

Abstract
Chronic alcohol consumption leads to liver inflammation and cirrhosis. Alcoholic liver disease patients have increased levels of hepatic RANTES/CCL5. However, less is known about the molecular mechanisms for ethanol-induced RANTES up-regulation. In this study, we observed that liver sinusoidal endothelial cells derived from ethanol-fed rats (E-rLSECs) showed severalfold increases in RANTES and hypoxia-inducible factor 1α (HIF-1α) mRNAs compared with control rLSECs (C-rLSECs). Similar effects were seen in acute ethanol treatment of isolated rLSECs and human dermal microvascular endothelial cells. Ethanol-induced RANTES mRNA expression required ethanol metabolism, p38 MAPK, HIF-1α, and JNK-2, but not JNK-1. EMSA experiments showed increased HIF-1α binding to wild-type hypoxia response elements (HREs; −31 to −9 bp) within the RANTES promoter in response to ethanol. RANTES promoter analysis showed that cis elements proximal to the transcription start site, HRE-1 (nt −22 to −19), HRE-2 (nt −32 to −29), and AP-1 (nt −250 to −244) were required for ethanol-mediated RANTES expression. These results were corroborated by chromatin immunoprecipitation assays showing augmented HIF-1α binding to HRE-1. Additionally, promoter analysis revealed c-Jun, c-Jun/c-Fos, and JunD, but not JunB, bound to the AP-1 site of the RANTES promoter. Ethanol-mediated activation of NF-κB led to HIF-1α activation and concomitant RANTES expression. Plasma of ethanol-fed c-Junflox/flox-Mx-1-Cre mice showed attenuated levels of RANTES compared with ethanol-fed control mice, supporting the role of c-Jun in ethanol-induced RANTES expression. Our studies showed that ethanol-mediated RANTES/CCL5 expression occurs via HIF-1α activation independently of hypoxia. The identification of HIF-1α and AP-1 in ethanol-induced RANTES expression provides new strategies to ameliorate ethanol-induced inflammatory responses.
Long-term consumption of alcohol is a major etiologic factor in liver disease, culminating in liver failure and death (1). The mortality rate ascribed to alcoholic liver disease in the United States is estimated to be ~5–6% of the population, making this a serious public health problem. The earliest stage of liver injury is fatty liver (steatosis), progressing to liver inflammation (steatohepatitis), cirrhosis, and hepatocellular carcinoma (2–5). The role of Kupffer cells activated by gut-derived endotoxin has been demonstrated in alcohol-induced liver injury (6, 7). Among various cytokines, TNF-αproduced by activated Kupffer cells has emerged as one of the key factors in liver disease (5, 8). Studies show that plasma levels of cytokines, including TNF-α, are higher in patients with alcoholic liver disease (9–13). Ethanol-activated Kupffer cells generate an inflammatory response that results in the recruitment of inflammatory cells such as neutrophils, monocytes, and T and B lymphocytes into the liver (3). In patients with alcoholic cirrhotic livers there is increased accumulation of neutrophils (3) and T lymphocytes (14, 15) in portal tracts that contributes to hepatic necrosis and liver injury.
RANTES/CCL5 is a member of the superfamily of proinflammatory cytokines referred to as the CC chemokine family (16). The RANTES protein is a chemoattractant for T lymphocytes, monocytes/macrophages (17), and eosinophils (18) and is produced by T lymphocytes, monocytes, endothelial cells, and fibroblasts (12). Studies have shown increased levels of hepatic RANTES in patients with alcoholic hepatitis (19), suggesting that RANTES has a role in ethanol-induced liver injury. However, relatively little is known about the cellular signaling mechanisms for ethanol-mediated RANTES up-regulation. TNF-α, LPS, and IL-1β have all been shown to augment the expression of RANTES in T lymphocytes, pulmonary endothelial cells, and hepatocytes (9–13). TNF-α-induced RANTES transcription involves cis-regulatory promoter elements, i.e., NF-κB, C/EBPβ, NF-IL-6, NF-AT, and the cAMP response element, CRE (11, 13, 16). LPS-induced RANTES promoter activity is mediated by interactions between C/EBP proteins and Rel proteins (10).
In this report, we show that liver sinusoidal endothelial cells (LSECs)3 derived from ethanol-fed rats (E-rLSECs) exhibited a ~5-fold increase in RANTES/CCL5 mRNA expression and a 3-fold increase in hypoxia-inducible factor (HIF)-1αmRNA compared with control rat LSECs (C-rLSECs). Treatment of C-rLSECs with ethanol also increased RANTES mRNA expression in addition to that of HIF-1α; expression of the former was attenuated by silencing HIF-1αwith small interfering RNA (siRNA). Because the rLSEC does not maintain its phenotype in cell culture (20), we instead used a dermal human microvascular endothelial cell (HMEC) line, HMEC-1, as a model system; the latter cells are characterized for ease of culture, transfection efficiency, and the availability of human RANTES promoter constructs. Our studies showed that ethanol-mediated signaling in HMEC-1, leading to increased RANTES mRNA expression, involved the activation of p38 MAPK, JNK-2, and HIF-1α, but not PI3K or NADPH oxidase. Analysis of the 3.5-kb RANTES promoter revealed the presence of five hypoxia response elements (HREs), a single AP-1 site, and several putative NF-κB sites. Our studies showed that two of these HREs (−32 to −29 and −22 to −19 bp) and the AP-1 site (−250 to −244 bp) in the proximal promoter of RANTES were essential for ethanol-mediated RANTES expression. Among members of the AP-1 transcription complex, JunD but not JunB activated RANTES transcriptional activity following ethanol treatment. Moreover, ethanol-mediated activation of NF-κB led to crosstalk with the HRE binding site within the HIF-1αpromoter and to its activation. Plasma from ethanol-fed c-Junflox/flox-Mx-1-Cre mice showed attenuated levels of RANTES compared with ethanol-fed control mice, supporting the role of c-Jun in ethanol-induced RANTES expression. These studies, to our knowledge, show for the first time the involvement of HIF-1α, independent of hypoxia, in the regulation of RANTES/CCL5 expression.
Materials and Methods
Animal experiments
The Institutional Animal Care and Use Committee of the University of Southern California (USC; Los Angeles, CA) approved the use of animals for this study. Male Wistar rats were implanted with gastrostomy catheters for chronic alcohol administration as described previously (21). The rats were given a diet high in fat (35% calories as corn oil) infused with either an increasing dose of ethanol (9–16 g/kg/day) or isocaloric dextrose solutions (22), along with weekly enteral LPS (5 mg/kg) for 9 wk (Alcoholic Liver and Pancreatic Diseases Animal Core, USC) (23).
Because the c-Jun-null mutation is embryonic lethal, c-Jun floxed mice (c-Junflox/flox) (24) were originally obtained from Dr. B. D. Carter (Vander-bilt University Medical School, Nashville, TN). To knock out c-Jun in sinusoidal endothelial cells and immune cells, heterozygous c-Junflox/wt mice (where wt is wild type) were bred to Mx-1-Cre mice to produce c-Junfl/wt-Mx-1-Cre(+) mice, and these were bred to c-Junflox/flox mice to generate c-Junflox/flox-Mx-1-Cre(+) mice. Mice were bred to homozygosity based on PCR analysis of tail DNA. Polyinosinic:polycytidylic acid (poly(I:C)) was injected to knock out c-Jun. For c-Junflox/flox-Mx-1-Cre mice, Cre expression in the liver was induced by i.p. injection of poly(I:C) (GE Healthcare). Three-hundred microliters of poly(I:C) solution (1 mg/ml in PBS) was injected three times at 48-h intervals. These mice were used for ethanol challenge studies in which they were pair-fed with a Lieber-DeCarli control liquid diet or a liquid ethanol (36% of total calories) diet (25). Both ethanol and control diets also contained 18% calories from protein and 8% from fat, and part of the carbohydrates were isocalorically substituted with ethanol (26). Plasma samples were obtained from wt mice and c-Junflox/flox mice fed ethanol or control diet for analysis of RANTES protein levels by ELISA (R&D Systems).
Isolation of LSECs
C-rLSECs and E-rLSECs were isolated as previously described (27). Briefly, livers were perfused with 0.05% type 1a collagenase, pressed through polypropylene mesh, and centrifuged. rLSECs were isolated from the digest by density gradient centrifugation and centrifugal elutriation as described (27, 28). The rLSECs were positively stained for acetylated low density lipoproteins and exhibited >95% purity. The absence of contaminating Kupffer cells was assessed by peroxidase staining. The viability of rLSECs was >95% as ascertained by trypan blue. The freshly isolated rLSECs displayed fenestrae and sieve plates (27) but lost this phenotypic characteristic and begin to express PECAM-1 (CD31) in culture (20). Thus, we used the cells within 48 h of isolation.
Endothelial cell culture
rLSECs were cultured in DMEM supplemented with 10% FBS for 2 days and exhibited cobblestone morphology, which is characteristic of endothelial cells. Originally developed by Dr. E. Ades and F. J. Candall of the Centers for Disease Control and Prevention (CDC; Atlanta, GA) and Dr. T. Lawley of Emory University (Atlanta, GA), the immortalized dermal HMEC-1 cell line was obtained from the CDC. HMEC-1 was cultured in RPMI 1640 supplemented with 10% FBS, 5 mM HEPES, 1 mM sodium pyruvate, 1 mM glutamine, 50 μg/ml endothelial cell mitogen (Biomedical Technologies), MEM vitamins and nonessential amino acids (1×), and heparin (20 U/ml). Unless otherwise indicated, HMEC-1 was cultured under serum-free conditions overnight, before treatment.
