

Abstract
Compelling recent experimental results make clear that sub-cellular structures are altered in ventricular myocytes during the development of heart failure, in both human samples and diverse experimental models. These alterations can include, but are not limited to, changes in the clusters of sarcoplasmic reticulum (SR) Ca2+-release channels, ryanodine receptors, and changes in the average distance between the cell membrane and ryanodine receptor clusters. In this review, we discuss the potential consequences of these structural alterations on the triggering of SR Ca2+ release during excitation–contraction coupling. In particular, we describe how mathematical models of local SR Ca2+ release can be used to predict functional changes resulting from diverse modifications that occur in disease states. We review recent studies that have used simulations to understand the consequences of sub-cellular structural changes, and we discuss modifications that will allow for future modelling studies to address unresolved questions. We conclude with a discussion of improvements in both experimental and mathematical modelling techniques that will be required to provide a stronger quantitative understanding of the functional consequences of changes in sub-cellular structure in heart disease.
Keywords: Transverse tubule, Ca2+ spark, Mathematical modelling, Ryanodine receptor, Ventricular myocyte
1. Introduction
The sub-cellular structure of cardiac myocytes is well suited to enable the function of these cells. As the ventricles are responsible for pumping blood, ventricular myocytes must be able to contract quickly and powerfully, which requires rapid and large increases in intracellular Ca2+ concentration. The increase in [Ca2+] that occurs with each heartbeat is initiated by Ca2+ entry through L-type Ca2+ channels and amplified by Ca2+ release from the sarcoplasmic reticulum (SR). Given the large size of these cells, however, diffusion would not be sufficient to rapidly transmit Ca2+ from the cell periphery to the cell interior. Thus, transverse tubules (T-tubules) penetrate into the cell and ensure that, in healthy cells, virtually all locations in the cytoplasm are close (<1 µm) to the cell membrane. Moreover, T-tubules bring together in close proximity the essential elements of excitation–contraction (EC) coupling: membrane L-type Ca2+ channels and Ca2+-release channels, ryanodine receptors (RyRs), in the SR membrane. Despite this optimized sub-cellular structure that allows for efficient function in healthy cells, recent evidence, discussed below and in other contributions to this special issue, indicates that deleterious structural changes occur in several disease states.1–3
Mathematical modelling has been used to illuminate several important aspects of EC coupling. For instance, the local control hypothesis, the idea that SR Ca2+ release consists of many independent functional units, originated from simulations with models,4 and the predictions of these simulations were subsequently verified experimentally.5–7 Similarly, computational studies helped to develop the hypothesis that release termination relies on substantial local depletion of SR [Ca2+],8 and the conceptual framework established by the modelling helped to interpret later experimental data that supported the hypothesis.9–13
Most modelling studies performed to date, however, have focussed on questions related to channel gating. For instance, simulations have been used to explore the mechanisms that allow for robust termination of Ca2+ release,8,14,15 and have examined how RyR sensitivity to Ca2+ influences the triggering of release by Ca2+ current.16–18 This has been a natural starting point, as these relevant questions can be addressed through approaches that follow the tradition established in studies of cellular action potentials, electrophysiology, and arrhythmias.19,20 It is clear, however, that to understand changes in EC coupling that occur in heart failure, simulation studies must consider not only changes in channel gating, but also changes in the sub-cellular structure.
In this brief review, we describe the structural changes to heart cells that occur in cardiac disease, and we discuss how simulation approaches will be an important complement to experiments for understanding dysfunctional changes to physiology. However, since the effects of structural alterations have been relatively less explored compared with changes in channel gating, many of the most important studies in this area have not yet been performed, and much of what follows will be prospective rather than retrospective.
2. Experimental evidence for T-tubule and dyad remodelling in disease
Numerous experimental studies have now shown that T-tubules and cardiac dyads become remodelled in disease states.21–32 Details differ depending on species and the particular disease model examined, but several observations appear to be robust and consistent between studies: (i) T-tubules become more disorganized and lose their regular structure as disease progresses; (ii) an increased fraction of T-tubules appears to be oriented longitudinally rather than transversely; and (iii) RyR clusters are more likely than T-tubules to maintain a regular structure, which means that many RyR clusters become ‘orphaned’ as the L-type Ca2+ channels in the cell membrane either move away or disappear along with the T-tubules. More recently, research has suggested that cardiomyopathy may also lead to changes in the morphology of the RyR clusters themselves, with smaller RyR clusters reported in a rat model of pressure overload-induced heart failure.33
Several of these established and suggested structural alterations are illustrated schematically in Figure 1. In healthy cells (left), RyR clusters are relatively evenly spaced, and most RyR clusters sit in close proximity to T-tubule membranes. In disease (right), the following changes may occur: (i) local SR volumes and RyR clusters may become smaller than normal33 (top); (ii) RyR clusters may move away from T-tubules34 (middle); (iii) pinching off of T-tubules or changes in the regular T-tubule structure may lead to RyR clusters that lose contact with L-type Ca2+ channels in the T-tubule membrane, thereby becoming orphaned23 (bottom).
Figure 1.
