

Summary
Progranulin (GRN) mutations cause frontotemporal dementia (FTD), but GRN’s function in the CNS remains largely unknown. To identify the pathways downstream of GRN, we used weighted gene co-expression network analysis (WGCNA) to develop a systems-level view of transcriptional alterations in a human neural progenitor model of GRN-deficiency. This highlighted key pathways such as apoptosis and ubiquitination in GRN deficient human neurons, while revealing an unexpected major role for the Wnt signaling pathway, which was confirmed by analysis of gene expression data from postmortem FTD brain. Furthermore, we observed that the Wnt receptor Fzd2 was one of only a few genes up-regulated at 6 weeks in a GRN knockout mouse, and that FZD2 reduction caused increased apoptosis, while its upregulation promoted neuronal survival in vitro. Together, these in vitro and in vivo data point to an adaptive role for altered Wnt signaling in GRN deficiency-mediated FTD, representing a potential therapeutic target.
Keywords: Progranulin, Frontotemporal Dementia, Wnt, Fzd2, WGCNA
Introduction
Frontotemporal dementia (FTD), the second most common cause of presenile dementia (Ratnavalli et al., 2002), is also highly heritable (Chow et al., 1999; Rohrer et al., 2009). Several classes of dominant causal mutations have been identified in the genes for MAPT, CHMP2B, and most recently, GRN (Baker et al., 2006; Cruts et al., 2006), which codes for the protein progranulin (GRN). The pathology of GRN+ FTD is characterized by ubiquitin positive TDP-43 inclusions and absence of tau pathology (Eriksen and Mackenzie, 2008; Josephs et al., 2007; Mackenzie et al., 2006; Neumann et al., 2006). GRN mutations are dominantly inherited and the disease mechanism is postulated to be haploinsufficiency (Ahmed et al., 2007; Cruts and Van Broeckhoven, 2008), as most GRN mutations lead to an approximately 50% reduction in GRN levels (Baker et al., 2006; Coppola et al., 2008; Cruts et al., 2006).
Unlike MAPT, GRN’s role in CNS function was previously not well-recognized prior to the identification of mutations in the GRN gene. GRN was not a candidate gene based on hypothesis driven work; it was found following exhaustive search by classic positional cloning following linkage (Baker et al., 2006; Cruts et al., 2006). GRN was first identified as a gene that was over-expressed in epithelial tumors and it was further found to be a player in wound healing and inflammation (Zhu et al., 2002). Progranulin and granulins have been known to function as growth factors, exerting opposing effects on cell growth and neurite outgrowth (Van Damme et al., 2008). But, the function of GRN in the CNS is poorly understood and it has only been sparsely studied (Neumann et al., 2009; Rohrer et al., 2009). Given its previously unknown role in neurodegenerative processes and disease, a broader understanding of its function in the nervous system would be of great value as a starting point for future therapeutic development.
Here we use a step-wise approach to gain a systems level view of the molecular consequences of GRN deficiency. Given the neuronal specificity of GRN deficiency, we developed an inducible in vitro model of GRN haploinsufficiency using shRNA in primary human neural progenitor cells (hNPCs) (Svendsen et al., 1998) and their differentiated progeny to faithfully model GRN deficiency. We next performed genome-wide transcriptome analysis to provide an unbiased and broad view of pathways directly downstream of GRN loss. By applying weighted gene co-expression network analysis (WGCNA), we visualized the network structure of acutely dysregulated genes downstream of GRN loss. We validate the in vitro results using expression data from postmortem FTD brain, identifying Wnt signaling as one of the major signaling pathways altered both during acute GRN loss in cell culture, and in human brain samples from patients with GRN mutations (GRN+). Functional analysis in human neural progenitors confirmed the predicted relationship between altered Wnt signaling and apoptosis observed in vitro. These data suggest that the pro-apoptotic effect of GRN knockdown may be mitigated by an alteration in Wnt signaling, which may represent a possible target for treatment of FTD.
Results
GRN Knockdown in Human Neural Progenitors and Progeny
The major effect of GRN deficiency in humans primarily involves the loss of neurons, despite nearly ubiquitous GRN expression in most cells and tissues (Daniel et al., 2000). We therefore developed an in vitro model of GRN deficiency using primary human neural stem cells in which shRNA was used to diminish GRN levels by at least 50%, as is observed in cells from patients with GRN mutations (Baker et al., 2006; Cruts et al., 2006; Finch et al., 2009; Ghidoni et al., 2008). We observed robust gene expression changes with GRN knockdown, including enrichment of gene ontology categories pertaining to the cell cycle and ubiquitination (Table S1). This was encouraging, given the presence of ubiquitinated inclusions including TDP-43 as a major pathological feature in patient brains (Liu-Yesucevitz et al., 2010; Neumann et al., 2006). However, we also observed that viral infection itself (Supplemental Methods) led to acute changes in GRN levels, consistent with its upregulation during the acute phase of inflammation (Guerra et al., 2007; He et al., 2003). So, to avoid the potential confound of the acute GRN changes associated with infection, we developed a tetracycline inducible system (Gossen and Bujard, 1992) that enables the study of GRN loss in NHNPs during proliferation, differentiation, and post-differentiation. To control for off-target effects, two hairpins against GRN were used, and a scrambled hairpin was used as a control.
hNPC expression of shRNA was verified by robust RFP expression (Fig. 1A). GRN knockdown was confirmed by western blotting (Fig. 1B), and at the RNA level via analysis of the GRN probes on the microarray (Fig. S1). The two GRN probes on the array demonstrated robust and statistically significant knockdown (60–74%, p<10E-6). Although the two hairpins had slightly different efficacy of knockdown, each resulted in GRN levels equivalent with GRN levels in patients, which typically range between a 50 and 75% reduction (Finch et al., 2009). Furthermore the cohort of genes differentially expressed with both hairpins was highly significantly overlapping (see below), providing further confidence in the robustness of the results.
Figure 1.

An in vitro model of GRN deficiency using shRNA in NHNP cells. A. Bright-field and fluorescent images of the NHNP cells before and after differentiation in the presence of doxycycline. hNPCs were infected, selected with puromycin for 2 weeks, then differentiated for 4 weeks. Expression of hairpin was verified by visualization of robust RFP expression. No leakage of the inducible promoter is visible. B. Confirmation of GRN knockdown by Western blot. Protein was extracted from the same samples on which the arrays were run, only one band was observed for GRN and for β-actin. C. Differential expression analysis was carried out and we observed hundreds of genes changing. Genes were determined to be changing only if they were differentially expressed in the overlap of both GRN hairpins versus control. Using the strict criterion of p<0.005 and computing this for both hairpins individually against control, the overlap contains 153 genes (hypergeometric probability = 10E-67). Depicted here is the heatmap of all samples for these 153 genes. Most genes (152/153) common to both hairpins are differentially expressed in the same direction relative to controls, further confirming that this list of genes is robustly related to GRN knockdown. See also figures S1 and S2 and table S1.
