

Abstract
In the midbrain, dopamine neurons can release dopamine somatodendritically. This results in an inhibitory postsynaptic current (IPSC) within adjacent dopamine cells that occurs by the activation of inhibitory D2 autoreceptors. Kappa, but not mu/delta, opioid receptors inhibit this IPSC. The aim of the present study was to determine the mechanism by which κ-opioid receptors inhibit the dopamine IPSC. In both the ventral tegmental area (VTA) and substantia nigra compacta (SNc) the κ-receptor agonist U69593 inhibited the IPSC, but not the current induced by the exogenous iontophoretic application of dopamine. The endogenous peptide dynorphin A (1–13) also inhibited IPSCs in the VTA and SNc, but also the dopamine iontophoretic current in the VTA. Although both kappa agonists induced a postsynaptic outward current in the VTA, the current induced by dynorphin was dramatically larger. This suggests that the decrease in iontophoretic dopamine current was the result of occlusion. Occlusion alone, however, could not completely account for suppression of the IPSC. The kappa opioid inhibition of the IPSC was not affected by global increases or decreases in dopamine cell activity within the slice. These findings suggest that, although kappa opioid receptors can hyperpolarize dopamine neurons, they also suppress dopamine release by direct actions at the release site. The results thus demonstrate both pre- and postsynaptic actions of kappa receptor agonists. The actions of dynorphin indicate that VTA dopamine cells are selectively regulated by kappa receptors.
INTRODUCTION
Dopamine neurons of the ventral tegmental area (VTA) and substantia nigra compacta (SNc) project to target areas, including the basal ganglia, striatum, nucleus accumbens (NAc), and prefrontal cortex (Albanese and Minciacchi 1983; Swanson 1982). Through their dopaminergic projections these cells play important roles in movement and reward and are involved in mediating the rewarding properties of nearly all drugs of abuse, including opioids (Spanagel and Weiss 1999; Williams et al. 2001).
Opioids, acting at both μ- and κ-receptors, exert a powerful but opposing influence on dopamine neuron activity (Williams et al. 2001). This opposing action of μ- and κ-receptors has consequences for governing behavior. Microinjections of μ-opioid receptor agonists in the VTA are rewarding and elicit place preference (Phillips and LePiane 1980). Injections of κ-opioid agonists produce the opposite effect and elicit conditioned place aversion (Bals-Kubik et al. 1993). By inhibiting GABAergic interneurons, μ-opioids disinhibit dopamine neurons within the SNc and VTA (Johnson and North 1992a,b). This leads to increased dopamine cell activity and increased dopamine release at terminal sites (Gysling and Wang 1983; Kalivas and Duffy 1990). κ-Opioid receptors, present on the axon terminals of dopamine neurons, mediate the inhibition of dopamine transmission at their target sites (Spanagel et al. 1990; Svingos et al. 1999); however, κ-receptors are also present on cell bodies of dopamine neurons, where they function to directly hyperpolarize the membrane potential (Margolis et al. 2003). Thus κ-opioids may inhibit dopamine release by two mechanisms: directly at terminal release sites or by hyperpolarization of the cell bodies.
Using either in vivo microdialysis or in vitro [3H]-dopamine efflux from slices or cell cultures, κ-opioid agonists were previously reported to reduce dopamine release at somatodendritic and axonal release sites (Dalman and O’Malley 1999; Heijna et al. 1990; Schoffelmeer et al. 1997; Smith et al. 1992; You et al. 1999). These techniques measure primarily the net efflux of dopamine outside of synapses (i.e., “spillover”). A D2-receptor inhibitory postsynaptic current (IPSC) mediated by the somatodendritic release of dopamine was recently identified (D2 IPSC) (Beckstead et al. 2004). We previously showed that this IPSC is inhibited by κ-opioid receptors (Ford et al. 2006). Here, we further examined this suppression, attempting to determine the mechanism of action of the exogenous agonist U69593 and the endogenous peptide agonist dynorphin.
The κ-opioid receptor system controls the tonic activity of dopamine neurons as well as the behavioral and neurochemical responses to psychostimulants such as cocaine (Chefer et al. 2005). Thus determining the actions of κ-receptors on dopamine neuron activity and dopamine release has important therapeutic potential. The aim of this investigation was first to determine whether κ-receptors act pre- or postsynaptically to inhibit the dopamine-mediated somatodendritic IPSC. The second aim was to address the intracellular mechanism of action by which these receptors mediate their inhibitory effects.
METHODS
Male 5- to 6-wk-old DBA/2J mice (Jackson Laboratories, Bar Harbor, ME) were used in all experiments. Acute midbrain slices containing the VTA and SNc were prepared as previously described (Ford et al. 2006). Briefly, 220 μM horizontal midbrain slices were cut in ice-cold saline containing (in mM) 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.4 NaH2PO4, 25 NaHCO3, 11 d-glucose, 0.4 ascorbate, and 1 kynurenic acid using a vibratome (Leica, Nussloch, Germany). Slices were transferred to 35°C oxygenated (95% O2-5% CO2) saline containing MK-801 (10 μM) for a minimum of 30 min. For electrophysiological recordings, slices were transferred to a recording chamber and perfused with 35°C oxygenated (95% O2-5% CO2) saline at 1.5 ml/min. Neurons were visualized with an Olympus BX51WI (Olympus America, San Diego, CA) microscope equipped with custom-built “Dodt”-gradient contrast infrared optics (Dodt et al. 2002).