Reagents
Diphenyleneiodonium chloride (DPI), GF109203X, and 4-methylpyrazole hydrochloride were obtained from Sigma-Aldrich. LY294002, SP600125, SB203580, PD98059, and sulfasalazine were obtained from Tocris. R59949 was purchased from Calbiochem. Protein phosphatase 1 (PP1) was obtained from Biomol. The following pharmacological inhibitors were used at the indicated concentrations as deemed optimal from the literature: DPI (10 μM); GF109203X (25 μM); 4-methylpyrazole hydrochloride (1 mM); LY294002 (15 μM); SP600125 (100 nM); SB203580 (1 μM); PD98059 (10 μM); sulfasalazine (2 μM); R59949 (30 μM); and PP1 (10 μM). Primary Abs for phospho-JNK-2 and JNK, and secondary Abs conjugated to HRP, were purchased from Santa Cruz Biotechnology. Unless specified otherwise, all other reagents were purchased from Sigma-Aldrich. HIF-1αsiRNA, HIF-1αscrambled RNA (scRNA), prolylhydroxylase (PHD)-2 siRNA, and PHD-2 scRNA were synthesized at the Microchem-ical Core Facility of the USC Comprehensive Cancer Center as previously described (29). p47phox siRNA, p47 scRNA, p38 siRNA, JNK-1 siRNA, JNK-2 siRNA, and control siRNA were obtained from Santa Cruz Bio-technology. The dominant-negative PI3K and JNK-2 expression plasmids were generously provided by Dr. D. Johnson (USC). The AP-1 luciferase construct was graciously provided by Dr. K. Machida (USC). The wt RANTES luciferase construct (−975 bp) (30) was generously provided by Dr. R. N. Taylor (Emory University).
RNA isolation and quantitative RT-PCR (qRT-PCR)
LSECs were isolated from the livers of control and ethanol-fed rats, and total RNA was extracted using TRIzol reagent (Invitrogen). Cultured C-rLSECs were treated with ethanol (100 mM) for the indicated time period followed by RNA extraction. HMEC-1 cells were stimulated with ethanol in the presence or absence of the specified pharmacological inhibitors followed by RNA extraction. mRNA expression was determined and quantified using the specific mRNA primers outlined in Table I. Using the iScript One-Step RT-PCR kit with SYBR Green (Bio-Rad), real-time qRT-PCR of mRNA templates (100 ng) was performed using the Applied Bio-systems ABI Prism 7900HT version 2.3 sequence detection system under the following conditions: cDNA synthesis at 50°C for 10 min, iScript reverse transcriptase inactivation at 95°C for 5 min, and PCR for 40 cycles entailing 95°C for 10 s, followed by annealing at 60°C for 30 s and detection. Values are expressed as the relative expression of mRNA normalized to the housekeeping GAPDH mRNA.
Table I.
Oligonucleotide primers used in this studya
| Organism/Gene | Method | Forward Sequence (5′→3′) | Reverse Sequence (5′→3′) |
|---|---|---|---|
| R, GAPDH | PCR | TTCAATGGCACAGTCAAGGC | TCACCCCATTTGATGTTAGCG |
| R, HIF-1α | PCR | CGAGCTGCCTCTTCGACAAG | CCCAGCCGCTGGAGCTA |
| R, RANTES | PCR | TGCCCACGTGAAGGAGTATTT | TTCTTCTCTGGGTTGGCACAC |
| H, GAPDH | PCR | AACCTGCCAAGTACGATGACATC | GTAGCCCAGGATGCCCTTGA |
| H, HIF-1α | PCR | CTCAAAGTCGGACAGCCTCA | CCCTGCAGTAGGTTTCTGCT |
| H, RANTES | PCR | TGCCCACGTGAAGGAGTATTT | TTCTTCTCTGGGTTGGCACAC |
| H, RANTES-HRE-M3 | SDM | CTCAAAGACATTAAGATCTTTTCCCAAAGGTCGCTTAGCAAG | CTTGCTAAGCGACCTTTGGGAAAAGATCTTAATGTCTTTGAG |
| H, RANTES-HRE-M2 | SDM | CCTGCAGAGGATCAAGACAGCGTCTGGACCTCGC | GCGAGGTCCAGACGCTGTCTTGATCCTCTGCAGG |
| H, RANTES-HRE-M1 | SDM | GCACGTGGACCTCGATCAGCCTCTCCCACA | CATGGTACCTGTGGGAGAGGCTGATCGAGGTCC |
| H, RANTES-AP-1-M | SDM | GTGCTTGGTCAAAGAGGAAACTGATACAGCCACTCTAGATGAGAGAGCAGTG | CACTGCTCTCTCATCTAGAGTGGCTGTATCAGTTTCCTCTTTGACCAAGCAC |
| H, RANTES wt HRE1 consensus oligonucleotides | EMSA | GTGGACCTCGCACAGCCTCTCC | GGAGAGGCTGTGCGAGGTCCAC |
| H, RANTES mutant HRE1 consensus oligonucleotides | EMSA | GTGGACCTCGATCAGCCTCTCC | GGAGAGGCTGATCGAGGTCCAC |
| H, RANTES wt AP1 consensus oligonucleotides | EMSA | GAAACTGATGAGCTCACTCTAG | CTAGAGTGAGCTCATCAGTTTC |
| H, RANTES mutant AP1 consensus oligonucleotides | EMSA | GAAACTGATACAGCCACTCTAG | CTAGAGTGGCTGTATCAGTTTC |
| H, RANTES | ChIP | AAAGAGGAAACTGATGAGCT | GTACCTGTGGGAGAGGCT |
H, Human; R, rat; SDM, site-directed mutagenesis. Site-specific mutations are depicted in bold.
Preparation of cytosolic and nuclear extracts
Cytosolic and nuclear fractions of HMEC-1 cells were prepared according to the modified procedure of Dignam et al. (31) as described previously (32). In brief, 5 ×106 cells were washed with cold PBS, lysed in cell lysis buffer followed by centrifugation for 1 min at 10,000 ×g (33). The supernatant (cytosolic extract) was collected, the pellet was resuspended in nuclear extraction buffer for lysis, and the nuclear extracts were obtained by centrifugation at 10,000 × g (33).
Western blot analysis
HMEC-1 (5 × 106 cells) in serum-free medium was treated with ethanol (100 mM) for the indicated time periods. Nuclear extracts were prepared, subjected to electrophoresis, and transferred to nitrocellulose membranes. Membranes were probed with Abs to HIF-1α and phosphorylated JNK-2 (1/20). Immunoblots were stripped and reprobed with either a β-actin or an unphosphorylated JNK (1/250) Ab as indicated. The protein bands were detected using Immunobilon Western reagents (Millipore).
Transient transfection of HMEC-1 cells with promoter constructs and siRNA
Cultured HMEC-1 (1 ×106 cells) were grown to 80% confluence in medium supplemented with 5% FBS. The cells were resuspended in 100 μl of RPMI 1640 (1×) with 1 ×g of expression plasmid and 0.5 μg of β-galac-tosidase reporter plasmid or siRNA (50 nM) (34) for nucleofection using an Amaxa nucleofection device (Lonza) as described previously (34). Cells were transferred to 6-well plates in complete medium for growth overnight, followed by 3 h of incubation in serum free medium before treatment with ethanol for the indicated time period. After treatment, cells were rinsed once with PBS. Cells were either lysed using reporter lysis buffer (Pro-mega) followed by centrifugation for 5 min at 10,000 μg or extracted with TRIzol for RNA. The supernatants from the lysed cells were collected and assayed for luciferase activity using a luminometer (Lumat LB 9501; Berthold), and β-galactosidase activity was measured using a Promega kit. For transfection efficiency, values of firefly luciferase were normalized to β-galactosidase values. Data are expressed relative to the promoter-less pGL3 basic vector.
EMSA for RANTES
The double-stranded oligonucleotides for RANTES containing its proximal AP-1 site corresponding to nt −258 to −237 of the promoter region, RANTES with a mutation in the same AP-1 site, RANTES containing its proximal HRE site corresponding to nt −31 to −9 of the promoter region, or RANTES with a mutation in the same HRE site (Table I) were biotin-labeled using a LightShift chemiluminescent EMSA kit (Pierce Chemical). The nuclear extract (10 μg), 5 mM MgCl2, 5% glycerol, 0.05% Nonidet P-40, 50 ng/μl poly(deoxyinosinic:deoxycytidylic), and 0.5 ng of biotin-ylated RANTES oligonucleotides were incubated at room temperature for 20 min. After the samples were subjected to nondenaturing, 6% PAGE, they were transferred to Hybond-N+ nylon membranes (Amersham). DNA-protein complex bands were detected with streptavidin-HRP chemiluminescence (Pierce). Specificity of the DNA-protein interaction was con-firmed using a 50-fold excess of unlabeled probe (33).
Chromatin immunoprecipitation (ChIP) assays
In serum-free medium, HMEC-1 (5 ×106 cells) was treated with ethanol (100 mM), in the absence or presence of inhibitors for the indicated time period. ChIP analysis was performed using a HIF-1α Ab as previously described (29). Briefly, 5 μl of the DNA sample was amplified by PCR with primers for the complete RANTES promoter region containing the AP-1, HRE-1, and HRE-2 sites (Table I). PCR amplification was performed under the following conditions for 35 cycles: denaturation at 95°C for 30 s, annealing at 55°C for 60 s, and extension at 72°C for 2 min. PCR products were subjected to electrophoresis on a 2% agarose gel and visualized with ethidium bromide staining. Densitometric analysis was performed using the Alpha Imager 2000 documentation and analysis system (Alpha Innotech).
Quantification of RANTES release
In serum-free medium, rLSEC (1 ×106 cells/ml) and HMEC-1 (1 ×106 cells/ml) were stimulated with ethanol (100 mM) in the presence or absence of inhibitors for 24 and 48 h, respectively. The supernatants of these cells were assayed for the RANTES protein using an ELISA kit (R&D Systems). RANTES release was also measured in plasma from ethanol-fed c-Junflox/flox and control mice in the same manner.
Chemotaxis assay
A chemotaxis assay was performed as previously described (35) using 96-well plates with Transwell pore-size inserts of 5 μm (Neuro Probe). In brief, THP-1 monocytic cells (1 ×105 cells) were washed and resuspended in 50 μl of serum-free medium and loaded into the Boyden chamber insert. Chemotaxis medium, comprised of 30 μl of serum-free medium from ethanol-treated (100 mM for 24 h) HMEC-1 cells containing −20 ng/ml che-mokine RANTES was loaded into the bottom compartment. Where indicated, a RANTES Ab (R&D Systems) or CCR5 Ab (BD Biosciences) was used to antagonize chemotaxis. After incubation for 1–4 h at 37°C, THP-1 cells that had migrated into the lower chamber were counted, as previously described (35).