Given its role as a mitogen, we first explored GRN’s effects on NHNP cells in their proliferative state. We found that GRN has little effect on progenitors, as only 6 genes were differentially expressed between the targeting and scrambled hairpin conditions (Table S1, Bayesian t-test, p<0.05, log ratio>0.2). We next investigated cell number and proliferation, observing no change in cell number or in proliferation rate in the face of GRN knockdown (Fig. 2A). This is consistent with the absence of an obvious developmental phenotype in GRN knockout mice (Ahmed et al., 2010; Yin et al., 2010) and the phenotype of patients with GRN mutations, who suffer loss of post-mitotic neurons after several decades of life.
Figure 2.
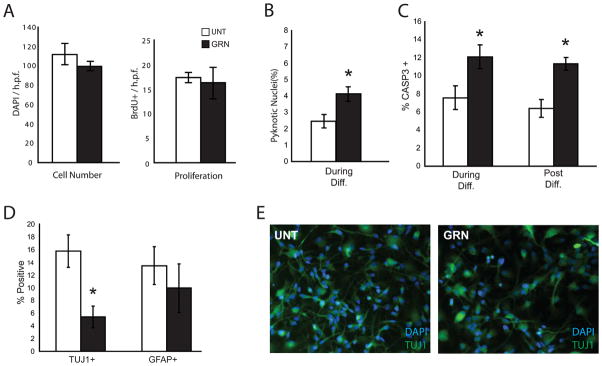
Neuronal cell death occurs in differentiating NHNP cells with GRN knockdown. A. GRNi cells demonstrate no difference in cell number or proliferation. B. GRNi cells demonstrate significantly increased pyknotic nuclei. C. There is a statistically significant increase in activated CASP3 staining in GRNi cells whether GRN is lost during differentiation (left) or post-differentiation (right).
D–E. There are significantly (P <0.05) fewer TUJ1+ cells but similar number of GFAP+ cells (P = 0.50) in cells lacking GRN. Cells were only considered to be TUJ1+ if they had a neuronal morphology. Asterisks indicate P ≤ 0.05 and error bars are ± s.e.m. (unpaired two-tailed Student’s t-test, n=3). See also figure S3.
As neurodegeneration primarily affects mature cells, we then studied the effects of GRN knockdown in non-dividing, differentiated hNPC cells. hNPCs were differentiated for four weeks in the presence of doxycycline, which induced shRNA expression. We confirmed cellular differentiation, first showing that nestin staining one month post-differentiation is lost (Fig. S2A–B). We then confirmed the upregulation of markers of early neuronal differentiation and maturation. Differential expression analysis showed clear upregulation of neuronal markers (Table S1) such as DCX (p<10E-12) and TUBB3 (also named Tuj1, p<1E-2), and also of glial markers such as GFAP (p<5E-3) at the RNA level and MAP2, TUJ1, and GFAP staining at the protein level (Fig. S2C–D). GO analysis provides a more global view of the changes, yielding obvious enrichment in DE categories such as neuron differentiation, axonogenesis and neural projection development, apoptosis, and regulation of cell size (Table S1, EASE Score, p < 10E-6), further confirming neuronal differentiation.
Given that the ultimate consequence of GRN insufficiency (GRNi) in humans is neuronal death, we determined the effect of GRN deficiency on neuronal survival. In GRNi neural progenitor cultures, we observed increased pyknotic nuclei (Fig. 2B) relative to the scrambled hairpin control condition, consistent with increased cell death. To confirm this observation, we assessed activated CASP3 staining, which confirmed the initial observations of increasing apoptosis with GRN reduction (Fig. 2C). To show that this apoptotic phenotype is not an artifact of progenitor proliferation or differentiation state, we also reduced GRN levels in differentiated cells via doxycycline application after differentiation of the progenitors over a course of three weeks, so that there were no remaining mitotically active cells. We saw a similar increase in CASP3 staining in this condition as well (Fig. 2C).
To determine whether GRN loss preferentially affects neurons or glia, we performed immunostaining, observing loss of Tuj1+ positive cells, but similar numbers of GFAP+ cells, indicating that sub-acute GRN loss leads to neuronal apoptosis (Fig. 2D–E). Additionally, when inducing GRNi post-differentiation, GRNi cultures demonstrated fewer, but not statistically significantly less MAP2 positive cells at a 6 week time point (Fig. S3), further confirming that the loss of neurons is not due to an effect on progenitor cell differentiation. Thus, GRN down-regulation in primary neuronal cells in vitro also leads to reduced neuronal survival, as it does in vivo.
Differential Gene Expression Analysis Highlights the Wnt Pathway
We next assessed gene expression changes associated with GRNi that might contribute to the apoptotic phenotype. We took the union of the changes observed in both GRN hairpins relative to the scrambled condition, so as to identify a conservative and highly robust group of 58 upregulated and 95 down-regulated genes (Bayesian t-test, p<0.005; Fig. 1C, Table S3). The intersection of differentially expressed genes between the two hairpins is highly significant (hypergeometric probability of 10E-67). Furthermore, virtually all (>99%) of those identified in these two independent experiments with different targeting hairpins were differentially expressed in the same direction relative to control, a high degree of internal consistency. These data provide evidence of robust alterations in gene expression specifically caused by GRN loss.
As a first step in organizing and categorizing the function of the differentially expressed genes with GRN loss, we conducted gene ontology (GO) analysis using David (http://david.abcc.ncifcrf.gov/) to determine enrichment of GO categories (Table S2). Several potentially important observations emerged (Fig. 3A), including cell death and apoptosis, consistent with the cell death observed by GRN loss in differentiating neural progenitor cells. To further confirm these gene ontology categories, we create a custom network via Ingenuity, in which the top network is significant for cell death (Fig. S4).
Figure 3.

A. Gene ontology analysis was carried out using DAVID on the set of differentially expressed genes. B. qRT-PCR was performed on some of the important players in the Wnt pathway to verify these gene expression changes. The left two bars show expression changes on array, and the right bar shows expression changes as measured by qPCR. qPCR was performed by comparing the second GRN hairpin versus the scrambled hairpin. C. KEGG Pathway for Wnt Signaling, with differentially expressed genes during GRNi (p<0.05) circled. Upregulated genes are colored red and downregulated genes are colored green. See also figures S4 and S5 and table S2.
Notably, within the GO analysis there was only one KEGG signaling pathway whose members were over-represented with GRN knockdown, the Wnt signaling pathway. Members of the Wnt signaling pathway with significant alterations in expression included: CD24, WNT1, SFRP1, NKD2 and the Wnt receptor FZD2. Other Wnt genes that were nominally significant include GSK3B, PPP2R2B, APC2, and CER1. To provide independent validation, gene expression changes of key Wnt signaling pathway members are additionally validated by qRT-PCR (Fig. 3B). These changes follow a clear pattern: genes that typically activate canonical Wnt signaling are upregulated (WNT1, FZD2, APC2), whereas genes that normally inhibit Wnt signaling are down-regulated (GSK3B, SFRP1, NKD2, CER1) (Fig. 3C). This indicates that an early consequence of GRN loss in vitro in human neural cells is an increase in Wnt signaling components that increases pathway activity. To test this prediction, we performed a direct experimental assay of Wnt activity in this model using the canonical LEF/TCF reporter (Methods). LEF/TCF signaling was increased in the GRN knockdown condition (Fig. S5), confirming that indeed Wnt signaling is altered. Moreover, non-canonical Wnt signaling pathways AP1, cJun, and NFAT assayed by the same reporter system (Methods; Fig. S5) show no significant changes, indicating that the alterations in Wnt signaling converge on the canonical pathway.
