

Abstract
Background:
The ideal management of thalassemia involves a multidisciplinary therapeutic team approach and should be preferably done at a comprehensive thalassemia care center with all sorts of specialists and the backup of a well-equipped blood bank. However, in developing country like ours, these facilities are not available in rural set up. So, a situation where conservative therapy with regular blood transfusion is the only choice left to innumerable thalassemic children.
Objective:
To evaluate the existing conservative management protocol of Beta-thalassemia major patients in the setup of a subdivision level Government Hospital of rural West Bengal, India.
Materials and Methods:
The study was performed between December 2009 and December 2011. Beta-thalassemia major patients, registered in blood bank for moderate transfusion regimen, were taken in study. All the patients were screened for Transfusion Transmittable Infections at the time of registration and thereafter periodically every six months. Iron chelation therapy was given simultaneously with transfusion at a dose of 20 to 40 mg/kg/day for six days. The patients were advised to follow up with chelation therapy at home by daily infusion with a goal of maintaining serum ferritin level below 1000 ng/ml. Over this long period of study, the patients were periodically evaluated for complications.
Results:
The average blood requirement (ml/kg/year) in 1-5 years, 6-10 years, and 11-15 years were 110, 150, and 180, respectively. Incidence of Hepatitis C Virus infection in 1-5 years and 6-10 years were 1.75% and 2.08%, respectively. It is well seen that serum ferritin level increase with ascending age as does the blood consumption.
Conclusion:
Conservative management may be the best alternative and at times the only hope for patients in developing country like ours. However, in order to decrease the disease load, steps need to be taken to introduce preventive measures.
Keywords: Beta-thalassemia major, blood transfusion, conservative management, iron chelation, prevention program
INTRODUCTION
Thalassemia is an autosomal recessive disease. As per WHO estimate, 4.5% of world populations are carriers of hemoglobinopathies.[1] The largest concentration of thalassemia patients are seen in South East Asia, Sri Lanka, Bangladesh, North West India, Pakistan, Middle East Countries, Greece, and Italy.[2] In South East Asia, there are 40 million carriers of thalassemia gene, 50% of which are in India alone.[3] The mean prevalence of the carrier status in India is 3.3% (ranging from 1 to 17% in various communities). If a line is drawn between Mumbai and Kolkata on the Map of India, the region above the line has an incidence of 3 to 17%, whereas region below the line has incidence of less than 3%.[2] It is estimated that every year approximately 1 00 000 children with thalassemia major are born all over the world.[4] With the birth rate of 22.8/1000 in India, it is estimated that there are about 9 000 to 10 000 cases being added every year.[2]
The thalassemia syndrome are a heterogeneous group of hereditary disorders of reduced hemoglobin synthesis.[5] If the synthesis of beta globin chain is suppressed because of mutation in beta gene, then production of adult hemoglobin is suppressed and it is called beta-thalassemia.[6,7] The ideal management of thalassemia involves a multidisciplinary therapeutic team approach[8] and should be preferably done at a comprehensive thalassemia care center having a team of pediatric hematologist, pediatrician, transfusion medicine specialist, endocrinologist, psychologist, social worker, and the backup of a well-equipped blood bank.[5] But, in developing country like ours, these facilities are not available in any block, subdivision, or district-level Hospital.[9] The management of thalassemia major in a developing country poses a major challenge to the Government health services. Lack of resources and coordination hampers the treatment of this multidisciplinary problem in a variety of ways. Awareness and availability of antenatal diagnosis is limited. The decision to terminate pregnancy is a difficult choice in our society. Premarital counseling, especially amongst the uneducated, is unheard of. The curative treatment of marrow transplantation is prohibitively expensive. The resultant impact of all these factors combined has led to a situation where conservative management of the child born with this disease is, perhaps not the best but, the only choice. The mainstay of managing the beta-thalassemia major cases is by repeated blood transfusion and associated iron chelation therapy to reduce complication.[2,6] Cost of current management of thalassemia varies between 2 500 and 4 000 dollars per children per year, depending upon the weight of child and form of chelation therapy.[10] As major share of this cost is to be borne by the state itself, the management of thalassemia major in developing country like ours poses a major challenge to the Government Health Service.
In this background, it was thought pertinent to evaluate the existing conservative management protocol of Beta-thalassemia major patients in the setup of a subdivision level Government Hospital of rural West Bengal, India.
