

Abstract
NKp30 is a natural cytotoxicity receptor (NCR) that is expressed on natural killer (NK) cells and recognizes B7-H6, which is expressed on several types of tumors but few normal cells. To target effector T cells against B7-H6-positive tumors, we have developed several chimeric antigen receptors (CAR) based on NKp30, which contain the CD28 and/or CD3ζ signaling domains with the transmembrane domains from CD3ζ, CD28 or CD8α, respectively. The data show that chimeric NKp30-expressing T cells responded to B7-H6-positive tumor cells. The NKp30 CAR expressing T cells produced IFN-γ and killed B7-H6 ligand-expressing tumor cells, and this response was dependent upon ligand expression on target cells but not on MHC expression. PBMC-derived dendritic cells (DCs) also express NKp30 ligands, including immature DCs (iDCs), and they can stimulate NKp30 CAR-bearing T cells to produce IFN-γ, but to a lesser extent. The addition of a CD28 signaling domain significantly enhanced the activity of the NKp30 CAR in a PI3-kinase-dependent manner. Adoptive transfer of T cells expressing a chimeric NKp30 receptor containing a CD28 signaling domain inhibited the growth of a B7-H6-expressing murine lymphoma (RMA/B7-H6) in vivo. Moreover, mice that remained tumor-free were resistant to a subsequent challenge with the wildtype RMA tumor cells, suggesting the generation of immunity against other tumor antigens. Overall, this study demonstrates the specificity and therapeutic potential of adoptive immunotherapy with NKp30 CAR-expressing T cells against B7-H6+ tumor cells in vivo.
Keywords: Immunotherapy, CD8 T cell, NKp30, lymphoma, B7-H6, CARs, NK cell, NCR
INTRODUCTION
T cells play important roles in anti-tumor immunity. Genetic modification of T cells with tumor-targeting chimeric antigen receptors (CARs) and adoptive transfer of CAR-modified T cells have emerged as a promising way of treating cancers (1–5). One advantage of this strategy over other approaches is the ability to expand large number of tumor-specific T cells (>1010 cells) in a relatively short time (<4 weeks) (6, 7). In addition, the functional activities of CAR-modified T cells, such as production of Th1 cytokines, cytotoxicity and in vivo persistence can be enhanced by integrating the signaling domains of co-stimulatory molecules (such as CD28, 4-1BB and OX40) to CARs (3, 5). To date, most of CARs are derived from single chain antibody fragment (scFv) against antigens on tumor cells, such as CD19 in B cell lymphoma (3, 8). However, there is a need for more targeting receptors against tumors, and it would helpful to generate pan-tumor targeting CARs that could be used to treat multiple types of cancer.
Natural killer (NK) cells attack tumor and virally infected cells in the absence of MHC restriction, utilizing a combination of signals from activating and inhibitory receptors (9). One group of activating NK cell receptors are natural cytotoxicity receptors (NCRs), which includes NKp46 (NCR1 and CD335), NKp44 (NCR2 and CD336) and NKp30 (NCR3 and CD337) (9–11). These receptors are expressed on NK cells, some subsets of γδ T cells, and they play important roles in NK-mediated tumor cell-killing (9, 12). NKp30 is a type I transmembrane protein that contains a single Ig-like extracellular domain followed by a short stalk region connected to a transmembrane segment and intracellular domain (13). In mice, NKp30 is not expressed since it is a pseudogene (14). When expressed on NK cells, NKp30 receptor as a monomer associates with CD3ζ and FcRγ for signal transduction (13, 15). Two cellular ligands for NKp30 receptor have been identified: BAT3 and B7-H6 (16, 17). BAT3 is a nuclear protein, which is involved in the interaction with P53 and induction of apoptosis after stress such as DNA damage (17, 18). BAT3 can also be released by immature dendritic cells (iDCs) on the surfaces of exosomes to stimulate NK cells (19). B7-H6 is a newly identified B7 family member. Unlike BAT3, B7-H6 is expressed on the surface of tumor cells, but not most normal cells (16). Recently, the structure of NKp30 receptor in complex with B7-H6 has been deciphered (20, 21). NKp30 uses both front and back β-sheets to engage the Ig-like V region of B7-H6 via predominantly hydrophobic interactions (20, 21). NKp30 receptor has been shown to be important in mediating anti-tumor effects in gastrointestinal stromal tumors and lymphoid leukemia (12, 15).
To develop a new strategy for T cells to recognize and attack tumor cells and induce host anti-tumor immunity, we genetically modified primary T cells with a CAR based on the NKp30 receptor (chNKp30). We have developed several chNKp30 receptors, which contain the CD3ζ and/or CD28 chain signaling domains. The transmembrane (TM) domain of NKp30 receptor was replaced with the TM from CD3ζ, CD8α, or CD28 for optimal surface expression. Our hypothesis was that the chimeric NKp30-modified T cells would react to NKp30 ligand-positive tumor cells and become fully activated resulting in elimination of the tumor and induction of host anti-tumor immunity. Many types of tumor cells (including leukemia, lymphoma, and gastrointestinal stromal tumors) and iDCs (which can induce immune tolerance and suppression) express NKp30 ligands (15, 16). Therefore, NKp30 ligand-targeting provides a fairly specific means to engage T cells against multiple types of tumors and to promote anti-tumor immunity. In this study, we determined the function and efficacy of these NKp30-based CARs.
MATERIALS AND METHODS
Mice
C57BL/6 (B6, wild-type, wt) mice were purchased from the National Cancer Institute (Frederick, MD). Perforin deficient mice C57BL/6-Prf1tm1Sdz/J (Pfp−/−) were obtained from the Jackson Laboratory (Bar Harbor, ME). All experiments were conducted according to protocols approved by Dartmouth College’s Institutional Animal Care and Use Committee.
Cell lines and cell culture
Cell lines Bosc23, GP+E86, PT67, K652, U937, Hela, U266 and Jurkat were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). Breast cancer cell lines MCF-7 and T47D were provided by Dr. James Direnzo (The Geisel School of Medicine at Dartmouth). Pancreatic cancer cell line Panc-1 was provided by Dr. Murray Korc (Indiana University, School of Medicine). Prostate cancer cell line DU145 and melanoma cell line A375 were provided by Dr. Marc Ernstoff (The Geisel School of Medicine at Dartmouth). A RMA subline RMA/B7-H6 that expresses a NKp30 ligand, B7-H6, was generated by retroviral transduction using dualtropic retroviral vectors containing the B7-H6 gene according to our previous protocol (22). Packaging cells Bosc23, GP+E86 and PT67 were grown in Dulbecco’s modified Eagle medium (DMEM) with a high glucose concentration (4.5 g/liter) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA), 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mM pyruvate, 10 mM Hepes, 0.1 mM non-essential amino acids and 50 μM 2-mercaptoethanol. All other cell lines were cultured in RPMI plus the same supplements as in DMEM. Human DCs were generated from blood mononuclear cells that were obtained from cell cones from the DHMC Blood Donor Center from leukopheresis cell donations. CD14+ cells were selected using magnetic beads (Miltenyi Biotec) and were then cultured in 6-well plates (5×105/ml) in 2ml of complete RPMI1640 media with rhIL-4 (100ng/ml, Peprotech, Rocky Hill, NJ) and rhGM-CSF (100ng/ml, Peprotech). On day 4 and 6, 2ml of fresh media with IL-4 and GM-CSF were added to the cultures. On day 8, the non-adherent cells were collected and used as iDCs. To generate mDCs, the media were replaced with fresh media containing LPS (1μg/ml, Sigma, St. Louise, MO) and CD40L (200ng/ml, Peprotech) on day 6 for 2 days.