WGCNA highlights Cell Cycle, Ubiquitin, and Wnt Signaling Pathways
Although the GO analysis points to several potential key alterations in biological and molecular functions coupled with GRN deficiency, GO analysis is considered only a first step, since the function of many genes is not well-annotated. Recently, we have shown that WGCNA (Zhang and Horvath, 2005) provides a system level framework for the understanding of transcriptional profiles in many distinct cellular and tissue contexts (Geschwind and Konopka, 2009; Miller et al., 2008; Oldham et al., 2008; Winden et al., 2009; Voineagu et al., 2011). WGCNA has the power to reveal the underlying organization of the transcriptome of a system under study based on the degree of gene neighborhood sharing, which is defined based on co-expression relationships. This facilitates the identification of modules of functionally related, highly co-expressed genes, as well as the most central or hub genes that are of prime importance to module function (Geschwind and Konopka, 2009; Miller et al., 2008; Oldham et al., 2008; Winden et al., 2009). We condense the gene expression pattern within a module to a “module eigengene”(ME) which is a weighted summary of gene expression in the module (Oldham et al., 2008). Correlation between the ME and a given state, such as GRNi is indispensible as a metric to determine how a module may be relevant to that state.
We performed WGCNA (Methods and Supplemental Methods) identifying 24 modules, five of which (correlation > 0.50, p<0.05) were significantly correlated with GRN knockdown (Table S3). Two of these modules were of particular interest: the green module that contained GRN and the yellow module whose module eigengene was most correlated with GRN knockdown.
The green module contains 902 genes, and this module was then further divided into submodules (Supplemental Methods). We focused on the submodule containing GRN (Fig 4A–B, left). GO of this submodule, containing 167 genes, revealed that it is primarily comprised of genes related to mitochondrial function (Table S5, EASE Score p<0.001), the majority of which decrease with GRN loss. Mitochondria have been implicated as pivotal organelle in many neurodegenerative diseases, including AD and PD (Morais and De Strooper, 2010; Swerdlow, 2009). These data indicate that alteration in mitochondrial function is a primary effect of GRN deficiency in the CNS and support a role for mitochondrial dysfunction in GRN-related FTD as well.
Figure 4.

WGCNA Analysis of GRN deficiency in cell culture. WGCNA analysis (see Methods) was carried out on the cell-culture data, and we chose to focus on two modules of importance: one containing GRN, and on another that is most correlated with GRN knockdown samples. A. Module eigengenes (ME) are computed from the first principle component of the gene expression data to summarize the major trend in gene expression within each module. Samples are plotted along the x-axis where each bar represents one sample, and the value of the ME for each sample is displayed on the y-axis. These plots show that these two modules are specific to the samples containing GRN knockdown. Genes in the green module are generally upregulated with GRN loss, whereas genes in the yellow module are downregulated. B. The strongest connections based on topological overlap are plotted for ease of analysis. This allows for easy visualization of highly connected genes within each module. See also figure S6 and tables S3 and S4.
The key module that may represent a cellular response to GRN loss is the yellow module, which contains 517 genes and was most correlated with GRN deficiency. We observed that this module contained a submodule with even higher correlation to GRN deficiency, so we chose to analyze this submodule as it represents the group of genes with highest correlation to GRNi (Fig. 4A–B, right). The yellow submodule contains 241 genes, and more than 95% of these 241 genes increase with GRNi (Table S4). GO analysis of this module (Table S4) demonstrated that it was enriched in the categories of ubiquitin-mediated proteolysis (p<0.03), Wnt signaling (p<0.05), and apoptosis (p<0.02). Notable highly connected (“hub”) genes in this module include Wnt signaling genes, such as FZD2, but also upregulation of pro-apoptotic genes like CASP9 and MGRN1, the latter a ubiquitin ligase whose depletion has been shown to cause neurodegeneration (Chakrabarti and Hegde, 2009). Other Wnt signaling genes such as WNT1, CTNNBL1, and VANGL2 are also highly connected. Ubiquitin positive inclusions containing TDP-43 are a hallmark of GRN positive FTD (Neumann et al., 2006), and upregulated genes within this module that are related to this pathway include the ubiquitin conjugating enzymes, UBE2C and UBE2D3. To test for up-regulation of ubiquitination within this model we performed western blotting with an anti-polyubiquitin polyclonal antibody, demonstrating a significant increase in poly-ubiquitinated proteins with GRN knockdown (Fig. S6). The up-regulation of these pathways here further confirms an early increase in the ubiquitin protein stress pathways concomitant with GRN loss.
This analysis clearly connects GRN function to mitochondria and protein degradation pathways, and places multiple members of the Wnt signaling pathway in central roles downstream of GRN deficiency. These network data also bolster the suggestion, based on the initial analysis of differential expression, that Wnt signaling may be integral to GRN function and regulation, as both a supervised analysis using differential expression and an unsupervised analysis using WGCNA highlight Wnt signaling as a major pathway associated with GRN loss.
Gene expression related to postmortem human brain
Next, we sought to extend these in vitro observations to in vivo human data and provide independent validation of their relevance to human FTD. Although in vitro derived expression data have the power to demonstrate which expression changes observed in brain are direct effects of GRN loss, and not postmortem confounders (Mirnics and Pevsner, 2004), the optimal translational value lies in extending these observations to human patient material (Karsten et al., 2006). In this framework, first we determine causality in the absence of post-mortem confounders in vitro, and we then use the postmortem tissue to confirm the relevance of the in vitro findings to human FTD pathophysiology (Karsten et al., 2006).
We performed WGCNA on a dataset provided by Chen-Plotkin et al. (Chen-Plotkin et al., 2008) where microarrays were run on three brain regions (cerebellum, hippocampus, and frontal cortex) from three subject groups (controls, sporadic FTD, and GRN+ FTD). Following quality control to remove technical outliers (Methods; Oldham et al., 2006), 52 arrays remained. WGCNA identified 29 modules (Fig. 5A), 14 of which were related to brain region (e.g. cortex or cerebellum; Oldham et al., 2006), 8 were significantly correlated with GRN+ FTD (correlation>0.50, p<0.05, Table 1), and 2 of which were significantly correlated with sporadic FTD (Table S5, Methods). Lastly, some modules are driven by individuals, and may be related to factors such as cause of death or agonal state, as has been previously reported (Oldham et al., 2008). The eight modules whose ME is significantly correlated with GRN+ FTD show that there is a specific gene network associated with GRN+ disease state. Given the similarity in pathology of GRN+ and sporadic FTD, this is remarkable in showing that despite the chronic inflammation and microgliosis present in both forms of FTD, GRN loss produces a specific set of altered gene networks that is preserved even late in disease (Figs. S7, A–G). Of note, there is nearly total agreement between the ME correlations observed in two brain regions, hippocampus and frontal cortex, consistent with the notion that the ME is a robust measure, as has been previously demonstrated in several settings (Konopka et al., 2009; Oldham et al., 2008; Winden et al., 2009; Voineagu et al., 2011).