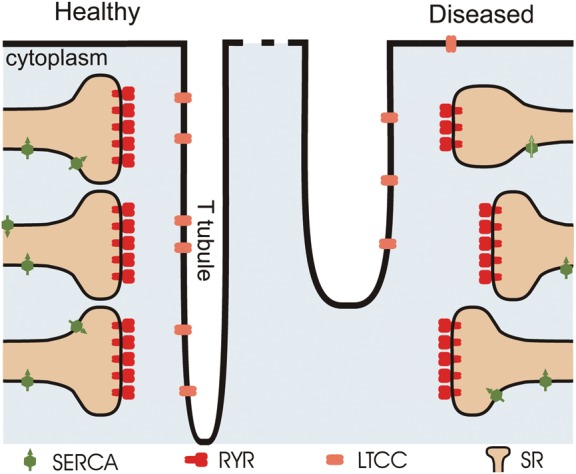
Schematic diagram of dyadic junctions in healthy and diseased cardiac cells. The left side of the diagram shows the regular organization of RyR clusters in healthy ventricular myocytes. In diseased cells (right), several possible forms of remodelling have been proposed: (i) RyR clusters and associated local SR volumes may become smaller (top)33; (ii) the gap between the T-tubule membranes and RyR clusters may become larger as SR moves away from the Z-line (middle); and (iii) gross alterations in T-tubule structure may lead to ‘orphaning’ of RyR clusters (bottom).23 The schematic is based on similar diagrams presented elsewhere.33,36
The structural changes observed in disease occur in parallel with several alterations to SR Ca2+ release. Two early studies by Gomez et al. demonstrated reduced ‘gain’, or an impaired ability for L-type Ca2+ current to trigger SR Ca2+ release in cells from failing hearts.35,36 An additional important early study by Litwin and Bridge37 in rabbits after myocardial infarction used confocal imaging to demonstrate a decrease in the synchrony of Ca2+ spark initiation, as several subsequent studies in a variety of heart failure models have confirmed.23–26 More recent studies have extended these initial observations. For instance, decreased ‘coupling fidelity’, or the capacity for L-type Ca2+ channel openings to trigger local Ca2+ release in the form of Ca2+ sparks, has been shown at the level of individual dyads in a rat model of pressure overload-induced hypertropy.38 Additionally, in pigs after myocardial infarction, spontaneous Ca2+ sparks in sub-cellular regions near T-tubules have been shown to exhibit an increased frequency but decreased duration compared with sparks seen in regions far from T-tubules.39
One of the functions of mathematical modelling is to explain these disparate experimental observations within a unified framework. As will be described below, this has been accomplished to a limited extent, but considerable additional research needs to be performed to fully understand the functional consequences of sub-cellular structural changes that occur in cardiac disease.
3. The importance of geometry in numerical simulations of Ca2+ sparks
Stochastic simulations of Ca2+ sparks provide an initial framework for understanding how changes in sub-cellular structure in disease states influence SR Ca2+ release and EC coupling in ventricular myocytes. Many studies have used Monte-Carlo methods to simulate local SR Ca2+ release,8,14,15,17,40,41 and contemporary models generally share the following features (Figure 3A): (i) a cluster of stochastically gating RyRs; (ii) a local dyadic space between the SR and T-tubule membranes; (iii) a local junctional SR (JSR) connected to a larger network SR (NSR); and (iv) Ca2+ buffering in both the dyadic and JSR spaces. Models differ in the assumptions made regarding geometry and RyR gating. Nonetheless, from simulations performed in several studies in the context of healthy myocytes, some general conclusions seem to apply, and these can help us to predict the quantitative consequences of various alterations that may occur in disease states.
Figure 3.

Simulations of altered Ca2+ sparks due to structural rearrangements in disease. (A) Compartments and fluxes in an established stochastic mathematical model of the cardiac Ca2+ spark.8,41 Arrows describes Ca2+ fluxes between compartments. DS, dyadic space; cyto, cytoplasm; TT, T-tubule; JSR, junctional sarcoplasmic reticulum; NSR, network sarcoplasmic reticulum. (B) Simulation of a Ca2+ spark from a healthy dyad (left) and from a diseased dyad (right). Compared with control, we simulated disease by 50% decreases in the number of RyRs, the dyadic space volume, and the JSR volume; a 25% increase in the myoplasmic [Ca2+]; and a 20% decrease in NSR and initial JSR [Ca2+]. In separate simulations, we examined the effects of orphaning by doubling the dyadic space volume. The first set of changes caused the changes shown in the figure; orphaning led to a dramatic decrease in the probability that an individual RyR opening would trigger a Ca2+ spark, from 73 to 2%. Nopen = number of open RyRs as a function of time. F/F0 = normalized simulated fluorescence. [Ca2+]JSR = free Ca2+ concentration in the local JSR compartment.
An increase in SR [Ca2+] will lead to larger Ca2+ sparks, and will increase the probability that a spontaneous RyR opening will trigger neighbouring channels to produce a spark. This latter effect results from two complementary factors: (i) RyRs become sensitized at higher SR [Ca2+] and (ii) single RyR openings carry more current, and this larger current is more effective at raising dyadic [Ca2+] and inducing neighbouring channels to open.42 A change in the number of RyRs in each cluster will alter spark morphology in expected ways: larger RyR clusters produce larger sparks. Changes in RyR quantity have relatively minor effects on spark amplitude, however, if JSR volume is kept constant.8,40 This is because more RyRs in a cluster will lead to faster depletion of local SR [Ca2+], which will mitigate the effects of having more channels in the cluster. An increase in either the local JSR volume or the JSR buffering power (concentration of calsequestrin, CSQ) will augment the amount of local Ca2+ available for release, and will also increase Ca2+ spark amplitude. Finally, changes in dyadic space volume are predicted to have little effect on spark amplitude or spark duration. This volume, however, can significantly influence the probability that individual RyR openings are able to trigger sparks. When a single RyR opens, the dyadic space volume is one of the main factors that determines the local dyadic [Ca2+], and this variable in turn determines the probability that neighbouring RyRs open. Thus, the effective cytosolic volume associated with a cluster of RyRs can determine whether that cluster primarily generates Ca2+ sparks or mostly produces isolated RyR openings, so-called ‘invisible’ SR Ca2+ leak.
