jr, Freudenberger R, Colucci WS, et al. Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J Am Coll Cardiol. 2008;51:2301–2309. doi: 10.1016/j.jacc.2008.01.068. [DOI] [PubMed] [Google Scholar]
- 74.Miyamoto MI, del Monte F, Schmidt U, DiSalvo TS, Kang ZB, Matsui T, et al. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci USA. 2000;97:793–798. doi: 10.1073/pnas.97.2.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.del Monte F, Harding SE, Dec GW, Gwathmey JK, Hajjar RJ. Targeting phospholamban by gene transfer in human heart failure. Circulation. 2002;105:904–907. doi: 10.1161/hc0802.105564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124:304–313. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lyon AR, Bannister ML, Collins T, Pearce E, Sepehripour AH, Dubb SS, et al. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ Arrhythm Electrophysiol. 2011;4:362–372. doi: 10.1161/CIRCEP.110.961615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tilemann L, Ishikawa K, Weber T, Hajjar RJ. Gene therapy for heart failure. Circ Res. 2012;110:777–793. doi: 10.1161/CIRCRESAHA.111.252981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Janczewski AM, Zahid M, Lemster BH, Frye CS, Gibson G, Higuchi Y, et al. Phospholamban gene ablation improves calcium transients but not cardiac function in a heart failure model. Cardiovasc Res. 2004;62:468–480. doi: 10.1016/j.cardiores.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 80.O'Donnell JM, Fields A, Xu X, Chowdhury SA, Geenen DL, Bi J. Limited functional and metabolic improvements in hypertrophic and healthy rat heart overexpressing the skeletal muscle isoform of SERCA1 by adenoviral gene transfer in vivo. Am J Physiol Heart Circ Physiol. 2008;295:H2483–H2494. doi: 10.1152/ajpheart.01023.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]