Whole cell recordings were made with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA) using 1.5- to 2.5-MΩ pipettes. Pipette internal solution contained (in mM) 115 K-methylsulfate, 20 NaCl, 1.5 MgCl2, 5 HEPES, 10 BAPTA, 2 ATP, 0.3 GTP, and 10 phosphocreatine (pH 7.3, 270 mOsm). BAPTA (10 mM) was used to chelate intracellular Ca2+ to prevent metabotropic glutamate receptor IPSCs from interfering with the D2 IPSC. Cells were voltage clamped at −60 mV (except for experiments involving 10 mM KCl, in which case Vh = −70 mV) and series resistance, ranging from 3 to 15 MΩ, was compensated by 80%. In the current-clamp experiments, cells were held at their resting membrane potential (Vm = −55 to −65 mV). Based on our previously published study (Ford et al. 2006), VTA and SNc dopamine neurons were physiologically identified by the presence of pacemaker (1–5 Hz) firing with spikes exhibiting action potential (AP) widths of ≥1.2 ms (measured in cell-attached voltage-clamp mode from the initial inward current to the peak of the outward current). We previously showed this to be a marker of tyrosine hydroxylase (TH) +, principal cells (Ford et al. 2006). Consistent with our previously published study (Ford et al. 2006), hyperpolarizing steps to −120 mV elicited large h-currents (Ih: 300–1,200 pA) from SNc neurons and smaller Ih (20–200 pA) from VTA dopamine cells. However, because of the variability in Ih (Ford et al. 2006; Liss et al. 2005; Margolis et al. 2006; Neuhoff et al. 2002) Ih was not the only measure used to identify neuronal populations. Rather, populations were additionally identified anatomically, with dopamine neurons medial to the medial lemniscus being classified within the VTA and dopamine cells lateral to the medial terminal nucleus of the accessory optic tract (MT) being classified as within the SNc (Ford et al. 2006).
To elicit D2 IPSCs, a monopolar saline-filled glass electrode (2–8 MΩ) was placed 100–200 μM from the neuron being recorded. The stimulation protocol consisted of a train of five stimuli (0.5-ms duration, 40 Hz) every minute. IPSCs were isolated pharmacologically with picrotoxin (100 μM), 6,7-dinitroquinoxaline-2,3-dione (DNQX, 10 μM), CGP 56999a (100 nM), and MK-801 (10 μM). To elicit D2 iontophoretic currents, dopamine hydrochloride was ejected as a cation with a single pulse (25–100 nA, 5–75 ms) from thin-walled iontophoretic electrodes (70 to >100 MΩ) containing dopamine (1M). Iontophoretic electrodes were placed about 10 μm from the cell and a retention current of 1–5 nA was applied to prevent passive leakage. Dopamine-mediated IPSCs and iontophoretic currents were recorded at 5 kHz and filtered at 2 kHz. Whole cell outward currents were sampled at 100 Hz and filtered at 50 Hz.
Values listed are means ± SE. Statistical significance was assessed using either Student’s unpaired t-test or one-way ANOVA (Tukey). P < 0.05 was considered a significant difference.
Dynorphin A (1–13), N-d-phe-cys-tyr-d-trp-agr-thr-pen-thr-nh2 (CTAP), and (d-pen2, pen5)-enkephalin (DPDPE) were from Bachem (King of Prussia, PA). (5α,7α,8α)-(+)-N-methyl-N-[7-(pyrrolidinly)-1-oxaspiro[4,5]dec-8-yl]-benzeneacatamide (U69593), baicalein, staurosporine, and H-89 were from Biomol (Plymouth Meeting, PA). ω-Agatoxin IVA and ω-Conotoxin GVIA were from Alomone Labs (Jerusalem, Israel). Cocaine was obtained from National Institute on Drug Abuse (NIH, Bethesda, MD). All other chemicals were from Sigma–Aldrich (St. Louis, MO). If bath applied, dynorphin, CTAP, and DPDPE were combined with the endopeptidase inhibitors bestatin hydrochloride (10 μM) and thiorphan (1 μM).
RESULTS
κ-Opioids inhibit somatodendritic dopamine release in the VTA
To mimic the “burst firing mode” of dopamine neurons, a train of electrical stimuli (five stimuli of 0.5-ms duration at 40 Hz once a minute) was delivered to evoke the somatodendritic release of dopamine. This induced a slow outward IPSC in the recorded neuron (Beckstead et al. 2004) (Fig. 1, A and B). The IPSC was completely blocked by the D2 receptor antagonist sulpiride (200 nM; n = 14, Supplementary Fig. 11), confirming that the IPSC was mediated by dopamine acting by the D2 receptor. Receptor antagonists were used to block γ-aminobutyric acid types A and B (GABAA- and GABAB-), N-methyl-Daspartate (NMDA-), and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)–mediated synaptic currents. A D2 IPSC could be recorded from every VTA dopamine cell sampled, although to ensure accurate measurement of IPSC amplitudes only neurons with IPSCs >30 pA were included in this study.
FIG. 1.

