

Abstract
Aims
Female gender is a risk factor for long QT-related arrhythmias, but the underlying mechanisms remain uncertain. Here, we tested the hypothesis that gender-dependent function of the post-depolarization ‘late’ sodium current (INa-L) contributes.
Methods and results
Studies were conducted in mice in which the canonical cardiac sodium channel Scn5a locus was disrupted, and expression of human wild-type SCN5A cDNA substituted. Baseline QT intervals were similar in male and female mice, but exposure to the sodium channel opener anemone toxin ATX-II elicited polymorphic ventricular tachycardia in 0/9 males vs. 6/9 females. Ventricular INa-L and action potential durations were increased in myocytes isolated from female mice compared with those from males before and especially after treatment with ATX-II. Further, ATX-II elicited potentially arrhythmogenic early afterdepolarizations in myocytes from 0/5 male mice and 3/5 female mice.
Conclusion
These data identify variable late INa as a modulator of gender-dependent arrhythmia susceptibility.
Keywords: Mouse, Late sodium current, Gender, Arrhythmias
1. Introduction
Congenital long QT syndrome (LQTS) is characterized by QT interval prolongation and susceptibility to sudden cardiac death (SCD) due to a morphologically distinctive ventricular tachycardia (VT) termed torsades de pointes (TdP).1 Mutations in 13 genes encoding cardiac ion channels or accessory regulatory proteins have been reported to underlie congenital LQTS.2 In addition, QT prolongation and SCD due to TdP can occur in an ‘acquired’ form of the long QT syndrome, often due to drugs or heart block,3 and QT interval prolongation has been identified as a risk factor for SCD in the general population.4,5
Mutations in the canonical cardiac sodium channel gene SCN5A cause type 3 LQTS.6 Cardiac sodium channel currents ordinarily activate to initiate the action potential in atrium and ventricle, and then rapidly inactivate.7 However, in type 3 LQTS, channels fail to inactivate normally, resulting in persistent inward sodium current (INa),8 often termed a ‘gain of function’, during the action potential plateau. This increased ‘late’ INa (INa-L) in turn is postulated to generate prolonged action potentials and increased QT interval.9 In this form of LQTS, as in others, arrhythmogenic early afterdepolarizations (EADs) are thought to play a role in initiating TdP when action potentials prolong.10 A small INa-L has also been recognized in normal ventricular myocytes, and enhanced INa-L has been suggested as a mechanism underlying longer action potentials in mid-myocardial cells.11 More generally, INa-L is enhanced by oxidant stress,12,13 and block of this increase has been suggested as the major mechanism of action of the new antianginal ranolazine.14
Female gender is a risk factor for TdP in both congenital and acquired forms of LQTS,15–17 and a number of studies have implicated gender-dependent expression of cardiac potassium channels in this heightened arrhythmic sensitivity.18–22 In the present study, we demonstrate striking gender-dependent differences in INa-L. These differences translate directly to dysregulated action potential duration, EADs, QT interval prolongation, and polymorphic VT and thus for the first time implicate gender-dependent differences in INa-L as a risk factor for long QT-related arrhythmia.
2. Methods
2.1. Generating H/H mice
We have previously reported the successful implementation of the technique of recombinase-mediated cassette exchange (RMCE)23–25 to target exon 2 of the murine Scn5a locus.26 The targeting ablates expression of the mouse gene and allows substitution at the locus of full-length wild-type or mutated human SCN5A cDNAs. When we used this technique to generate mice homozygous for the wild-type human SCN5A at the murine locus, ECGs and ventricular INa were not different from those observed in unmodified mice.26
The first step in these RMCE experiments was homologous recombination in mouse 129/Sv ES cells to insert an acceptor cassette flanked by loxP/inverted loxP sites into the targeted site, Scn5a exon 2, and flanking intronic regions.26 This region was chosen because it includes the translation start site in exon 2 and previous studies had shown that exon 2 knockout eliminated Scn5a expression,27 indicating that the gene does not include other translation start sites. The second step was to generate exchange vectors encoding the desired insertion at the targeted site also flanked by loxP/inverted loxP sites: cDNAs for full-length wild-type (H). The exchange vector was then co-electroporated with a Cre recombinase vector into acceptor cassette-positive ES cells and cells were positively selected by gancyclovir and negatively selected by hygromycin as previously described.26 Clones were validated for the recombination event and strand orientation using previously described PCR strategies and then expanded for C57BL/6 blastocyst microinjections. Mice were then propagated by crossing male offspring from H, and previously described H injections with 129/Sv females. Backcrosses resulted in mice with the H/H genotype studied here.
All experiments involving animals conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 2011). Our animal protocol was approved by the Vanderbilt Institutional Animal Care and Use Committee. The protocol number is M/06/522.
