

Abstract
Background:
New Delhi metallo-β-lactamase-1 (NDM-1)-producing Gram-negative bacteria are today's major worldwide health concern. The enzyme NDM-1 provides bacterial resistance by its hydrolytic activity against the β-lactam ring of antibiotics. Inhibition of NDM-1 may prevent the hydrolysis of β-lactam ring of the antibiotics, and therefore, plays an important role against antibacterial resistance.
Materials and Methods:
Here we made an attempt to design suitable inhibitors against NDM-1 from different natural antibacterial compounds using molecular docking approach.
Results:
We observed that natural compounds such as Nimbolide and Isomargololone are showing an appreciable IC50 value as well as significant binding energy value for NDM-1. We further observed these compounds showing better affinity to NDM-1 on comparison with 14 β-lactam antibiotics.
Conclusion:
Finally, our study provides a platform for the development of a potent inhibitor of NDM-1, which may be considered as a potential drug candidate against bacterial resistance.
Keywords: New Delhi metallo-beta-lactamase 1, β-lactam antibiotics, energy minimization, ligand, virtual screening
INTRODUCTION
Antibiotic resistance in bacterial strains has become a major clinical concern.[1] Bacteria have several strategies to combat against β-lactam antibiotics[2,3] including production of β-lactamases enzyme which hydrolyze the β-lactam ring of lactam antibiotics.[4–6] The β-lactamases are classified into four groups (1, 2, 3 and 4) on the basis of function. Moreover, based on molecules (nucleotides and amino acid sequences) β-lactamases are classified into four groups (A, B, C and D) comprising two families: Serine-β-lactamases (SBLs) and metallo-β-lactamases (MBLs).[7,8]
New Delhi metallo-β-lactamase-1 (NDM-1) belongs to the subclass B1 of MBL, in which two zinc ion (s) are required for the activity, where the tightly bound zinc is referred to as Zn1 and the loosely bound zinc is called as Zn2.[9,10] In Zn1, a tetrahedral geometry is coordinated by three histidine residues and one solvent molecule in the crystal structure. While, a distorted trigonal bipyramidal geometry is the coordination of Zn2 by three amino acids (His, Cys, Asp), a water molecule and a solvent molecule (glycerol) that serves as a ligand to Zn1 too.[11]
The MBL was first identified in a Swedish patient of Indian origin, from New Delhi, who was suffering from urinary tract infection, caused by carbapenem-resistant Klebsiella pneumoniae (CRKP). The multi-drug-resistant bacteria K. pneumoniae containing NDM-1 was named as ‘superbug’.[12,13] NDM-1 is mainly found in K. pneumoniae, but recently NDM-1 activity is also observed in many other bacteria such as Enterobacteriaceae and Acinetobacter baumannii,[14] which causes antibiotic resistance due to its hydrolyzing tendency for β-lactam antibiotics. β-lactam antibiotics resistance was restricted geographically, and restricted to specific bacterial species. However, such species-specific barrier was maintained due to the gene coding for NDM-1 is present as mobile genes on plasmids, therefore, such gene can readily spreaded through bacterial populations.[15]
There is still a lack of clinically potent inhibitors of NDM-1. Hence, it would be a promising choice to block the NDM-1 with suitable inhibitor. To reveal the resistance mechanism of bacteria to β-lactam antibiotics, we selected a series of 35 different natural antibacterial compounds from various literary sources, and successively docked with the active site of NDM-1 [Table 1]. Some of the selected natural compounds show a good binding affinity at the active site of NDM-1, particularly Nimbolide and Isomargololone, produce lower binding energy than the β-lactam antibiotics with NDM-1 and also have appreciable IC50 value. These findings may provide useful insights for designing new potent drugs to fight against the antibiotic resistance of NDM-1.
Table 1.