Figure 5.

WGCNA analysis on the postmortem brain dataset. A. Dendrogram showing relationship of topological overlap of genes and their relation to modules. B. First module eigengene is plotted of the blue module, a measure of the general expression of the genes in a given module. Green samples are controls, red samples are FTD with GRN mutations, and blue samples are sporadic FTD. The module containing GRN, the blue module, is positively correlated with diseased GRN+ samples. This provides further evidence that GRN+ FTD has a distinct molecular phenotype even in late-stage FTD as compared to sporadic FTD. C. A graphic depiction of the blue module using VisANT (http://visant.bu.edu). For ease of viewing, pairs of genes with the highest intramodular topological overlap are depicted, with each link corresponding to a TO value between the connected nodes. This module is notable because it contains GRN. Its hub genes are ANXA5 and LRP10, indicating that this module is likely related to apoptosis and Wnt signaling. See also figure S7 and tables S5 and S6.
Table 1. Table of modules specific to GRN+ FTD disease state.
Correlation of the first module eigengene with diseased state is listed here. This table characterizes the modules by gene ontology, listing the gene ontology category that is most characteristic of the module in question, and by top 3 connected genes by Kme. See also table S6.
| Module | Correlation | P-Value | Top three hub genes by Kme | Notable Gene ontology (P-value) |
|---|---|---|---|---|
| Blue | 0.66 | 7.95E-08 | PARP4, FOXO1, RIN2 | Wnt Signaling Pathway (<0.02), Apoptosis (<0.02), UbI Conjugation (<0.005) |
| Cyan | −0.53 | 4.66E-05 | DNM1L, SYNJ1, NBEA | Protein Kinase (<10e-4), Nucleotide Binding (<0.002) |
| darkolivegreen | −0.65 | 1.60E-07 | NAP1L3, AAK1, CAMTA1 | Synaptic Transmission (<10e-4), Calcium Ion Binding (<0.01) |
| Green | −0.56 | 1.51E-05 | BSCL2, ACOT7, GNG3 | Oxidative Phosphorylation (<10e-18), Mitochondria (<10e-15) |
| lightgreen | 0.58 | 7.86E-06 | PARP12, XAF1, SP100 | Antiviral Defense (<10e-17), Immun Response (<10e-9) |
| orange | 0.52 | 6.88E-05 | C1QA, HCLS1, VSIG4 | Response to Wounding (<10e-21), Immune Response (<10e-20) |
| steelblue | 0.67 | 7.31E-08 | FRMD4B, PIP4K2A, RASGRP3 | Myelination (<10e-7), Gliogenesis (<0.003) |
| tan | −0.60 | 2.60E-06 | NSF, GLRB, AAK1 | Nucleus (<3e-7), RNA Splicing (<3e-5) |
GO analysis of the GRN+ associated modules (Methods) revealed some pathways previously linked to neurodegeneration, such as those relating to inflammation, mitochondria, synaptic transmission, neural development, and cell loss. At the same time, other modules with significant correlation to GRN+ disease status revealed unexpected GO categories, such as myelination, gliogenesis, and protein kinase and nucleotide binding (Table 1). These data complement the data already provided in (Chen-Plotkin et al., 2008), providing a systems level framework in which to delineate the GRN+ FTD molecular signature identified using differential expression analysis in the original paper. WGCNA allows for separation of distinct factors that may be related to GRN+ FTD, and facilitates the focus on the gene expression changes most relevant to disease pathogenesis.
The GRN Co-expression Module Implicates Apoptosis and Wnt Signaling Alterations
To further explore the relationship of the genes identified in vitro with GRN downregulation in vivo, we analyzed the GRN containing module, the blue module. The blue ME is highly specific to GRN+ FTD affected brain regions (Fig. 5B), indicating that genes in this module are specifically upregulated in these brain areas. GO analysis identified Wnt signaling to be significantly enriched within this module (Table S6), including canonical Wnt pathway transcription factors LEF1, TCF7L1, and TCF7L2. To probe the genes most associated with chronic GRN deficiency in vivo, we again examined the submodule containing GRN within this larger module. Remarkably, this module is centered around two hub genes that are both upregulated in disease (Fig. 5C): Annexin-V (ANXA5), a known mediator of apoptosis (Vermes et al., 1995), and LRP10, a newly discovered inhibitor of the canonical Wnt signaling pathway (Jeong et al., 2010). This module also contains FZD2, which is up-regulated and negatively correlated with GRN levels in vivo, consistent with the in vitro data. Analysis of these human brain samples revealed that FZD2 is significantly up-regulated only in frontal cortex of GRN+ FTD samples, underscoring its potential role in disease pathogenesis.
Target Validation in Mouse
The up-regulation of multiple Wnt pathway activating components and down-regulation of negative regulators both in vitro and in vivo showed a remarkable degree of consistency. These data not only support the relevance of the Wnt pathway changes observed in cell-culture and in human FTD in vivo, but conversely indicate which of the changes observed in brain are a direct effect of GRN loss, and are not due to postmortem confounders, such as a change in cell composition (due to inflammation or cell loss) during the neurodegenerative process. We were particularly intrigued by the consistent upregulation of FZD2, since it is one of the most proximal pathway members, acting as a Wnt receptor (Chan et al., 1992; Slusarski et al., 1997). To follow Fzd2 in vivo at a time prior to neuropathological alterations or overt neurodegeneration, we analyzed independent gene expression data from cerebellum, cortex, and hippocampus of 6-week old GRN knockout mice, at a time point before overt cell loss or neuropathology. This analysis demonstrated only 25 differentially expressed genes in cortex (Table S7, p<0.005, log ratio > 0.2) and no significantly enriched KEGG pathways. A comparison to the human data showed that human GRNi stem cells have a greater number of genes in common with patient GRN+ brain than the knockout mice at this time point (Fig. 6A). But, of the few changes observed in mice, Fzd2 upregulation is one of the most consistently up-regulated targets at this early stage in all brain regions (Fig. 6B). Fzd2 is also the second most upregulated gene in cortex (Table S7) and it is upregulated in proportion to GRN loss; Grn −/− mice have twice the Fzd2 increase as the Grn +/− mice.
Figure 6.