In a recent study (Y.S. Lee, in preparation) we attempted to place these and related simulation results into a coherent quantitative context by performing a thorough parameter sensitivity analysis of an established Ca2+ spark model.8,41 We randomized parameters to generate a population of models, each with slightly different characteristics. After running simulations with each candidate model, we used statistical regression methods43,44 to relate the parameters to the simulation results. Results from this study (Y.S. Lee, in preparation) reproduced in Figure 2, show the following predictions: first, increases in the JSR Ca2+ capacity, through either CSQ concentration (parameter CSQ) or JSR volume (parameter VJSR), increase both Ca2+ spark amplitude and Ca2+ spark duration. Secondly, changes in the number of RyRs per cluster (parameter NRyR) or the Ca2+ flux through each open RyR (parameter DRyR) have opposite effects on spark amplitude and duration. This occurs because an increase in the parameter causes faster JSR depletion, and hence shorter sparks. Local depletion also explains why the effects of DRyR and NRyR on amplitude are smaller than the effects of CSQ or VJSR. Thirdly, the parameters controlling Ca2+ spark probability largely differ from those that determine spark morphology (amplitude and duration). These general rules, established qualitatively through simulations in many studies and more quantitatively here, help us to understand the changes in Ca2+ sparks that may occur in disease states.
Figure 2.

Sensitivity analysis of a Ca2+ spark model. Plots are reproduced with permission from a study (Y.S. Lee, in preparation) that used recently developed methods43,44 to analyse parameter sensitivities in a stochastic Ca2+ spark model. (A) Parameter sensitivities indicating how 18 model parameters affect: (i) the probability that an individual RyR opening will trigger a Ca2+ spark (top); (ii) Ca2+ spark amplitude (middle); (iii) Ca2+ spark duration (bottom). (B) Validation of two of the parameter sensitivities shown in (A, top). An increase in the volume of the dyadic space (parameter Vds, red) causes a significant decrease in Ca2+ spark probability, whereas an increase in the local JSR volume (parameter VJSR, green) causes only a small increase in spark probability.
4. How should one simulate a Ca2+ spark arising from a diseased dyad?
Given the structural alterations that have been observed in multiple studies as described above, what changes should one make in a mathematical model to simulate Ca2+ sparks arising from a diseased dyad? We present results and discuss such a provisional simulation in Figure 3. As our baseline mathematical model we used identical parameters to those presented in Ramay et al.41 To simulate a diseased myocyte, we made the following alterations: (i) the number of RyRs per cluster was reduced by 50%, consistent with the recent finding of smaller dyads in disease33; (ii) along with the decrease in the number of RyRs, local JSR and dyadic space volumes were also reduced by 50%33; (iii) diastolic network SR and initial JSR [Ca2+] were each reduced by 20%; (iv) initial cytosolic and dyadic space [Ca2+] were each increased by 25% to simulate impaired relaxation and elevated diastolic [Ca2+]; and (v) in a separate set of simulations dyadic space volume was increased by a factor of 2 to simulate orphaning of RyR clusters.23 Results shown in Figure 3 demonstrate that Ca2+ sparks originating from diseased RyR clusters may be expected to exhibit the following characteristics: (i) smaller amplitude due to the reduced Ca2+ capacity of the smaller dyads33; (ii) slightly longer duration due to an enhanced ability for the network SR to refill the smaller JSR volume during the spark13,41,45; (iii) in simulations that include the effect of orphaning, a substantially reduced ability for individual RyR openings to trigger sparks by activating neighbouring RyRs within the cluster.
Several alterations that are thought to take place in heart failure are not included in this simulation, and these excluded elements suggest avenues for future work (see also Sections 6 and 7). For instance, SERCA activity, frequently reported as decreased in heart failure,46 is not explicitly included in this particular model. This alteration is included indirectly through the decrease in [Ca2+]NSR, but more complete models that simulate the balance between leak and SR Ca2+ reuptake17,18,47 can explore more thoroughly how SERCA influences SR [Ca2+], and how this in turn influences Ca2+ sparks. Secondly, intracellular [Na+] is not included as a variable in this Ca2+ spark model, although it is likely to be elevated in heart failure due to changes in Na+ current48 and/or Na+–H+ exchange.49 Changes in intracellular [Na+] may influence Ca2+ sparks indirectly, although any such effect will also depend on the localization of RyR clusters relative to Na+–Ca2+ exchangers, a topic that is currently being researched actively (see Sections 6 and 7).
5. What are the consequences of T-tubule remodelling at the cellular level?
In the previous section, we discussed the expected effects of sub-cellular structural changes on Ca2+ sparks, and we described how simulations can assist with predicting and understanding the quantitative effects of each alteration. At the cellular level, however, different categories of models must be used to understand the cellular-level consequences of the structural changes.50 For instance, a few recent cellular models can simulate both stochastic triggering of sparks and Ca2+ flux balance between the cytosol and the SR.17,18,47 These new models can consider not only some of the structural changes observed in HF such as RyR orphaning, but also changes to cell-wide Ca2+ fluxes, such as increased NCX activity and decreased activity of the SR Ca2+ ATPase (SERCA). Although these whole-cell models have been used to understand SR Ca2+ leak in quiescent cells from diseased myocytes, most simulations performed to date have focussed on changes in RyR gating or SERCA function rather than on structural changes.18,47 Future studies will be expected to directly compare the functional effects of altered channel gating vs. altered sub-cellular structures.
In simulations at the cellular level, we would expect the following changes to be observed: (i) in quiescent cells, since orphaning makes spontaneous RyR openings less likely to trigger sparks (Figure 3), we would expect a greater proportion of the SR Ca2+ leak to be invisible rather than in the form of Ca2+ sparks; (ii) during triggered release, orphaning should lead to the initiation of fewer Ca2+ sparks early in each action potential (AP) and reduced synchrony, as recently demonstrated in a cellular simulation17; (iii) prolonged APs will likely allow for increased Ca2+ entry through L-type Ca2+ channels, which may partially offset the lower SR Ca2+ load caused by reduced SERCA activity; (iv) NCX, although perhaps up-regulated at the cellular level, may be less efficient at extruding Ca2+ if fewer exchangers are located close to RyRs51 and if intracellular Na+ is elevated48,49; and (v) the altered AP shape may influence not only overall Ca2+ entry, but also the triggering of Ca2+ sparks early in the AP.52–54 Because of electrophysiological differences between species, this last effect will be expected to depend greatly on the specific disease model examined.