List of natural compounds taken for docking with NDM-1

MATERIALS AND METHODS
Data set and molecule preparation
In order to find the NDM-1 inhibitors, we selected 35 different antibacterial natural compounds for docking against NDM-1 [Table 1, Figure 1a]. We further selected β-lactam antibiotics from PubChem (http://pubchem.ncbi.nlm.nih.gov/) and subsequently created a representative set of 14 β-lactam antibiotics, which interact with NDM-1 [Table 2, Figure 1b]. The molecular structures of antibacterial natural compounds and β-lactam antibiotics were drawn by the Chemdraw,[16] and molecules were converted into protein data bank (pdb) format from MDL Mol by using online MN molecular format convertor (http://www.molecular-networks.com). High-resolution structure of NDM-1 was already reported, and its atomic coordinates were taken from the protein data bank (PDB code: 3Q6X) for docking.[17] Visualization of all docked structure was performed on PyMol molecular graphics program, a comprehensive software package for rendering and animating 3D-structures.[18]
Figure 1.

Alignment of all (a) Natural compounds and (b) Antibiotic molecules (stick model) in the lowest-energy confirmation in the NDM-1 structure shown in cartoon model (light grey). Chemical structural representation of (c) Nimbolide and (d) Isomargolonone. Three-dimensional structure of (e) Nimbolide and (f) Isomargolonone are shown in ball and stick model drawn in PyMOL
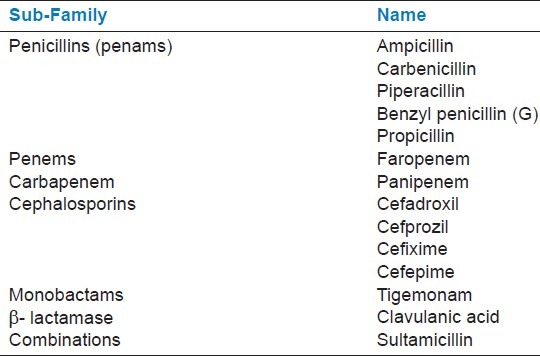
Table 2.
List of antibiotics from the β-lactam family used in docking study
Molecular docking
We used AutoDock 4.2.3 program for docking simulations of ligands shown in Table 1 and Table 2 with NDM-1.[19] We applied Lamarckian genetic algorithm (LGA) to analyze protein-ligand interactions.[20] Further, we added polar hydrogen atoms and performed Kollman united atom charge assignment to NDM-1 molecules followed by the generation of PDBQ file. AutoGrid program was used to generate 3D affinity grid fields with grid map of 40 × 50 × 40 points. The default settings were used for all other parameters. AutoDock tools utility was used to generate both grid and docking parameter files (i.e., gpf and dpf).
We performed 50 independent runs with the step sizes of 0.2 Ε for translations and 8° for orientations and torsions for docking of NDM-1 with various ligands. For LGA, pseudo-solids and wets local search methods were used. The possibility of performing local search on an individual in the population was 0.06 and the termination criterion for the local search was 0.01. van der Waals interaction, hydrogen bonding, and the Coulombic electrostatic potential for charges were used for binding energy calculations. Electrostatic grid maps were calculated by distance-dependent dielectric permittivity.
The docking simulations with multiple runs and cluster analysis of ligands were performed with their corresponding docked energy. Docking solutions with ligand all-atom root mean square deviation (rmsd) within 1.0 Ε of each other were clustered together and ranked by the lowest energy. The lowest-energy solution of the lowest ligand all-atom rmsd cluster was accepted as the calculated binding energy. The whole system was minimized to convergence. Although the solvation energies could not be explicitly considered during the minimization, the energy calculations were performed with a distance-dependent dielectric constant of 5 to mimic the solvation effect of the inhibitors in the protein environment.[21]
Energy minimization
Energy minimization of the docked complex of Nimbolide and Isomargolonone with NDM-1 were performed by Ammp molecular dynamics software of VEGA OpenGL package version VEGA ZZ 2.4.0 using the SP4 force-field.[22] Firstly, the correct bond types were assigned to the NDM-1 complex. Moreover, using the default parameters in the VEGA program, force fields and charges were assigned according to AMBER and Gasteiger algorithms, respectively. Then, NDM-1 complex with Nimbolide and Isomargolonone was embedded in a box solvated with 2613 water molecules. To neutralize the system 52 Cl- ions were added and arranged randomly in the system. Thus, the entire system was composed of 12302 atoms in total. Then, the entire system underwent energy minimization relaxation in the two steps procedure without any restraints. First 500 steps of steepest decent method followed by 2000 steps of conjugate gradient minimization method.