Statistical analysis
Data are presented as means ± SD. Significance differences in mean values among multiple groups were analyzed with a parametric one-way ANOVA and then a Turkey-Kramer test using the Instat 2 software program (Graph-Pad). Samples treated with ethanol were used for multiple comparisons with Student’s t test to evaluate significance differences between untreated and ethanol-treated samples. Values of p < 0.05 were considered statistically significant.
Results
E-rLSECs show increased mRNA expression of RANTES and HIF-1α
As shown in Fig. 1A, E-rLSECs, compared with C-rLSECs, showed a ~5-fold increase in RANTES mRNA expression as determined by qRT-PCR. Additionally, there was a ~3.5-fold increase in HIF-1α mRNA expression in E-rLSECs compared with C-rLSECs (Fig. 1A).
FIGURE 1.

Ethanol augments RANTES expression in rLSECs via JNK kinase, p38 MAPK, NF-κB, and HIF-1α. A, Ethanol-induced mRNA expression of HIF-1α and RANTES in E-rLSECs. The data represent fold increases following ethanol feeding compared with isocaloric-fed wt (control) mice. B, Ethanol-induced HIF-1α and RANTES mRNA expression in ethanol-treated rLSECs. C, Ethanol-induced RANTES mRNA expression in rLSECs that were preincubated with the inhibitors DPI (NADPH oxi-dase inhibitor), LY294002 (PI3K inhibitor), SP600125 (JNK kinase), or SB203580 (p38 MAPK inhibitor) for 30 min before ethanol treatment. qRT-PCR data of ethanol-treated rLSECs represent fold increases in mRNA expression resulting from ethanol treatment (100 mM) for 4 h compared with untreated cells. All mRNA expression was normalized to GAPDH mRNA levels, and the data shown represent three independent experiments (mean ± SD). D, ELISA of RANTES release from rLSECs in response to ethanol treatment (100 mM) for 48 h expressed as fold change compared with untreated cells. The basal levels of RANTES release in untreated rLSECs was 23.3 ± 5 ng/ml (n =4). Values of p are denoted as follows: ***, p < 0.001; **, p <0.01; and ns, p > 0.05.
Ethanol treatment of rLSEC induces mRNA expression of RANTES and HIF-1α
As shown in Fig. 1B, treatment of rLSECs with ethanol (100 mM) showed a 2.3- and a 5-fold increase in HIF-1α and RANTES mRNA, respectively, as assessed by qRT-PCR. Preincubation of rLSECs with pharmacological inhibitors of flavin oxidases such as NADPH-oxidase (DPI) and PI3K (LY294002) did not affect ethanol-mediated RANTES mRNA expression (Fig. 1C). However, inhibitors of JNK (SP600125) and p38 MAPK (SB203580) reduced ethanol-induced RANTES mRNA expression by 63 ±3 and 73 ± 2%, respectively. RANTES released from rLSEC (Fig. 1D) increased 2.3-fold in response to ethanol stimulation, which was reduced below control (122 ± 1%) by preincubation with inhibitors of both NF-κB (sulfasalazine) and HIF-1α (R59949). Furthermore, the inhibitor of p38 MAPK (SB203580) attenuated ethanol-mediated RANTES release from rLSEC by 89 ± 4% compared with control. However, DPI did not have any effect on RANTES release (Fig. 1D). Taken together, these data indicate that ethanol-induced RANTES mRNA and protein expression in rLSEC involves activation of JNK, p38 MAPK, NF-κB, and HIF-1α, but not flavin oxidases (NADPH oxidase) or PI3K.
Ethanol augments mRNA expression of RANTES in dermal HMEC-1 cell line via activation of p38 MAPK and JNK-2
Because ethanol, in vivo and in vitro, induced RANTES mRNA expression in rLSECs, we examined whether human endothelial cells from vascular beds other than the liver sinusoid exhibited a similar response as that seen in rLSECs. We used the HMEC-1 cell line as a model system for rLSECs in delineating ethanol-induced cell signaling pathways because of ease of culture, transfection efficiency, and availability of human RANTES promoter constructs. HMEC-1 treated with 100 mM ethanol showed a time-dependent increase in RANTES mRNA levels (2–8 h), with a maximal and sustained increase at 4 h (data not shown). Treatment of HMEC-1 with ethanol (100 mM) for 4 h, compared with control HMEC-1, showed a ~5-fold increase in RANTES mRNA expression (Fig. 2A) similar to that observed in rLSECs treated acutely with ethanol. It is pertinent to mention that lower concentrations of ethanol (25 and 50 mM) increased RANTES mRNA expression by 2- and 3-fold, respectively (data not shown). The viability of HMEC-1 cells was unaffected by ethanol (100 mM) treatment for 4 h. These results indicate that ethanol exhibits its optimal effect at 100 mM, corresponding to a concentration of 0.46% of alcohol, which can be found in chronic alcoholics.
FIGURE 2.

Ethanol augments RANTES expression in the HMEC-1 cell line via p38 MAPK, JNK-2, NF-κB, and HIF-1α. A, HIF-1α and RANTES mRNA levels in HMEC-1 treated with ethanol compared with untreated cells. B, Ethanol-induced HIF-1α and RANTES mRNA expressions in HMEC-1 that was preincubated with the inhibitors DPI, LY294002, SP600125, or SB203580 for 30 min before ethanol treatment. qRT-PCR data represent fold increase in mRNA expression following treatment with ethanol (100 mM) for 4 h compared with absence of ethanol treatment. All mRNA expressions were normalized to GAPDH mRNA levels, and the data shown represent three independent experiments (mean ± SD). C, ELISA of RANTES release from HMEC-1 in response to ethanol (100 mM) treatment for 48 h expressed as fold-change compared with no ethanol treatment. Where indicated, HMEC-1 was pretreated with the inhibitors DPI, sulfasalazine, or R59949. The basal level of RANTES released from untreated HMEC-1 were 36.3 ± 6.3 ng/ml. The data represent three independent experiments in duplicate (mean ± SD). D, Chemotaxis assay of ethanol-treated HMEC-1 medium on THP-1 monocyte migration for the indicated time period. Where noted, HMEC-1 medium was treated with RANTES Ab and THP-1 was treated with an Ab to CCR5. Results are represented as numbers of migrated cells per high-powered (×40 original magnification) microscopic field. Data are expressed as means ± SD of experiments done in triplicate. E, Ethanol-induced RANTES expression in HMEC-1 that were transiently transfected with p38 siRNA, JNK-1 siRNA, JNK-2 siRNA, or mock siRNA as control before ethanol stimulation (100 mM) for 4 h. Where indicated, HMEC-1 transfected with a JNK-2 expression plasmid were without ethanol treatment. F, RANTES mRNA levels in HMEC-1 cell treated with ethanol, acetaldehyde, or isopropanol at the indicated concentrations for 4 h, compared with no ethanol treatment. Where indicated, HMEC-1 cells were also preincubated with 4-methylpyrazole (1.0 mM) for 30 min before ethanol treatment. qRT-PCR data represent fold increases in RANTES mRNA expressions. RANTES mRNA levels were normalized to GAPDH mRNA levels, and the data shown represent three independent experiments (mean ± SD). Values of p are denoted as follows: ***, p < 0.001; ** , p < 0.01; and ns, p > 0.05.
Treatment of HMEC-1 with pharmacologic inhibitors of flavin oxidases (DPI) and PI3K (LY294002) did not affect ethanol-induced RANTES mRNA expression (Fig. 2B). However, pharmacological inhibitors of JNK (SP600125) and p38 MAPK (SB203580) reduced RANTES mRNA expression by 62 ± 1 and 72 ± 2%, respectively, similar to that seen in ethanol-treated rLSEC. Furthermore, RANTES released from HMEC-1 (Fig. 2C) increased ~8.6-fold in response to ethanol treatment, which was inhibited by 66 ± 3 and 90 ± 3% after preincubation with inhibitors of NF-κB (sulfasalazine) and HIF-1α (R59949), respectively. Note that DPI did not show any effect on RANTES release. As shown in Fig. 2D, RANTES released from HMEC-1 was functional in a chemotaxis assay wherein conditioned medium from ethanol-treated HMEC-1 cells increased the migration of THP-1 monocytes in a time-dependent manner. Furthermore, the effect was specific for RANTES, as an Ab to RANTES reduced chemo-taxis by >80%. Additionally, blockage of the RANTES receptor CCR5 on THP-1 cells also reduced chemotaxis by >80% (Fig. 2D). Taken together, these data indicated that ethanol-induced RANTES mRNA and protein expression in HMEC-1 involves activation of Jun N-terminal kinase, p38 MAPK, NF-κB, and HIF-1α, but not flavin oxidases or PI3K as observed for rLSEC. Furthermore, these results indicated that ethanol caused activation of the same pathways in rLSEC and HMEC-1 for the expression of RANTES and that the RANTES released from these cells was biologically functional in promoting chemotaxis of monocytes.
Because pharmacological inhibitors can act in a nonspecific manner, HMEC-1 cells were transfected with siRNAs for p38 MAPK, JNK-1, JNK-2, and mock siRNA, the latter being a scrambled sequence that is not specific for any known cellular mRNA. As shown in Fig. 2E, siRNAs for both p38 MAPK and JNK-2 completely inhibited ethanol-mediated RANTES mRNA expression compared with control ( p < 0.001). However, siRNA for JNK-1 had no effect. The role of JNK-2 in RANTES expression was further supported by increased RANTES mRNA expression in HMEC-1 cells transfected with a JNK-2 expression plasmid in the absence of ethanol treatment (Fig. 2E). These results suggested the involvement of p38 MAPK and JNK-2 in ethanol-mediated RANTES expression.