A. Overlap of differential expression between NHNP cells, mouse, and FTD GRN+ patients, showing that there is greater overlap between NHNP cells and affected patient brain (p < 0.05, Bayesian t-test). B. Fzd2 is upregulated in proportion to Grn gene dosage in mouse brain. (n=5, unpaired Student’s t-test, p<0.05). Plots depict log fold change of Fzd2. C. Differential expression of FZD2 in GRN knockout mice at 6 weeks, 6 months, and 9 months (n=3–5, unpaired Student’s t-test, p<0.05). Plots depict fold change of Fzd2 in Grn −/− versus Grn +/+ mice. D, E. Differentiated NHNP cells in culture with FZD2 knockdown demonstrate a significantly larger percentage (n=3, error bars ± s.e.m., unpaired Student’s t-test, p<0.05) of dying cells (D) and lower total cell number (E). However, dual FZD2/GRN knockdown demonstrate similar numbers of apoptotic cells relative to FZD2 alone (D). F. Staining showing increased activated CASP3 staining (green) in FZD2 knockdown (right) as opposed to control (left). Numerous pyknotic nuclei are evident in FZD2 knockdown cells as well. See also figure S8 and tables S7 and S8.
In mouse, Fzd2 remains upregulated at 6 and 9 months of age (Fig. 6C). But far more gene expression changes are detected at these later ages, consistent with progression of the disease (Table S7, 1203 and 813 genes, respectively, Bayesian t-test p<0.05, log ratio > 0.2). At 9 months “lysosome” is the most statistically significant gene ontology category, which is notable because progranulin is endocytosed and delivered to lysosomes by sortilin (Hu et al., 2010). Additionally, at 6 and 9 months the Wnt signaling pathway becomes enriched in cortex of Grn −/− mice (Table S8, p<0.05). These mouse data confirm that Fzd2 is upregulated with GRN loss, but that it is one of the earliest features of GRN deficiency, confirming the human data that it is not an in vitro artifact, or the result of chronic degenerative or postmortem changes.
Experimental Validation of FZD2 In Vitro
Given the role of both the canonical and non-canonical Wnt signaling pathway in cell death and survival in many contexts from cancer to the nervous system, we set out to provide a proof of principle experimental test of our analyses. Since FZD2 is a Wnt receptor and its expression showed early and robust upregulation with GRN knockdown, we sought to manipulate FZD2 expression in vitro (Fig. S8), and to test one of two models based on our data. The first is that FZD2 upregulation reflects a protective or compensatory response, and the second that it is part of the neurodegenerative process. In the first case, knocking down FZD2 would be pro-apoptotic and in the second knocking down FZD2 would be protective. FZD2 deficient differentiating neuronal progenitors (Methods; Fig. 6D, F; Fig. S7) had fewer total cells (Fig. 6E) relative to the control condition and demonstrated an increase in activated CASP3 staining, indicating apoptotic cell death. Dual FZD2/GRN knockdown cells demonstrated similar numbers of apoptotic cells relative to FZD2 knockdown alone (Fig. 6D), suggesting that upregulation of FZD2 is directly downstream of GRN down-regulation, since there was no additive effect.
We next verified that upregulation of FZD2 increases cell survival in the context of GRN loss in post-differentiated, non-proliferating GRNi cells that subsequently overexpress FZD2. Upregulation of FZD2 in these GRNi cells increases total cell number (Fig. 7A), and decreases cell death (Fig. 7B). Overexpression of FZD2 does not cause an increase in proliferation as measured by PCNA immunoblotting (Fig. 7C) and by brdU immunocytochemistry (Fig. 7D), indicating that this is not via an effect on proliferation, but rather cell survival. The induction of cell death by FZD2 down-regulation suggests that the observed consistent upregulation of FZD2 in neurons in vitro and in vivo in patient brain reflects a potential neuroprotective response to GRN loss.
Figure 7.

A–B. GRNi cells were differentiated and after 4 weeks, and subsequently FZD2 was overexpressed for 4 weeks concurrently with GRN loss. The cells overexpressing demonstrate greater cell number (A), and fewer dying cells (B), suggesting overexpression of FZD2 is neuroprotective in the context of GRN loss (n=3, error bars ± s.e.m., unpaired two-tailed Student’s t-test, p<0.05). C–D. In normal NHNP cells, FZD2 overexpression does not cause a change in proliferation as measured by PCNA immunoblotting (C) or by brdU immunostaining (D).
Discussion
The present study was designed to identify the systems-level changes that accompany GRN reduction in human neurons. Initially, we examined the effects of progranulin deficiency in human fetal neural progenitors, a well-controlled in vitro model of GRN-haploinsufficiency. An important strength of this model is that it avoids potential confounding effects, such as chronic inflammation, that plague postmortem studies of dementia. Using this model, we demonstrate that progranulin deficiency selectively compromises neuronal survival and engages the canonical Wnt signaling pathway, the latter by way of both gene expression and a direct measure of signaling activity. The canonical Wnt signaling pathway is classically understood to increase cell survival, which is likely its role here in the context of GRN loss. Moreover, this association with Wnt is robustly preserved in the higher-order network architecture of transcriptional changes in the frontal cortex of FTD patients with progranulin mutations, and in Grn −/− mouse, proving its in vivo relevance. These data show that the combination of in vitro data, which can prove causality, and in vivo data, which confirms relevance to human disease, is a powerful approach.
Given the potential divergence of mouse and human transcriptional networks, this parallel between mouse and human systems was not a forgone conclusion (Miller et al., 2010), but provides another key line of evidence supporting the role of Wnt signaling and FZD2. Since these changes are evident well before the onset of observable inflammation, microgliosis, or overt neurodegeneration, these data suggest that FZD2 may prove useful as an early biomarker of disease progression. We show that loss of FZD2 is sufficient to cause cell death, but gain of FZD2 increases cell survival, suggesting that the upregulation of FZD2 is likely neuroprotective, consistent with its role as a growth factor. Together these data suggest that FZD2 plays a compensatory, potentially neuroprotective role in GRN deficient cells.
Dual Role of Wnt Signaling in Neurodegeneration
A general association exists between Wnt-Frizzled signaling and cell survival in many cellular systems (Chen et al., 2001; Rawal et al., 2009). A more specific link between aberrant Wnt signaling and FTD or Alzheimer’s Disease (AD) was first postulated because the canonical messenger GSK-3β is responsible for phosphorylation of tau, an initial step in the formation of neurofibrillary tangles (Behrens et al., 2009; Boonen et al., 2009; Jackson et al., 2002; Karsten et al., 2006). Subsequently, animal models of AD and FTD further implicated Wnt signaling in the etiology of neurodegeneration by showing (1) disease causing mutated presenilin alters Wnt signaling, increasing apoptosis (Soriano et al., 2001; Weihl et al., 1999), and (2) Aβ can both directly and indirectly interfere with canonical Wnt signaling and that this interference compromises neuronal survival (Caricasole et al., 2004; Scali et al., 2006). Wnt signaling therefore may provide a bridge between neurodevelopment and neurodegeneration (Geschwind et al., 2001).
Implications for Disease Pathophysiology
It is still an open question as to whether loss of GRN causes FTD pathology through a cell autonomous or non-cell autonomous mechanism. Loss of GRN may increase neuronal vulnerability, conferring an intrinsic property in which neurodegeneration is more likely. Alternatively, microglia lacking GRN may become hyperactive, creating a poor extrinsic environment that leads to neuronal death. This is a complex issue because GRN is a secreted protein, the form of GRN that is clinically relevant to FTD is currently unknown, and GRN transcripts are decreased in blood, but are paradoxically increased in GRN+ diseased brain (Chen-Plotkin et al., 2009).