MATERIALS AND METHODS
The study was performed over a period of 2 years between December 2009 and December 2011 after obtaining usual permission from ethical committee. A cohort of 124 patients of Beta-thalassemia major at the Subdivision Hospital (SDH) of rural West Bengal diagnosed by complete blood count and hemoglobin electrophoresis by High-performance liquid chromatography, HPLC, technique was taken as subjects. The inclusion criteria were patients registered in blood bank for moderate transfusion regimen that aimed at maintaining pretransfusion hemoglobin level at 9 ± 0.5 g/dl were taken in study.[8,11] The subjects were divided into three age groups: 1-5 years, 6-10 years, and 11-15 years. Patients were managed conservatively with blood transfusion, aimed at maintaining pretransfusion hemoglobin level of 9 ± 0.5 g/dl as per the recommendation of thalassemia international federation.[8,11] Parameters studied were requirement of transfused blood, any seroconversion for transfusion transmitted infections (TTI) such as Human Immunodeficiency Virus (HIV), Hepatitis B Virus (HBV), and Hepatitis C Virus (HCV) infections and serum ferritin level, and occurrence of complications such as marked splenomegaly, heart diseases, skeletal abnormality, and delay in onset of puberty.
Blood transfusion carried out at SDH by providing packed cell @ 10 to 15 ml/kg body weight of the child. Mostly, patients were transfused once in every four weeks with an aim of maintaining pretransfusion hemoglobin level at 9 ± 0.5 g/dl.[11]
All the patients were screened for TTI infection at the time of registration in the study and thereafter periodically every six months. All the HBV negative patients were given three doses of Hepatitis B vaccination. Only four patients in the age group of 11 to 15 years underwent splenectomy due to massive splenomegaly and hypersplenism.
Iron chelation therapy was given with desferroxamine simultaneously with transfusion at a dose of 20 to 40 mg/kg/day for six days. The patients were advised to follow up with chelation therapy at home by daily infusion with a goal of maintaining serum ferritin level below 1000 ng/ml. Serum ferritin was evaluated three times per year and average serum ferritin level was calculated for each group.
Over this long period of study, the patients were periodically evaluated for development of complication of the disease – once a month in the blood bank counseling room of SDH. This was performed keeping in mind mainly the following complications – splenomegaly, heart diseases, skeletal abnormality, and delay in onset of puberty.
RESULTS
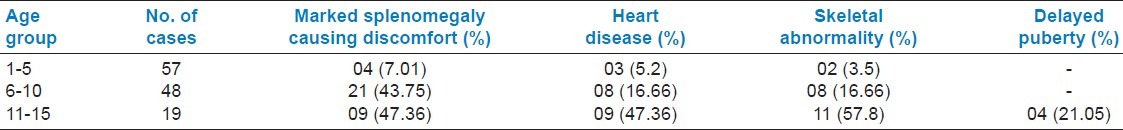
Blood transfusion requirement of thalassemic patients are depicted in Table 1. TTI status of thalassemic patients is depicted in Table 2. Average serum ferritin level in the study group is shown in Table 3. It is well seen that serum ferritin level increases with ascending age as does the blood consumption. Complications of the disease age group wise are depicted in Table 4.
Table 1.
Requirement of blood by thalassemia patients
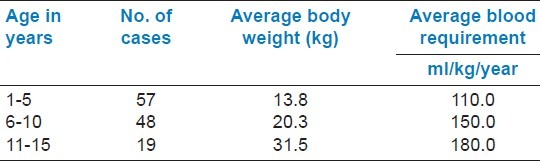
Table 2.
Transfusion transmittable infections status of thalassemia patients
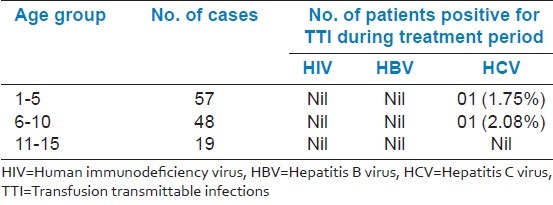
Table 3.
Average serum ferritin level in the study group
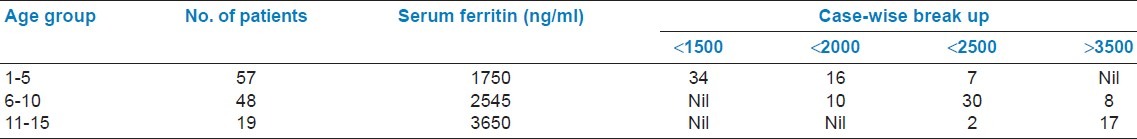
Table 4.
Complications of thalassemia patients under study
DISCUSSION
Long-term transfusion support is the backbone of conservative management in Beta-thalassemia major patients. These patients require regular transfusions to alleviate anemia. The hemoglobin level is utilized as an indicator for transfusion; as the hemoglobin falls, patients experience discomfort and attempts are made to elevate the hemoglobin level through transfusion to provide acceptable relief of symptoms.[11] It seems logical and predictable that lower endpoint target hemoglobin will allow a rational use of blood. The objective in employing the moderate transfusion regimen in our country is to keep the blood consumption to a minimum in order to curtail the iron overload.