Construction of chimeric NKp30 receptors
The full-length human NKp30, CD28 and CD8α cDNAs were purchased from Open Biosystems (Huntsville, AL). Human CD3ζ chain signaling domain and full-length B7-H6 cDNAs was cloned by RT-PCR using RNAs from Jurkat cells as templates. The chNKp30 constructs used in this study are illustrated in Fig. 1A. Wild-type (WT) NKp30 contains the full-length human NKp30. Chimeric receptor NKp30-3ζ comprises the extracellular domain (amino acids 1–139) of human NKp30 fused to the TM domain (amino acids 31–51) and signaling domains of human CD3ζ chain (amino acids 52–164). The NKp30-CD8-3ζ receptor was constructed by joining the extracellular domain of NKp30 to the TM domain of human CD8α (amino acids 183–203) and signaling domain of CD3ζ chain. The NKp30-CD28-3ζ receptor contains the TM and signaling domains of CD28 (amino acids 153–220) and the signaling domain of the CD3ζ chain. As a control receptor, NKp30-28(TM)-3ζ is similar to NKp30-CD28-3ζ, except the CD28 signaling domain was removed. All PCR reactions were performed using a high-fidelity DNA polymerase Phusion™ (New England Biolabs, Ipswich, MA). All oligos were synthesized by Sigma-Genosys (Woodsland, TX). All genes were cloned into a retroviral vector pFB-neo (Stratagene, Palo Alto, CA).
Figure 1. Structure and expression of NKp30 CARs.

Schematic diagram of wild type (wt) and chimeric NKp30 proteins are shown in (A). The extracellular (EC), transmembrane (TM) and cytoplasmic regions (CYP) are indicated. In the NKp30-3ζ, the CD3ζ chain (TM plus CYP domains) was fused to the EC domain of the NKp30 molecule. The TM domains of CD8α and CD28 were inserted between NKp30 EC and CD3ζ CYP domains to make NKp30-CD8(TM)-3ζ and NKp30-CD28(TM)-3ζ, respectively. NKp30-CD28-3ζ receptor contains the human CD28 TM and CYP domains in between the NKp30 EC and CD3ζ CYP domains. (B) NKp30 expression on human T cells 7 days after transduction with NKp30 CARs. NKp30 expression was measured using the PE-conjugated anti-NKp30 mAbs in combination with anti-CD4-FITC mAbs. More than 99% of cells were CD3+ T cells (data not shown). CD4- T cells are CD8+ T cells. The data are representative of three experiments.
Retroviral transduction
Production of retroviral vectors and retroviral transduction were performed according to modified protocols as described previously (22, 23). In brief, transduction of murine primary T cells was conducted using ecotropic viruses collected from vector-transfected GP+E86 cells, whereas dualtropic retroviral viruses generated from vector-transfected PT67 cells were used to infect human primary T cells. Primary T cells from spleens of B6 mice were infected 18~24 hr after concanavalin A (ConA, 1μg/ml, Sigma) stimulation. Two days after infection, transduced primary T cells (0.5~1×106/ml) were selected in RPMI-10 media containing G418 (1 mg/ml) plus 25 U/ml rHuIL-2 for additional 3 days. Viable cells were isolated using Histopaque-1083 (Sigma), washed extensively, and expanded for 2 days without G418 before functional analyses or intravenous injection. Primary human cells were stimulated with anti-CD3 mAb OKT3 (40 ng/ml, eBioscience) for 3 days before retroviral transduction. G418 selection of retrovirally transduced human T cells is the same as the procedures for selecting CAR transduced murine T cells.
Production of soluble human NKp30-mIgG2a fusion protein
To make a soluble human NKp30-mIgG2a fusion protein, the extracellular portion of human NKp30 (amino acids 1–262) was fused to the mouse IgG2a hinge-CH2-CH3 portion. NKp30-mIgG2a gene was cloned into pFB-neo. NKp30-mIgG2a fusion protein was expressed in retroviral vector-stably transduced B16F10 cells. Production and purification of NKp30-mIgG2a protein was performed according to previous protocols (22, 23).
Generation of anti-B7-H6 mAbs
Eight- to 12-week-old B6 mice were immunized (i.p.) with mitomycin-C-treated RMA/B7-H6 cells (5×106). Two weeks after initial immunization, mice were boosted with 50 μg recombinant B7-H6 (extracellular domain) prepared from E. Coli and Freund’s incomplete adjuvant (Sigma) three times at weekly intervals. Three days after last boosting immunization, mice splenocytes were fused to NS1 cells (provided by Dr. William Wade, The Geisel School of Medicine at Dartmouth) using standard techniques and hybridoma supernatants were screened for reactivity to both B7-H6-negative and –positive cell lines by flow cytometry. Two clones 47.39 and 127.4 were isolated (both were of the IgG2a subclass).
Flow cytometry
For flow cytometry analysis of NKp30 ligand expression, cells were stained with either NKp30-mIgG2a or anti-B7-H6 mAb followed by DyLight ™649 conjugated-goat anti-mouse IgG (Biolegend, CA). Cell-surface phenotyping of transduced primary T cells was determined by staining with FITC conjugated-anti-CD4 (clone OKT4, Biolegend), PE conjugated-anti-NKp30 (clone P30-15, Biolegend) mAbs. To analyze the cell surface phenotype of DCs, the following mAbs were used: APC-conjugated anti-CD86 (Biolegend), PE-conjugated anti-CD11c (Biolegend), FITC-conjugated anti-CD83 (Biolegend), FITC-conjugated anti-HLA-DR (eBioscience, CA). Intracellular staining of Bcl-XL in T cells was performed using anti-Bcl-XL-FITC (SouthernBiotech, Birmingham, AL) based on the protocol described previously (24). All samples were pre-incubated with either FcR block antibody (anti-mouse CD16/CD32, 2.4G2, BioXcell, Lebanon, NH) for mouse cells staining or human γ globins (Cohn’s fraction, G4386, Sigma) for human cell staining. Cell fluorescence was monitored using an Accuri C6 cytometer (Ann Arbor, MI). Flow cytometry analysis was performed using either Accuri or FlowJo software (Ashland, OR).
Reverse transcription-PCR and quantitative PCR
Extraction of total RNA and preparation of cDNAs from human tumor cell lines and PBMCs were performed as described (23). The resulting cDNA, corresponding to 50 ng of total RNA, was subjected to PCR amplification in a total volume of 20 μL, including 0.5 μmol/L of each primer, 0.2 mmol/L of each deoxynucleotide triphosphate, and 1 unit Taq DNA polymerase (New England Biolabs). The primers used for amplification are shown in Supplemental Table 1. The PCR conditions were as follows: 95°C for 5 minutes followed by 30 cycles of 95°C for 30 seconds (denaturation), 60°C for 30 seconds (annealing), and 72°C for 30 seconds (extension), with 3-minute incubation at 72°C at the end. The PCR products were run on agarose gels and visualized by staining with SYBR safe (Invitrogen). For quantitative real-time PCR of human IL-2 mRNA, Triplicates of cDNA samples from T cells were mixed with the SYBR Green Master Mix (Applied Biosystems) and IL-2 specific primers (Supplemental Table 1) in a total volume of 25 μL. The reactions were run on a Bio-Rad icycler (Bio-Rad). The relative gene expression was calculated as described (25). GAPDH gene was used as an internal control. The mean value of relative IL-2 expression in control mAb-treated T cell samples was set as 1.
Cytotoxicity assay
Cytotoxicity of T cells against target cells was determined by a LDH release assay using the CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega, WI). Specific lysis was determined by the following equation: % Specific lysis = ((experimental − effector spontaneous − target spontaneous)/(target maximum − target spontaneous)) × 100. In the cytotoxicity blocking experiments, K562 target cells were pre-incubated with a soluble NKp30 receptor, NKp30-mIgG2a (10μg/ml), for 30 min before co-culture with T cells in a LDH-release assay.