Figure 4 shows results from an important recent study that demonstrates the state-of-the-art in our quantitative understanding of how changes in T-tubules influence cellular Ca2+ handling.55 These investigators used super-resolution fluorescence imaging (stimulated emission depletion microscopy, or STED) to examine the T-tubule system in cells from mouse hearts several weeks after myocardial infarction. Measurements with this advanced technique confirmed the increased longitudinal and oblique T-tubule elements in disease and provided the novel finding that T-tubule cross-sectional area is increased in this disease model. The experiments were complemented by numerical simulations of how structural changes, alterations in Ca2+ fluxes (SERCA and NCX) and AP prolongation conspire to affect triggered Ca2+ release in diseased myocytes. As expected, the simulations confirmed that longer APs in disease lead to greater Ca2+ entry during each EC coupling cycle, and that orphaning will decrease the synchrony of spark triggering early in the AP.17 The computations also generated the novel prediction that orphaning per se can increase total Ca2+ entry during each AP, thereby increasing SR Ca2+ load and partially offsetting the reduced synchrony. This study55 therefore demonstrates that structural remodelling can lead to compensatory as well as deleterious effects, and that mathematical modelling can be helpful for predicting the quantitative consequences of various alterations.
Figure 4.

Experimental results demonstrating changes in T-tubule structure in disease. Three-dimensional reconstructions of T-tubules in healthy (left) and diseased mouse ventricular myocytes (right), obtained using STED microscopy. T-tubules in the diseased heart show increased cross-sections and more longitudinal elements running along the x-dimension. Scale bar = 1 µm. 8pMI = 8 weeks post myocardial infarction. Reproduced from Wagner et al.55 with permission.
6. Future directions: experimental studies
The discussion above indicates several unresolved issues relevant to disease-induced structural changes that will require additional experimental study. One obvious question to address is: how do RyR clusters themselves change in disease, and are these alterations specific to particular forms of cardiomyopathy? A recent study33 found that dyads are smaller in failing rat myocytes after aortic constriction, but it is not clear whether these results are generally applicable across different experimental models. It will be especially important in future studies to apply super-resolution optical techniques that allow researchers to determine the fine structure of RyR clusters.56 When these techniques have been applied to healthy cells,57 they have revealed an RyR cluster structure more complex and irregular than previously anticipated. It is not currently known, however, whether disease alters not only cluster size, but also the fine structure of these complexes.
It will also be important to determine how disease changes the localization of auxiliary proteins that normally reside close to RyR clusters and L-type Ca2+ channels and can indirectly modulate their function. One prominent example previously mentioned is the Na+–Ca2+ exchanger. Intriguing recent results39 suggest that NCX can reduce the duration of Ca2+ sparks in healthy myocytes, and that this modulation may be altered in disease. Structural studies, however, indicate that NCX is generally not co-localized with RyRs,58 although a significant percentage of NCX proteins may reside within a few hundred nanometres of RyR clusters.59 More precise localization of these proteins relative to one another, and possible alterations in disease, will help allow these issues to be explored more quantitatively through mathematical modelling. Other proteins clearly important for the regulation of Ca2+ sparks and EC coupling are CSQ and SERCA. Recent simulation studies suggest that localization of either protein can affect the characteristics of localized Ca2+ sparks or cellular Ca2+ waves.60,61 Although SERCA is generally down-regulated during heart failure whereas overall CSQ levels are thought to remain constant,46,62 at present there is only limited information regarding whether these proteins significantly change their nanoscale localization.
Although much has been learned in recent years about the changes in T-tubule membranes during heart failure, much less is known about whether disease causes remodelling of the SR itself. SR alterations have been demonstrated in two genetically modified mice,63,64 but it is not clear whether changes in the SR structure occur in other disease states. Improved methods to directly image the SR will be required to determine whether this is the case, but this is an important issue to address because Ca2+ diffusion within the SR is predicted to have significant functional consequences.45,61,65,66 Finally, the next several years are expected to bring significant new insight into the biochemical signalling mechanisms underlying changes in the T-tubule structure. Candidates identified to date include junctophilin34,67 and PI3-kinase,68 but further mechanistic insight will certainly be uncovered in future studies.
7. Future directions: improvements to mathematical models
Besides the new experimental results that are likely to be obtained, the next several years will undoubtedly bring improvements to mathematical models, and the combination of experiments and simulations should advance our understanding and help to address unresolved questions regarding the functional effects of altered sub-cellular structure in cardiac disease.
To understand the functional consequences of altered sub-cellular structures, it seems clear that stochastic models of local SR Ca2+ release,18,41,47 which usually assume idealized geometries, will need to be combined with models that simulate Ca2+ movements in complex and realistic geometries.60,69,70 As discussed in more detail elsewhere,50 these two categories of models have generally been developed separately, but the strengths of the two approaches must be combined to address how alterations to T-tubules and dyads, which can sometimes be subtle, might affect EC coupling. Along these lines, an interesting question that has not yet been addressed through simulations concerns the functional consequences of irregularities in the structure of RyR clusters.57 Similarly, mathematical modelling has not yet been used extensively to address how localization of mitochondria near RyR clusters affects either Ca2+ sparks or mitochondrial function. Models have integrated SR Ca2+ release with mitochondrial energetics at the cellular level,71 but stochastic gating of RyRs and greater spatial detail must be included to address the effects of changes in localization.