The data that we present here, in which GRN loss is sufficient to produce cell death in the absence of microglia, shows that neuronal GRN deficiency is sufficient to significantly reduce neuronal survival, a finding important for potential therapeutics development. While these data argue that GRN loss can indeed increase neuronal vulnerability, they do not preclude the compounding involvement of microglia in the pathophysiology of FTD in patient brain, which may form a vicious cycle (Pickford et al., 2011; Yin et al., 2010).
FZD2 and Non-Canonical Wnt Signaling, and Clinical Significance
Here we provide data from multiple systems showing that FZD2 expression increases with GRN loss. We then performed a proof of principle experiment, indicating that this increase may be protective. Classically, signaling through the FZD2 receptor activates the non-canonical Wnt signaling pathway (Oishi et al., 2003). This suggests that modulation of this pathway may have therapeutic relevance; previous work has shown that non-canonical Wnt agonists can be protective in other forms of dementia (Inestrosa and Toledo, 2008). As opposed to canonical Wnt signaling, which has already been established as a major player in neurodegeneration (Hooper et al., 2008; Toledo et al., 2008), these data indicate a role for Wnt signaling independent of GSK-3β and β-catenin in neurodegenerative pathology. Additionally, FZD2 is the initial change that precedes alterations in other Wnt pathway members in mouse in vivo at stages well prior to neurodegeneration or neuronal loss. Thus, FZD2 represents a primary target in that it is a consistent and early upregulated gene in the context of GRN loss.
Neuroprotection via upregulation of FZD2 is consistent with the characteristically understood role of Wnt signaling as pro-growth and anti-apoptosis in multiple systems (Chen et al., 2001; Lie et al., 2005; Willert et al., 2003; You et al., 2002). Further, disease onset and rate of progression may correlate with endogenous baseline activity of neuroprotection provided by the Wnt signaling pathway (De Ferrari et al., 2007). The manipulation of FZD2 and other members of its pathway is thus a potential therapeutic target worthy of further investigation. Thus it is reasonable to consider that small molecule agonists of the non-canonical Wnt signaling pathway are potential new targets in GRN+ FTD. Since Wnt signaling is cell context and receptor dependent, gaining a full understanding of the other players in the pathway that may interact with FZD2 is crucial. This work provides the impetus for further in depth exploration of Wnt signaling in FTD and suggests the potential use of Wnt agonists to assuage the neurodegenerative phenotype of GRN+ FTD.
Experimental Procedures
Microarray Data and WGCNA Analysis
Human neural progenitor cells were infected, selected with puromycin, and differentiated for 4 weeks. Four replicates of each condition were analyzed as previously described on Illumina (San Diego, CA) human version 3 microarray chips (Coppola et al., 2009); see Supplemental Methods for details). Postmortem expression data from FTD patients and controls was obtained from (Chen-Plotkin et al., 2008) (GSE13162:GSM329660-GSM329715). Network analysis was performed using previously published methods (Oldham et al., 2006; Oldham et al., 2008; Winden et al., 2009). Gene ontology (GO) analysis was performed using the DAVID functional annotation tool (David (http://david.abcc.ncifcrf.gov/) (Huang da et al., 2009a, b), see Supplemental Methods for more details.
Plasmids
Initially, the pLCIR plasmid was used containing CAG promoter, chosen because of its robust expression in neurons, driving shRNA against GRN. The control was a GFP hairpin used previously (Matsuda and Cepko, 2007). To knock down GRN in an inducible system, pTRIPZ vector was purchased from Open Biosystems (Huntsville, AL). Their scrambled pTRIPZ vector was used as a control, and their TRIPZ GRN hairpin was used to knock down GRN. The sequence of the other GRN hairpin was obtained from (Zhang et al., 2007), and was cloned into TRIPZ using the PCR shagging protocol (http://katahdin.cshl.org:9331/RNAi/html/rnai.html). FZD2 knockdown was accomplished by purchasing FZD2 hairpins in vector pLKO (Open Biosystems), and pLUIP was used to overexpress FZD2, containing a U6 promoter driving expression of FZD2 with PuroR driven by an IRES. More detailed material on plasmids and sequences can be found in Supplemental Materials and Methods.
Cell lines and cell culture
Human neural progenitor cells (NHNPs) were generated from a 17-week gestation aborted female fetus. Tissue was homogenized, plated, and cultured as previously described (Svendsen et al., 1998; Wexler et al., 2008), and further elaborated in the Supplemental Methods.
Antibodies, Reagents, and Assays
The following antibodies were used for either immunoblotting (IB) or immunofluorescence (IF): anti-GRN (rabbit polyclonal, Invitrogen; 1:500 IB), anti-β-actin (mouse monoclonal, Sigma;1:50,000 IB), anti-HA (mouse monoclonal, Covance; 1:2,000 IB), anti-polyubiquitinated conjugates (mouse monoclonal, Biomol, 1:2000 IB), anti-PCNA (rabbit polyclonal, Santa Cruz Biotechnology, 1:200), anti-cleaved-caspase-3 (rabbit polyclonal, Cell Signaling; 1:1,000 IF), anti-GFAP (goat polyclonal, Abcam; 1:1,000 IF), anti-TUJ1 (rabbit polyclonal, Millipore, 1:1,000 IF), anti-BrdU (rat monoclonal, Abcam, 1:1,000 IF), goat anti-rabbit horseradish peroxidase (Cell Signaling; 1:2,000), goat anti-mouse horseradish peroxidase (Sigma; 1:5,000), goat anti-rat Alexa Fluor 488 (1:1,1000), donkey anti-rabbit Alexa Fluor 488 (1:1,1000), donkey anti-mouse Alexa Fluor 647 (1:1,000), donkey anti-rabbit Alexa Fluor 633 (1:1,000).
Immunocytochemistry(ICC): Cells were fixed using 4% paraformaldehyde (Sigma). Cells were permeabilized by incubating with 0.5% Triton-X for 10 minutes. Antigen retrieval for brdU staining was performed using 100 units/mL of DNase I (Worthington, Lakewood, NJ) supplemented with 1 mM MgCl2. Cells were blocked for 30 minutes in 3% donkey serum, 0.2% Triton-X, diluted in TBS. Primary antibodies were diluted in blocking buffer and were incubated overnight at 4°C. Secondary antibodies were incubated for 1 hour at room temperature in the dark. All cells were counterstained with DAPI (1:5000 diluted in TBS) and coverslips were mounted using ProLong Antifade (Invitrogen). 3 biological replicates were performed per experiment. 6 high-power fields were counted per coverslip, except in relation to FZD2 manipulation experiments, in which 4 high-power fields were counted per coverslip.