2.2. Electrocardiograms and drug challenge
Electrocardiograms (ECGs) were recorded during inhaled administration of isoflurane vapour titrated to maintain light anaesthesia.28 Mice were anaesthetized initially under a concentration of 2% isoflurane and then held under a constant flow of roughly 1%, ventilated with O2. The heart rate was continuously monitored and isoflurane levels were adjusted to keep the heart rate between 350 and 450 bpm. Baseline ECG (leads I and II) was recorded for 5 min following which the animals were injected intraperitoneally with ATX-II (0.03 mg/kg) dissolved in water and measurements were acquired for 20min post-drug administration. ATX-II is a sea anemone toxin that interferes with sodium channel inactivation29 and that has been used to exaggerate the type 3 LQTS phenotype.30
Heart rate was measured as the average during a 30 s interval at baseline when a steady state was reached during anaesthesia. For measurement of all other ECG parameters, 30 s of data in each lead were signal averaged using a custom-built LabVIEW program (National Instruments, Austin, TX, USA) and the resultant waveform was analysed using an electric calliper by an investigator blind to the genotype.31 The larger value from each lead was used. QRS duration was measured from the first deflection of the Q-wave (or R-wave when the Q-wave was absent) and the end of the S-wave defined as the point of minimum voltage in the terminal phase of the QRS complex. QT interval was measured from the beginning of the QRS complex to the end of the T-wave defined as the point where the T-wave merges with the isoelectric line. Heart rate-corrected QT interval (QTc) was calculated using a formula developed for mice: QTc = QT/(RR/100)1/2.32
2.3. Isolation of mouse cardiac ventricular myocytes
Adult wild-type and H/H mouse ventricular myocytes were isolated by a modified collagenase/protease method.33 After intraperitoneal injection of 500 IU of heparin, adult mice were anaesthetized using inhaled 2% isoflurane/oxygen mixture, hearts excised, and their aortae rapidly cannulated and perfused with modified Tyrode's solution (MTS) for 3 min followed by MTS containing collagenase (Liberase Blendzyme-4, Roche, 0.04 mg/mL) for 5–7 min at a constant pressure of 80 mmHg and temperature of 34°C. The MTS contained (in mmol/L) NaCl 130, HEPES 10, glucose 10, KCl 5.4, MgCl2 1.2, NaH2PO4, 2,3-butanedione monoxime 10, pH of 7.2. The digested ventricles were minced in MTS containing 1 mg/mL bovine serum albumin and 0.2 mmol/L CaCl2 and triturated by gently pipetting. The resulting solution was strained and the myocytes allowed to sediment in MTS of increasingly higher Ca2+ concentrations (0.2, 0.5, and 1 mmol/L). This resulted in rod-shaped, Ca2+-tolerant myocytes that were used for the electrophysiology studies.
2.4. Peak and late sodium current recording
To allow recording of sodium current (INa) in mouse ventricular myocytes, external Na2+ concentration was lowered to 5 mM, wide-tip electrodes with tip resistance <1 MΩ were used, and experiments were conducted at 18°C. INa was recorded using whole-cell voltage clamp. The pipette-filling (intracellular) solution contained (in mmol/L): NaF 5, CsF 110, CsCl 20, EGTA 10, HEPES 10, with a pH of 7.4 adjusted with CsOH. The bath (extracellular) solution had (in mmol/L): NaCl 5, CsCl 110, TEA-Cl 5, CaCl2 0.1, MgCl2 1, HEPES 10, and glucose 10, with a pH of 7.4 adjusted by CsOH. To eliminate L- and T-type inward calcium currents, 1 µM nisoldipine, and 200 µM NiCl2 were added to the bath solution.
Data acquisition was carried out using an Axopatch 200B patch-clamp amplifier and pCLAMP version 9.2 software (MDS Inc., Mississauga, Ontario, Canada). Currents were filtered at 5 kHz (−3 dB, four-pole Bessel filter) and digitized using an analog-to-digital interface (DigiData 1322A, MDS Inc.). To minimize capacitive transients, capacitance and series resistance were corrected 70–85%. The holding potential was −120 mV for all experiments. INa densities mouse ventricular myocytes were compared with pA/pF after normalization to cell sizes, which were generated from the cell capacitance calculated by Membrane Test (OUT 0) in pClamp 9.2. Late current levels were measured in a time window of 3 ms (195–198 ms after the pulse) before the capacity transient at the end of the 200 ms pulse. Electrophysiological data were analysed using Clampfit version 9.2 (Axon Instruments), and the figures were prepared by using Origin 7.0 (OriginLab Corp., Northampton, MA, USA). Current–voltage relations for steady-state activation and inactivation were fit with the Boltzmann equation I/Imax = (1 + exp[(V - V1/2)/k])−1 to determine the membrane potentials for half-maximal activation (V1/2-activation) and inactivation (V1/2-inactivation).