Actions of κ-opioids on ventral tegmental area (VTA) dopamine neurons. A: (5α,7α,8α)-(+)-N-methyl-N-[7-(pyrrolidinly)-1-oxaspiro[4,5]dec-8-yl]-benzeneacatamide (U69593, 200 nM) inhibited D2 inhibitory postsynaptic currents (IPSCs) but had no effect on currents generated by the iontophoretic application of dopamine. B: dynorphin (Dyn, 200 nM) inhibited both the IPSC and the dopamine iontophoretic current. C and D: summary of the effects of U69593 and Dyn on VTA dopamine cells. Effects of U69593 and dynorphin were reversed by the opioid antagonist naloxone (NLX, 1 μM). E and F: Dyn produces a greater whole cell current than U69593 in VTA dopamine cells. Top traces: examples of the effects of U69593 and dynorphin on single cells. Bottom graphs: summarized data.
In randomly sampled VTA neurons, the κ-opioid agonist U69593 induced a 52.9 ± 4.9% (Fig. 1, A and C; n = 10) suppression in the IPSC. Because the effect of U69593 is slow to wash out, we applied the general opioid antagonist naloxone (NLX, 1 μM) to induce a rapid reversal of the effect. We hypothesized that the mechanism responsible for the U69593-induced suppression of the IPSC was by the presynaptic reduction in transmitter release. To test this dopamine was iontophoresed onto these same neurons. With iontophoretic application of dopamine it was possible to induce an outward current of similar magnitude and duration as the IPSC (Fig. 1, A and B). U69593 (200 nM) had no effect on the current induced by iontophoretically applied dopamine (Fig. 1, A and C). Because the dopamine current induced by iontophoresis is exclusively postsynaptic, these results demonstrate that within the VTA, the κ-opioid agonist U69593 acts presynaptically to reduce the somatodendritic release of dopamine. Next the effects of the putative endogenous peptidergic κ-agonist dynorphin (Chavkin et al. 1982) were examined. Dynorphin (200 nM) induced a suppression of the IPSC of 55.1 ± 2.6% (n = 8; Fig. 1, B and D) and decreased the outward current induced by iontophoretically applied dopamine by 37.1 ± 9% (n = 7, P < 0.05; Fig. 1, B and D). These effects were rapidly terminated on the cessation of dynorphin application and the initiation of NLX application (1 μM; Fig. 1, C and D). Thus within the VTA, dynorphin and U69593 suppressed the D2 IPSC, although only dynorphin suppressed the current induced by the iontophoretic application of dopamine.
Kappa opioid agonists directly activate an outward G-protein–activated inwardly rectifying K+ (GIRK) conductance in midbrain dopamine cells (Margolis et al. 2003). Application of U69593 (200 nM) induced a 30.6 ± 9 pA (Fig. 1E; n = 8) outward current in cells randomly sampled from the VTA. Application of dynorphin (200 nM) in this same population of cells induced a large outward current of 101.9 ± 17 pA (Fig. 1F; n = 8) that was greater than the U69593-induced current (P < 0.05). Thus one reason that the current induced by iontophoretically applied dopamine was reduced could be occlusion of GIRK activation.
Kappa opioids inhibit somatodendritic dopamine release in the SNc
To date, studies examining the effects of κ-opioid agonists on dopamine cells focused mainly on cells within the VTA (Ford et al. 2006; Margolis et al. 2003, 2006). Because differences are known to exist between dopamine cells within the VTA and SNc (Liss et al. 2005; Neuhoff et al. 2002), we next examined the cellular and synaptic effects of κ-agonists in SNc dopamine neurons. As in the VTA, a train of electric stimuli induced an IPSC (Fig. 2, A and B) that was completely blocked by the D2 receptor antagonist sulpiride (200 nM) (data not shown; Beckstead et al. 2004). U69593 and dynorphin induced a similar suppression of the IPSC [U69593: 36.9 ± 3.5% (n = 10), dynorphin: 39.3 ± 3.3% (n = 8) P > 0.05] (Fig. 2, A–D). The outward current induced by iontophoretically applied dopamine on neurons in the SNc was unaffected by U69593 (Fig. 2, C and D). Dynorphin also failed to suppress the iontophoretic current, causing instead a slight potentiation (109.8 ± 8.3%, n = 9) (Fig. 2, C and D). This differs from the large inhibition seen with dynorphin in VTA dopamine cells (P < 0.05; Fig. 1, B and D).
FIG. 2.

U69593 and dynorphin inhibit the D2 IPSC in substantia nigra compacta (SNc) cells by a presynaptic mechanism. A: U69593 (200 nM) inhibited D2 IPSCs but had no effect on currents generated by the iontophoretic application of dopamine in SNc cells. B: Dyn (200 nM) inhibited both IPSCs but caused a small potentiation of the iontophoretic current in SNc cells. C and D: summary of the effects of U69593 and dynorphin on SNc dopamine cells. E and F: Dyn produces a greater whole cell current than U69593 in SNc dopamine cells.
Whole cell currents induced by U69593 and dynorphin were also examined in SNc dopamine neurons. U69593 produced an outward current of 7 ± 4 pA (Fig. 2E; n = 7), which was significantly smaller than that produced by dynorphin (29.1 ± 5 pA; n = 9; P < 0.05; Fig. 2F).