Wnt Activity Assays: Dual GFP/luciferease reporters were purchased from System Biosciences (Mountain View, CA) for TCF/LEF, AP1, cJun, and NFAT. Lentivirus was produced to infect proliferating NHNP cells already harboring targeting and non-targeting GRN hairpins, producing eight different transduced lines. 6 biological replicates of each line were induced for 1 week with doxycycline and compared to its uninduced counterpart. GFP fluorescence was assessed using a Plate Reader (BioTek) to determine relative activity for each reporter in each cell type.
Mice
Progranulin knockout mice were created as reported previously (Kao et al., 2011). The mice used for the microarray study were backcrossed into the C57BL/6 strain twice.
Supplementary Material
Acknowledgments
This work was supported by the Consortium for Frontotemporal Dementia(CFR). This work was supported by NIA 5R01AG026938(DHG), by NIH/NINDS Neurobehavioral Genetics Training Grant 5T32NS048004-05(EYR), the John Douglas French Alzheimer’s Foundation and NIMH K08MH74362 (EMW), and by NIH AG034793 (LHM). We are also grateful to Jeremy Miller for critical reading of this manuscript and we would like to thank Lauren Kawaguchi for her expertise as laboratory manager.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed Z, Mackenzie IR, Hutton ML, Dickson DW. Progranulin in frontotemporal lobar degeneration and neuroinflammation. J Neuroinflammation. 2007;4:7. doi: 10.1186/1742-2094-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, Yu X, Hou H, Chiba S, Yamanouchi K, et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol. 2010;177:311–324. doi: 10.2353/ajpath.2010.090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Behrens MI, Lendon C, Roe CM. A common biological mechanism in cancer and Alzheimer’s disease? Curr Alzheimer Res. 2009;6:196–204. doi: 10.2174/156720509788486608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonen RA, van Tijn P, Zivkovic D. Wnt signaling in Alzheimer’s disease: up or down, that is the question. Ageing Res Rev. 2009;8:71–82. doi: 10.1016/j.arr.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Caricasole A, Copani A, Caraci F, Aronica E, Rozemuller AJ, Caruso A, Storto M, Gaviraghi G, Terstappen GC, Nicoletti F. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer’s brain. J Neurosci. 2004;24:6021–6027. doi: 10.1523/JNEUROSCI.1381-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti O, Hegde RS. Functional depletion of mahogunin by cytosolically exposed prion protein contributes to neurodegeneration. Cell. 2009;137:1136–1147. doi: 10.1016/j.cell.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SD, Karpf DB, Fowlkes ME, Hooks M, Bradley MS, Vuong V, Bambino T, Liu MY, Arnaud CD, Strewler GJ, et al. Two homologs of the Drosophila polarity gene frizzled (fz) are widely expressed in mammalian tissues. J Biol Chem. 1992;267:25202–25207. [PubMed] [Google Scholar]
- Chen-Plotkin AS, Geser F, Plotkin JB, Clark CM, Kwong LK, Yuan W, Grossman M, Van Deerlin VM, Trojanowski JQ, Lee VM. Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Hum Mol Genet. 2008;17:1349–1362. doi: 10.1093/hmg/ddn023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Xiao J, Geser F, Martinez-Lage M, Grossman M, Unger T, Wood EM, Van Deerlin VM, Trojanowski JQ, Lee VM. Brain progranulin expression in GRN-associated frontotemporal lobar degeneration. Acta Neuropathol. 2009;119:111–122. doi: 10.1007/s00401-009-0576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J, Wang CY. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J Cell Biol. 2001;152:87–96. doi: 10.1083/jcb.152.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe SE, Boutros M, Michelson AM, Church GM, Halfon MS. Preferred analysis methods for Affymetrix GeneChips revealed by a wholly defined control dataset. Genome Biol. 2005;6:R16. doi: 10.1186/gb-2005-6-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia. Arch Neurol. 1999;56:817–822. doi: 10.1001/archneur.56.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola G, Karydas A, Rademakers R, Wang Q, Baker M, Hutton M, Miller BL, Geschwind DH. Gene Expression Study on Peripheral Blood Identifies Progranulin Mutations. Ann Neurol. 2008;64:92–6. doi: 10.1002/ana.21397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola G, Winden K, Konopka G, Gao F, Geschwind DH. Expression and network analysis of Illumina microarray data. Nature Protocols. 2009 doi: 10.1038/nprot.2009.215. [DOI] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Cruts M, Van Broeckhoven C. Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet. 2008;24:186–194. doi: 10.1016/j.tig.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Daniel R, He Z, Carmichael KP, Halper J, Bateman A. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 2000;48:999–1009. doi: 10.1177/002215540004800713. [DOI] [PubMed] [Google Scholar]
- De Ferrari GV, Papassotiropoulos A, Biechele T, Wavrant De-Vrieze F, Avila ME, Major MB, Myers A, Saez K, Henriquez JP, Zhao A, et al. Common genetic variation within the low-density lipoprotein receptor-related protein 6 and late-onset Alzheimer’s disease. Proc Natl Acad Sci U S A. 2007;104:9434–9439. doi: 10.1073/pnas.0603523104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen JL, Mackenzie IR. Progranulin: normal function and role in neurodegeneration. J Neurochem. 2008;104:287–297. doi: 10.1111/j.1471-4159.2007.04968.x. [DOI] [PubMed] [Google Scholar]
- Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, Bisceglio G, Rovelet-Lecrux A, Boeve B, Petersen RC, et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009;132:583–591. doi: 10.1093/brain/awn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Konopka G. Neuroscience in the era of functional genomics and systems biology. Nature. 2009;461:908–915. doi: 10.1038/nature08537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Robidoux J, Alarcon M, Miller BL, Wilhelmsen KC, Cummings JL, Nasreddine ZS. Dementia and neurodevelopmental predisposition: cognitive dysfunction in presymptomatic subjects precedes dementia by decades in frontotemporal dementia. Ann Neurol. 2001;50:741–746. doi: 10.1002/ana.10024. [DOI] [PubMed] [Google Scholar]
- Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology. 2008;71:1235–1239. doi: 10.1212/01.wnl.0000325058.10218.fc. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra RR, Kriazhev L, Hernandez-Blazquez FJ, Bateman A. Progranulin is a stress-response factor in fibroblasts subjected to hypoxia and acidosis. Growth Factors. 2007;25:280–285. doi: 10.1080/08977190701781222. [DOI] [PubMed] [Google Scholar]
- He Z, Ong CH, Halper J, Bateman A. Progranulin is a mediator of the wound response. Nat Med. 2003;9:225–229. doi: 10.1038/nm816. [DOI] [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vaegter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Toledo EM. The role of Wnt signaling in neuronal dysfunction in Alzheimer’s Disease. Mol Neurodegener. 2008;3:9. doi: 10.1186/1750-1326-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34:509–519. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- Jeong YH, Sekiya M, Hirata M, Ye M, Yamagishi A, Lee SM, Kang MJ, Hosoda A, Fukumura T, Kim DH, Saeki S. The low-density lipoprotein receptor-related protein 10 is a negative regulator of the canonical Wnt/beta-catenin signaling pathway. Biochem Biophys Res Commun. 2010;392:495–499. doi: 10.1016/j.bbrc.2010.01.049. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS, Petersen RC, Davies P, Duara R, Graff-Radford NR, et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol. 2007;66:142–151. doi: 10.1097/nen.0b013e31803020cf. [DOI] [PubMed] [Google Scholar]