2.5. Action potential recordings
Action potentials from isolated ventricular myocytes in adult male and female H/H mice were elicited with injection of brief stimulus current (1–2 nA, 2–6 ms, variable stimulation frequencies) in current clamp mode (Axopatch 200A amplifier, Molecular Devices, Sunnyvale, CA, USA). The bath (extracellular) solution contained (in mmol/L): NaCl 140, KCl 5.4 , CaCl2 1.8, and MgCl2 1, HEPES 5, glucose 10, with a pH of 7.4 (adjusted by NaOH). The pipette-filling (intracellular) solution had (in mmol/L): KCl 110, K2-ATP 5, MgCl2 1, BAPTA 0.1, HEPES 10, with a pH of 7.2 (adjusted by KOH). Microelectrodes of 3–5 mΩ were used. To measure action potential durations at 50% (APD50) and 90% (APD90) repolarization, 10 successive traces were averaged for analysis. Action potential recordings were obtained prior to and following exposure to 100 nM ATX-II. In another set of experiments, 10 µM ranolazine was used (Supplementary material online, Figure S2). Water-soluble ATX-II and ranolazine were purchased from Sigma-Aldrich Co. (St Louis, MO, USA).
2.6. Western blotting
Lysates were generated by pulverizing flash frozen sections of ventricular tissue followed by homogenization in a Dounce apparatus with 1xRIPA (150 mM NaCl, 50 mM Tris, pH 7.5, 1% NP-40 (IGEPAL), 0.5% Sodium deoxycholate, and 0.1% sodium dodecyl sulphate in 1x DPBS pH7.5) buffer. The homogenates were centrifuged at 10 000g for 5min at 4°C and the supernatant lysates were transferred to new chilled micro-centrifuge tubes. Lysate protein concentration was analysed using a bicinchoninic acid assay (Pierce Biochemicals) following the manufacturer's instructions. Twenty to 80 µg of lysate from each cardiac preparation was separated on NuPage 8% Tris-Acetate gels (Invitrogen). Separated proteins were transferred to 0.2 μm nitrocellulose membranes (Amersham Biosciences) and were blocked overnight in blocking buffer comprised of 0.05% Tween-20 Tris–buffered saline (TTBS) plus 5% non-fat dry milk at 4°C, and then were incubated with immunoglobulins against NaV1.5 (pAb 1:200, Alomone Labs) and calnexin (pAb 1:1000, Stressgen BioReagents) at room temperature for 2 h. Membranes were washed three times with TTBS for 10 min each and incubated with secondary anti-rabbit horseradish peroxidase-linked antibodies (Amersham Biosciences) in TTBS at room temperature for 1 h. Blots were then washed four times for 10 min each in TTBS. We visualized antibody interactions after transfer of light images to autoradiography film using the ECL system (Amersham Biosciences). Blot images were then scanned using a BioRad ImageOne processor and subjected to image densitometry using the ImageJ software (http://rsb.info.nih.gov/ij/) and averaging applications were done in Microsoft Excel.
2.7. Statistical analysis
Results are presented as mean ± standard error, and statistical comparisons were made using the unpaired or paired Student's t-test. A value of P< 0.05 was considered statistically significant.
3. Results
3.1. Electrocardiograms and drug challenge
Initial experiments examined the role of late current in generation of LQTS-related arrhythmias. We assessed the effect of a range of doses of ATX-II, which impairs inactivation of the channel on QT interval in mice.29 We found that at baseline, female mice exhibited normal ECG intervals that were not significantly different from that of males. However, exposure to ATX-II (0.03 mg/kg) elicited striking QT prolongation, changes in QT morphology, sustained ventricular arrhythmias, and death in female mice (Figure 1) while the same dose produced no effect on QT interval or heart rhythm in male animals. At this dose of ATX-II, 6/9 female animals showed polymorphic ventricular tachycardia similar to that shown in Figure 1, associated with long QT intervals that often approached the duration of the RR interval, and thus resemble human TdP (Figure 1B and C) All male mice (9/9) injected with this dose of ATX-II survived and no arrhythmias were observed.
Figure 1.

ECG tracings prior to and following ATX-II injection in one female mouse. (A) Baseline recording showing normal sinus rhythm and no discernable QT interval. (B) Following ATX-II injection, there is marked deformity of the T wave. (C) This is followed by development of polymorphic ventricular tachycardia.
We quantified the effect of ATX-II by measuring QT interval, T wave amplitude, and T wave area. At baseline, the end of the QT interval in mice is often difficult to time precisely, even with signal averaging. After 0.03 mg/kg ATX-II, QT intervals were deformed (as in Figure 1B) and readily measured: post-ATX-II QT was similar in male (79.2 ± 6.7 ms) and female mice (84.8 ± 5.0 ms). However, females exhibited dramatic differences in T wave amplitude (60.8 ± 32.3 vs. 250.7 ± 48.9 µV; Figure 2A) and area (5396 ± 2874 vs. 25529 ± 6259; Figure 2B). These differential responses to a potent and specific sodium channel opener immediately raised the hypothesis that the late sodium current plays a role in gender-specific development of TdP. An alternative explanation of the difference between male and female mice with respect to the arrhythmic potential of INa-L would be the relative expression of the primary cardiac sodium channel. However, we found no significant difference in a series of immunoblots (Supplementary material online, Figure S1).