Dynorphin acts on kappa receptors
U69593 binds with high specificity to the κ-opioid receptor (Raynor et al. 1994). Dynorphin, although showing greater affinity to κ-receptors, can bind in the nanomolar range to μ-opioid receptors (Raynor et al. 1994). The observation that dynorphin was more efficacious than U69593 at activating whole cell GIRK currents and suppressing the current induced by exogenous dopamine may result from the activation of a receptor other than the κ-opioid receptor. To test this we applied dynorphin (200 nM) in the presence of the μ-opioid antagonist CTAP (500 nM). In the continued presence of CTAP, dynorphin produced a degree of inhibition of the IPSC in VTA dopamine neurons similar to that observed in the absence of CTAP (50.8 ± 6%, n = 5; P > 0.05) (Fig. 3A). Likewise in the presence of CTAP, dynorphin still produced large outward currents, similar to those seen in the absence of CTAP (82.4 ± 4 pA, n = 5; P > 0.05) (Fig. 3A). To confirm that the action of dynorphin was being mediated by the κ-receptor, we next applied dynorphin (200 nM) in the presence of the specific κ-receptor antagonist norBNI (100 nM). In the presence of norBNI, dynorphin failed to suppress the D2 IPSC (4 ± 5%, n = 4; P > 0.05; Fig. 3B) or induce an outward whole cell current (2 ± 12 pA, n = 4; P > 0.05; Fig. 3B), confirming that the inhibition mediated by dynorphin resulted from the activation of κ-receptors. The specific δ-opioid agonist DPDPE (200 nM) was also examined but failed to inhibit the IPSC or produce a measurable outward current (P > 0.05; n = 5; data not shown). These results suggest that the actions of dynorphin did not arise from the recruitment of μ/δ-opioid receptors but instead from the selective activation of κ-receptors.
FIG. 3.
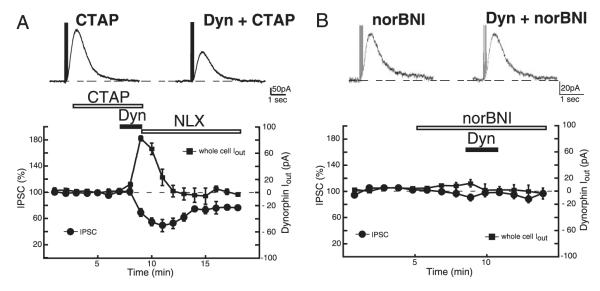
Effects of dynorphin arise from activation of κ-opioid receptors. A: in the presence of the μ-opioid antagonist N-d-phe-cys-tyr-d-trp-agr-thr-pen-thr-nh2 (CTAP, 500 nM), dynorphin still suppressed the IPSC and produced outward currents. B: application of the κ-opioid antagonist norBNI (100 nM) completely prevented dynorphin (200 nM) from either activating an outward current or suppressing the IPSC.
Dual actions of kappa agonists
Regardless of the agonist used, application of U69593 or dynorphin induced a similar suppression of the D2 IPSC (P > 0.05; Fig. 4A). However, both agonists induced a greater suppression of the IPSC in the VTA than in the SNc (P < 0.05; Fig. 4A). This suggests a greater efficacy or amount of κ-receptors that couple to suppression of the IPSC in the VTA than the SNc.
FIG. 4.

Summary of effects of dynorphin and U69593 in the VTA and SNc. A: summary of the inhibition produced by U69593 and dynorphin on the D2 IPSC. Note that dynorphin and U69593 caused a similar degree of inhibition of the IPSC and both induced a greater suppression in VTA dopamine cells. B: summary of amplitudes of whole cell outward currents generated by dynorphin and U69593. Note the range of currents produced by either agonist does not match the range of IPSC suppression observed in A. C: correlation between the whole cell current produced by dynorphin vs. the degree of IPSC suppression and the whole cell current produced by dynorphin vs. the degree of suppression of the iontophoretic response. A stronger correlation existed for the iontophoretic response than for the IPSC. D: effect of U69593 on the current produced by iontophoretically applied dopamine or on the IPSC failed to correlate with the outward current induced. Only inhibitory responses to U69593 or dynorphin are plotted in C and D.
Unlike the suppression of the IPSC, U69593 and dynorphin differed in their abilities to induce whole cell outward currents. Dynorphin induced significantly greater outward currents in both the VTA and SNc than U69593 (P < 0.05; Fig. 4B). These results show that, although both κ-agonists suppress the IPSC to the same degree, they do not induce outward currents to the same extent. This suggests that separate pools of receptors or different mechanisms of action account for the two separate actions (IPSC suppression and outward current generation) of κ-agonists on dopamine neurons.
To confirm that these effects were not the result of a lack of maximal concentrations of agonists used we also tested higher concentrations. Increasing dynorphin to 2 μM did not produce a greater inhibition of the IPSC or induce larger whole cell outward currents than dynorphin (200 nM) (data not shown; n = 5; P > 0.05). Likewise U69593 (2 μM) had no greater effect than U69593 (200 nM) (data not shown; n = 10; P > 0.05). Thus the difference between U69593 (200 nM) and dynorphin (200 nM) was not the result of submaximal activation of κ-receptors.