- Kao AW, Eisenhut RJ, Martens LH, Nakamura A, Huang A, Bagley JA, Zhou P, de Luis A, Neukomm LJ, Cabello J, et al. A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc Natl Acad Sci U S A. 2011;108:4441–4446. doi: 10.1073/pnas.1100650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsten SL, Sang TK, Gehman LT, Chatterjee S, Liu J, Lawless GM, Sengupta S, Berry RW, Pomakian J, Oh HS, et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron. 2006;51:549–560. doi: 10.1016/j.neuron.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Konopka G, Bomar JM, Winden K, Coppola G, Jonsson ZO, Gao F, Peng S, Preuss TM, Wohlschlegel JA, Geschwind DH. Human-specific transcriptional regulation of CNS development genes by FOXP2. Nature. 2009;462:213–217. doi: 10.1038/nature08549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderwyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One. 2010;5:e13250. doi: 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, Gass J, Cannon A, Rademakers R, Hutton M, Feldman HH. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129:3081–3090. doi: 10.1093/brain/awl271. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A. 2007;104:1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Horvath S, Geschwind DH. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci U S A. 2010;107:12698–12703. doi: 10.1073/pnas.0914257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Oldham MC, Geschwind DH. A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. J Neurosci. 2008;28:1410–1420. doi: 10.1523/JNEUROSCI.4098-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirnics K, Pevsner J. Progress in the use of microarray technology to study the neurobiology of disease. Nat Neurosci. 2004;7:434–439. doi: 10.1038/nn1230. [DOI] [PubMed] [Google Scholar]
- Morais VA, De Strooper B. Mitochondria dysfunction and neurodegenerative disorders: cause or consequence. J Alzheimers Dis. 2010;20(Suppl 2):S255–263. doi: 10.3233/JAD-2010-100345. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Neumann M, Tolnay M, Mackenzie IR. The molecular basis of frontotemporal dementia. Expert Rev Mol Med. 2009;11:e23. doi: 10.1017/S1462399409001136. [DOI] [PubMed] [Google Scholar]
- Oishi I, Suzuki H, Onishi N, Takada R, Kani S, Ohkawara B, Koshida I, Suzuki K, Yamada G, Schwabe GC, et al. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells. 2003;8:645–654. doi: 10.1046/j.1365-2443.2003.00662.x. [DOI] [PubMed] [Google Scholar]
- Oldham MC, Horvath S, Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc Natl Acad Sci U S A. 2006;103:17973–17978. doi: 10.1073/pnas.0605938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickford F, Marcus J, Camargo LM, Xiao Q, Graham D, Mo JR, Burkhardt M, Kulkarni V, Crispino J, Hering H, Hutton M. Progranulin is a chemoattractant for microglia and stimulates their endocytic activity. Am J Pathol. 2011;178:284–295. doi: 10.1016/j.ajpath.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. doi: 10.1212/wnl.58.11.1615. [DOI] [PubMed] [Google Scholar]
- Rawal N, Corti O, Sacchetti P, Ardilla-Osorio H, Sehat B, Brice A, Arenas E. Parkin protects dopaminergic neurons from excessive Wnt/beta-catenin signaling. Biochem Biophys Res Commun. 2009;388:473–478. doi: 10.1016/j.bbrc.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, Isaacs AM, Authier A, Ferrari R, Fox NC, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73:1451–1456. doi: 10.1212/WNL.0b013e3181bf997a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scali C, Caraci F, Gianfriddo M, Diodato E, Roncarati R, Pollio G, Gaviraghi G, Copani A, Nicoletti F, Terstappen GC, Caricasole A. Inhibition of Wnt signaling, modulation of Tau phosphorylation and induction of neuronal cell death by DKK1. Neurobiol Dis. 2006;24:254–265. doi: 10.1016/j.nbd.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Slusarski DC, Yang-Snyder J, Busa WB, Moon RT. Modulation of embryonic intracellular Ca2+ signaling by Wnt-5A. Dev Biol. 1997;182:114–120. doi: 10.1006/dbio.1996.8463. [DOI] [PubMed] [Google Scholar]
- Soriano S, Kang DE, Fu M, Pestell R, Chevallier N, Zheng H, Koo EH. Presenilin 1 negatively regulates beta-catenin/T cell factor/lymphoid enhancer factor-1 signaling independently of beta-amyloid precursor protein and notch processing. J Cell Biol. 2001;152:785–794. doi: 10.1083/jcb.152.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen CN, ter Borg MG, Armstrong RJ, Rosser AE, Chandran S, Ostenfeld T, Caldwell MA. A new method for the rapid and long term growth of human neural precursor cells. J Neurosci Methods. 1998;85:141–152. doi: 10.1016/s0165-0270(98)00126-5. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH. The neurodegenerative mitochondriopathies. J Alzheimers Dis. 2009;17:737–751. doi: 10.3233/JAD-2009-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo EM, Colombres M, Inestrosa NC. Wnt signaling in neuroprotection and stem cell differentiation. Prog Neurobiol. 2008;86:281–296. doi: 10.1016/j.pneurobio.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Van Hoecke A, Lambrechts D, Vanacker P, Bogaert E, van Swieten J, Carmeliet P, Van Den Bosch L, Robberecht W. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–4. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl CC, Ghadge GD, Kennedy SG, Hay N, Miller RJ, Roos RP. Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J Neurosci. 1999;19:5360–5369. doi: 10.1523/JNEUROSCI.19-13-05360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler EM, Geschwind DH, Palmer TD. Lithium regulates adult hippocampal progenitor development through canonical Wnt pathway activation. Mol Psychiatry. 2008;13:285–292. doi: 10.1038/sj.mp.4002093. [DOI] [PubMed] [Google Scholar]
- Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, 3rd, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- Winden KD, Oldham MC, Mirnics K, Ebert PJ, Swan CH, Levitt P, Rubenstein JL, Horvath S, Geschwind DH. The organization of the transcriptional network in specific neuronal classes. Mol Syst Biol. 2009;5:291. doi: 10.1038/msb.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Dumont M, Banerjee R, Ma Y, Li H, Lin MT, Beal MF, Nathan C, Thomas B, Ding A. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. 2010;24:4639–4647. doi: 10.1096/fj.10-161471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Saims D, Chen S, Zhang Z, Guttridge DC, Guan KL, MacDougald OA, Brown AM, Evan G, Kitajewski J, Wang CY. Wnt signaling promotes oncogenic transformation by inhibiting c-Myc-induced apoptosis. J Cell Biol. 2002;157:429–440. doi: 10.1083/jcb.200201110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Dickey CA, Buratti E, Baralle F, Bailey R, Pickering-Brown S, Dickson D, Petrucelli L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27:10530–10534. doi: 10.1523/JNEUROSCI.3421-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, Ding A. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111:867–878. doi: 10.1016/s0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.