Figure 2.

QT measurement in male and female mice after ATX-II injection. (A) T wave amplitude measurement is marked with a double-headed arrow. (B) Quantification of the T wave is performed by marking the T90 area and counting pixels within the region. †P < 0.05, *P < 0.01. ATX-II treatment produced much greater changes in both indices of repolarization in female compared with male animals.
3.2. Late current and action potential measurements
To test this hypothesis, we studied isolated ventricular myocytes from male and female mice before and after exposure to ATX-II. Peak sodium current was similar (in pA/pF, n = 10–12 each): 45.2 ± 2.6 (males) vs. 39.4 ± 2.7 (female, P > 0.05). However, there was a clear gender difference in the amplitude of non-inactivating (‘late’) current, which was approximately five-fold larger in females. After ATX-II, late current increased 3.8-fold (0.13 ± 0.01% in control vs. 0.32 ± 0.02% in ATX-II) in cells from male animals and 4.5-fold (0.52 ± 0.03% in control vs. 2.98 ± 0.44% in ATX-II) in cells from females (Figure 3C; Table 1). These differences translated into functional effects at the level of cardiac action potentials (Figure 4). At baseline, action potential durations measured at 50 and 90% repolarization were significantly longer in cells from female animals: at APD50, 9.3 ± 0.8 vs. 6.2 ± 0.5 ms (male) and at APD90, 35.2 ± 2.7 vs. 23.3 ± 1.8 ms (male); P < 0.01 (Figure 4A and B). However, with ATX-II, action potentials remained still markedly longer in the female animals: at APD50, 32.8 ± 2.7 vs. 20.0 ± 1.3 ms (male); at APD90, 116.4 ± 8.7 vs. 49.5 ± 3.8 ms (male); P < 0.01 (Figure 4C and D).
Figure 3.
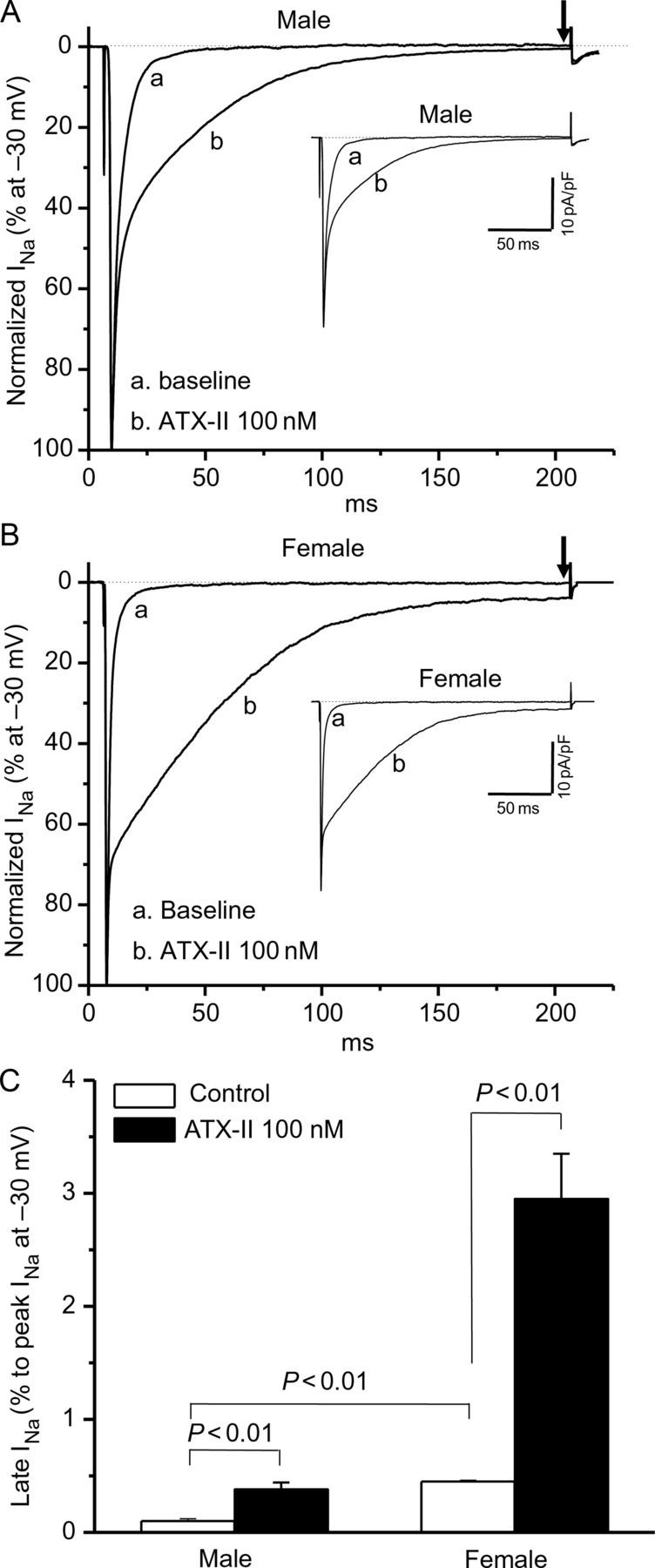
Late sodium current (INa-L) in male and female ventricular myocytes. Late sodium current in male (A) or female (B) myocytes with or without the addition of ATX-II. Female myocytes have a significantly larger late current at baseline that is further increased by ATX-II treatment. Late current was measured as a percentage of peak current before the ending of 200 ms pulsing after peak INa (indicated by arrows), while non-normalized raw current traces are shown in insets. Late current data (n = 6 each) are summarized in (C).