Because ethanol is metabolized to acetaldehyde by cellular alcohol dehydrogenase, we examined whether ethanol metabolism is required for RANTES expression. HMEC-1 cells treated with ethanol in the presence of 1.0 mM 4-methylpyrazole reduced ethanol-mediated RANTES expression by 113 ± 5%, i.e., below the basal level (Fig. 2F). Moreover, acetaldehyde (0.5–1.0 mM) dose-dependently increased RANTES expression to the same level as was observed with ethanol (Fig. 2F). The expression of RANTES was specific for ethanol, as nonmetabolizing isopropanol (2-propanol) did not augment RANTES expression (Fig. 2F). Furthermore, cyanamide, an inhibitor of aldehyde dehydrogenase, attenuated ethanol-induced and acetaldehyde-induced RANTES mRNA expression to basal levels (data not shown). Although these concentrations of ethanol and acetaldehyde may be physiologically high, such a situation likely exists in chronic alcoholics. Taken together, our results showed that ethanol-induced RANTES expression required ethanol metabolism, p38 MAPK, and JNK-2, but not JNK-1.
Ethanol-induced RANTES expression in HMEC-1 and rLSEC involves activation of HIF-1α
To determine whether HIF-1α plays a role in ethanol-mediated RANTES expression, HMEC-1 was transfected with siRNA for HIF-1α. As shown Fig. 3A, transfection with HIF-1α siRNA reduced RANTES and HIF-1α mRNA expression to the basal level. However, transfection with scrambled HIF-1α siRNA (HIF-1α scRNA) had no effect on the ethanol-induced expression of HIF-1α and RANTES mRNA (Fig. 3A). Moreover, transfection with siRNA for HIF-2α had no effect on RANTES mRNA expression (Fig. 3A) indicating that HIF-1α, but not HIF-2α, is involved in ethanol-mediated RANTES expression. Furthermore, transfec-tion with PHD-2 siRNA, which stabilizes the HIF-1α protein, augmented RANTES expression in the absence of ethanol treatment (Fig. 3A). Transfection of scrambled PHD-2 siRNA (PHD-2 scRNA) did not augment RANTES mRNA expression (Fig. 3A).
FIGURE 3.
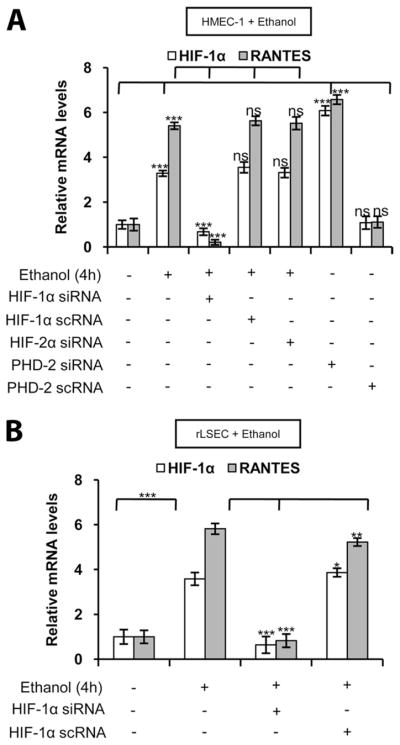
Ethanol-mediated RANTES mRNA expression in HMEC-1 cells and rLSECs involves HIF-1α. A, Ethanol-induced HIF-1α and RANTES mRNA expression in HMEC-1 cells that were transiently transfected with HIF-1α siRNA, scrambled HIF-1α scRNA, or HIF-2α siRNA before treatment with ethanol (100 mM) for 4 h. HMEC-1 was transfected with PHD-2 siRNA or scrambled PHD-2 scRNA in the absence of ethanol treatment. B, Ethanol-induced RANTES expression in rLSECs that were transiently transfected with HIF-1α siRNA or scrambled HIF-1α scRNA, before ethanol treatment (100 mM) for 4 h. qRT-PCR data represent fold increase in mRNA expression in ethanol-treated cells vs untreated cells. mRNA expression data were normalized to GAPDH mRNA levels, and the data shown represent three independent experiments (mean ± SD). Values of p are denoted as follows: ***, p < 0.001; **, p < 0.01; *, p < 0.05; and ns, p > 0.05.
To determine whether ethanol-mediated RANTES expression in rLSEC also involves HIF-1α, rLSEC cells were transfected with HIF-1α siRNA. As shown in Fig. 3B, rLSECs transfected with HIF-1α siRNA attenuated RANTES induction (104 α 4%), whereas HIF-1α scRNA had no effect on ethanol-induced RANTES expression. These results indicated that, in both rLSEC and HMEC-1, ethanol-mediated RANTES expression required activation of HIF-1α.
Ethanol-induced RANTES expression requires HREs and AP-1 motif in its promoter
Previous studies (16) have shown that the 5′ flanking region of the RANTES gene contains potential NF-κB, C/EBP, NF-IL6, AP-1, and GATA motifs. An in silico analysis of the RANTES promoter element (nt −974 to −1; GenBank accession no. AB023652) revealed the presence of a HRE motif (RCGTG) at nt −32 to −29, two inverted HREs (RGCAC) at nt −22 to −19 and nt −720 to − 717, and an AP-1 binding site at nt −250 to −244.
As shown in Fig. 4A, treatment of HMEC-1 with ethanol (25–100 mM) resulted in a dose-dependent increase in full-length RANTES promoter-driven luciferase activity, with an optimal increase of 10-fold activity by 100 mM ethanol. Furthermore, ethanol metabolism was essential, as 4-methylpyrazole reduced RANTES promoter activity to the basal level (Fig. 4B). Moreover, acetaldehyde (1.0 mM) showed the same levels of increase in RANTES promoter activity as was seen with ethanol (100 mM). As shown in Fig. 4C, ethanol-induced RANTES promoter lucif-erase activity was not affected by flavin oxidase inhibitor (DPI) and PI3K inhibitor (LY294002). However, ethanol-induced RANTES promoter luciferase activity was reduced by 74 ± 3% with p38 MAPK inhibitor (SB203580) and to basal level by R59949 (Fig. 4C). As shown in Fig. 4D, p38 MAPK siRNA, JNK-2 siRNA, and HIF-1α siRNA, but neither p47 siRNA nor dominant negative PI3K, attenuated ethanol-induced RANTES promoter activity by >65%. Taken together, these results indicated that ethanol-induced RANTES promoter activation requires p38 MAPK, JNK-2, and HIF-1α and does not involve NADPH oxidase and PI3K, as was seen for the expression of RANTES mRNA.
FIGURE 4.

Ethanol metabolism augments full-length wt RANTES promoter (nt −975 to −1) activity in HMEC-1 via involvement of p38 MAPK, HIF-1α, and JNK-2. A, Dose-response effect of ethanol (25–100 mM) for 4 h on RANTES promoter (−975bp) activity. B, RANTES promoter activity in response to ethanol (50–100 mM) or acetaldehyde (0.5–1.0 mM) treatment. Where indicated, cells were preincubated with 4-methylpyrazole (1.0 mM) for 30 min before ethanol treatment. C, Effect of inhibitors on RANTES promoter activity in response to ethanol (100 mM) treatment. Where indicated, cells were preincubated with DPI, LY294002, SB203580, or R59949 for 30 min before ethanol addition. D, Effect of silencing with siRNAs for p38 MAPK, JNK-2, p47phox, and HIF-1α, and dominant negative (Dn) PI-3K on ethanol-induced RANTES promoter activity. Luciferase assay data are expressed as fold change and have been normalized to the change in luciferase activity of untreated controls and for transfection efficiency with β-galactosidase activity. The data shown represent three independent experiments in duplicate (means ± SD). Values of p are denoted as follows: ***, p < 0.001; **, p < 0.01; *, p < 0.05; and ns, p > 0.05.
To determine which HREs in the RANTES proximal promoter were essential for ethanol-induced RANTES promoter activity, we generated HRE mutant constructs of the RANTES promoter designated as HRE-M1, HRE-M2, and HRE-M3 as illustrated in the schematics of Fig. 5A. Additionally, a mutant construct of the AP-1 site (AP-1-M) in the RANTES promoter was generated and is also depicted in Fig. 5A. As shown in Fig. 5B, ethanol induction of the HRE-M1 and HRE-M2 promoters showed reduced activation by 76 ± 1 and 43 ± 2%, respectively. However, HRE-M3 retained ethanol-induced RANTES promoter activity. Furthermore, mutation of the AP-1 site also led to a 69 ± 1.4% reduction in activity. Taken together, these results indicated that the HRE-1, HRE-2, and AP-1 sites are the essential functional elements required for ethanol-mediated induction of the RANTES promoter.
FIGURE 5.

Ethanol enhances RANTES promoter activity via its two proximal HRE sites (HRE-1 and HRE-2) and its AP-1 site. A, Schematic of the human RANTES (975 bp) promoter region, containing three HRE sites and one AP-1 site. The mutations of specific nucleotides in the HRE and AP-1 sites of the RANTES promoter are denoted by asterisks (*). B, Effect of HREs and AP-1 mutations on RANTES (975 bp) promoter activity in HMEC-1 in response to ethanol (100 mM) treatment for 4 h. Mutant site promoters were constructed in the context of the full-length (975 bp) RANTES promoter. The RANTES HRE-M3 luciferase construct has a mutation in the HRE at nt −720 to −717. The RANTES HRE-M2 and HRE-M1 luciferase constructs have mutations in their HREs at nt −32 to −29 and nt −22 to −19, respectively. The RANTES AP-1-M luciferase construct has a mutation in the AP-1 binding site at nt −250 to −244. C, Effect of c-Jun, c-Fos, JunD, and JunB expression plasmids on RANTES promoter activity. HMEC-1 was transiently transfected with 1 μg of each expression plasmid for c-Jun, c-Fos, c-Jun and c-Fos, JunB, JunD, c-Jun and JunD, and a combination of c-Jun, c-Fos, and JunD. D, Effect of ethanol (100 mM) on AP-1 promoter luciferase activity in cells preincubated with the inhibitors GF109203X (PKC inhibitor), PP1 (Src kinase inhibitor), SB203580, or SP600125 for 30 min before ethanol treatment. Luciferase activity was normalized as described above. The data shown represent three independent experiments (means ± SD). E, ELISA of RANTES in plasma isolated from ethanol-fed mice expressed as fold-changes compared with control. Where indicated, ethanol-fed c-Junflox/flox mice were compared with ethanol-fed wt mice. The basal levels of RANTES in the plasma of control wt mice were 44.6 ± 6.1 ng/ml. The data represent plasma from eight mice in each category, and the assay was performed in duplicate (mean ± SD). Values of p are denoted as follows: ***, p < 0.001; **, p < 0.01; *, p < 0.05; and ns, p > 0.05.