Moderate transfusion regimen of 2005 as recommended by thalassemia international federation is utilized in our study group.[11] The most ideal way to transfuse thalassemics is using group-and type-specific packed red cells that are compatible by direct antiglobulin test. As per literature review,[9] 150 to 180 ml/kg/year of blood transfusion is required in non-splenectomized, non-sensitized patient to maintain the hemoglobin level above 10 gm%, whereas splenectomized patient require 130 to 135 ml/kg/year.[11] Even without hypersplenism, the requirement is 30% higher in non-splenectomized patients.[11] In our study, the blood consumptions varied from 110 to 180 ml/kg/year, which is similar as found in other studies.[9,11]
In our blood bank, we provided ABO, Rh-matched Coomb's cross-matched blood to the patients. Whenever blood requirement crossed 250 ml/kg/year and other signs of hypersplenism found, we recommended splenectomy.[9] Due to inadequate resources, only four patients of age group 11 to 15 years underwent splenectomy.
Multiple transfusions in thalassemia major patients are at a risk of developing TTI including HIV, HBV, and HCV.[12] Strict criteria of safe donor selection have to be adopted in order to minimize risk of TTI.[1] We have found no HIV- and HBV-positive patients in this study group. However, incidence of Hepatitis C is 1.75% in the age group of 1 to 5 years and 2.08% in the age group of 6 to 10 years which is relatively lower than the incidence in other countries of South East Asia.[2]
HCV infection has gained importance as one of the major complication in multiple transfused patients during last two decades. The prevalence rate of seropositivity increases with the numbers of transfusion. This post-transfusion hepatitis has significantly contributed to the morbidity in thalassemia. It has been found that HCV hepatitis is more threatening than HBV hepatitis due to a greater risk of developing chronicity of liver diseases.[1,12]
In our study, during an interview of the parents of thalassemic children affected with Hepatitis C, it was revealed that 4% children got blood transfusion from outside due to short supply of particular blood group in our blood bank. Seronegativity for Hepatitis B in all the cases of our study group can be attributed to Hepatitis B vaccination schedule which we managed to implement within our hospital setup.[7] However, TTI screening in our blood bank was formidable and both internal audit and external quality control with random sampling of true positives and negatives in referral center, Medical College, proved our good performance in TTI screening.
A major problem encountered in the management of thalassemia is iron overload.[6] Regular red cell transfusion to maintain hemoglobin as well as increased iron absorption from GI tract due to ineffective erythropoiesis and consequent low hemoglobin in irregularly transfused children is accountable for iron over blood. The goal of iron chelation is to reduce the iron overload and subsequently maintain ferritin levels below 1 000 ng/ml.[6,7] But, our patients even in the younger age group showed high serum ferritin level. In 1-5 years age group, average serum ferritin is 1 750 ng/ml, and this rises to 3 650 ng/ml in older patients, 11-15 years. The serum ferritin level could not be controlled well as only few patients fully complied with recommended regimen at home.[11] The major reason for poor compliance to chelation therapy is that it is painful, costly, and the method of administration is time consuming. It appears in our study that perhaps chelation therapy is the most deficient area of conservative thalassemia management. The oral preparation, Deferiprone, might help to change this scenario, but high cost of the oral medicine is another problem in this case.
Splenectomy should be considered if the patient has already developed splenomegaly that is causing physical discomfort, presenting with signs of hypersplenism and the age of patient is above 5 years.[6,7] In our study, 7.01%, 43.7%, and 47.3% are the occurrence rate of splenomegaly in the three study groups, as complication which are in agreement with previous studies.[7,8]
Cardiac failure and arrhythmias are important causes of mortality in thalassemic children. Holter monitoring, 2D-Echo can assist in evaluation of cardiac function. T2-weighted cardiac MRI is the best method[7] of assessing severity of cardiac iron overload. However, for all practical purpose in our study, we used ECG and ejection fraction as complication monitoring device.[6] Cardiac complication is seen in age group of 1-5 years - 5.2%, 6-10 years - 16.6%, and in 11-15 years - 31.57%. These findings of our study are similar to the findings of few national-level studies done previously.[1,7] Thalassemia patients show a variety of bone diseases including bone pain, bone deformity, bone age delay, growth failure, rickets, scoliosis, spinal deformities, pathological fracture, osteoporosis, and osteopenia.[6] Osteoporosis is emerging as an important cause of morbidity in thalassemia patients with increasing age. In our study, we found skeletal abnormality of 3.5% in 1-5 years age group and 16.6% in 6-10 years age group. It increased up to 57.8% in 11-15 years age group. Only few similar studies had the same observations.[7] Iron overload in thalassemia is responsible for dysfunction of many endocrine glands like thyroid, pituitary, and gonads leading to development of stunted growth and delayed puberty. In our study, we found growth failure and delayed puberty in 21.05% cases in the age group 11-15 years. However, more extensive studies are needed along with exclusion of other causes of endocrine failure to establish these findings.[6,7]
So, our study highlights the very need of conservative management of Beta-thalassemia major patients at the SD level Hospital and block Primary Health Centre Level (BPHC) in the Government set up with limited financial and manpower support and without unnecessary referral to higher centers. Nevertheless, sub-division (SD) level non-Enzyme-Linked Immunosorbent Assay (ELISA) blood bank of Government Sector is reasonably dependable to cater thalassemia patients of rural districts to a certain extent. It is worth mentioning that developing countries like ours face a host of organizational, logistic, and funding problems. The experience we acquired in this background points out multiple deficiencies in the conservative management in our country. Thalassemia prevention program is very much necessary to reduce the disease load of the country.