The D2 IPSC is mediated by GIRK2 channels (Beckstead et al. 2004). The outward current induced by U69593 is also mediated by a GIRK conductance (Margolis et al. 2003). This raised the possibility that a postsynaptic occlusion of available GIRK channels contributed to the κ-receptor induced suppression of the IPSC. Dynorphin caused a marked reduction in the current induced by iontophoretic dopamine in VTA cells and a potentiation in SNc cells (Figs. 1B and 2B). The decrease in the current produced by the iontophoretic application of dopamine was most likely the result of an occlusion of available GIRK channels because there was a strong correlation between suppression of the iontophoretic response with the amplitude of outward current (R2 = 0.91; P < 0.05; Fig. 4C). In contrast, the amount of suppression of the IPSC produced by dynorphin, although still correlating with the amplitude of outward current produced (R2 = 0.42; P < 0.05; Fig. 4C), did so to a much lesser extent. This confirms that additional mechanisms at the release site are recruited by dynorphin to suppress the IPSC. These results suggest that dynorphin, which activates a GIRK-mediated postsynaptic conductance, also reduces the presynaptic release of dopamine.
In contrast there was no correlation between the amplitude of the whole cell outward current produced by U69593 and the amount of suppression of the current induced by the iontophoretic application of dopamine (R2 = 0.26; P > 0.05; Fig. 4D) or the suppression of the IPSC (R2 = 0.04; P > 0.05; Fig. 4D). This lack of correlation suggests that, whereas U69593 does induce outward currents, this mechanism does not account for the suppression of the IPSC. This again suggests that the generation of the outward current and suppression of the IPSC are separate events. Furthermore this strongly suggests that the mechanism by which U69593 suppresses the IPSC is by a mechanism at the “presynaptic” release site.
κ-Opioid suppression of the IPSC is not the result of membrane hyperpolarization
The suppression of the IPSC by U69593 appears to be separate from its actions in generating whole cell currents (Fig. 4C). However, the bath application of U69593 will cause an overall dampening of neuronal excitability arising from its hyperpolarizing effects on adjacent dopamine neurons. By reducing neuronal excitability, U69593 may alter background dopamine release. This reduction in background release may partially account for the reduction in the IPSC we observe on application of U69693. To test this hypothesis, we first applied the nonspecific monoamine transport blocker cocaine (1 μM). By blocking the activity of dopamine uptake transporter (DAT) cocaine prevents the reuptake of dopamine released by adjacent tonically active neurons. We predicted that this block of reuptake would lead to “spillover” of dopamine out of dendrodendritic “synapses,” thus increasing the basal level of dopamine in the slice. Recording in current-clamp mode from VTA and SNc dopamine cells, application of cocaine hyperpolarized cells by 8 ± 2 mV (P < 0.05; n = 6; Fig. 5, A and C). This hyperpolarization was sufficient to silence the spontaneous “pacemaker” AP firing of five of six dopamine cells (Fig. 5A). Thus cocaine (1 μM) was able to alter the background level of dopamine that was sufficient to induce a significant hyperpolarization of dopamine cells. In voltage-clamp mode, as previously noted (Beckstead et al. 2004; Ford et al. 2006), by preventing the reuptake of dopamine, cocaine (1 μM) potentiated the D2 IPSC and substantially slowed the kinetics of decay (Fig. 5D). In the presence of cocaine, however, U69593 was still able to suppress the IPSC to an extent similar to that in control (41 ± 6%; n.s. vs. control; n = 8; Fig. 5, D and F). Thus reducing background neuronal activity did not prevent U69593 from suppressing the IPSC.
FIG. 5.

Effects of U69593 are not dependent on alterations in membrane potential of adjacent neurons. A and B: current-clamp recordings from representative midbrain dopamine cells illustrating the hyperpolarizing effect of cocaine (1 μM) (A) and the depolarizing effect of 10 mM KCl (B). Note the suppression of spontaneous action potentials (APs) on application of cocaine and the increase in frequency of APs on application of KCl. C: summary of change in membrane potential of dopamine cells resulting from cocaine (1 μM) and KCl (10 mM). D and E: effect of cocaine (1 μM) (D) or KCl (10 mM) (E) on dopamine D2 IPSCs. Application of cocaine or KCl failed to alter the U69593-induced inhibition of the IPSC (E, note: Vh = −70 mV for E only). Application of 10 mM KCl shifts equilibrium potential for K+ (EK) to −56 mV, causing D2 IPSCs to become inward currents. F: summary of U69593-induced inhibition for control cells, cells in the presence of cocaine (1 μM), or cells in the presence of KCl (10 mM).
We next tested the effects of increasing cellular activity. Increasing the extracellular K+ concentration will shift the equilibrium potential for K+ (EK) to a more depolarized potential, thus depolarizing the cell membrane. In current-clamp mode, the application of KCl (10 mM) induced a 14 ± 2-mV depolarization of VTA and SNc dopamine cells (P < 0.05; n = 6; Fig. 5, B and C). This depolarization was sufficient to increase the frequency of spontaneous APs in six of six cells (Fig. 5B). To test the effects of increased cellular activity on the ability of U69593 to suppress the IPSC, we applied KCl (10 mM) in voltage-clamp mode while evoking D2 IPSCs. Using K+ internal solution (115 mM), the calculated EK in Krebs external solution is −96 mV. Application of KCl (10 mM) will shift EK to −56 mV. To increase our visualization of D2 GIRK-mediated IPSCs we changed the Vhold from −60 to −70 mV. At −70 mV, as a result of the shift in EK the IPSCs were inward (Fig. 5E). In the presence of KCl (10 mM), U69593 was still able to suppress the IPSC to an extent similar to that in control (37 ± 4%; n.s. vs. control; n = 6; Fig. 5, E and F). These results suggest that neither increasing nor decreasing neuronal activity affects the ability of U69593 to suppress dopamine-mediated IPSCs. This confirms that κ-receptors reduce dopamine release independently of membrane potential. These findings again suggest that κ-opioid receptors inhibit somatodendritic D2 IPSCs by a presynaptic inhibition, at the site of dopamine release.