As representations of local geometry in mathematical models become more realistic, we expect that more simulation studies will address the consequences of changes in the localization of NCX proteins. Since this transporter can either import or extrude Ca2+, depending on conditions, it can potentially influence both Ca2+ spark triggering and the decay of [Ca2+] after the initiation of release. NCX activity depends on [Na+] and [Ca2+] in addition to membrane potential, so simulations of local concentration changes are necessary to predict the consequences of altered NCX localization relative to L-type Ca2+ channels and RyRs. Although several studies, both experimental39,72,73 and computational,74–77 have addressed the potential importance of NCX, we expect that these issues will be examined in more detail in coming years, especially as novel experimental data regarding NCX localization in disease states become available.
More broadly, mathematical modellers will need to make a conceptual leap and become comfortable comparing the functional consequences of structural changes with the effects of altered ion channel expression and gating. Sensitivity analysis represents one method to consider biological changes within the same quantitative framework so that the effects of altered channel gating can be compared directly with the effects of remodelled structures (Y.S. Lee, in preparation).50 This type of approach is only straightforward to implement, however, when simple geometries are assumed and a parameter such as compartment volume can easily be scaled. Novel methods will need to be developed in order to use such a strategy with models that consider complex geometries.
8. Conclusions
In this review, we have summarized alterations to T-tubules and dyads that occur in ventricular myocytes in disease, and we have described how mathematical modelling has provided important insight into the functional effects of these changes. Although work performed to date has improved our quantitative understanding of changes that occur with disease, further improvements, both experimental and computational, will be required to address unresolved questions. These include the effects of alterations in RyR cluster structure, and the nanoscale localization of auxiliary proteins such as NCX, CSQ, and SERCA. We expect that over the next several years, experimental measurements combined with simulations will provide significant new insights into these issues.
Funding
This work was supported by grants from the National Institutes of Health (HL076230, GM071558 to E.A.S.).
Acknowledgments
Conflict of interest: none declared.
References
- 1.Bito V, Heinzel FR, Biesmans L, Antoons G, Sipido KR. Crosstalk between L-type Ca2+ channels and the sarcoplasmic reticulum: alterations during cardiac remodelling. Cardiovasc Res. 2008;77:315–324. doi: 10.1093/cvr/cvm063. doi:10.1093/cvr/cvm063. [DOI] [PubMed] [Google Scholar]
- 2.Orchard CH, Pasek M, Brette F. The role of mammalian cardiac t-tubules in excitation-contraction coupling: experimental and computational approaches. Exp Physiol. 2009;94:509–519. doi: 10.1113/expphysiol.2008.043984. doi:10.1113/expphysiol.2008.043984. [DOI] [PubMed] [Google Scholar]
- 3.Louch WE, Sejersted OM, Swift F. There goes the neighborhood: pathological alterations in T-tubule morphology and consequences for cardiomyocyte Ca2+ handling. J Biomed Biotechnol. 2010;2010:503906. doi: 10.1155/2010/503906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. doi:10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. doi:10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 6.Cannell MB, Cheng H, Lederer WJ. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophys J. 1994;67:1942–1956. doi: 10.1016/S0006-3495(94)80677-0. doi:10.1016/S0006-3495(94)80677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wier WG, Egan TM, Lopez-Lopez JR, Balke CW. Local control of excitation-contraction coupling in rat heart cells. J Physiol (Lond) 1994;474:463–471. doi: 10.1113/jphysiol.1994.sp020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sobie EA, Dilly KW, Dos Santos CJ, Lederer WJ, Jafri MS. Termination of cardiac Ca2+ sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J. 2002;83:59–78. doi: 10.1016/s0006-3495(02)75149-7. doi:10.1016/S0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL, Gyorke S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res. 2002;91:414–420. doi: 10.1161/01.res.0000032490.04207.bd. doi:10.1161/01.RES.0000032490.04207.BD. [DOI] [PubMed] [Google Scholar]
- 10.Terentyev D, Viatchenko-Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: mechanism for hereditary arrhythmia. Proc Natl Acad Sci USA. 2003;100:11759–11764. doi: 10.1073/pnas.1932318100. doi:10.1073/pnas.1932318100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szentesi P, Pignier C, Egger M, Kranias EG, Niggli E. Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+-induced Ca2+ release refractoriness in heart muscle. Circ Res. 2004;95:807–813. doi: 10.1161/01.RES.0000146029.80463.7d. doi:10.1161/01.RES.0000146029.80463.7d. [DOI] [PubMed] [Google Scholar]