Effect of overexpression of Jun and Fos proteins on RANTES promoter activity
Because the active AP-1 complex comprises dimers of c-Jun and a heterodimer of c-Fos/c-Jun and combines with other candidate Jun proteins, cotransfection studies were performed using c-Fos, c-Jun, JunB, and JunD expression plasmids and the 975-bp RANTES luciferase promoter construct. As shown in Fig. 5C, ethanol treatment of HMEC-1 increased RANTES promoter activity by ~14-fold compared with untreated cells. c-Fos overexpression did not change promoter activity, but c-Jun alone and c-Jun with c-Fos augmented RANTES promoter activity by ~21- and ~28-fold, respectively. JunD, but not JunB, also augmented RANTES promoter activity by ~20-fold (Fig. 5C). c-Jun with JunD enhanced RANTES promoter activity by ~25-fold, and c-Jun with c-Fos and JunD increased the promoter activity of RANTES to its greatest extent at ~30-fold (Fig. 5C). Taken together, these results indicated that c-Jun homodimers, c-Fos/c-Jun heterodimers, JunD dimer, JunD/c-Jun, and JunD/c-Jun/c-Fos form an AP-1 transcriptional complex that interacts with the AP-1 site in the RANTES promoter to augment its activity. Significantly, JunB had no role in ethanol-induced RANTES promoter activity.
To examine the upstream signaling events in ethanol-mediated AP-1 promoter activation, HMEC-1 cells were transfected with a consensus AP-1 promoter luciferase construct followed by treatment with ethanol in the presence or absence of pharmacological inhibitors. As shown in Fig. 5D, ethanol increased AP-1 promoter activity by ~9.3-fold in HMEC-1, which was attenuated 36 ± 5 and 49 ± 5% when the cells were pretreated with inhibitors for protein kinase C (PKC) (GF109203X) and Src kinase (PP1), respectively. Additionally, HMEC-1 cells pretreated with inhibitors for p38 MAPK (SB203580) and JNK kinase (SP600125) showed reduced (78 ± 5 and 86 ± 5%, respectively) ethanol-induced AP-1 promoter activation compared with ethanol-treated cells. Thus, ethanol-induced AP-1 promoter activity in HMEC-1 involves PKC, Src kinase, p38 MAPK, and JNK kinase.
RANTES protein expression in the plasma of ethanol-fed c-Junflox/flox mice
Because our in vitro studies showed the involvement of c-Jun in ethanol-mediated expression of RANTES, we determined whether c-Jun played a role in vivo. Because the c-Jun-null mutation is embryonic lethal, we used Mx-1-Cre-Lox mice for conditional deletion of c-Jun in the sinusoidal endothelial cells and immune cells of the liver. For these experiments, c-Jun floxed mice, c-Junflox/flox (24), were used. The plasma was obtained from ethanol-fed mice and ethanol-fed c-Junflox/flox mice, and RANTES release was measured by ELISA. Ethanol feeding of wt (control) mice resulted in ~3.4-fold increased levels of RANTES protein in the plasma compared with isocaloric-fed wt mice (Fig. 5E). However, there was no difference in RANTES protein in the plasma of ethanol-fed c-Junflox/flox mice compared with ethanol-fed wt mice (Fig. 5E). Also, plasma from c-Jun floxed mice fed an isocaloric diet showed modestly reduced levels of RANTES protein (20 ± 4%) when compared with isocaloric-fed control mice. These results support the role of c-Jun and, thus, the AP-1 complex in ethanol-mediated RANTES protein expression in vivo.
Ethanol augments protein expression of HIF-1α and RANTES
Because ethanol (100 mM) caused an increase in HIF-1α mRNA expression in HMEC-1 cells, we determined whether ethanol affected HIF-1α protein levels in the nuclear extracts of HMEC-1 cells. As shown in Fig. 6A, ethanol treatment of HMEC-1 resulted in a time-dependent (1–4 h) increase in the HIF-1α protein, which was optimal at 4 h. We have also shown that ethanol treatment of HMEC-1 resulted in increased RANTES mRNA expression. We determined whether ethanol affected RANTES protein levels. As shown in Fig. 6B, treatment of HMEC-1 with ethanol led to a time dependent (2–24 h) increase in RANTES protein expression. There was a ~4.1-fold increase in RANTES protein expression at 24 h. These results complement the increase in HIF-1α and RANTES mRNA seen in response to ethanol.
FIGURE 6.

Ethanol augments HIF-1α protein and AP-1 complex binding to HREs and AP-1 site in the RANTES promoter. A, Time course of 100 mM ethanol-induced HIF-1α protein expression in nuclear extracts of HMEC-1. B, Time course of ethanol-induced RANTES protein expression in HMEC-1. The data were normalized to β-actin as a loading control. C, EMSA of nuclear extracts from HMEC-1 using oligonucleotide probes for the AP-1 and HRE-1 sites in the RANTES promoter. Where indicated, HMEC-1 was transfected with siRNA followed by ethanol treatment for 4 h and isolation of nuclear extracts. As indicated, oligonucleotide probes with mutations in either the AP-1 or HRE-1 sites were used. Where indicated, 50-fold excess of cold probe was added to the nuclear extracts. The data are representative of three independent experiments. Vertical lines indicate repositioned gel lanes. D, Ethanol augments HIF-1α binding to HRE-1 in the RANTES promoter as assessed by ChIP analysis. HMEC-1 was preincubated for 30 min with inhibitors before ethanol treatment for 4 h. Soluble chromatin was immunoprecipitated with either HIF-1α Ab (top panel) or control rabbit IgG (bottom panel). Primers used to amplify the products flanking the HRE-1 site in the RANTES promoter are indicated in Table I. The middle panel represents the amplification of input DNA before immunoprecipitation. Densitometric analysis was performed and is depicted in the graph. Data are representative of three independent experiments.
Ethanol augments HIF-1α protein binding to HREs in HMEC-1 as demonstrated by EMSA and ChIP
Next, we performed an EMSA to corroborate the involvement of AP-1 complex proteins and HIF-1α protein in ethanol-dependent induction of RANTES. Nuclear extracts from ethanol treated HMEC-1 cells were assayed with both wt and mutant oligonucleotides that flanked the AP-1 site (nt −250 to −244 region) and the wt and mutant oligonucleotides that flanked the HRE-1 site (nt −22 to −19 region) in the RANTES promoter as probes. As shown in Fig. 6C, nuclear extracts of ethanol-treated HMEC-1 showed a DNA-protein complex with the AP-1 probe compared with control (Fig. 6C, lane 2) that was abrogated when the cells were transfected with c-Jun siRNA before ethanol treatment (Fig. 6C, lane 3). A 50-fold excess of unlabeled AP-1 oligonucleotide probe competed out the DNA-protein complex, demonstrating the specificity of this complex (Fig. 6C, lane 4). Moreover, the mutant AP-1 probe showed complete lack of protein-DNA binding (Fig. 6C, lane 5) compared with the wt probe (Fig. 6C, lane 1). Also shown in Fig. 6C, nuclear extracts of ethanol-treated HMEC-1 showed increased HIF-1α protein-DNA binding (Fig. 6C, lane 7) compared with control (Fig. 6C, lane 6) that was absent when cells were transfected with HIF-1α siRNA before ethanol treatment (Fig. 6C, lane 8). Furthermore, a 50-fold excess unlabeled HRE probe competed out protein- DNA binding (Fig. 6C, lane 9). Moreover, the mutant HRE oligonucleotides showed reduced protein-DNA binding (Fig. 6C, lane 10), compared with the wt probe (Fig. 6C, lane 6). These results indicated the importance of both AP-1 complex and HIF-1α protein binding to the promoter region of RANTES in response to ethanol.
These results were also confirmed by ChIP analysis, which showed HIF-1α binding to the RANTES promoter in HMEC-1 chromatin (Fig. 6D). Ethanol treated HMEC-1 showed a ~1.5-fold increase in the expected PCR product size of 264 bp, corresponding to the RANTES promoter region (nt −263 to −1) containing AP-1, HRE-2, and HRE-1 sites using the primers listed in Table I. As shown in Fig. 6D, pretreatment of HMEC-1 cells with SB203580 and R59949 reduced the expected PCR product by ~84 and ~96%, respectively, as determined by densitometry, which is shown in the panel as fold change, whereas LY294002 exhibited no effect. The amplification of input DNA before immunoprecipitation was equal in all samples (Fig. 6D). Immunoprecipitation of chromatin samples with rabbit IgG as a control did not show any amplification of the product. These data indicate that ethanol increased HIF-1α binding to the RANTES promoter to up-regulate the expression of RANTES in vivo.
Ethanol induced signaling for activation of JNK and AP-1 involves PKC, Src kinase, and p38 MAPK
Because ethanol-mediated mRNA expression and promoter activity of RANTES was attenuated by the pharmacological inhibitor SP600125 and the siRNA for JNK-2, we examined the upstream signaling events involved with the activation of JNK-2. Previous studies (36) show that thrombin-mediated activation of AP-1 in tubular epithelial cells involves both PKC-Src-JNK-1 and AP-1 axis. As shown in Fig. 7A, ethanol treatment of HMEC-1 cells resulted in a time-dependent (10–60 min) increase in JNK-2 phos-phorylation with optimal phosphorylation at 10 min. Pretreatment of HMEC-1 with pharmacological inhibitors of PKC (GF109203X), JNK kinase (SP600125) and Src kinase (PP1) reduced ethanol-induced JNK phosphorylation, whereas the p38 MAPK inhibitor (SB203580) and the HIF-1 inhibitor R59949 had no effect (Fig. 7B). These data indicate the involvement of PKC, Src kinase, and JNK kinase in the activation of JNK-2.