Table 1.
Gender and ATX-II-dependent effects on late sodium current (INa-L) in isolated ventricular myocytes from male and female mice
| N | INa-L at −30 mV (% vs. peak INa) | INa-L at −30 mV (pA/pF) | |
|---|---|---|---|
| Male | |||
| Control | 6 | 0.13 ± 0.01 | 0.24 ± 0.07 |
| ATX-II | 6 | 0.32 ± 0.02* | 0.71 ± 0.16* |
| Female | |||
| Control | 6 | 0.52 ± 0.03# | 1.05 ± 0.16# |
| ATX-II | 6 | 2.98 ± 0.44*§ | 5.60 ± 0.43*§ |
Mean ± SEM; *P < 0.01 vs. control in individual gender group; #P < 0.01 vs. control in male; §P < 0.01 vs. ATX-II in male. Cell sizes were not different between genders: 138.4 ± 3.6 pF (males) vs. 137.3 ± 3.1 pF (females, P > 0.05).
Figure 4.

Comparisons of ventricular action potentials (APs) in male and female mice with and without ATX-II at a stimulation frequency of 2 Hz. Differences in action potentials recorded from myocytes from male (M) or female (F) mice can be detected at baseline in (A) and after ATX-II treatment in (B). Measurement of APD50 (C) and APD90 (D) demonstrate increased action potential durations in female mice at baseline (*P < 0.01) and with ATX-II treatment (#P < 0.01).
In settings of prolonged and disordered repolarization, EADs are a major arrhythmia trigger. Since EADs are known to be bradycardia-dependent, we also assessed the effect of slow stimulation rate on action potentials. In this setting, ATX-II prolonged APD90 further in myocytes from male animals but no EADs were observed (0/5, Figure 5A and D). In contrast, EADs were observed in 3/5 cells from females (Figure 5B and D). This finding demonstrates a gender-dependent sensitivity to increased late current that potentiates arrhythmia generation.
Figure 5.

Comparisons of ventricular action potentials from male and female mice at slow stimulation frequencies in the presence of ATX-II. Action potentials were recorded from isolated cardiomyocytes in male (A) and (B) female mice. With slowed frequency (0.5 Hz), alterations in the trajectory of late repolarization (arrows) were not observed in cells from the male mice (0/5), but were recorded in cells from female mice (3/5). (C) At an even slower rate, 0.1 Hz, a more prominent discontinuity of late repolarization was seen in female myocytes (3/5). At slow stimulation rate, ATX-II-induced APD prolongations were greater in female mice, as summarized in (D) (*P < 0.01).
3.3. Ranolazine did not differentially affect the action potential durations in males and females
Another set of experiments were conducted to address the effects of the anti-anginal ranolazine on action potentials in females compared with males. In these experiments, we found that ranolazine slightly prolonged action potentials in both male and female ventricular myocytes. This effect may reflect the combined actions of late sodium channel block and potassium channel block by the drug. However, there was no statistically significant difference detected between males and females (Supplementary material online, Figure S2).
4. Discussion
Congenital or acquired long QT syndrome leads to a lengthening of action potential duration and increased risk of potentially fatal arrhythmias. In the course of examining the arrhythmogenic potential of late sodium current using ATX-II in mice, we uncovered striking gender-specific differences in drug sensitivity. No male mice exhibited arrhythmias or died, whereas 6/9 females injected with the same dose developed QT prolongation and fatal polymorphic ventricular tachycardia, the TdP syndrome. In humans and large animal models, a common mechanism for this syndrome is block of specific potassium current, IKr. However, IKr does not play a major role in repolarization in the mouse heart and TdP-like arrhythmias are correspondingly less common. In contrast, the major roles for cardiac sodium channel opening in atrium and ventricle—to initiate the action potential and to support fast conduction—is preserved from the mouse to human; indeed, Scn5a is >89% identical at the amino acid level to its human ortholog.34 Thus, while the mouse has some drawbacks as a model to study potassium channel-related TdP, our data demonstrate that disordered sodium channel physiology leading to TdP is readily replicated in mice.