Cytokine production by T cells
To determine whether CAR T cells responded to tumor cells with production of IFN-γ, T cells (105) were co-cultured with suspension tumor cells at an E:T ration of 1:1 or with adherent tumor cells at an E:T ratio of 1:0.25 (105:2.5×104) in 96-well v-bottom or flat-bottom plates, respectively, for 24 h. Cell-free supernatants were assayed for IFN-γ by ELISA using Duoset ELISA kits (R&D systems).
Treatment of lymphoma-bearing mice with chimeric NKp30-modified T cells
As a systemic mouse lymphoma model, B6 mice were injected with 105 of RMA/B7-H6 cells via tail veins in 400μl of HBSS. For treatment with T cells, mice were administered i.v. with 5×106 wildtype NKp30 (wtNKp30) or chNKp30-modified T cells starting on day 5 post-tumor inoculations. T-cell transfer was repeated on days 7 and 9 using T cells from the same T cell preparation that were expanded for additional days in vitro. Mice were monitored closely and sacrificed when they became moribund.
Tumor rechallenge
Mice that survived for 60 days after initial tumor inoculation without signs of disease were regarded as tumor free and were inoculated s.c. with 104 wild-type RMA tumor cells on the shaved right flank. Naïve B6 mice were used as controls. Tumor size was monitored every two days, and mice were sacrificed when tumor burden became excessive.
Statistical analysis
Differences between groups were analyzed using a Student’s t-test or ANOVA. P values < 0.05 were considered significant. Kaplan-Meier survival curves were plotted and analyzed using Prism software (GraphPad Software, San Diego, CA).
RESULTS
Construction and expression of chimeric antigen receptors based on the human NKp30 receptor
A series of chNKp30 receptor-expressing retroviral vectors were generated in which all receptors share the common extracellular domain of NKp30. The structures of the chNKp30 and wtNKp30 receptors used are diagramed in Fig. 1A. In NKp30-3ζ, the transmembrane (TM) domain and cytoplasmic tail of NKp30 was replaced by the corresponding region of CD3ζ chain. To facilitate surface expression and dimerization of chimeric receptors, the human CD8α TM domain was used instead to make NKp30-CD8(TM)-3ζ. To determine whether CD28 signaling domain can enhance the overall activation signal upon engagement of chNKp30 to its ligand, the human CD28 TM and cytoplasmic domains were inserted between the extracellular domain of NKp30 and CD3ζ cytoplasmic tail. The resulting receptor is referred to as NKp30-CD28-3ζ. A control receptor, NKp30-CD28(TM)-3ζ, has the CD28 TM portion but not the cytoplasmic region of the CD28 molecule included.
Surface expression of chNKp30 receptors on human T cells was analyzed by flow cytometry using anti-NKp30 and anti-CD4 mAbs. As shown in Fig. 1B, retroviral transduction of human T cells with wtNKp30 gene did not lead to significant surface expression. Insufficient expression of wtNKp30 on the cell surface may be due to an absence of FcγR expression on T cells, which is associated with CD3ζ and NKp30 on human NK cells. Replacement of NKp30 TM domain with CD3ζ TM domain improved CAR expression. The hinge and TM domains of CD8α have been widely used for expressing CARs on T cells (7, 26). Our results showed that CD8α TM domain efficiently allows surface expression of chNKp30 receptor on T cells, which is consistent with these previous studies. Similar to the CD8α TM domain, human CD28 TM domain resulted in higher surface expression of chNKp30 with or without the CD28 cytoplasmic (CYP) domain.
Chimeric NKp30-expressing T cells produce IFN-γ upon co-culture with NKp30 ligand-positive tumor cells
A panel of human tumor cell lines was screened for NKp30 ligand expression using a soluble human NKp30-mIgG2a fusion protein and an anti-B7-H6 mAb (called 47.39). As shown in Fig. 2A, K562, A375, Hela and T47D cells expressed high amounts of NKp30 ligands, whereas U937, RPMP8226, DU145 and Panc-1 cells expressed low amounts of NKp30 ligands. Some tumor cells (IM9, U266 and MCF-7) did not express NKp30 ligands on the cell surface. All tested human tumor cells have detectable levels of BAT3 mRNA, as detected by RT-PCR (Fig. 2B). In contrast, B7-H6 mRNA levels were correlated to surface expression of NKp30 ligands, suggesting that B7-H6 is the major surface ligand of NKp30 in tumors. The result was also consistent with the fact that BAT3 is a nuclear protein, which is usually not expressed on cell surface. In addition, RT-PCR results showed that PBMCs lacked the mRNAs of B7-H6 and Bat3 (Fig. 2B)
Figure 2. Human tumor cell lines express varying amounts of NKp30 ligands on the cell surface.

(A) NKp30 ligand expression on the surface of human tumor cell lines was measured by flow cytometry using anti-B7H6 mAbs (solid line) or a soluble NKp30 receptor fused to a mouse IgG2a Fc region (NKp30-mIgG2a, dashed line) followed by staining with APC-conjugated goat anti-mouse IgG. Isotype controls are shown as a dotted line. (B) Detection of NKp30 ligands B7-H6 and BAT3 by RT-PCR. mRNA expression of human NKp30 ligands B7-H6 and BAT3 in human cell lines and human PBMCs were determined by RT-PCR. GAPDH was used as an internal control.
To determine whether chimeric NKp30-transduced human T cells were able to recognize NKp30 ligand-positive tumor cells, the chNKp30 CAR-bearing T cells were cultured with different tumor cells and IFN-γ responses measured by ELISA. As shown in Fig. 3, NKp30-3ζ+, NKp30- CD8(TM)-3ζ+, NKp30-CD28(TM)-3ζ+ or NKp30-CD28-3ζ+ T cells produced significant amounts of IFN-γ after co-culture with NKp30 ligand-positive cells but not when cultured with ligand-negative cells indicating that these NKp30 CAR-modified T cells could functionally recognize NKp30 ligand-bearing tumor cells. In contrast, wtNKp30-modified T cells did not show any significant response to the stimulation by NKp30–ligand positive cells. NKp30-CD28-3ζ expressing T cells produced significantly more IFN-γ than T cells expressing NKp30-3ζ+, NKp30- CD8(TM)-3ζ+, or NKp30-28(TM)-3ζ+ in the presence of NKp30 ligand-positive tumor cells, especially when the ligand expression was low. The reason was likely due to better surface expression of this CAR and/or the presence of the CD28 co-stimulatory signaling domain in NKp30-CD28-3ζ.
Figure 3. NKp30 CAR-modified T cells respond to NKp30 ligand positive cells by producing IFN-γ.

Five to seven days after retroviral transduction, NKp30 CAR-modified T cells (105 cells) were co-cultured with either irradiated or mitomycin-C treated tumor cells for 24 h. (A) Suspension tumor cells (105 cells) and (B) adherent tumor cells (2.5×104 cells) were used. RMA and MCF-7 cells were used as negative controls. IFN-γ amounts in the supernatants were analyzed with ELISA. Results are shown in mean + SD. *: p<0.05
Human DCs express NKp30 ligands and can stimulate chimeric NKp30-expressing T cells to produce IFN-γ
It has been known that NKp30 plays an important role in human NK-DC interactions (27, 28). At low NK:DC ratios, interaction with DCs promote IFN-γ production and cytotoxicity by NK cells in NKp30-dependent manner (28, 29), which suggests that DCs express NKp30 ligands. With the use of NKp30-mIg2a, we confirmed that both immature DCs (iDCs) and mature DCs (mDCs) can bind to soluble NKp30, which is consistent with DCs expressing ligands for NKp30 (Fig. 4A). The level of cell surface staining on iDCs was higher than in mDCs. However, there was no significant expression of B7-H6 on DCs as determined with mAb 47.39, a specific anti-B7-H6 mAb. To determine whether the NKp30 CAR-modified T cells can respond to DCs, T cells were co-cultured with either iDCs or mDCs at a ratio of 5:1(T:DC) for 24h. IFN-γ production (200–800 pg/ml) was observed by NKp30-28-3ζ-modified T cells after co-culture with DCs (Fig. 4B). Compared with mDCs, iDCs induced higher amounts of IFN-γ, which reflected their higher binding to soluble NKp30.