Mechanism of inhibition of the IPSC
Having shown that kappa opioids are capable of inducing an inhibition of dopamine release, we next attempted to determine the mechanism by which they do so. Classically the actions of opioid receptors involve the inhibition of Ca2+ conductance, activation of K+ conductance, inhibition of adenylyl cyclase, and inhibition of transmitter release (Williams et al. 2001). Any of these mechanisms could explain the κ-opioid suppression of dopamine release. In that the effects of dynorphin involved both pre- and postsynaptic actions, U69593 was used because the inhibition of the IPSC involved a purely presynaptic mechanism (Figs. 1, A and C; 2, A and C; and 4D). Cells from both the SNc and VTA were sampled.
The role of voltage-gated Ca2+ channels (VGCCs) was tested first. To test whether U69593 inhibits dopamine release by suppression of N-type VGCCs, ω-Conotoxin GVIA (Ctx, 1 μM) was applied. This toxin suppressed the IPSC by 36.4 ± 2% (n = 3; P < 0.05; Fig. 6A), confirming that N-type Ca2+ channels are involved in mediating evoked somatodendritic dopamine release. After blocking N-type channels, U69593 was still able to suppress the IPSC to the same degree seen in control cells (n = 4; P > 0.05; Fig. 6, A and B). The P/Q Ca2+ channel blocker ω-Agatoxin IVA (Atx, 100 nM) had no effect on the IPSC (P > 0.05; Fig. 6A), confirming the lack of P/Q-type channels in mediating the IPSC (Beckstead et al. 2004). After application of ω-Agatoxin IVA, U69593 was still able to suppress the IPSC to the same extent as control (P > 0.05 Fig. 6, A and D). The R/T-type channel blocker mibefradil (10 μM) reduced the IPSC by 8.8 ± 4% (n = 8; P < 0.05), indicating that R/T-type channels mediate a small component of the Ca2+ influx responsible for the IPSC. However, U69593 was again able to suppress the IPSC after application of the blocker (n = 8; P > 0.05). The L-type Ca2+ channel blocker nimodipine (10 μM) had no effect on the IPSC (P > 0.05; n = 3; data not shown), illustrating the lack of involvement of these channels in mediating the IPSC. After application of nimodipine, U69593 was still able to suppress the IPSC to the same extent as control (P > 0.05; n = 3; data in Fig. 6B). These data suggest that κ-opioid receptors do not mediate a reduction in dopamine release by selective inhibition on one class of VGCCs.
FIG. 6.

Mechanism of suppression of the D2 IPSC by U69593. A: Ca2+ entry by voltage-gated Ca2+ channel blockers is necessary to evoke D2 IPSCs, but does not prevent the U69593-mediated suppression. N-type channel blocker ω-Conotoxin GVIA (ω-CTX, 1 μM) and the R/T-type channel blocker mibefradil (10 μM) both caused an inhibition of the IPSC, illustrating the involvement of these channels in mediating the IPSC. Neither blocker nor the P/Q-type channel blocker ω-Agatoxin IVA (100 nM) prevented the U69593-mediated suppression of the IPSC. B: summary of results showing the effects of N-, P/Q-, R/T-, and L-type Ca2+ channel blockers (ω-Conotoxin GVIA, 1 μM, ω-Agatoxin IVA, 100 nM, mibefradil 10 μM, nimodipine 10 μM), K+ channel blocker [4-aminopyridine (4-AP), 10 μM], 12-lipoxygenase inhibitor (baicalein, 5 μM), kinase inhibitor (staurosporine, 1 μM), and the protein kinase A (PKA) inhibitor (H-89, 10 μM) on the U69593-induced IPSC suppression.
To test whether U69593 mediates suppression of somatodendritic dopamine release in the midbrain by activation of voltage-dependent K+ conductances, we applied the inhibitor of voltage-gated K+ channels, 4-aminopyridine (4-AP). High concentrations (1–10 mM) inhibited the IPSC (data not shown), presumably through a direct block of GIRK channels. At lower concentrations, 4-AP (10 μM) induced a potentiation of the IPSC (data not shown) that was likely mediated through depolarization of adjacent dopamine dendrites. In the presence of 4-AP, U69593 caused the same degree of inhibition as that seen in control neurons (Fig. 6B; P > 0.05; n = 6). This demonstrates that 4-AP–sensitive K+ channels are not involved in mediating the κ-induced reduction in dopamine release. In the periaqueductal gray (PAG) μ-opioid–induced inhibition of GABA release upstream, which involves 4-AP–sensitive K+ channels, also involves the enzyme 12-lipoxygenase (Vaughan et al. 1997). To confirm that 12-lipoxygenase was not involved in meditating the actions of U69593, baicalein (5 μM) was applied. In the presence of the inhibitor, U69593 caused inhibition of the IPSC to the same extent as control (Fig. 6B; P > 0.05; n = 6), confirming that this pathway does not mediate the actions of U69593 in suppressing the D2 IPSC.