- 12.Sobie EA, Song LS, Lederer WJ. Local recovery of Ca2+ release in rat ventricular myocytes. J Physiol. 2005;565:441–447. doi: 10.1113/jphysiol.2005.086496. doi:10.1113/jphysiol.2005.086496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zima AV, Picht E, Bers DM, Blatter LA. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ Res. 2008;103:e105–e115. doi: 10.1161/CIRCRESAHA.107.183236. doi:10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huertas MA, Smith GD. The dynamics of luminal depletion and the stochastic gating of Ca2+-activated Ca2+ channels and release sites. J Theor Biol. 2007;246:332–354. doi: 10.1016/j.jtbi.2007.01.003. doi:10.1016/j.jtbi.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Liang X, Hu XF, Hu J. Dynamic interreceptor coupling contributes to the consistent open duration of ryanodine receptors. Biophys J. 2009;96:4826–4833. doi: 10.1016/j.bpj.2009.03.042. doi:10.1016/j.bpj.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stern MD, Song LS, Cheng H, Sham JS, Yang HT, Boheler KR, et al. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J Gen Physiol. 1999;113:469–489. doi: 10.1085/jgp.113.3.469. doi:10.1085/jgp.113.3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaur N, Rudy Y. Multiscale modeling of calcium cycling in cardiac ventricular myocyte: macroscopic consequences of microscopic dyadic function. Biophys J. 2011;100:2904–2912. doi: 10.1016/j.bpj.2011.05.031. doi:10.1016/j.bpj.2011.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato D, Bers DM. How does stochastic ryanodine receptor-mediated Ca leak fail to initiate a Ca spark? Biophys J. 2011;101:2370–2379. doi: 10.1016/j.bpj.2011.10.017. doi:10.1016/j.bpj.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winslow RL, Cortassa S, O'Rourke B, Hashambhoy YL, Rice JJ, Greenstein JL. Integrative modeling of the cardiac ventricular myocyte. Wiley Interdiscip Rev Syst Biol Med. 2011;3:392–413. doi: 10.1002/wsbm.122. doi:10.1002/wsbm.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fink M, Niederer SA, Cherry EM, Fenton FH, Koivumaki JT, Seemann G, et al. Cardiac cell modelling: observations from the heart of the cardiac physiome project. Prog Biophys Mol Biol. 2011;104:2–21. doi: 10.1016/j.pbiomolbio.2010.03.002. doi:10.1016/j.pbiomolbio.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 21.He J, Conklin MW, Foell JD, Wolff MR, Haworth RA, Coronado R, et al. Reduction in density of transverse tubules and L-type Ca2+ channels in canine tachycardia-induced heart failure. Cardiovasc Res. 2001;49:298–307. doi: 10.1016/s0008-6363(00)00256-x. doi:10.1016/S0008-6363(00)00256-X. [DOI] [PubMed] [Google Scholar]
- 22.Balijepalli RC, Lokuta AJ, Maertz NA, Buck JM, Haworth RA, Valdivia HH, et al. Depletion of T-tubules and specific subcellular changes in sarcolemmal proteins in tachycardia-induced heart failure. Cardiovasc Res. 2003;59:67–77. doi: 10.1016/s0008-6363(03)00325-0. doi:10.1016/S0008-6363(03)00325-0. [DOI] [PubMed] [Google Scholar]
- 23.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci USA. 2006;103:4305–4310. doi: 10.1073/pnas.0509324103. doi:10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I, et al. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol Lond. 2006;574:519–533. doi: 10.1113/jphysiol.2006.107227. doi:10.1113/jphysiol.2006.107227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, et al. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res. 2008;102:338–346. doi: 10.1161/CIRCRESAHA.107.160085. doi:10.1161/CIRCRESAHA.107.160085. [DOI] [PubMed] [Google Scholar]
- 26.Lyon AR, Macleod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, et al. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci USA. 2009;106:6854–6859. doi: 10.1073/pnas.0809777106. doi:10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mork HK, Sjaastad I, Sejersted OM, Louch WE. Slowing of cardiomyocyte Ca2+ release and contraction during heart failure progression in postinfarction mice. Am J Physiol Heart Circ Physiol. 2009;296:H1069–H1079. doi: 10.1152/ajpheart.01009.2008. doi:10.1152/ajpheart.01009.2008. [DOI] [PubMed] [Google Scholar]
- 28.Ibrahim M, Al MA, Navaratnarajah M, Siedlecka U, Soppa GK, Moshkov A, et al. Prolonged mechanical unloading affects cardiomyocyte excitation-contraction coupling, transverse-tubule structure, and the cell surface. FASEB J. 2010;24:3321–3329. doi: 10.1096/fj.10-156638. doi:10.1096/fj.10-156638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, et al. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. doi: 10.1161/CIRCRESAHA.109.212324. doi:10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crossman DJ, Ruygrok PN, Soeller C, Cannell MB. Changes in the organization of excitation-contraction coupling structures in failing human heart. PLoS One. 2011;6:e17901. doi: 10.1371/journal.pone.0017901. doi:10.1371/journal.pone.0017901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kemi OJ, Hoydal MA, MacQuaide N, Haram PM, Koch LG, Britton SL, et al. The effect of exercise training on transverse tubules in normal, remodeled, and reverse remodeled hearts. J Cell Physiol. 2011;226:2235–2243. doi: 10.1002/jcp.22559. doi:10.1002/jcp.22559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sachse FB, Torres NS, Savio-Galimberti E, Aiba T, Kass DA, Tomaselli GF, et al. Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy. Circ Res. 2012;110:588–597. doi: 10.1161/CIRCRESAHA.111.257428. doi:10.1161/CIRCRESAHA.111.257428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu HD, Xu M, Li RC, Guo L, Lai YS, Xu SM, et al. Ultrastructural remodelling of Ca2+ signalling apparatus in failing heart cells. Cardiovasc Res. 2012;95:430–438. doi: 10.1093/cvr/cvs195. doi:10.1093/cvr/cvs195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, et al. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. doi: 10.1161/CIRCULATIONAHA.110.006437. doi:10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, et al. Defective excitation–contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. doi:10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 36.Gomez AM, Guatimosim S, Dilly KW, Vassort G, Lederer WJ. Heart failure after myocardial infarction: altered excitation–contraction coupling. Circulation. 2001;104:688–693. doi: 10.1161/hc3201.092285. doi:10.1161/hc3201.092285. [DOI] [PubMed] [Google Scholar]