Figure 4. Human DCs bind to NKp30 and can stimulate autologous NKp30-CD28-3ζ-modified T cells to produce IFN-γ.

(A) The cell surface phenotype and binding to NKp30 of PBMC-derived human DCs (both iDCs and mDCs) was determined by flow cytometry. Specific mAb or NKp30-Ig as indicated (solid line) or an isotype control Ab staining (dashed line) are shown. (B) Five to seven days after retroviral transduction, NKp30 CAR-modified T cells (105 cells) were co-cultured with either iDCs or mDCs cells at a ratio of 5:1 (T: DC) for 24 h. IFN-γ amounts in the supernatants were determined by ELISA. Results shown (mean + SD) are representative of two experiments shown. *: p<0.05
Chimeric NKp30-bearing human T cells kill NKp30-ligand positive tumor cells
The cytotoxic activity of chNKp30-modified human T cells against various tumor cell lines was determined. As shown in Fig. 5A, NKp30 CAR-bearing T cells were able to lyse NKp30 ligand-positive target cells (RMA/B7-H6, T47D, Panc-1, A375, K562 and RPMI8226) but not the ligand-negative cell lines RMA and MCF-7 in vitro. Similar to cytokine production, no significant killing was observed when wtNKp30-modified T cells were used. NKp30-CD28-3ζ receptor bestowed T cells with significantly higher lytic activity than NKp30-3ζ, NKp30-CD8(TM)-3ζ, or NKp30-CD28(TM)-3ζ. Since RMA/B7-H6 tumor cells lack expression of human MHC class I and II molecules, these data indicate that the chNKp30 receptor-modified T cells mediated killing of these tumor cells was ligand dependent and MHC-independent. To confirm that chNKp30-mediated killing is dependent on the interactions between NKp30 and its ligands, soluble NKp30 (NKp30-mIgG2a) was pre-incubated with K562 target cells prior to the co-culture with T cells. As shown in Fig. 5B, NKp30-mIgG2a significantly reduced NKp30-28-3ζ-bearing T cell-mediated cytotoxicity. These results demonstrated that chNKp30-bearing T cells killed ligand-positive tumor cells, and interaction between chNKp30 receptors and NKp30 ligands was essential for chNKp30-mediated T cell function.
Figure 5. NKp30 CAR-modified T cells lyse NKp30 ligand-positive tumor cells in vitro.

(A) Effector T cells derived from human PBMCs were modified with either wtNKp30(grey), NKp30-3ζ (black) or NKp30-CD28-3ζ(thatch) were cocultured with tumor cells at a ratio of 5:1 in 5-hr LDH release assays. The data are presented as mean + SD of triplicates from 2 independent experiments. (B) Effector T cells modified with wtNKp30 or NKp30-CD28-3ζ were cocultured with target cells K562 in the presence of 10μg/ml NKp30-mIgG2a (■) or mouse IgG (□) at a ratio of 5:1, and percent specific lysis determined after a 5-hr LDH release assay. The data are presented as mean + SD and are representative of 2 independent experiments. (C) PI3-kinase is involved in NKp30-CD28-3ζ receptor-mediated cytotoxicity. NKp30-modified effector T cells were preincubated with a PI3-kinase inhibitor LY294002 (10 μM) at 37°C for 1 h before co-culture with K562 target cells at a E:T ratio of 5:1 in 5-hr LDH release assays. Vehicle controls are 0.1% DMSO. The data shown are presented as mean + SD of triplicates and are representative of two independent experiments. *: p<0.05
Activation of PI3-kinase (PI3-K) pathway is involved in enhanced IFN-γ production as well as cytotoxicity of NKp30-CD28-3ζ+ T cells
Crosslinking of CD28 leads to activation of the PI3-K pathway. Therefore, we hypothesized that significantly enhanced IFN-γ production and cytotoxicity by T cells after engagement of the NKp30-CD28-3ζ receptor might be due to activation of PI3-K. To test this hypothesis, a PI3-K inhibitor, LY294002, was added to the co-culture of CAR T cells with irradiated Hela cells. Compared with vehicle control, the presence of LY294002 (10 μM) inhibited NKp30-CD28-3ζ T cell-mediated IFN-γ production and cytotoxicity, indicating that incorporation of a CD28 signaling domain into chimeric NKp30 receptors can activate T cells via the PI3-K pathway (Fig 5C).
Integration of a CD28 signal into the NKp30 CAR promotes in vitro T-cell proliferation
It is known that CD28 signals enhance T-cell survival and proliferation. To investigate whether integration of CD28 signaling in the chNKp30 receptor resulted in similar outcomes upon engagement of NKp30 receptor, CFSE-labeled T cells were cocultured with Hela cells (NKp30 ligand-positive) in the presence of a low amount of IL-2 (25 U/ml) for 3 days. As shown in Fig. 6A, engagement of NKp30-CD28-3ζ-bearing T cells led to more T-cell proliferation than NKp30-3ζ-bearing T cells. Two important mechanisms through which CD28 signaling promotes T-cell survival are through up-regulation of IL-2 and an anti-apoptotic protein, Bcl-XL. To determine whether NKp30-CD28-3ζ+ T cells up-regulated IL-2 and Bcl-XL in response to cross-linking of the chimeric receptor, T cells were cultured in anti-NKp30 mAb-coated wells for 24 hours. IL-2 mRNA and Bcl-XL protein were determined using real-time PCR, and intracellular staining, respectively. The results show that NKp30-CD28-3ζ+ T cells increased IL-2 expression by 25-fold after receptor cross-linking as compared to 10-fold induction in NKp30-3ζ+ T cells (Fig. 6B). No significant up-regulation of IL-2 was observed in wtNKp30-modified T cells cultured under these conditions. Similarly, we observed higher expression of Bcl-XL in NKp30-CD28-3ζ+ T cells as compared to NKp30-3ζ+ T cells (Fig. 6C). These data suggest that NKp30-CD28-3ζ+ T cells can receive a co-stimulatory signal through CD28 that leads to increased IL-2 and Bcl-XL expression.
Figure 6. Engagement of NKp30-CD28-3ζ receptor led to increased T cell proliferation and up-regulation of IL-2 and Bcl-XL.

(A) NKp30 receptor (either wtNKp30 or chNKp30)-modified human T cells were labeled with CFSE as described in Methods and Materials and were cocultured with Hela cells (105 cells, NKp30 ligand-positive) in the presence of a low amount of IL-2 (25 U/ml) for 3 days. Analysis of T cell proliferation (i.e. CFSE dilution) was performed on both NKp30+ (FL4) and NKp30− cells within the same mixed T cell population by flow cytometry. (B) NKp30-modified T cells (2.5×105 cells) were cultured in anti-NKp30 mAb (4μg)-coated 24-well plates for 24 h. Mouse IgG was used as a negative control. IL-2 gene expression was determined by real-time PCR as described in Methods and Materials. Results are shown as fold increase, in which the IL-2 gene expression in the control mAb-treated T cells was normalized to 1. Data are presented as mean + SD from two independent experiments. *: p<0.05 (C) Twenty-four hours after cross-linking with immobilized anti-NKp30 mAbs, as described above, T cells were collected. Bcl-XL expression was determined by flow cytometry after intracellular staining with anti-Bcl-XL-FITC (solid line) or isotype control mAbs (broken line).