A prominent effect of opioids is suppression of adenylyl cyclase. Neither the broad-spectrum kinase inhibitor staurosporine (1 μM; n = 6), nor the PKA inhibitor H-89 (1–10 μM; n = 4) prevented U69593 from inhibiting the IPSC (Fig. 6B; P > 0.05 for both). This suggests that kinases, and specifically protein kinase A (PKA), are not involved in κ-opioid–induced inhibition of somatodendritic dopamine release.
Throughout our experiments we were not able to observe miniature IPSC events in the presence of either Cd2+ or tetrodotoxin (data not shown). This prevented us from using this alternate technique to examine the pathways involved in mediating the U69593-induced inhibition of dopamine release. In conclusion, our results show that, although κ-receptors are capable of reducing the somatodendritic release of dopamine, they do so by an unknown mechanism.
DISCUSSION
Activation of κ-receptors
In both the VTA and SNc, IPSCs were inhibited by the κ-receptor agonist U69593. U69593 failed to inhibit the current activated by exogenously applied dopamine, suggesting that U69593 acted at presynaptic sites to inhibit somatodendritic dopamine release. Application of the putative endogenous peptide dynorphin (Chavkin et al. 1982) also inhibited IPSCs to a similar extent as U69593; thus both dynorphin and U69593 inhibit dopamine release to a similar extent. Additionally, dynorphin caused a large outward current and reduced the current induced by iontophoretically applied dopamine in cells in the VTA. Thus both dynorphin and U69593 maximally inhibited the IPSC, but U69593 did not cause a maximal outward current. This indicates that either separate receptor subtypes or different mechanisms could explain these two actions. The results suggest that the coupling efficiency is better at sites that activate the outward GIRK current.
The D2 IPSC is most likely mediated by the local somatodendritic release of dopamine and not by axon collaterals from distant dopamine cells (Beckstead et al. 2004; Kalivas and Duffy 1991; Nirenberg et al. 1996, 1997; yet see Deutch et al. 1988). Using our local stimulation protocol, the IPSC is generated only from dopamine released from adjacent cells onto the neuron being recorded. The two κ-agonists produced a wide range of outward currents ranging from 7 pA produced by U69593 in the SNc to 102 pA by dynorphin in the VTA, which did not match with the range of inhibition of the IPSCs (roughly 40% in the SNc and 55% in the VTA). If local hyperpolarization of cell bodies accounted for the reduction in dopamine release, one would expect a strong correlation between outward currents (i.e., a measure of local neuronal hyperpolarization) and IPSC inhibition (i.e., reduction in dopamine release). No correlation existed for U69593 and only a relatively poor correlation (R2 = 0.42) for dynorphin. Thus it appears that U69593 acts to inhibit release by a mechanism that is not dependent on neuronal hyperpolarization. Furthermore, it is unlikely that U69593 mediates the suppression of the IPSC by altering excitability of adjacent cells because increasing neuronal activity in adjacent cells by applying KCl (10 mM), or reducing activity by applying cocaine (1 μM), failed to affect the ability of U69593 to suppress the IPSC. These findings suggest that κ-receptors are capable of two separate actions: activation of a GIRK-mediated outward current and inhibition of dopamine release. Presumably, dynorphin uses similar mechanisms to suppress dopamine release and, in addition, may mediate suppression of dopamine signaling by the silencing of presynaptic neurons, arising from the activation of a large outward current. It is this large GIRK-mediated current that occluded the dopamine iontophoretic response and produced the inhibition.
Dynorphin is less selective for κ-receptors than U69593 (Raynor et al. 1994). Thus conceivably the increased outward currents caused by dynorphin could be attributable to activation of other (i.e., μ or δ) opioid receptors. This did not appear to be the case because the effect of dynorphin was not blocked by the μ-antagonist CTAP nor mimicked by the δ-agonist DPDPE, but was blocked by the selective κ-antagonist norBNI. Furthermore, increasing the concentration of dynorphin or U69593 failed to produce greater effects than the 200 nM concentration used. Thus the additional effects of dynorphin could be explained by an increased efficacy at activating GIRK-mediated outward currents. These results suggest that the endogenous κ-opioid may mediate a powerful suppression of dopaminergic transmission through multiple signaling pathways.
Mechanism of inhibition
Inhibition of GABA release in the NAc by U69593 involves N-type Ca2+ channels (Hjelmstad and Fields 2003). Because the D2 IPSC is also dependent on N-type channels (Beckstead et al. 2004), we examined whether U69593 suppressed dopamine release by Ca2+ channel inhibition. We found that both N- and R/T-type channels are required for evoked dopamine release, confirming that Ca2+ is required for somatodendritic release (Cheramy et al. 1981; Geffen et al. 1976). However, blocking N-, P/Q-, R/T-, or L-type channels failed to prevent U69593 from inhibiting dopamine release. Thus the κ-opioid induced inhibition of somatodendritic dopamine release occurs either at a site downstream of Ca2+ entry or involves a mechanism that inhibits multiple classes of VGCCs. Inhibiting voltage-gated K+ channels or the enzymes 12-lipoxygenase, PKA, PKC, and CAMKII with 4-AP, cocaine, baicalein, H-89, and staurosporine also failed to prevent the U69593-mediated inhibition of the D2 IPSC. Thus unlike chronic administration of κ-opioid agonists (Thompson et al. 2000) or opioid inhibition of glutamatergic and GABAergic transmission in the VTA and the PAG (Bergevin et al. 2002; Manzoni and Williams 1999; Vaughan et al. 1997) none of these pathways is involved in mediating the acute effects of κ-agonists on suppressing somatodendritic dopamine release.