RESULTS
The crystal structure of NDM-1 has determined recently.[17] It is evident from the crystal structure analysis that the NDM-1 has an open active site with a unique electrostatic profile, which essentially provides broader substrate specificity. Moreover, the NDM-1 undergoes important conformational changes upon substrate binding.[23] The antibiotic binding domain consists of bivalent zinc metal ion as a cofactor. Zinc ion catalyses the hydrolysis of the lactam ring of the antibiotics and in turn inactivates. We performed the flexible docking of NDM-1 with selected 35 antibacterial natural compounds [Table 1 and Figure 1a] and 14 β-lactam-containing antibiotics [Table 2 and Figure 1b]. We have used two parameters, IC50 and binding energy to screen the best compound out of the 35-compound library.
As evident from Table 3 and Table 4 Nimbolide (Molecule 15), Margolone (Molecule 18), Marganone (Molecule 19), Isomargolonone (Molecule 20), Acetyl Aleuritolic acid (Molecule 27) and Harmane (Molecule 33) have lower IC50 value than the β-lactam antibiotics. We further screened all these molecules on the basis of binding energy, in which only Nimbolide and Isomargolonone qualify, showing significant binding in proximity of the active site [Table 3] [Figure 1c–f]. Since all the selected compounds were natural, we did not perform Lipinski's filter analysis.[24]
Table 3.
Binding energy of natural compounds with NDM-1 calculated using AutoDock-4.2.3
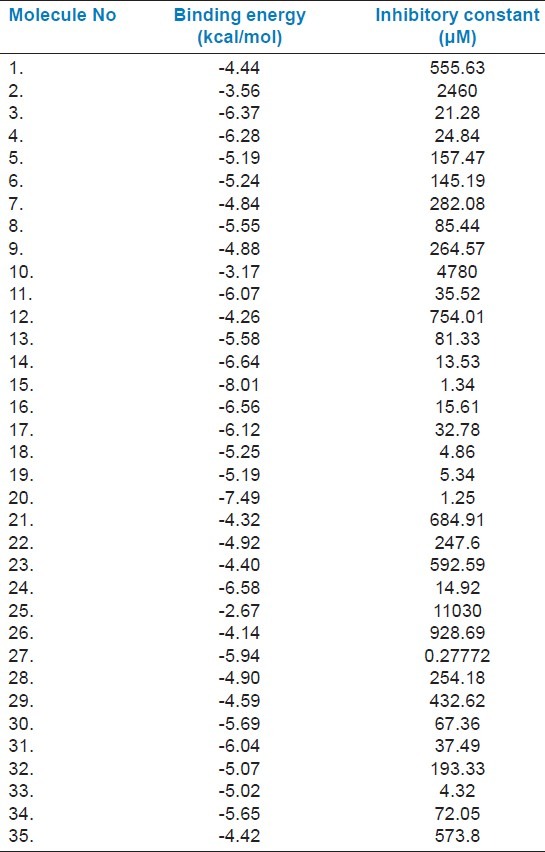
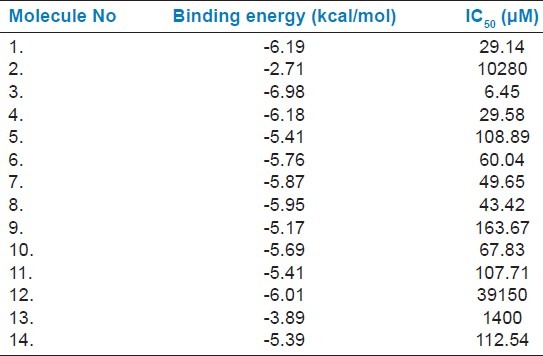
Table 4.