Adoptive transfer of NKp30-CD28ζ+ T cells significantly improves the survival of RMA/B7- H6 tumor-bearing mice and induces the generation of immunological memory
Because NKp30 is a pseudogene in inbred mice, we determined whether human NKp30 CARs could be expressed on mouse T cells, which allows the testing of in vivo efficacy of chimeric NKp30–modified T cells against NKp30 ligand-positive tumor cells in immunocompetent mouse tumor models. Similar to the expression profile observed on human T cells, NKp30 CARs were expressed on mouse T cells (Fig. 7A). All chNKp30-modified murine T cells responded to co-culture with RMA/B7-H6 cells but not RMA cells by producing IFN-γ, and NKp30-CD28-3ζ bearing T cells produced more IFN-γ compared to NKp30-3ζ-expressing T cells (Fig. 7B). WtNKp30-modified mouse T cells also produced low level of IFN-γ in response to B7-H6 (Fig. 7B). In addition, chNKp30-modified mouse T cells were highly cytotoxic. Even at an E:T ratio of 1:1, T cells expressing either NKp30-CD28-3ζ or NKp30-3ζ receptor killed RMA/B7-H6 cells at an efficiency of 80% (Fig. 7C). No significant killing of NKp30 ligand-negative RMA cells was observed. Perforin (Pfp) had a role in the killing process because NKp30 CAR-modified T cells deficient in pfp showed a significantly reduced (p <0.05) ability to kill RMA/B7-H6 tumor cells.
Figure 7. Human NKp30 CARs can be expressed and function on murine T cells.
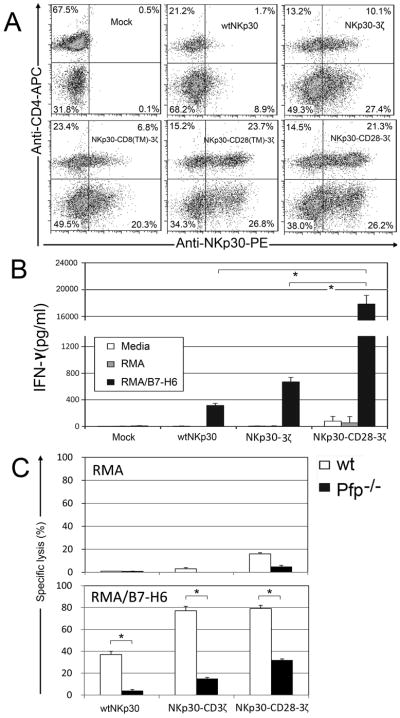
(A) Human NKp30 expression on mouse T cells 7 days after transduction. NKp30 expression was detected using the PE-conjugated anti-NKp30 mAb in combination with anti-mouse CD4-FITC mAb. CD4-T cells are CD8+ T cells. The data are representative of three experiments. (B) Seven days after retroviral transduction, NKp30-modified T cells (105 cells) were co-cultured with irradiated RMA/B7-H6 cells (105 cells) for 24 h. Mouse lymphoma cell line RMA used as a negative control. IFN-γ amounts in the supernatants were analyzed by ELISA. Results are shown in mean + SD. (C) Effector T cells derived from WT B6 (open bar) and perforin deficient (Pfp−/−, filled) mice that were modified with either wtNKp30, NKp30-3ζ or NKp30-CD28-3ζ were cocultured with RMA or RMA/B7-H6 cells, respectively, at a ratio of 1:1 in 5-hr LDH release assays. The data are presented as mean + SD of triplicates from a representative experiment of two independent experiments. *: p<0.05
Previous results show that intravenous injection of RMA cells leads to systemic lymphoma in B6 mice (30). Therefore, we tested whether RMA/B7-H6 cells could develop systemic lymphoma after i.v. inoculation into immunocompetent B6 mice. Although B7-H6 is a human molecule, expression of B7-H6 did not significantly alter the growth of RMA cells. Intravenous administration of 105 RMA/B7-H6 cells led to development of systemic lymphoma, with a median survival of 18 days, which is the typical growth for a similar dose of RMA tumor cells in B6 mice (30). To determine the efficacy of NKp30 CAR-bearing T cells in eliminating established tumors in this model, 5×106 T cells (transduced with either wtNKp30 or chNKp30 genes) were administered i.v. on days 5, 7 and 9 post-tumor inoculations. As shown in Fig. 8A, treatment with NKp30-CD28-3ζ+ T cells significantly improved median survival from 18 days to 30 days, and four of twenty-three mice (17%) became long-term survivors (Fig. 8A). Surprisingly, although wtNKp30, NKp30-3ζ, NKp30-CD8(TM)-3ζ allowed T cells to respond to RMA/B7-H6 cells in vitro, these murine T cells modified with these receptors showed little effect on survival of tumor-bearing mice in this aggressive lymphoma model.
Figure 8. Adoptive transfer of NKp30-CD28-3ζ-modified T cells promotes the survival of RMA/B7-H6 tumor-bearing mice and induces memory responses.
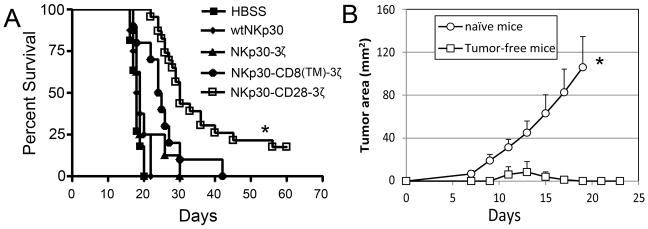
(A) B6 mice were inoculated with RMA/B7-H6 (105 cells, i.v., day 0). Five days after tumor inoculation, mice were treated with either wtNKp30-(◆, n=8), NKp30-3ζ-( ▲, n=8), NKp30-CD8(TM)-3ζ-( ●, n=10) or NKp30-CD28-3ζ (□, n=19)-modified T cells (5×106 cells). T-cell transfer was repeated on days 7 and 9. HBSS (■, n=11) treatment was used as negative control. Data pooled from two independent experiments are presented in Kaplan-Meier survival curves. *: p<0.01. (B) Tumor free mice (□) in the NKp30-CD28-3ζ-treated RMA lymphoma model (shown in panel A) and age-matched naïve mice (○) were re-challenged with wild type RMA cells (104 cells, s.c.) into the left flank. The tumor areas are represented as Mean + SEM. *: p<0.01.
Because ligand-negative tumor cells could selectively grow out after CAR T cell therapy, it would be beneficial if treatment with NKp30-CD28-3ζ+ T cells induced host immunity against other tumor antigens. The four mice that remained tumor-free after 60 days (from Fig. 8A) were re-challenged s.c. with wild-type RMA tumor cells, which do not express any NKp30 ligands. These tumor-free mice were resistant to a subsequent challenge of RMA cells, whereas all control naive mice had aggressive tumors after 18 days (Fig. 8B). These data are consistent with the idea that adoptive transfer of NKp30-CD28-3ζ+ T cells allowed hosts to generate immunological memory against RMA tumor antigens. In addition, we observed that NKp30-CD28-3ζ+ T cells persisted longer than either NKp30-3ζ+ or NKp30-CD8(TM)-3ζ+ T cells (data not shown), which correlates with their enhanced anti-tumor efficacy.