- 37.Litwin SE, Zhang D, Bridge JH. Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ Res. 2000;87:1040–1047. doi: 10.1161/01.res.87.11.1040. doi:10.1161/01.RES.87.11.1040. [DOI] [PubMed] [Google Scholar]
- 38.Xu M, Zhou P, Xu SM, Liu Y, Feng X, Bai SH, et al. Intermolecular failure of L-type Ca2+ channel and ryanodine receptor signaling in hypertrophy. PLoS Biol. 2007;5:e21. doi: 10.1371/journal.pbio.0050021. doi:10.1371/journal.pbio.0050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biesmans L, MacQuaide N, Heinzel FR, Bito V, Smith GL, Sipido KR. Subcellular heterogeneity of ryanodine receptor properties in ventricular myocytes with low T-tubule density. PLoS One. 2011;6:e25100. doi: 10.1371/journal.pone.0025100. doi:10.1371/journal.pone.0025100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laver DR, Kong CH, Imtiaz MS, Cannell MB. Termination of calcium-induced calcium release by induction decay: an emergent property of stochastic channel gating and molecular scale architecture. J Mol Cell Cardiol. 2013;54:98–100. doi: 10.1016/j.yjmcc.2012.10.009. doi:10.1016/j.yjmcc.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 41.Ramay HR, Liu OZ, Sobie EA. Recovery of cardiac calcium release is controlled by sarcoplasmic reticulum refilling and ryanodine receptor sensitivity. Cardiovasc Res. 2011;91:598–605. doi: 10.1093/cvr/cvr143. doi:10.1093/cvr/cvr143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo T, Gillespie D, Fill M. Ryanodine receptor current amplitude controls Ca2+ sparks in cardiac muscle. Circ Res. 2012;111:28–36. doi: 10.1161/CIRCRESAHA.112.265652. doi:10.1161/CIRCRESAHA.112.265652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sobie EA. Parameter sensitivity analysis in electrophysiological models using multivariable regression. Biophys J. 2009;96:1264–1274. doi: 10.1016/j.bpj.2008.10.056. doi:10.1016/j.bpj.2008.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarkar AX, Christini DJ, Sobie EA. Exploiting mathematical models to illuminate electrophysiological variability between individuals. J Physiol. 2012;590:2555–2567. doi: 10.1113/jphysiol.2011.223313. doi:10.1113/jphysiol.2011.223313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Picht E, Zima AV, Shannon TR, Duncan AM, Blatter LA, Bers DM. Dynamic calcium movement inside cardiac sarcoplasmic reticulum during release. Circ Res. 2011;108:847–856. doi: 10.1161/CIRCRESAHA.111.240234. doi:10.1161/CIRCRESAHA.111.240234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lipskaia L, Chemaly ER, Hadri L, Lompre AM, Hajjar RJ. Sarcoplasmic reticulum Ca2+ ATPase as a therapeutic target for heart failure. Expert Opin Biol Ther. 2010;10:29–41. doi: 10.1517/14712590903321462. doi:10.1517/14712590903321462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams GS, Chikando AC, Tuan HT, Sobie EA, Lederer WJ, Jafri MS. Dynamics of calcium sparks and calcium leak in the heart. Biophys J. 2011;101:1287–1296. doi: 10.1016/j.bpj.2011.07.021. doi:10.1016/j.bpj.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. doi:10.1161/01.CIR.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 49.Baartscheer A, Schumacher CA, Belterman CN, Coronel R, Fiolet JW. [Na+]i and the driving force of the Na+/Ca2+-exchanger in heart failure. Cardiovasc Res. 2003;57:986–995. doi: 10.1016/s0008-6363(02)00848-9. doi:10.1016/S0008-6363(02)00848-9. [DOI] [PubMed] [Google Scholar]
- 50.Lee YS, Liu OZ, Sobie EA. Decoding myocardial Ca2+ signals across multiple spatial scales: a role for sensitivity analysis. J Mol Cell Cardiol. doi: 10.1016/j.yjmcc.2012.09.009. Advance Access published September 28, 2012, doi: 10.1016/j.yjmcc.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quinn FR, Currie S, Duncan AM, Miller S, Sayeed R, Cobbe SM, et al. Myocardial infarction causes increased expression but decreased activity of the myocardial Na+–Ca2+ exchanger in the rabbit. J Physiol. 2003;553:229–242. doi: 10.1113/jphysiol.2003.050716. doi:10.1113/jphysiol.2003.050716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sah R, Ramirez RJ, Oudit GY, Gidrewicz D, Trivieri MG, Zobel C, et al. Regulation of cardiac excitation-contraction coupling by action potential repolarization: role of the transient outward potassium current (Ito) J Physiol. 2003;546:5–18. doi: 10.1113/jphysiol.2002.026468. doi:10.1113/jphysiol.2002.026468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cannell MB, Crossman DJ, Soeller C. Effect of changes in action potential spike configuration, junctional sarcoplasmic reticulum micro-architecture and altered t-tubule structure in human heart failure. J Muscle Res Cell Motil. 2006;27:297–306. doi: 10.1007/s10974-006-9089-y. doi:10.1007/s10974-006-9089-y. [DOI] [PubMed] [Google Scholar]
- 54.Louch WE, Hake J, Jolle GF, Mork HK, Sjaastad I, Lines GT, et al. Control of Ca2+ release by action potential configuration in normal and failing murine cardiomyocytes. Biophys J. 2010;99:1377–1386. doi: 10.1016/j.bpj.2010.06.055. doi:10.1016/j.bpj.2010.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wagner E, Lauterbach MA, Kohl T, Westphal V, Williams GS, Steinbrecher JH, et al. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ Res. 2012;111:402–414. doi: 10.1161/CIRCRESAHA.112.274530. doi:10.1161/CIRCRESAHA.112.274530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soeller C, Baddeley D. Super-resolution imaging of EC coupling protein distribution in the heart. J Mol Cell Cardiol. doi: 10.1016/j.yjmcc.2012.11.004. Advance Access published November 13, 2012, doi: 10.1016/j.yjmcc.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 57.Baddeley D, Jayasinghe ID, Lam L, Rossberger S, Cannell MB, Soeller C. Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc Natl Acad Sci USA. 2009;106:22275–22280. doi: 10.1073/pnas.0908971106. doi:10.1073/pnas.0908971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scriven DR, Dan P, Moore ED. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J. 2000;79:2682–2691. doi: 10.1016/S0006-3495(00)76506-4. doi:10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jayasinghe ID, Cannell MB, Soeller C. Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophys J. 2009;97:2664–2673. doi: 10.1016/j.bpj.2009.08.036. doi:10.1016/j.bpj.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hake J, Edwards AG, Yu Z, Kekenes-Huskey PM, Michailova AP, McCammon JA, et al. Modelling cardiac calcium sparks in a three-dimensional reconstruction of a calcium release unit. J Physiol. 2012;590:4403–4422. doi: 10.1113/jphysiol.2012.227926. doi:10.1113/jphysiol.2012.227926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramay HR, Jafri MS, Lederer WJ, Sobie EA. Predicting local SR Ca2+ dynamics during Ca2+ wave propagation in ventricular myocytes. Biophys J. 2010;98:2515–2523. doi: 10.1016/j.bpj.2010.02.038. doi:10.1016/j.bpj.2010.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Faggioni M, Knollmann BC. Calsequestrin 2 and arrhythmias. Am J Physiol Heart Circ Physiol. 2012;302:H1250–H1260. doi: 10.1152/ajpheart.00779.2011. doi:10.1152/ajpheart.00779.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Swift F, Franzini-Armstrong C, Oyehaug L, Enger UH, Andersson KB, Christensen G, et al. Extreme sarcoplasmic reticulum volume loss and compensatory T-tubule remodeling after Serca2 knockout. Proc Natl Acad Sci USA. 2012;109:3997–4001. doi: 10.1073/pnas.1120172109. doi:10.1073/pnas.1120172109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sobie EA, Lederer WJ. Dynamic local changes in sarcoplasmic reticulum calcium: physiological and pathophysiological roles. J Mol Cell Cardiol. 2012;52:304–311. doi: 10.1016/j.yjmcc.2011.06.024. doi:10.1016/j.yjmcc.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Swietach P, Spitzer KW, Vaughan-Jones RD. Ca2+-mobility in the sarcoplasmic reticulum of ventricular myocytes is low. Biophys J. 2008;95:1412–1427. doi: 10.1529/biophysj.108.130385. doi:10.1529/biophysj.108.130385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu M, Wu HD, Li RC, Zhang HB, Wang M, Tao J, et al. Mir-24 regulates junctophilin-2 expression in cardiomyocytes. Circ Res. 2012;111:837–841. doi: 10.1161/CIRCRESAHA.112.277418. doi:10.1161/CIRCRESAHA.112.277418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu CY, Jia Z, Wang W, Ballou LM, Jiang YP, Chen B, et al. PI3Ks maintain the structural integrity of T-tubules in cardiac myocytes. PLoS One. 2011;6:e24404. doi: 10.1371/journal.pone.0024404. doi:10.1371/journal.pone.0024404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheng Y, Yu Z, Hoshijima M, Holst MJ, McCulloch AD, McCammon JA, et al. Numerical analysis of Ca2+ signaling in rat ventricular myocytes with realistic transverse-axial tubular geometry and inhibited sarcoplasmic reticulum. PLoS Comput Biol. 2010;6:e1000972. doi: 10.1371/journal.pcbi.1000972. doi:10.1371/journal.pcbi.1000972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hatano A, Okada J, Hisada T, Sugiura S. Critical role of cardiac t-tubule system for the maintenance of contractile function revealed by a 3D integrated model of cardiomyocytes. J Biomech. 2012;45:815–823. doi: 10.1016/j.jbiomech.2011.11.022. doi:10.1016/j.jbiomech.2011.11.022. [DOI] [PubMed] [Google Scholar]
- 71.Cortassa S, O'Rourke B, Winslow RL, Aon MA. Control and regulation of mitochondrial energetics in an integrated model of cardiomyocyte function. Biophys J. 2009;96:2466–2478. doi: 10.1016/j.bpj.2008.12.3893. doi:10.1016/j.bpj.2008.12.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Torres NS, Larbig R, Rock A, Goldhaber JI, Bridge JH. Na+ currents are required for efficient excitation–contraction coupling in rabbit ventricular myocytes: a possible contribution of neuronal Na+ channels. J Physiol. 2010;588:4249–4260. doi: 10.1113/jphysiol.2010.194688. doi:10.1113/jphysiol.2010.194688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sobie EA, Cannell MB, Bridge JHB. Allosteric activation of Na+-Ca2+ exchange by L-type Ca2+ current augments the trigger flux for SR Ca2+ release in ventricular myocytes. Biophys J. 2008;94:L54–L56. doi: 10.1529/biophysj.107.127878. doi:10.1529/biophysj.107.127878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lines GT, Sande JB, Louch WE, Mork HK, Grottum P, Sejersted OM. Contribution of the Na+/Ca2+ exchanger to rapid Ca2+ release in cardiomyocytes. Biophys J. 2006;91:779–792. doi: 10.1529/biophysj.105.072447. doi:10.1529/biophysj.105.072447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sher AA, Noble PJ, Hinch R, Gavaghan DJ, Noble D. The role of the Na+/Ca2+ exchangers in Ca2+ dynamics in ventricular myocytes. Prog Biophys Mol Biol. 2008;96:377–398. doi: 10.1016/j.pbiomolbio.2007.07.018. doi:10.1016/j.pbiomolbio.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 76.Sato D, Despa S, Bers DM. Can the sodium–calcium exchanger initiate or suppress calcium sparks in cardiac myocytes? Biophys J. 2012;102:L31–L33. doi: 10.1016/j.bpj.2012.03.051. doi:10.1016/j.bpj.2012.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kekenes-Huskey PM, Cheng Y, Hake JE, Sachse FB, Bridge JH, Holst MJ, et al. Modeling effects of L-type Ca2+ current and Na+–Ca2+ exchanger on Ca2+ trigger flux in rabbit myocytes with realistic T-tubule geometries. Front Physiol. 2012;3:351. doi: 10.3389/fphys.2012.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]