Binding energy of β-lactam antibiotics with NDM-1 calculated using AutoDock- 4.2.3
We further minimized the energy of both protein-ligand complexes. The energy minimized model of NDM-1-Nimbolide and NDM-1-Isomargolonone complexes showed Rmsd of 0.246 and 0.367 Ǻ2 respectively. The result of energy-minimized structure showed a significant number of hydrogen-bonded as well as van der Waals interactions formed between NDM-1 residues and Nimbolide or Isomargolonone [Figure 2]. The protein-ligand complexes are highly stable and complex formation occurs with high affinity.
Figure 2.
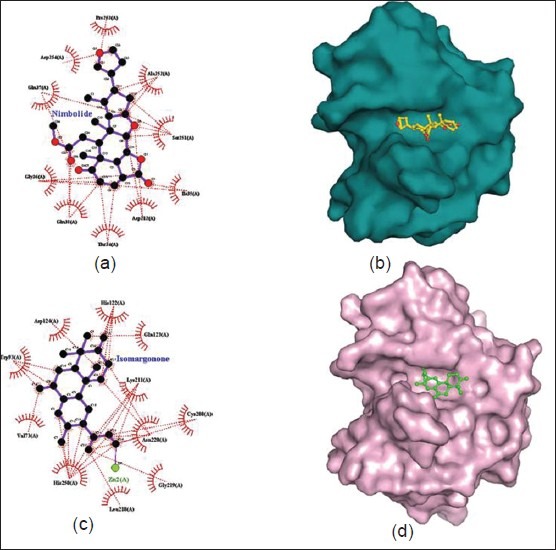
(a) Ligplot showing residues involved in interactions between NDM-1and Nimbolide (stick model). Interactions between protein and ligands are shown in dotted line. (b) Surface model of the docked NDM-1 with nimbolide where protein molecules are shown as surface (cyan) and nimbolide structure is shown in ball and stick (yellow). (c) Ligplot showing residues involved in interactions between NDM-1 and Isomargolonone (stick model). Interactions between protein and ligands are shown in dotted line. (d) Presence of Isomargolonone in the NDM-1 structure protein molecules are shown as surface (light pink) and Isomargolonone structure is shown in ball and stick (green)
Figure 2a and b show the mode of interaction of Nimbolide with the NDM-1 as evident from Ligplot[25] and surface diagram. Figure 2a clearly indicates that Nimbolide forms a large number of interactions with NDM-1 residues such as Pro263, Asp124, Gln37, Gly36, Gln38, Asp212, Ile35, Ser251 and Ala252. The Nimbolide was found in close proximity of the Zn1 of active site. Interestingly, interaction of Zn1 has been also observed with His120, His122 and His189 of active site (not shown in Figure). Therefore, the binding of Nimbolide to NDM-1 may affect the coordination of the Zn1 atom which further influences the catalytic activity of the enzyme. All these evidences suggest that Nimbolide may be a potent inhibitor for the NDM-1.
Figure 2c and 2d show the mode of interaction of Isomargololone with the NDM-1. We can observe in Figure 2c that Isomargololone forms a large number of interactions with NDM-1 residues such as His122, Asp124, Trp93, Val73, His250, Leu218, Gly219, Lys211, Cys208 and Gln123. Moreover, Isomargololone has shown ionic interaction with the Zn2 [Figure 2c] and affects the coordination of Zn1 with its binding residues. Furthermore, the surface diagram clearly indicates the presence of Isomargololone in the active site cavity of NDM-1 [Figure 2d]. A tight mode of binding observed between Isomargololone and NDM-1 suggests that the activity of the enzyme may be highly affected by this ligand.
DISCUSSION
In order to identify the novel classes of NDM-1 inhibitors by means of structure-based drug design, prediction of protein-ligand interaction is essential for virtual screening approaches.[26] This process requires docking tools to produce suitable conformations of a ligand within a protein-active site. Furthermore, a reliable energy evaluation can easily indicate the quality of a receptor-ligand putative complex and provide insights for biomedical science and drug development. Since the active site of NDM-1 may bind to different classes of the carbapenem family of antibiotics, it can be useful to consider the differences in binding affinity of the selected inhibitors and their natural substrate, that is carbapenem antibiotics. Therefore, we performed the docking analysis of 14 β-lactam antibiotics with NDM-1. We observed that the Piperacillin (antibiotic 3) had lowest binding energy (-6.98 Kcal/mol) and IC50 (6.45 μM) value as well.