DISCUSSION
The specificity of a therapeutic agent is a key factor in cancer immunotherapy. T cells use their highly specific TCR to recognize a specific antigenic peptide in the context of MHC. However, T cell-mediated anti-tumor immunity is hindered by many factors, such as (1) low frequency of tumor-specific T cells; (2) low affinity of TCR to its ligand due to central deletion of T cells with high affinity to self-antigens in the thymus; and (3) down-regulation of MHC or antigen presentation on tumor cells (31). As an important component of innate immunity, NK cells respond to cancers with broader specificities using receptors such as the natural cytotoxic receptors (NCRs) (9, 32). One promising NCR that can be harnessed for T-cell-based cancer immunotherapy is NKp30, since its primary ligand B7-H6 is expressed on tumor cells but not 48 different normal tissues (33). Thus, the NKp30 receptor-B7-H6 interaction provides a relatively specific system for T cells to recognize tumor cells and become fully activated irrespective of tumor cell MHC/tumor antigen expression. Several human hematopoietic and non-hematopoietic tumor cells have been found to express B7-H6 (34). Genetic modification of human T cells with chimeric NKp30 receptors provides a rapid means for generating anti-tumor T-cell immunity against multiple human tumors. Although NK cells are able to recognize tumors using a variety of activating receptors, NK activities are also restricted by expression of inhibitory receptors (34). NK cells have demonstrated improved clinical outcomes when in AML patients when there has been a mismatch between donor NK cells and host MHC molecules (35, 36). However, adoptive therapy with NK cells has shown little or no clinical benefit in patients with solid tumors or NK insensitive tumors even when high amounts of NK cells are present in the blood after transfer (37, 38). In comparison to NK cells, T cells are much more abundant, proliferate faster in response to stimulation and have a longer half-life. Therefore, combination of broad spectrum of recognizing tumor cells by NK cell receptors, such as NKp30, with these advantageous biological properties of T cells are expected to translate into enhanced anti-tumor efficacy.
When expressed on NK cells, NKp30 associates with CD3ζ and FcRγ chains as a monomer (13). Retroviral transduction of wtNKp30 gene into human T cells does not lead to significant surface expression, which may be due to the absence of FcRγ chain expression in human αβ T cells. In NKp30-3ζ, the NKp30 TM and CYP domains were replaced with the counterparts of CD3ζ chain to minimize the need for association with FcRγ chain and improved surface expression of chimeric NKp30 was observed. Whether the NKp30-3ζ receptor expresses as a homodimer or hybridimer with CD3ζ chain is unclear and will be determined in future studies. Since both CD8α and CD28 can form homodimers without the need to association with other proteins, inclusion of either CD8α or CD28 TM domains into chimeric NKp30 receptors gave rise to high surface expression (>50% of cells) with these chimeric receptors. Higher expression of CD28-containing chimeric receptors has been shown to correlate with better functional activity (such as cytokine production and cytotoxicity) (39). CD28 TM–containing CARs often show higher surface expression than CARs with CD3ζ TM (40). The reason may be that CD28 TM-containing CARs tend to form predominantly homodimers independent of TCR-CD3 complex, whereas CD3ζ TM-containing CARs can form heterodimers of CAR with the endogenous CD3ζ chain that may be limited by TCR-CD3 complex expression (40, 41)
Chimeric NKp30-modified T cells responded to NKp30 ligand-positive tumor cells by producing IFN-γ. In anti-tumor immunity, IFN-γ production is often essential to mediate anti-tumor effects by adoptively transferred T cells in vivo because IFN-γ coordinates a diverse array of cellular programs (42, 43). In some models, IFN-γ has been shown to be critical in inhibiting tumor growth by inducing apoptosis or inhibiting angiogenesis (44, 45). Corthay et al. showed that IFN-γ was critical for CD4+ T cell-mediated macrophage activation and tumor rejection (46). NKp30-3ζ+ T cells showed a similar in vitro cytotoxicity against RMA/B7-H6 cells as NKp30-CD28-3ζ+ T cells. However, the IFN-γ production by NKp30-CD28-3ζ+ T cells in response to the stimulation of RMA/B7-H6 cells was much higher, suggesting that the threshold for chimeric NKp30 receptors to exert cytotoxicity is lower than IFN-γ production. There was a general correlation between B7-H6 expression and IFN-γ production, which was not observed for cytotoxicity. Cytotoxicity was observed by NKp30 CAR T cells if B7H6 was expressed on the cell surface and did not differ within the variation of ligand expression of the tumor cells tested. Faroudi et al. reported a similar observation that the threshold for CTL-mediated cytotoxicity was 100-fold less than that for IFN-γ production, and the differences were likely due to the formation of distinct immunological synapses (lytic synapse vs stimulatory synapse) (47). Much more IFN-γ was produced when CAR T cells were cultured with suspension tumor cells than adherent tumor cells, which may reflect differences in the assay conditions. Differences in responses against individual tumor cell lines may also reflect variations in the tumor cells production of inhibitory or stimulatory factors that may also affect CAR T cell function. In addition, only NKp30-CD28-3ζ+ bearing T cells conferred significant in vivo anti-tumor efficacy, suggesting that IFN-γ production, in addition to direct killing, by chimeric NKp30-modified T cells may be critical for in vivo efficacy.
Besides B7-H6, HLA-B–associated transcript 3 (BAT3) has also been identified as a ligand for NKp30 receptor (17, 18). However, BAT3 is a nuclear protein, which is not normally expressed on cell surface. BAT3 mRNA-positive tumor cells (such as MCF-7, DU145 and IM9) lack surface staining of NKp30 ligands, which is consistent with BAT3 being a nuclear protein. In addition, the responsiveness of NKp30-CD28-3ζ+ T cells to tumor cells was correlated with the expression of B7-H6 mRNA in the target cells, suggesting that B7-H6 is the major NKp30 ligand being recognized on the tested tumor cells. In this study, freshly isolated human PBMCs were deficient in B7-H6 based on flow cytometry and RT-PCR data. However, PBMC-derived DCs (both iDCs and mDCs) that were generated from in vitro culture (7–10 days) with GM-CSF and IL-4 expressed NKp30 ligands, although B7-H6 expression was not expressed by DCs. BAT3 has been shown to be expressed on the surface of iDC-released exosomes, which can stimulate NK cells to produce IFN-γ (19). Whether BAT3 is directly expressed on the surface of iDCs is not clear. NKp30 plays a role in NK cell-mediated killing of iDCs, but NK cell killing of monocytes and mDCs is minimal (28). In this study, NKp30-CD28-3ζ+ T cells produced significantly less IFN-γ upon co-culture with mDCs compared with iDCs, which is in agreement with previous findings. Lower levels of NKp30 ligands on mDCs may contribute to reduced stimulatory effects on NKp30-CD28-3ζ+ T cells. To further minimize the reactivity of CAR-modified T cells against normal cells, such as DCs, B7-H6-specific CARs can be developed.
Heparan sulfate epitope(s) on tumor cells may also bind to NKp30 (48, 49). Removal of heparan sulfate from the surface proteins on some tumor cells with heparin lyases resulted in ~50% reduction in the binding to an NKp30-Ig fusion protein (48). The glycosylation status of NKp30 receptor affects its binding to some of the ligands (49). NKp30 receptor has several potential N-linked glycosylation sites. Treatment of NKp30 receptor with protein N-glycanase F (PNGase), which removes the N-glycans from glycoproteins, significantly reduces the binding of NKp30 to its ligands (49). However, N-linked glycosylation may not affect the NKp30-B7-H6 interaction because the glycosylation sites of NKp30 are located outside the interface of NKp30 with B7-H6 (21).