FIGURE 7.

Ethanol-induced cellular signaling leading to RANTES expression involves activation of JNK-2, NF-κB, and HIF-1α. A, Time course of JNK-2 phosphorylation (p-JNK-2) in nuclear extracts from ethanol-treated HMEC-1 cells. The data was normalized to unphosphorylated JNK as a loading control. B, Ethanol-mediated JNK-2 phosphorylation in nuclear extracts of HMEC-1 preincubated with the inhibitors GF109203X, SP600125, PP1, SB203580, or R59949 for 30 min before ethanol treatment. The data are representative of three independent experiments. C, Ethanol augments the interaction of NF-κB proteins with the HIF-1α promoter. HMEC-1 cells were transfected with a HIF-1α luciferase (luc) promoter construct. Where indicated, cells were preincubated with the inhibitors LY294002, SB203580, sulfasalazine, or PD98059 for 30 min before ethanol treatment for 2 h. Luciferase assay data are expressed as fold change and have been normalized as described above. The data represent duplicate assays from three independent experiments (means ± SD). D, Illustration of the ethanol-mediated signaling pathway showing that ethanol metabolism leads to activation of two independent signaling pathways. In one pathway, activation of AP-1 via PKC, Src kinase, and JNK-2 leads to up-regulation of RANTES. This involves an AP-1 complex consisting of c-Jun, c-Fos, and JunD, but not JunB. The second pathway involves activation of HIF-1α via crosstalk with NF-κB, the latter involving activation of MEK. The activation of HIF-1α leads to HRE-binding in the RANTES promoter followed by increased gene transcription. We suggest that ethanol-mediated RANTES generated from LSECs augments the infiltration of monocytes/polymorphonuclear neutrophils and T and B cells from circulation into the liver, contributing to inflammation.
Ethanol-induced HIF-1α promoter activation involves p38 MAPK, MAPK, and NF-κB
Because the proximal promoter of HIF-1α has been shown to have a binding site for NF-κB (37), we determined whether ethanol-mediated activation of MAPK and NF-κB could lead to the activation of HIF-1α promoter activity. As shown in Fig. 7C, trans-fection of HMEC-1 with a HIF-1α luciferase promoter construct (p9HIF1-Luc) followed by ethanol treatment led to a large (−27-fold) increase in luciferase activity (Fig. 7C). Ethanol-induced HIF-1α luciferase activity was significantly attenuated to the extent of −45–75 − 3–7% by SB203580, PD98059, sulfasalazine, and p65 siRNA, but not by LY294002 or mock siRNA. These results suggest that ethanol-mediated activation of p38 MAPK, MAPK, and NF-κB regulated HIF-1α promoter activity.
Discussion
In the present study, we show that ethanol in vivo and in vitro increased RANTES/CCL5 expression by severalfold in liver sinusoidal endothelial cells isolated from rats. Similarly, ethanol induced the expression of RANTES in a human dermal microvascular endothelial cell line, HMEC-1. HMEC-1 cells were used as a model system to delineate the ethanol-induced cellular signaling mechanisms. The metabolism of ethanol was essential for RANTES expression, as inhibitors of alcohol dehydrogenase (4-meth-ylpyrazole) and aldehyde dehydrogenase (cyanamide) attenuated RANTES mRNA levels in response to ethanol induction. Ethanol-mediated RANTES expression was dose dependent (25–100 mM), with maximal expression at 100 mM ethanol. Although this concentration (0.46%) of ethanol may seem physiologically high, recent studies (38) have shown that the metabolism of ethanol increases to accommodate increased alcohol intake in chronic alcoholics. It was shown that as alcohol consumption increased in mice, alcohol dehydrogenase 1 activity in the liver decreased whereas alcohol dehydrogenase 3 (high Km for ethanol) activity increased (38).
We showed that in inhibitor experiments, ethanol-induced expression of RANTES in HMEC-1 involved activation of p38 MAPK and JNK, but not activation of PI3K and flavin oxidases. Previous studies (11) show that TNF-α-induced RANTES expression in hepatocytes involves activation of p38 MAPK and NF-κB. Because pharmacological inhibitors can be nonspecific, as they may target multiple sites, we used RNA interference-mediated gene knockdown to delineate the signaling pathway. Gene silencing with p38 MAPK siRNA and JNK-2 siRNA attenuated ethanol-induced RANTES mRNA expression. However, dominant negative PI-3K, p47 siRNA (NADPH oxidase gene knockdown), and JNK-1 siRNA did not affect ethanol-induced RANTES expression, thus supporting the role of p38 MAPK and JNK-2 in ethanol-mediated RANTES expression. Moreover, overexpression of JNK-2, in the absence of ethanol treatment, increased RANTES expression. These results indicated that selective activation of JNK-2 by ethanol among ubiquitously expressed JNK-1 and JNK-2 up-regulates the expression of RANTES.
At present, 13 MAPK kinases (MKKs) have been identified that regulate JNKs, and how these MKKs differentially regulate JNKs is not completely understood (39). Previous studies (40) show that MKK4 is important for stress-induced activation of JNK, whereas MKK7 is involved in inflammatory cytokine-induced activation of JNK. More recent studies from Czaja and coworkers (41) have revealed differential roles of JNK-1 and JNK-2 in murine steato-hepatitis and insulin resistance using knockdowns of JNK-1 and JNK-2 in mice. These studies show that both JNK-1 and JNK-2 mediated insulin resistance in high fat diet-fed mice. However, JNK isoforms show differential effects in steatohepatitis, wherein JNK-1 promotes steatosis and hepatitis, whereas JNK-2 inhibits hepatocyte cell death (41). We suggest that ethanol-mediated JNK-2 activation plays a role in the expression of chemoattractant RANTES/CCL5.
We identified upstream signaling events involved in the ethanol-mediated activation of JNK by phosphorylation that included activation of PKC, Src kinase and p38 MAPK. These signaling events are similar to that observed for thrombin-mediated activation of AP-1 in tubular epithelial cells, which has been shown to involve activation of the PKC-Src-JNK-1 and AP-1 axis (36). The family of AP-1 transcriptional factors mainly consists of three groups: the Jun proteins (c-Jun, JunB, and JunD), the Fos proteins (c-Fos, Fos B, Fra1, and Fra2), and the activating transcription factors (ATF2, ATF3, and B-ATF) (42). Members of these families can form homodimers or heterodimers that constitute the active AP-1 complex. Numerous studies have identified JNK-mediated phosphorylation of c-Jun as influencing the transcriptional activation of the AP-1 complex (43). We observed that overex-pression of c-Jun, c-Jun with c-Fos, and JunD greatly increased RANTES promoter activity, whereas overexpression of either c-Fos or JunB had no effect. These data suggest that c-Jun forms a homodimer or heterodimer with c-Jun, c-Fos, or JunD to augment RANTES promoter activity. Moreover JunD, but not JunB, bound to the RANTES promoter to increase its activity. Recent studies (44) have examined the roles of JunD and JunB in the regulation of hemeoxygenase (HO)-1 promoter activity. These studies show that JunB activates whereas JunD represses HO-1 expression, demonstrating the opposing roles of JunB and JunD in regulating HO-1 (44). JunD has been shown to protect the liver from ischemia/reperfusion injury by dampening AP-1 transcriptional activation (45). In contrast, our studies showed that JunD activated while JunB had no effect on RANTES expression, suggesting differential roles of JunD in gene expression. These results are in consonance with previous studies (46) wherein JunD is suggested to exert its role in cellular growth control through both transcriptional activation and repression of target genes.
We showed that silencing of HIF-1α with siRNA attenuated ethanol-induced RANTES expression. Furthermore, transfection with PHD-2 siRNA, which stabilizes HIF-1α protein, augmented RANTES expression in the absence of ethanol treatment. However, transfection with siRNA for HIF-2α did not affect ethanol-mediated RANTES mRNA expression. These studies suggested that HIF-1α, but not HIF-2α, is involved in ethanol-mediated RANTES expression in both rLSEC and HMEC-1. This was further supported by analysis of the RANTES promoter. Our studies showed that ethanol or its metabolite, acetaldehyde, augmented full-length (nt −975 to −1) RANTES promoter activity, indicating that the metabolism of ethanol is involved in RANTES gene expression. Moreover, ethanol-induced RANTES promoter reporter activity was inhibited by the same pharmacological inhibitors and siRNAs that affected native RANTES mRNA expression. An in silico analysis of the RANTES promoter element (−974 to −1 bp) revealed the presence of an HRE on the sense strand (5′-CGTG-3′) at nt −32 to −29, two HREs on the antisense strand (5′-GTGC-3′) at nt −22 to −19 and nt −720 to −717, and an AP-1 binding site at nt −250 to-244. The presence of inverted HREs (GCAC) in the sense strand of the proximal promoter of endothelin-1 has been previously shown to have a role in hypoxia-induced expression of the ET-1 gene (47). We examined the effect of mutations of the three HRE sites and single AP-1 site in the RANTES promoter to delineate the roles of these binding sites in promoter activation. Our studies showed that HRE-1, HRE-2, and AP-1 in the RANTES proximal promoter, but not HRE-3, were essential for ethanol-mediated RANTES promoter activity. Previous studies (16) indicated that the 5′ flanking region of the RANTES gene has potential NF-κB, C/EBP, NF-IL-6, AP-1, and GATA motifs. Cy-tokine-stimulated RANTES/CCL5 gene transcription in the pulmonary alveolar epithelium has been shown to involve NF-κB, AP-1, and NF-IL-6 promoter binding (48). LPS-induced RANTES/CCL5 transcription in monocytic cells involves C/EBP and NF-κB (10). IFN-γ-induced RANTES expression in macrophages was shown to involve the transcriptional activator IRF-1 (interfer-on-regulating factor 1) (49). The binding of HIF-1α to the HREs in the proximal promoter of RANTES was substantiated by EMSA, wherein we observed augmented binding of nuclear extract proteins, which contain HIF-1α and AP-1 complexes, to oli-gonucleotides containing bona fide HREs and AP-1 DNA sequences of the RANTES promoter. Moreover, HIF-1α siRNA and c-Jun siRNA abrogated protein-DNA binding in the nuclear extracts of ethanol-treated HMEC-1. Additionally, ChIP analysis confirmed the binding of HIF-1α to the HREs of the RANTES proximal promoter in the chromatin of ethanol-stimulated HMEC-1 cells. Together, our results showed that an ethanol-mediated increase of HIF-1α and AP-1 complex proteins (c-Jun/Fos) resulted in augmented binding to the HRE and AP-1 sites in the RANTES promoter, leading to increased transcription of RANTES/CCL5.