Enhanced late INa has been implicated as a contributor to especially long action potentials in mid-myocardial cells11 and has been implicated as an arrhythmogenic current with oxidant stress12 and activation of Ca/calmodulin-kinase II (CaMKII).13,35 Type 3 LQTS resulting from ‘gain of function’ mutations in Nav1.5 and other settings in which sodium channels fail to undergo normal fast inactivation are arrhythmogenic in both animal models and simulation studies.36,37 Further, drugs that activate sodium current have been examined as potential inotropic agents and small clinical trials with one such compound, DPI 201-106, reported QT prolongation and TdP.38 There are thought to be two mechanisms by which this persistent current contributes to an arrhythmia-prone substrate. The first is the lengthening of action potential duration and formation of EADs as a result of disrupted repolarization reserve.39 A second proposed mechanism is alteration of calcium homeostasis leading to delayed afterdepolarizations and myocardial remodelling as a consequence of calcium overload.40 Our studies indicate that augmentation of the already larger late current in female mice results in significantly longer action potentials as well as the generation of EADs, particularly at slow frequency rates. Thus, these data represent the first evidence of a gender-dependent disruption in repolarization due to an increase in late sodium current.
The mechanism underlying the gender-specific differences in late INa will require further study. Reduced potassium channel subunit function or expression in females has been described in both animal studies18,19 and human expression profiling,21 and has been implicated in gender-dependent susceptibility to long QT-related arrhythmias. In addition, in guinea pig myocytes, gender-dependent increased L-type calcium current has been reported.41 In mice, experiments timed to the oestrous cycle and in ovariectomized females indicated that oestrogen prolongs QT by down-regulating Kv4.3 and Kv1.5 expression.42 Arguing against a role for hormonal differences in gender effects are studies suggesting that gender differences in QT or its response to drugs can be identified in prepubertal animals.43–45 Few reports have addressed gender-specific differences in INa. One study noted smaller peak INa in epicardium and endocardium in female vs. male dogs, but similar values in mid-myocardium; the authors speculated that as a result, there was increased dispersion of repolarization in female hearts although data on the late current or arrhythmias were not reported.46
Gender differences in late sodium current have not been explored in humans; however, reduced net outward current by disease or genetic variation is thought to reduce repolarization ‘reserve’ and thus predispose to TdP on further repolarization stress, e.g. by drug administration.47 The common cause for reduced net outward current is decreased potassium current, and the present data support the idea that reduced repolarization reserve can also reflect increased late sodium current, in an unexpectedly strong gender-specific fashion.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the National Heart Lung and Blood Institute (HL49989).
Acknowledgements
We gratefully acknowledge W. Zhang for excellent animal care and M. Ryan for cell isolation.
Conflict of interest: none declared.
References
- 1.Webster G, Berul CI. Congenital long-QT syndromes: a clinical and genetic update from infancy through adulthood. Trends Cardiovasc Med. 2008;18:216–224. doi: 10.1016/j.tcm.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackerman MJ, Mohler PJ. Defining a new paradigm for human arrhythmia syndromes: phenotypic manifestations of gene mutations in ion channel- and transporter-associated proteins. Circ Res. 2010;107:457–465. doi: 10.1161/CIRCRESAHA.110.224592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roden DM, Viswanathan PC. Genetics of acquired long QT syndrome. J Clin Invest. 2005;115:2025–2032. doi: 10.1172/JCI25539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz PJ, Wolf S. QT interval prolongation as predictor of sudden death in patients with myocardial infarction. Circulation. 1978;57:1074–1077. doi: 10.1161/01.cir.57.6.1074. [DOI] [PubMed] [Google Scholar]