Incorporation of domains from co-stimulatory molecules (such as 4-1BB, OX40 and CD28) into chimeric receptors has also been shown to improve anti-tumor efficacy of T cells both in vitro and in vivo (26, 50–51). The signaling domain of CD28 in chimeric receptors is particularly important in IL-2 production, proliferation, and cell survival of lymphocytes (52–54). In this study, we have confirmed that upon cross-linking of NKp30 receptor by anti-NKp30 mAbs or engagement with NKp30 ligand-positive tumor cells, the CD28 signaling domain in NKp30- CD28-3ζ receptor enhanced T cell functions, such as up-regulation of IL-2 and Bcl-XL and cell proliferation.
This study showed that adoptive therapy of NKp30-CD28-3ζ+ T cells into RMA/B7-H6 tumor-bearing mice elicited hosts to generate memory responses against NKp30 ligand-negative RMA cells, suggesting the development of “epitope spreading”. Initial targeting of B7-H6 on RMA cells by NKp30-CD28-3ζ+ T cells is expected to lead to tumor cell death followed by efficient presentation of tumor antigens by professional APCs (such as DCs), probably due to the presence of proinflammatory cytokines (such as IFN-γ, TNF-α and GM-CSF) and chemokines. Cross-priming of host T cells by these APCs may further lead to expansion of polyclonal tumor-specific T cells (i.e., epitope spreading). In order for tumors to evade immune surveillance, tumor cells are selected with mutations or deletions of targeted antigens (55, 56). Therefore, induction of polyclonal tumor-specific T cells will minimize the chances of tumor cells to “escape”. Generation and maintenance of memory responses are important for reducing recurrent tumor disease.
In summary, a chNKp30 receptor can be utilized to redirect T cells against NKp30 ligand-expressing tumors. Incorporation of a CD28 signaling domain into chimeric NKp30 receptors can stimulate both primary and co-stimulatory signals for enhanced anti-tumor activities. NKp30 can recognize its ligands on several different types of tumor cells, and this study demonstrates a potential broad therapeutic utility of this chNKp30 CAR approach for the treatment of cancer.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the Hitchcock Foundation (250-4032), NIH (CA130911 and T32AR007576), and support from the Department of Microbiology & Immunology and the Norris Cotton Cancer Center.
We thank the NCI Biological Resource Branch for providing recombinant human IL-2, and the staff of the Animal Resources Center for assistance with animal care.
References
- 1.Rossig C, Brenner MK. Genetic modification of T lymphocytes for adoptive immunotherapy. Mol Ther. 2004;10:5–18. doi: 10.1016/j.ymthe.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 2.Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell. 2003;3:431–437. doi: 10.1016/s1535-6108(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 3.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced Leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park TS, Rosenberg SA, Morgan RA. Treating cancer with genetically engineered T cells. Trends Biotechnol. 2011;29:550–557. doi: 10.1016/j.tibtech.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21:215–223. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 8.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, Carroll RG, Riley JL, Pastan I, June CH. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 10.Moretta L, Bottino C, Pende D, Castriconi R, Mingari MC, Moretta A. Surface NK receptors and their ligands on tumor cells. Semin Immunol. 2006;18:151–158. doi: 10.1016/j.smim.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Correia DV, Fogli M, Hudspeth K, da Silva MG, Mavilio D, Silva-Santos B. Differentiation of human peripheral blood Vdelta1+ T cells expressing the natural cytotoxicity receptor NKp30 for recognition of lymphoid leukemia cells. Blood. 2011;118:992–1001. doi: 10.1182/blood-2011-02-339135. [DOI] [PubMed] [Google Scholar]
- 13.Pende D, Parolini S, Pessino A, Sivori S, Augugliaro R, Morelli L, Marcenaro E, Accame L, Malaspina A, Biassoni R, Bottino C, Moretta L, Moretta A. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J Exp Med. 1999;190:1505–1516. doi: 10.1084/jem.190.10.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hollyoake M, Campbell RD, Aguado B. NKp30 (NCR3) is a pseudogene in 12 inbred and wild mouse strains, but an expressed gene in Mus caroli. Mol Biol Evol. 2005;22:1661–1672. doi: 10.1093/molbev/msi162. [DOI] [PubMed] [Google Scholar]
- 15.Delahaye NF, Rusakiewicz S, Martins I, Menard C, Roux S, Lyonnet L, Paul P, Sarabi M, Chaput N, Semeraro M, Minard-Colin V, Poirier-Colame V, Chaba K, Flament C, Baud V, Authier H, Kerdine-Romer S, Pallardy M, Cremer I, Peaudecerf L, Rocha B, Valteau-Couanet D, Gutierrez JC, Nunes JA, Commo F, Bonvalot S, Ibrahim N, Terrier P, Opolon P, Bottino C, Moretta A, Tavernier J, Rihet P, Coindre JM, Blay JY, Isambert N, Emile JF, Vivier E, Lecesne A, Kroemer G, Zitvogel L. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med. 2011;17:700–707. doi: 10.1038/nm.2366. [DOI] [PubMed] [Google Scholar]
- 16.Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, Haldeman B, Ostrander CD, Kaifu T, Chabannon C, Moretta A, West R, Xu W, Vivier E, Levin SD. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med. 2009;206:1495–1503. doi: 10.1084/jem.20090681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pogge von Strandmann E, Simhadri VR, von Tresckow B, Sasse S, Reiners KS, Hansen HP, Rothe A, Boll B, Simhadri VL, Borchmann P, McKinnon PJ, Hallek M, Engert A. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity. 2007;27:965–974. doi: 10.1016/j.immuni.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 18.Sasaki T, Gan EC, Wakeham A, Kornbluth S, Mak TW, Okada H. HLA-B-associated transcript 3 (Bat3)/Scythe is essential for p300-mediated acetylation of p53. Genes Dev. 2007;21:848–861. doi: 10.1101/gad.1534107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simhadri VR, Reiners KS, Hansen HP, Topolar D, Simhadri VL, Nohroudi K, Kufer TA, Engert A, Pogge von Strandmann E. Dendritic cells release HLA-B-associated transcript-3 positive exosomes to regulate natural killer function. PLoS One. 2008;3:e3377. doi: 10.1371/journal.pone.0003377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joyce MG, Tran P, Zhuravleva MA, Jaw J, Colonna M, Sun PD. Crystal structure of human natural cytotoxicity receptor NKp30 and identification of its ligand binding site. Proc Natl Acad Sci U S A. 2011;108:6223–6228. doi: 10.1073/pnas.1100622108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Wang Q, Mariuzza RA. Structure of the human activating natural cytotoxicity receptor NKp30 bound to its tumor cell ligand B7-H6. J Exp Med. 2011;208:703–714. doi: 10.1084/jem.20102548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood. 2005;106:1544–1551. doi: 10.1182/blood-2004-11-4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang T, Barber A, Sentman CL. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res. 2006;66:5927–5933. doi: 10.1158/0008-5472.CAN-06-0130. [DOI] [PubMed] [Google Scholar]
- 24.Eriksson M, Meadows SK, Wira CR, Sentman CL. Unique phenotype of human uterine NK cells and their regulation by endogenous TGF-beta. J Leukoc Biol. 2004;76:667–675. doi: 10.1189/jlb.0204090. [DOI] [PubMed] [Google Scholar]