We have previously shown that amyloid peptide-induced CCR5 expression in THP-1 monocytes promotes RANTES-mediated chemotaxis (35). In the present study, we showed that RANTES released from ethanol-treated endothelial cells was biologically functional, as it promoted the chemotaxis of THP-1 monocytes. Additionally, RANTES-mediated chemotaxis was abrogated by an Ab to RANTES. Furthermore, an Ab to CCR5 prevented RANTES-dependent THP-1 migration toward the conditioned medium from ethanol-treated endothelial cells.
Our studies also show that plasma from c-Jun floxed mice (c-Junflox/flox) had lower levels of RANTES compared with control mice. Moreover, ethanol-fed c-Junflox/flox mice, compared with control mice that were fed ethanol for the same period, showed no ethanol-mediated induction of RANTES protein. It is important to note that the c-Jun floxed mice were generated using an Mx-1 promoter driven Cre recombinase. Mx-1 is present in sinusoidal endothelial cells, immune cells, and hepatocytes, all of which are IFN-inducible cells in the liver. Injections of these mice with poly(I:C) for induction of Mx-1-Cre expression also induced IFN activation. As measured by ELISA, reduced RANTES release in plasma from c-Junflox/flox ethanol-fed mice compared with wt mice of the same group suggested that ethanol-induced RANTES expression in endothelial cells involves c-Jun. However, this abrogation in RANTES release could also be due to a deletion of c-Jun in cells other than sinusoidal endothelial cells in the liver. Thus, further investigations into the role of c-Jun in RANTES expression by sinusoidal endothelial cells in vivo are warranted.
Because pharmacological inhibitors of MAPK (PD98059) and NF-κB (sulfasalazine) attenuated ethanol-mediated RANTES induction, we determined whether ethanol-induced activation of NF-κB led to crosstalk with the HIF-1α promoter for the subsequent up-regulation of RANTES. Our studies showed that ethanol-mediated HIF-1α promoter luciferase reporter activity was significantly attenuated by SB203580, PD98059, sulfasalazine, and p65 siRNA, indicating that ethanol-mediated activation of p38 MAPK, MAPK, and NF-κB regulates HIF-1α promoter activity. We propose that ethanol-mediated activation of NF-κB led to up-regulation of HIF-1α, which is another pathway through which RANTES expression can be regulated. These data are in accord with recent studies wherein investigators have identified NF-κB as a direct modulator of HIF-1α, as knockouts of NF-κB subunits lead to reduced HIF-1α mRNA expression (50). Additionally, studies have shown that NF-κB activation in response to hypoxia, bacterial infection, or inflammation occurs through the phosphorylation of IκB kinase-β, leading to IκB degradation and NF-κB nuclear translocation (48). Nuclear NF-κB can then bind to the promoter of HIF-1α, resulting in increased expression of HIF-1α mRNA and its target genes, such as vascular endothelial growth factor (37). IκB kinase-β knockout mice studies show reduced expression of HIF-1α mRNA and its regulated genes (37). Based on these studies and our results, we suggest that regulation of RANTES expression in response to ethanol likely occurs through NF-κB-mediated activation of HIF-1α.
In conclusion, our studies show that ethanol in vivo up-regulates the expression of RANTES/CCL5 in liver sinusoidal endothelial cells of ethanol-fed rats and a human cell model system, HMEC-1. Ethanol-mediated intracellular signaling involves the metabolism of ethanol, and downstream activation of the PKC-Src-p38 MAPK-JNK2-AP-1 axis, as illustrated in the schematic shown in Fig. 7D. More significantly, JunD, but not JunB, plays a role in the transcriptional activation of the AP-1 complex, leading to occupancy of the AP-1 binding site of the RANTES promoter with subsequent increase in RANTES gene expression. Additionally, we propose that ethanol-mediated activation of HIF-1α and its binding to the HRE sites HRE-1 and HRE-2 in the proximal promoter of RANTES are crucial to RANTES gene expression. The ethanol-mediated activation of HIF-1α likely occurs via crosstalk with NF-κB, which is activated via MAPKs. Overall, our studies show that ethanol-mediated activation of the AP-1 complex and HIF-1α regulates RANTES/CCL5 expression. Thus, ethanol-mediated increases in RANTES/CCL5 by liver sinusoidal endothelial cells can promote the infiltration of monocytes, polymorphonu-clear neutrophils, and memory T and B cells from the circulation into the liver via sinusoids (Fig. 7D), which can then activate hepa-tocytes and other nonparenchymal cells to cause inflammation and liver injury. We suggest that use of pharmacological inhibitors to attenuate AP-1 and HIF-1α activity can be of therapeutic use in ethanol-induced liver injury.
Acknowledgments
We thank Dr. Kinji Asahina and Jiaohong Wang for isolating and providing rat liver sinusoidal endothelial cells. We also thank Dr. Robert N. Taylor at Emory University, GA, for providing the RANTES luciferase construct. Furthermore, we acknowledge Dr. Debbie Johnson at the University of Southern California (Los Angeles, CA) for providing the dominant negative PI3K and JNK-2 expression plasmids, and we thank Dr. Stanley Tahara for critical review of the manuscript.
Footnotes
This work was supported by pilot project funding from National Institutes of Health Grants P50-AA011999 (to V.K.K.), P50-AA011999 (to H.T.), R24-AA012885 (to H.T. for support of animal and morphological core facilities, including the Non-Parenchymal Liver Cell Core), T32-AA07578 (predoctoral fellowship to S.Y.), CA108302 (to K.M.), and P30-DK048522 (for support of the Analytical-Metabolic-Instrumentation Core, University of Southern California Research Center for Liver Diseases).
Abbreviations used in this paper: LSEC, liver sinusoidal endothelial cell; ChIP, chromatin immunoprecipitation; C-rLSEC, control rat LSEC; DPI, diphenyleneiodo-nium chloride; E-rLSEC, ethanol-fed rat LSEC; HIF, hypoxia-inducible factor; HMEC, human microvascular endothelial cell; HO, hemeoxygenase; HRE, hypoxia response element; MKK, MAPK kinase; PHD, prolylhydroxylase; poly(I:C), polyi-nosinic:polycytidylic acid; PKC, protein kinase C; PP1, protein phosphatase 1; qRT-PCR, quantitative RT-PCR; rLSEC, rat LSEC; scRNA, scrambled RNA; siRNA, small interfering RNA; wt, wild type.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Thun MJ, Peto R, Lopez AD, Monaco JH, Henley SJ, Heath CW, Doll R. Alcohol consumption and mortality among middle-aged and elderly U.S. adults. N Engl J Med. 1997;337:1705–1714. doi: 10.1056/NEJM199712113372401. [DOI] [PubMed] [Google Scholar]
- 2.Vidali M, Stewart SF, Albano E. Interplay between oxidative stress and immunity in the progression of alcohol-mediated liver injury. Trends Mol Med. 2008;14:63–71. doi: 10.1016/j.molmed.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol. 2007;35:757–766. doi: 10.1080/01926230701584163. [DOI] [PubMed] [Google Scholar]
- 4.Tsukamoto H, Kaplowitz N. Pathogenesis of alcoholic liver disease. In: Kaplowitz N, editor. Liver and Biliary Diseases. Williams and Wilkins; Baltimore: 1996. pp. 121–137. [Google Scholar]
- 5.Pritchard MT, Nagy LE. Ethanol-induced liver injury: potential roles for Egr-1. Alcohol Clin Exp Res. 2005;29(Suppl 11):146S–150S. doi: 10.1097/01.alc.0000189286.81943.51. [DOI] [PubMed] [Google Scholar]
- 6.Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol induced liver injury. Hepatology. 1994;20:453–460. [PubMed] [Google Scholar]
- 7.Enomoto N, Ikejima K, Bradford BU, Rivera CA, Kono H, Brenner DA, Thurman RG. Alcohol causes both tolerance and sensitization of rat Kupffer cells via mechanisms dependent on endotoxins. Gastroenterology. 1998;115:443–451. doi: 10.1016/s0016-5085(98)70211-2. [DOI] [PubMed] [Google Scholar]
- 8.Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343:1467–1476. doi: 10.1056/NEJM200011163432007. [DOI] [PubMed] [Google Scholar]
- 9.Baggiolini M, Dewald B, Moser B. Human Chemokines: An Update. Annu Rev Immunol. 1997;15:675–705. doi: 10.1146/annurev.immunol.15.1.675. [DOI] [PubMed] [Google Scholar]
- 10.Fessele S, Boehlk S, Mojaat A, Miyamoto NG, Werner T, Nelson EL, Schloendorff D, Nelson PJ. Molecular and in silico characterization of a promoter module and C/EBP element that mediate LPS-induced RANTES/ CCL5 expression in monocytic cells. FASEB J. 2001;15:577–589. doi: 10.1096/fj.00-0459fje. [DOI] [PubMed] [Google Scholar]
- 11.Hirano F, Komura K, Fukawa E, Makino I. Tumor necrosis factor α (TNF-α)-induced RANTES chemokine expression via activation of NF-κB and p38 MAP kinase: roles of TNF-α in alcoholic liver diseases. J Hepatol. 2003;38:483–489. doi: 10.1016/s0168-8278(02)00456-7. [DOI] [PubMed] [Google Scholar]
- 12.Schall TJ. Biology of RANTES/SIS cytokine family. Cytokine. 1991;3:165–183. doi: 10.1016/1043-4666(91)90013-4. [DOI] [PubMed] [Google Scholar]