- 5.Chugh SS, Reinier K, Singh T, Uy-Evanado A, Socoteanu C, Peters D, et al. Determinants of prolonged QT interval and their contribution to sudden death risk in coronary artery disease: the Oregon Sudden Unexpected Death Study. Circulation. 2009;119:663–670. doi: 10.1161/CIRCULATIONAHA.108.797035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 7.Abriel H. Roles and regulation of the cardiac sodium channel Nav 1.5: recent insights from experimental studies. Cardiovasc Res. 2007;76:381–389. doi: 10.1016/j.cardiores.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 8.Bennett PB, Yazawa K, Makita N, George AL., Jr Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 9.Ackerman MJ. The long QT syndrome: ion channel diseases of the heart. Mayo Clin Proc. 1998;73:250–269. doi: 10.4065/73.3.250. [DOI] [PubMed] [Google Scholar]
- 10.Wang DW, Yazawa K, Makita N, George AL, Jr, Bennett PB. Pharmacological targeting of long QT mutant sodium channels. J Clin Invest. 1997;99:1714–1720. doi: 10.1172/JCI119335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol Heart Circ Physiol. 2001;281:H689–H697. doi: 10.1152/ajpheart.2001.281.2.H689. [DOI] [PubMed] [Google Scholar]
- 12.Song Y, Shryock JC, Belardinelli L. A slowly inactivating sodium current contributes to spontaneous diastolic depolarization of atrial myocytes. Am J Physiol Heart Circ Physiol. 2009;297:H1254–H1262. doi: 10.1152/ajpheart.00444.2009. [DOI] [PubMed] [Google Scholar]
- 13.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, et al. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555–565. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaza A, Belardinelli L, Shryock JC. Pathophysiology and pharmacology of the cardiac ‘late sodium current. Pharmacol Ther. 2008;119:326–339. doi: 10.1016/j.pharmthera.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: findings from the International LQTS Registry. Circulation. 1998;97:2237–2244. doi: 10.1161/01.cir.97.22.2237. [DOI] [PubMed] [Google Scholar]
- 16.Makkar RR, Fromm BS, Steinman RT, Meissner MD, Lehmann MH. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA. 1993;270:2590–2597. doi: 10.1001/jama.270.21.2590. [DOI] [PubMed] [Google Scholar]
- 17.Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–1874. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 18.Drici MD, Burklow TR, Haridasse V, Glazer RI, Woosley RL. Sex hormones prolong the QT interval and downregulate potassium channel expression in the rabbit heart. Circulation. 1996;94:1471–1474. doi: 10.1161/01.cir.94.6.1471. [DOI] [PubMed] [Google Scholar]
- 19.Wu Y, Anderson ME. Reduced repolarization reserve in ventricular myocytes from female mice. Cardiovasc Res. 2002;53:763–769. doi: 10.1016/s0008-6363(01)00387-x. [DOI] [PubMed] [Google Scholar]
- 20.Yang PC, Clancy CE. Effects of sex hormones on cardiac repolarization. J Cardiovasc Pharmacol. 2010;56:123–129. doi: 10.1097/FJC.0b013e3181d6f7dd. [DOI] [PubMed] [Google Scholar]
- 21.Gaborit N, Varro A, Le BS, Szuts V, Escande D, Nattel S, et al. Gender-related differences in ion-channel and transporter subunit expression in non-diseased human hearts. J Mol Cell Cardiol. 2010;49:639–646. doi: 10.1016/j.yjmcc.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 22.Jonsson MK, Vos MA, Duker G, Demolombe S, van Veen TA. Gender disparity in cardiac electrophysiology: implications for cardiac safety pharmacology. Pharmacol Ther. 2010;127:9–18. doi: 10.1016/j.pharmthera.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Seibler J, Schubeler D, Fiering S, Groudine M, Bode J. DNA cassette exchange in ES cells mediated by Flp recombinase: an efficient strategy for repeated modification of tagged loci by marker-free constructs. Biochemistry. 1998;37:6229–6234. doi: 10.1021/bi980288t. [DOI] [PubMed] [Google Scholar]
- 24.Long Q, Shelton KD, Lindner J, Jones JR, Magnuson MA. Efficient DNA cassette exchange in mouse embryonic stem cells by staggered positive-negative selection. Genesis. 2004;39:256–262. doi: 10.1002/gene.20053. [DOI] [PubMed] [Google Scholar]
- 25.Jones JR, Shelton KD, Magnuson MA. Strategies for the use of site-specific recombinases in genome engineering. Methods Mol Med. 2005;103:245–257. doi: 10.1385/1-59259-780-7:245. [DOI] [PubMed] [Google Scholar]
- 26.Liu K, Hipkens S, Yang T, Abraham R, Zhang W, Chopra N, et al. Recombinase-mediated cassette exchange to rapidly and efficiently generate mice with human cardiac sodium channels. Genesis. 2006;44:556–564. doi: 10.1002/dvg.20247. [DOI] [PubMed] [Google Scholar]