Here our aim is to identify a potent inhibitor to block the lactamase activity of NDM-1. We selected 35 natural compounds from various plant sources and subsequently docked in the active site of NDM-1. Binding affinity calculation suggests that sorted ligands strongly interact to the NDM-1. A representative set of 14 antibiotics of the β-lactam family was selected from different sub-families like Penicillins (penams), Penems, Carbapenem, Cephalosporins, Monobactams, β-lactamase inhibitors and combinations as shown in Table 2. The docking analysis of β-lactam antibiotic with NDM-1 revealed that Piperacillin (Antibiotic 3) antibiotic have lowest binding energy of -6.9 Kcal/mol and IC50 value (6.45 μM). On the other hand, the docking analysis of the antibacterial natural compounds showed that Nimbolide (1.34 μM), Margolone (5.25 μM), Margolonone (5.34 μM), Isomargolonone (1.25 μM), Acetyl Aleuritolic acid (0.2772 μM) and Harmane (4.32 μM), had IC50 value lower then β-lactam antibiotics. Interestingly, Isomargololone (-7.49 kcal/mol) and Nimbolide (-8.01 kcal/mol) showed lesser binding energy than the β-lactam antibiotics. Moreover, their IC50 values are quite good to consider as drug molecule [Table 2]. Furthermore, both these compounds are devoid of β-lactam ring therefore, they can easily resist the hydrolase activity of NDM-1. All these results clearly indicate that Isomargololone and Nimbolide, and their derivative compounds may block NDM-1, however, further work is needed to optimize these compounds for specificity, efficacy and experimental validation.
CONCLUSIONS
Recent studies showed that NDM-1 plays an essential role in the bacterial resistance to antibiotics. Here we showed that Isomargololone and Nimbolide may be considered as potent and suitable inhibitors for the antibiotic resistance against NDM-1. These compounds showed an appreciable binding affinity in comparison to other natural compounds as well as their substrate antibiotics. All these observations revealed that Isomargololone and Nimbolide and their derivative compounds may block NDM-1 activities and provide a significant basis for drug development for therapeutic intervention in bacterial resistance to antibiotics. However, experimental validations are needed to consider these molecules as a suitable drug against the superbug.
ACKNOWLEDGMENT
MIH is thankful to the Indian Council of Medical Research for financial assistance.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Levy SB, Marshall B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat Med. 2004;10:S122–9. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 2.Tipper DJ. Mode of action of beta-lactam antibiotics. Pharmacol Ther. 1985;27:1–35. doi: 10.1016/0163-7258(85)90062-2. [DOI] [PubMed] [Google Scholar]
- 3.Waxman DJ, Strominger JL. Penicillin-binding proteins and the mechanism of action of beta-lactam antibiotics. Annu Rev Biochem. 1983;52:825–69. doi: 10.1146/annurev.bi.52.070183.004141. [DOI] [PubMed] [Google Scholar]
- 4.Bush K. New beta-lactamases in gram-negative bacteria: Diversity and impact on the selection of antimicrobial therapy. Clin Infect Dis. 2001;32:1085–9. doi: 10.1086/319610. [DOI] [PubMed] [Google Scholar]
- 5.Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to beta-lactam antibiotics: Compelling opportunism, compelling opportunity. Chem Rev. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 6.Jarlier V, Nicolas MH, Fournier G, Philippon A. Extended broad-spectrum beta-lactamases conferring transferable resistance to newer beta-lactam agents in Enterobacteriaceae: Hospital prevalence and susceptibility patterns. Rev Infect Dis. 1988;10:867–78. doi: 10.1093/clinids/10.4.867. [DOI] [PubMed] [Google Scholar]