- 13.Casola A, Henderson A, Liu T, Garofalo RP, Brasier AR. Regulation of RANTES promoter activation in alveolar epithelial cells after cytokine stimulation. Am J Physiol. 2002;283:L1280–L1290. doi: 10.1152/ajplung.00162.2002. [DOI] [PubMed] [Google Scholar]
- 14.Chedid A, Mendenhall CL, Moritz TE, French SW, Chen TS, Morgan TR. Cell-mediated hepatic injury in alcoholic liver disease. Veterans Affairs Cooperative Study Group 275. Gastroenterology. 1993;105:254–266. doi: 10.1016/0016-5085(93)90034-a. [DOI] [PubMed] [Google Scholar]
- 15.Leevy CB, Elbeshbeshy HA. Immunology of alcoholic liver disease. Clin Liver Dis. 2005;9:55–66. doi: 10.1016/j.cld.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Nelson PJ, Kim HT, Manning WC, Goralski TJ, Krensky AM. Genomic organization and transcriptional regulation of the RANTES chemokine gene. J Immunol. 1993;151:2601–2612. [PubMed] [Google Scholar]
- 17.Schall TJ, Bacon K, Toy KJ, Goedell DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347:669–671. doi: 10.1038/347669a0. [DOI] [PubMed] [Google Scholar]
- 18.Kameyoshi Y, Dorshner AI, Mallet AI, Chrisophers E, Schroder JM. Cytokine RANTES released by thrombin-stimulated platelets is a potent chemoattractant for human eosinophils. J Exp Med. 1992;176:587–592. doi: 10.1084/jem.176.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maltby J, Wright S, Bird SNG. Chemokine levels in human liver homogenates: association between GROa and histopathological evidence of alcoholic hepatitis. Hepatology. 1996;24:1156–1160. doi: 10.1053/jhep.1996.v24.pm0008903391. [DOI] [PubMed] [Google Scholar]
- 20.DeLeve LD, Wang X, Hu L, McCuskey MK, McCuskey RS. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol. 2004;287:G757–G763. doi: 10.1152/ajpgi.00017.2004. [DOI] [PubMed] [Google Scholar]
- 21.Tsukamoto H, Mkrtchyan H, Dynnyk A. Intragastric ethanol infusion model in rodents. Methods Mol Biol. 2008;447:33–48. doi: 10.1007/978-1-59745-242-7_3. [DOI] [PubMed] [Google Scholar]
- 22.Xiong S, She H, Zhang AS, Wang J, Mkrtchyan H, Dynnyk A, Gordeuk VR, French SW, Enns CA, Tsukamoto H. Hepatic macrophage iron aggravates experimental alcoholic steatohepatitis. Am J Physiol. 2008;295:G512–G521. doi: 10.1152/ajpgi.90327.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxins in rats. Hepatology. 2000;32:1008–1017. doi: 10.1053/jhep.2000.19621. [DOI] [PubMed] [Google Scholar]
- 24.Palmada M, Kanwal S, Rutkoski NJ, Gustafson-Brown C, Johnson RS, Wisdom R, Carter BD. c-Jun is essential for sympathetic neuronal death induced by NGF withdrawal but not by p75 activation. J Cell Biol. 2002;158:453–461. doi: 10.1083/jcb.200112129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lieber CS, DeCarli LM. The feeding of ethanol in liquid diets. Alcohol Clin Exp Res. 1986;10:550–553. doi: 10.1111/j.1530-0277.1986.tb05140.x. [DOI] [PubMed] [Google Scholar]
- 26.Gukovsky I, Lugea A, Shahsahebi M, Cheng JH, Hong PP, Jung YJ, Deng QG, French BA, Lungo W, French SW, et al. A rat model reproducing key pathological responses of alcoholic chronic pancreatitis. Am J Physiol. 2008;294:G68–G79. doi: 10.1152/ajpgi.00006.2007. [DOI] [PubMed] [Google Scholar]
- 27.DeLeve LD, Wang X, McCuskey MK, McCuskey RS. Rat liver endothelial cells isolated by anti-CD31 immunomagnetic separation lack fenestrae and sieve plates. Am J Physiol. 2006;291:G1187–G1189. doi: 10.1152/ajpgi.00229.2006. [DOI] [PubMed] [Google Scholar]
- 28.Deleve LD. Dacarbazine toxicity in murine liver cells: a model of hepatic endothelial injury and glutathione defense. J Pharmacol Exp Ther. 1994;268:1261–1270. [PubMed] [Google Scholar]
- 29.Kim KS, Rajagopal V, Gonsalves C, Johnson C, Kalra VK. A novel role of hypoxia-inducible factor in cobalt chloride- and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J Immunol. 2006;177:7211–7224. doi: 10.4049/jimmunol.177.10.7211. [DOI] [PubMed] [Google Scholar]
- 30.Lebovic DI, V, Chao A, Taylor RN. Peritoneal macrophages induce RANTES (regulated on activation, normal T cell expressed and secreted) chemokine gene transcription in endometrial stromal cells. J Clin Endocrinol Metab. 2004;89:1397–1401. doi: 10.1210/jc.2003-031010. [DOI] [PubMed] [Google Scholar]
- 31.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giri RK, Selvaraj SK, Kalra VK. Amyloid peptide-induced cytokine and chemokine expression in THP-1 monocytes is blocked by small inhibitory RNA duplexes for early growth response-1 messenger RNA. J Immunol. 2003;170:5281–5294. doi: 10.4049/jimmunol.170.10.5281. [DOI] [PubMed] [Google Scholar]
- 33.Patel N, Gonsalves CS, Malik P, Kalra VK. Placenta growth factor augments endothelin-1 and endothelin -B receptor expression via hypoxia-inducible factor-1α. Blood. 2008;112:856–865. doi: 10.1182/blood-2007-12-130567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang J, Ramu S, Lee S, Aguilar B, Ganesan SK, Yoo J, Kalra VK, Koh CJ, Hong YK. Phosphate-buffered saline-based nucleofection of primary endothelial cells. Anal Biochem. 2009;386:251–255. doi: 10.1016/j.ab.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giri RK, Rajagopal V, Kalra VK. Curcumin, the active constituent of turmeric, inhibits amyloid peptide-induced cytochemokine gene expression and CCR5-mediated chemotaxis of THP-1 monocytes by modulating early growth response-1 transcription factor. J Neurochem. 2004;91:1199–1210. doi: 10.1111/j.1471-4159.2004.02800.x. [DOI] [PubMed] [Google Scholar]
- 36.Pontrelli P, Ranieri E, Ursi M, Ghosh-Choudhary G, Gesualdo L, Paulo SF, Grandaliano G. Jun-N-terminal kinase regulates thrombin-induced PAI-1 gene expression in proximal tubular epithelial cells. Kidney Int. 2004;65:2249–2261. doi: 10.1111/j.1523-1755.2004.00644.x. [DOI] [PubMed] [Google Scholar]
- 37.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haseba T, Duester G, Shimizu A, Yamamoto I, Kameyama K, Ohno Y. In vivo contribution of class III alcohol dehydrogenase (ADH3) to alcohol metabolism through activation by cytoplasmic solution hydrophobicity. Biochim Biophys Acta. 2006;1762:276–283. doi: 10.1016/j.bbadis.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 39.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 40.Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ. MKK7 is essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001;15:1419–1426. doi: 10.1101/gad.888501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNk1 and Jnk2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49:87–96. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chinenov Y, Kerppola TK. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene. 2001;20:2438–2452. doi: 10.1038/sj.onc.1204385. [DOI] [PubMed] [Google Scholar]
- 43.Minden A, Lin A, Smeal T, Derijard B, Cobb M, Davis R, Karin M. c-Jun N-terminal phosphorylation correlates with activation of the JNK subgroup but not the ERK subgroup of mitogen-activated protein kinases. Mol Cell Biol. 1994;14:6683–6688. doi: 10.1128/mcb.14.10.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hock TD, Liby K, Wright MM, McConnell S, Schorpp-Kistner M, Ryan TM, Agarwal A. JunB and JunD regulate human heme oxy-genase-1 gene expression in renal epithelial cells. J Biol Chem. 2007;282:6875–6886. doi: 10.1074/jbc.M608456200. [DOI] [PubMed] [Google Scholar]
- 45.Marden JJ, Zhang Y, Oakley FD, Zhou W, Luo M, Jia HP, McCray PB, Jr, Yaniv M, Weitzman JB, Engelhardt JF. JunD Protects the liver from ischemia/reperfusion injury by dampening AP-1 transcriptional activation. J Biol Chem. 2008;283:6687–6695. doi: 10.1074/jbc.M705606200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hernandez JM, Floyd DH, Weilbaecher KN, Green PL, Boris-Lawrie K. Multiple facets of JunD gene expressions are atypical among AP-1 family members. Oncogene. 2008;27:4757–4767. doi: 10.1038/onc.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu J, Discher DJ, Bishopric NH, Webster KA. Hypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strand. Biochem Biophys Res Commun. 1998;245:894–899. doi: 10.1006/bbrc.1998.8543. [DOI] [PubMed] [Google Scholar]
- 48.Wickremasinghe MI, Thomas LH, O’Kane CM, Uddin J, Friedland JS. Transcriptional mechanisms regulating alveolar epithelial cell-specific CCL5 secretion in pulmonary tuberculosis. J Biol Chem. 2004;279:27199–27210. doi: 10.1074/jbc.M403107200. [DOI] [PubMed] [Google Scholar]
- 49.Liu J, Guan X, Ma X. Interferon regulatory factor 1 is an essential and direct transcriptional activator for interferon κ-induced RANTES/CCl5 expression in macrophages. J Biol Chem. 2005;280:24347–24355. doi: 10.1074/jbc.M500973200. [DOI] [PubMed] [Google Scholar]
- 50.van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1α by NF-αB. Biochem J. 2008;412:477–484. doi: 10.1042/BJ20080476. [DOI] [PMC free article] [PubMed] [Google Scholar]