- 27.Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci USA. 2002;99:6210–6215. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.el-Sherif N, Fozzard HA, Hanck DA. Dose-dependent modulation of the cardiac sodium channel by sea anemone toxin ATXII. Circ Res. 1992;70:285–301. doi: 10.1161/01.res.70.2.285. [DOI] [PubMed] [Google Scholar]
- 30.Wu L, Shryock JC, Song Y, Li Y, Antzelevitch C, Belardinelli L. Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J Pharmacol Exp Ther. 2004;310:599–605. doi: 10.1124/jpet.104.066100. [DOI] [PubMed] [Google Scholar]
- 31.Casimiro MC, Knollmann BC, Ebert SN, Vary JC, Jr, Greene AE, Franz MR, et al. Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange-Nielsen Syndrome. Proc Natl Acad Sci USA. 2001;98:2526–2531. doi: 10.1073/pnas.041398998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol. 1998;274:H747–H751. doi: 10.1152/ajpheart.1998.274.3.H747. [DOI] [PubMed] [Google Scholar]
- 33.Mitra R, Morad M. A uniform enzymatic method for dissociation of myocytes from hearts and stomachs of vertebrates. Am J Physiol. 1985;249:H1056–H1060. doi: 10.1152/ajpheart.1985.249.5.H1056. [DOI] [PubMed] [Google Scholar]
- 34.Zimmer T, Bollensdorff C, Haufe V, Birch-Hirschfeld E, Benndorf K. Mouse heart Na+ channels: primary structure and function of two isoforms and alternatively spliced variants. Am J Physiol Heart Circ Physiol. 2002;282:H1007–H1017. doi: 10.1152/ajpheart.00644.2001. [DOI] [PubMed] [Google Scholar]
- 35.Maier LS. CaMKII regulation of voltage-gated sodium channels and cell excitability. Heart Rhythm. 2011;8:474–477. doi: 10.1016/j.hrthm.2010.09.080. [DOI] [PubMed] [Google Scholar]
- 36.Charpentier F, Bourge A, Merot J. Mouse models of SCN5A-related cardiac arrhythmias. Prog Biophys Mol Biol. 2008;98:230–237. doi: 10.1016/j.pbiomolbio.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 37.Cardona K, Trenor B, Rajamani S, Romero L, Ferrero JM, Saiz J. Effects of late sodium current enhancement during LQT-related arrhythmias. A simulation study. Conf Proc IEEE Eng Med Biol Soc. 2010;2010:3237–3240. doi: 10.1109/IEMBS.2010.5627184. [DOI] [PubMed] [Google Scholar]
- 38.Kühlkamp V, Mewis C, Bosch R, Seipel L. Delayed sodium channel inactivation mimics long QT syndrome 3. J Cardiovasc Pharmacol. 2003;42:113–117. doi: 10.1097/00005344-200307000-00017. [DOI] [PubMed] [Google Scholar]
- 39.Varro A, Baczko I. Cardiac ventricular repolarization reserve: a principle for understanding drug-related proarrhythmic risk. Br J Pharmacol. 2011;164:14–36. doi: 10.1111/j.1476-5381.2011.01367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. 2006;92(Suppl. 4):iv1–iv5. doi: 10.1136/hrt.2005.078782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mason SA, MacLeod KT. Cardiac action potential duration and calcium regulation in males and females. Biochem Biophys Res Commun. 2009;388:565–570. doi: 10.1016/j.bbrc.2009.08.050. [DOI] [PubMed] [Google Scholar]
- 42.Saito T, Ciobotaru A, Bopassa JC, Toro L, Stefani E, Eghbali M. Estrogen contributes to gender differences in mouse ventricular repolarization. Circ Res. 2009;105:343–352. doi: 10.1161/CIRCRESAHA.108.190041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu T, Choi BR, Drici MD, Salama G. Sex modulates the arrhythmogenic substrate in prepubertal rabbit hearts with Long QT 2. J Cardiovasc Electrophysiol. 2005;16:516–524. doi: 10.1046/j.1540-8167.2005.40622.x. [DOI] [PubMed] [Google Scholar]
- 44.Sims C, Reisenweber S, Viswanathan PC, Choi BR, Walker WH, Salama G. Sex, age, and regional differences in L-type calcium current are important determinants of arrhythmia phenotype in rabbit hearts with drug-induced long QT type 2. Circ Res. 2008;102:e86–e100. doi: 10.1161/CIRCRESAHA.108.173740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hreiche R, Morissette P, Zakrzewski-Jakubiak H, Turgeon J. Gender-related differences in drug-induced prolongation of cardiac repolarization in prepubertal guinea pigs. J Cardiovasc Pharmacol Ther. 2009;14:28–37. doi: 10.1177/1074248408331018. [DOI] [PubMed] [Google Scholar]
- 46.Barajas-Martinez H, Haufe V, Chamberland C, Roy MJ, Fecteau MH, Cordeiro JM, et al. Larger dispersion of INa in female dog ventricle as a mechanism for gender-specific incidence of cardiac arrhythmias. Cardiovasc Res. 2009;81:82–89. doi: 10.1093/cvr/cvn255. [DOI] [PubMed] [Google Scholar]
- 47.Roden DM, Abraham RL. Refining repolarization reserve. Heart Rhythm. 2011;8:1756–1757. doi: 10.1016/j.hrthm.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]