There are several limitations of the study; first, chelation therapy at home could not be ensured. Second, biochemical and hormonal parameters could not be studied due to lack of infrastructure and financial constraints. Lastly, blood bank equipped with ELISA and blood component separator were lacking in our study done at rural set up.
CONCLUSION
In conclusion, it can be said that conservative management of Beta-thalassemia major patients with regular blood transfusion and chelation therapy can be performed at SD Hospital and BPHC level with the backup of a Government sector blood bank, effectively in rural area of our country. Conservative management may be the best alternative and at times the only hope for patients in our country. However, in order to decrease the disease load, steps need to be taken to introduce preventive measures.
ACKNOWLEDGEMENTS
We would like to thank Ms. Mohua Bhattacharya, technical staff, for her active participation in our study. Special thanks to the management of Medical College, Kolkata, and SD Hospital, Bishnupur, for their active participation.
Footnotes
Source of Support: Medical College, Kolkata and SD Hospital, Bishnupur
Conflict of Interest: None declared
REFERENCES
- 1.World Health Organization. Weekly Epidemiological Record, No. 48. 2008;83:429–40. [Google Scholar]
- 2.World Health Organization. SEARO:(1999). Health situation in the South-East Asia Region, New Delhi 1994-97 [Google Scholar]
- 3.Weatherall DJ, Clegg JB. The Thalassemia Syndromes. 4th ed. Oxford Blackwell Science Ltd, Osney Mead, Oxford OX2 0EL; 2001. [Google Scholar]
- 4.Michael R, Baun DE, Vichinsky E. Hemoglobinopathies-Nelson Text Book of Pediatrics. 18th ed. Philadelphia: Saunders-An imprint of Elsevier; 2009. [Google Scholar]
- 5.Nadkarni A, Caraskar AC, Krishnamoorty R, Lu CY, Ghosh K, Colah R, et al. Molecular pathogenesis and clinical variability of β thalassemia syndrome among Indians. Am J Hematol. 2001;68:75–80. doi: 10.1002/ajh.1156. [DOI] [PubMed] [Google Scholar]
- 6.Lokeshwar MR, Nitin S. “Thalassemia”- IAP text book of Pediatrics. Editor in chief. In: Parthasarathy A, Lokeswar MR, editors. Pediatic Hematology. 4th ed. New Delhi: Jaypee Brothers; 2009. pp. 794–815. [Google Scholar]
- 7.Manglani M, Lokeshwar MR, Rao S. Defects in synthesis of hemoglobin Thalassemia syndrome-HbS, C, D, E. Current trends in Thalassemia therapy, Oncology (Under IAP President Action Plan 2006) :65–77. [Google Scholar]
- 8.Guide lines for the clinical management of thalassemia. 2nd ed. Nicosia Cyprus: Thalassemia International Federation; 2009. World Bank 2006, reported joint WHO – March of time meeting 2006-Quoted; p. 9. [Google Scholar]
- 9.Bouhass RA, Kabouya EA, Smahi C, Benaceur SM, Aguercif M. Management of Beta thalassemia in a developing country. Ann Pediatr (Paris) 1992;39:115–9. [PubMed] [Google Scholar]
- 10.WHO, The global burden of Diseases, 2004-2008, Updates. WHO Library Cataloguing-in-Publication DataThe global burden of disease: 2004 update1.Cost of illness.2.World Healthstatistics. 3.Mortality-trends.I. World Health Organization. ISBN 978 92 4 156371 0,(NLM classification: W 74) [Google Scholar]
- 11.Eleftheriou A. Thalassemia International Federation: Guidelines for the clinical management of thalassemia. 52. Nicosia Cyprus: TIF Magazine; 2008. Mar, [PubMed] [Google Scholar]
- 12.Agarwal MB, Malken GH, Bhave AA, Vishwanathan C, Billa V, Dube SR, et al. Antibody to Hepatitis C virus in multitransfused. Thalassemic-Indian expericnce. J Assoc Physicians India. 1993;41:195–7. [PubMed] [Google Scholar]