- 7.Bush K, Jacoby GA. Updated functional classification of beta-lactamases. Antimicrob Agents Chemother. 2010;54:969–76. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bush K, Jacoby GA, Medeiros AA. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother. 1995;39:1211–33. doi: 10.1128/aac.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas PW, Zheng M, Wu S, Guo H, Liu D, Xu D, et al. Characterization of Purified New Delhi Metallo-beta-lactamase-1. Biochemistry. 2011;50:10102–13. doi: 10.1021/bi201449r. [DOI] [PubMed] [Google Scholar]
- 10.Gibb AP, McCallum AK. New Delhi metallo-beta-lactamase 1. Lancet Infect Dis. 2010;10:751–2. doi: 10.1016/S1473-3099(10)70243-7. [DOI] [PubMed] [Google Scholar]
- 11.Green VL, Verma A, Owens RJ, Phillips SE, Carr SB. Structure of New Delhi metallo-beta-lactamase 1 (NDM-1) Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011;67:1160–4. doi: 10.1107/S1744309111029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hammerum AM, Toleman MA, Hansen F, Kristensen B, Lester CH, Walsh TR, et al. Global spread of New Delhi metallo-beta-lactamase 1. Lancet Infect Dis. 2010;10:829–30. doi: 10.1016/S1473-3099(10)70276-0. [DOI] [PubMed] [Google Scholar]
- 13.Koh TH, Khoo CT, Wijaya L, Leong HN, Lo YL, Lim LC, et al. Global spread of New Delhi metallo-beta-lactamase 1. Lancet Infect Dis. 2010;10:828. doi: 10.1016/S1473-3099(10)70274-7. [DOI] [PubMed] [Google Scholar]
- 14.Chihara S, Okuzumi K, Yamamoto Y, Oikawa S, Hishinuma A. First case of New Delhi metallo-beta-lactamase 1-producing Escherichia coli infection in Japan. Clin Infect Dis. 2011;52:153–4. doi: 10.1093/cid/ciq054. [DOI] [PubMed] [Google Scholar]
- 15.Poirel L, Hombrouck-Alet C, Freneaux C, Bernabeu S, Nordmann P. Global spread of New Delhi metallo-beta-lactamase 1. Lancet Infect Dis. 2010;10:832. doi: 10.1016/S1473-3099(10)70279-6. [DOI] [PubMed] [Google Scholar]
- 16.Li Z, Wan H, Shi Y, Ouyang P. Personal experience with four kinds of chemical structure drawing software: Review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. J ChemInfComputSci. 2004;44:1886–90. doi: 10.1021/ci049794h. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Hao Q. Crystal structure of NDM-1 reveals a common beta-lactam hydrolysis mechanism. FASEB J. 2011;25:2574–82. doi: 10.1096/fj.11-184036. [DOI] [PubMed] [Google Scholar]
- 18.Lill MA, Danielson ML. Computer-aided drug design platform using PyMOL. J Comput Aided Mol Des. 2011;25:13–9. doi: 10.1007/s10822-010-9395-8. [DOI] [PubMed] [Google Scholar]
- 19.Goodsell DS, Olson AJ. Automated docking of substrates to proteins by simulated annealing. Proteins. 1990;8:195–202. doi: 10.1002/prot.340080302. [DOI] [PubMed] [Google Scholar]
- 20.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–62. [Google Scholar]
- 21.Mehler EL, Solmajer T. Electrostatic effects in proteins: Comparison of dielectric and charge models. Protein Eng. 1991;4:903–10. doi: 10.1093/protein/4.8.903. [DOI] [PubMed] [Google Scholar]
- 22.Pedretti A, Villa L, Vistoli G. VEGA: A versatile program to convert, handle and visualize molecular structure on Windows-based PCs. J Mol Graph Model. 2002;21:47–9. doi: 10.1016/s1093-3263(02)00123-7. [DOI] [PubMed] [Google Scholar]
- 23.King D, Strynadka N. Crystal structure of New Delhi metallo-beta-lactamase reveals molecular basis for antibiotic resistance. Protein Sci. 2011;20:1484–91. doi: 10.1002/pro.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–49. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 25.Laskowski RA, Swindells MB. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J ChemInf Model. 2012;51:2778–86. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 26.Tang YT, Marshall GR. Virtual screening for lead discovery. Methods Mol Biol. 2011;716:1–22. doi: 10.1007/978-1-61779-012-6_1. [DOI] [PubMed] [Google Scholar]