- 25.Eriksson M, Meadows SK, Basu S, Mselle TF, Wira CR, Sentman CL. TLRs mediate IFN-gamma production by human uterine NK cells in endometrium. J Immunol. 2006;176:6219–6224. doi: 10.4049/jimmunol.176.10.6219. [DOI] [PubMed] [Google Scholar]
- 26.Varela-Rohena A, Carpenito C, Perez EE, Richardson M, Parry RV, Milone M, Scholler J, Hao X, Mexas A, Carroll RG, June CH, Riley JL. Genetic engineering of T cells for adoptive immunotherapy. Immunol Res. 2008;42:166–181. doi: 10.1007/s12026-008-8057-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moretta L, Bottino C, Pende D, Vitale M, Mingari MC, Moretta A. Human natural killer cells: Molecular mechanisms controlling NK cell activation and tumor cell lysis. Immunol Lett. 2005;100:7–13. doi: 10.1016/j.imlet.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 28.Vitale M, Della Chiesa M, Carlomagno S, Pende D, Arico M, Moretta L, Moretta A. NK-dependent DC maturation is mediated by TNFalpha and IFNgamma released upon engagement of the NKp30 triggering receptor. Blood. 2005;106:566–571. doi: 10.1182/blood-2004-10-4035. [DOI] [PubMed] [Google Scholar]
- 29.Ferlazzo G, Tsang ML, Moretta L, Melioli G, Steinman RM, Munz C. Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J Exp Med. 2002;195:343–351. doi: 10.1084/jem.20011149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang T, Barber A, Sentman CL. Chimeric NKG2D modified T cells inhibit systemic T-cell lymphoma growth in a manner involving multiple cytokines and cytotoxic pathways. Cancer Res. 2007;67:11029–11036. doi: 10.1158/0008-5472.CAN-07-2251. [DOI] [PubMed] [Google Scholar]
- 31.Gilboa E. The makings of a tumor rejection antigen. Immunity. 1999;11:263–270. doi: 10.1016/s1074-7613(00)80101-6. [DOI] [PubMed] [Google Scholar]
- 32.Zamai L, Ponti C, Mirandola P, Gobbi G, Papa S, Galeotti L, Cocco L, Vitale M. NK cells and cancer. J Immunol. 2007;178:4011–4016. doi: 10.4049/jimmunol.178.7.4011. [DOI] [PubMed] [Google Scholar]
- 33.Kaifu T, Escaliere B, Gastinel LN, Vivier V, Baratin M. B7-H6/NKp30 interaction: a mechanism of alerting NK cells against tumors. Cell Mol Life Sci. 2011;68:3531–3539. doi: 10.1007/s00018-011-0802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koh CY, Blazar BR, George T, Welniak LA, Capitini CM, Raziuddin A, Murphy WJ, Bennett M. Augmentation of antitumor effects by NK cell inhibitory receptor blockade in vitro and in vivo. Blood. 2001;97:3132–3137. doi: 10.1182/blood.v97.10.3132. [DOI] [PubMed] [Google Scholar]
- 35.Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ, Orchard PJ, Blazar BR, Wagner JE, Slungaard A, Weisdorf DJ, Okazaki IJ, McGlave PB. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 36.Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, Posati S, Rogaia D, Frassoni F, Aversa F, Martelli MF, Velardi A. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–2100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 37.Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. 2011;17:6287–6297. doi: 10.1158/1078-0432.CCR-11-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR, Burger SR, Panoskaltsis-Mortari A, Keever-Taylor CA, Zhang MJ, Miller JS. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplant. 2003;32:177–186. doi: 10.1038/sj.bmt.1704086. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen P, Moisini I, Geiger TL. Identification of a murine CD28 dileucine motif that suppresses single-chain chimeric T-cell receptor expression and function. Blood. 2003;102:4320–4325. doi: 10.1182/blood-2003-04-1255. [DOI] [PubMed] [Google Scholar]
- 40.Emtage PC, Lo AS, Gomes EM, Liu DL, Gonzalo-Daganzo RM, Junghans RP. Second-generation anti-carcinoembryonic antigen designer T cells resist activation-induced cell death, proliferate on tumor contact, secrete cytokines, and exhibit superior antitumor activity in vivo: a preclinical evaluation. Clin Cancer Res. 2008;14:8112–8122. doi: 10.1158/1078-0432.CCR-07-4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol. 2010;184:6938–6949. doi: 10.4049/jimmunol.0901766. [DOI] [PubMed] [Google Scholar]
- 42.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 43.Blankenstein T, Qin Z. The role of IFN-gamma in tumor transplantation immunity and inhibition of chemical carcinogenesis. Curr Opin Immunol. 2003;15:148–154. doi: 10.1016/s0952-7915(03)00007-4. [DOI] [PubMed] [Google Scholar]
- 44.Wigginton JM, Gruys E, Geiselhart L, Subleski J, Komschlies KL, Park JW, Wiltrout TA, Nagashima K, Back TC, Wiltrout RH. IFN-gamma and Fas/FasL are required for the antitumor and antiangiogenic effects of IL-12/pulse IL-2 therapy. J Clin Invest. 2001;108:51–62. doi: 10.1172/JCI10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beatty G, Paterson Y. IFN-gamma-dependent inhibition of tumor angiogenesis by tumor-infiltrating CD4+ T cells requires tumor responsiveness to IFN-gamma. J Immunol. 2001;166:2276–2282. doi: 10.4049/jimmunol.166.4.2276. [DOI] [PubMed] [Google Scholar]
- 46.Corthay A, Skovseth DK, Lundin KU, Rosjo E, Omholt H, Hofgaard PO, Haraldsen G, Bogen B. Primary antitumor immune response mediated by CD4+ T cells. Immunity. 2005;22:371–383. doi: 10.1016/j.immuni.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 47.Faroudi M, Utzny C, Salio M, Cerundolo V, Guiraud M, Muller S, Valitutti S. Lytic versus stimulatory synapse in cytotoxic T lymphocyte/target cell interaction: manifestation of a dual activation threshold. Proc Natl Acad Sci U S A. 2003;100:14145–14150. doi: 10.1073/pnas.2334336100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bloushtain N, Qimron U, Bar-Ilan A, Hershkovitz O, Gazit R, Fima E, Korc M, Vlodavsky I, Bovin NV, Porgador A. Membrane-associated heparan sulfate proteoglycans are involved in the recognition of cellular targets by NKp30 and NKp46. J Immunol. 2004;173:2392–2401. doi: 10.4049/jimmunol.173.4.2392. [DOI] [PubMed] [Google Scholar]
- 49.Hershkovitz O, Jarahian M, Zilka A, Bar-Ilan A, Landau G, Jivov S, Tekoah Y, Glicklis R, Gallagher JT, Hoffmann SC, Zer H, Mandelboim O, Watzl C, Momburg F, Porgador A. Altered glycosylation of recombinant NKp30 hampers binding to heparan sulfate: a lesson for the use of recombinant immunoreceptors as an immunological tool. Glycobiology. 2008;18:28–41. doi: 10.1093/glycob/cwm125. [DOI] [PubMed] [Google Scholar]
- 50.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia. N Engl J Med. 2011 doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yin Y, Li Y, Kerzic MC, Martin R, Mariuzza RA. Structure of a TCR with high affinity for self-antigen reveals basis for escape from negative selection. EMBO J. 2011;30:1137–1148. doi: 10.1038/emboj.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tammana S, Huang X, Wong M, Milone MC, Ma L, Levine BL, June CH, Wagner JE, Blazar BR, Zhou X. 4–1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum Gene Ther. 2010;21:75–86. doi: 10.1089/hum.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye Q, Loisiou M, Levine BL, Suhoski MM, Riley JL, June CH, Coukos G, Powell DJ., Jr Engineered Artificial Antigen Presenting Cells Facilitate Direct and Efficient Expansion of Tumor Infiltrating Lymphocytes. J Transl Med. 2011;9:131. doi: 10.1186/1479-5876-9-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-xL. Immunity 1995. 2010;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]; J Immunol. 185:3788–3799. [Google Scholar]
- 55.Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117:1137–1146. doi: 10.1172/JCI31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007;121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.