Whether κ-opioid receptors inhibit dopamine release at a process downstream of Ca2+ entry by direct inhibition of the dopamine release machinery or by another, unknown mechanism is not clear. Interestingly though, κ-receptor–induced inhibition of glutamate release in the NAc was also not sensitive to either 4-AP–sensitive K+ channel or VGCC blockers (Hjelmstad and Fields 2003). This suggests that this yet to be determined mechanism may be a common way that κ-receptors function to inhibit transmitter release.
Implications and conclusions
Past studies examining the inhibition of dopamine by κ-opioids relied on measuring dopamine in vivo with dialysis or measuring the electric- and/or glutamate-evoked [3H]-dopamine efflux from slices (Dalman and O’Malley 1999; Di Chiara and Imperato 1988; Heijna et al. 1990; Schoffelmeer et al. 1997; Smith et al. 1992; Werling et al. 1988; You et al. 1999). These studies concluded that κ-receptors are located on dopamine axon terminals, where they act to reduce dopamine release at the target site. Measuring dopamine efflux with dialysis in vivo or from slices in vitro examines only dopamine that has escaped from the synapse. Discovery of the D2 IPSC determined that dopamine somatodendritic transmission may be more analogous to classical synaptic transmission than previously believed (Beckstead et al. 2004). Recent work indicated that κ-opioids can directly hyperpolarize dopamine neurons (Margolis et al. 2003). This suggests that κ-opioids could inhibit dopamine release by two possible mechanisms: directly at the terminal or indirectly by the silencing of presynaptic neuronal activity. By examining the IPSC, the present work determined that κ-opioid receptors are capable of doing both. However, the presynaptic silencing of neuronal activity cannot fully explain the suppression of dopamine release that accounts for the inhibition of the IPSC.
Dynorphin mRNA levels are upregulated after exposure to cocaine and amphetamine (Spangler et al. 1993; Turchan et al. 1998). Furthermore κ-opioid receptors not only mediate the tonic activity of mesolimbic dopamine neurons, but also regulate the behavioral and neurochemical responses to cocaine (Chefer et al. 2005). These findings suggest that the activity specifically of the mesolimbic dopamine system and its regulation by the κ-opioid system may be directly affected by psychostimulants. Our results identifying the actions of κ-opioids in regulating the activity and somatodendritic release of dopamine may help to explain the behavioral effects induced by these drugs of abuse.
Recent work has begun to address important differences between dopamine neurons within the VTA and the SNc (Ford et al. 2006; Liss et al. 2005; Margolis et al. 2006; Neuhoff et al. 2002). This present work is a continuation of that work, illustrating another important example of how the regulations of these neurons differ. Identifying the pharmacological and physiological properties of subtypes of midbrain dopamine neurons provides a greater understanding for the role of signaling between different brain regions.
Supplementary Material
Acknowledgments
This work was supported by National Institute on Drug Abuse Grant DA-08163 to J. T. Williams. C. P. Ford is the Wyeth fellow of the Life Sciences Research Foundation and a fellow of the Alberta Heritage Foundation for Medical Research.
Footnotes
The online version of this article contains supplementary data.
REFERENCES
- Albanese A, Minciacchi D. Organization of the ascending projections from the ventral tegmental area: a multiple fluorescent retrograde tracer study in the rat. J Comp Neurol. 1983;216:406–420. doi: 10.1002/cne.902160406. [DOI] [PubMed] [Google Scholar]
- Bals-Kubik R, Ableitner A, Herz A, Shippenberg TS. Neuroanatomical sites mediating the motivational effects of opioids as mapped by the conditioned place preference paradigm in rats. J Pharmacol Exp Ther. 1993;264:489–495. [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron. 2004;42:939–946. doi: 10.1016/j.neuron.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Bergevin A, Girardot D, Bourque MJ, Trudeau LE. Presynaptic mu-opioid receptors regulate a late step of the secretory process in rat ventral tegmental area GABAergic neurons. Neuropharmacology. 2002;42:1065–1078. doi: 10.1016/s0028-3908(02)00061-8. [DOI] [PubMed] [Google Scholar]
- Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- Chefer VI, Czyzyk T, Bolan EA, Moron J, Pintar JE, Shippenberg TS. Endogenous kappa-opioid receptor systems regulate mesoaccumbal dopamine dynamics and vulnerability to cocaine. J Neurosci. 2005;25:5029–5037. doi: 10.1523/JNEUROSCI.0854-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheramy A, Leviel V, Glowinski J. Dendritic release of dopamine in the substantia nigra. Nature. 1981;289:537–542. doi: 10.1038/289537a0. [DOI] [PubMed] [Google Scholar]
- Dalman FC, O’Malley KL. kappa-Opioid tolerance and dependence in cultures of dopaminergic midbrain neurons. J Neurosci. 1999;19:5750–5757. doi: 10.1523/JNEUROSCI.19-14-05750.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutch AY, Goldstein M, Baldino F, Roth RH. Telencephalic projections of the A8 dopamine cell group. Ann NY Acad Sci. 1988;537:27–50. doi: 10.1111/j.1749-6632.1988.tb42095.x. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Opposite effects of mu and kappa opiate agonists on dopamine release in the nucleus accumbens and in the dorsal caudate of freely moving rats. J Pharmacol Exp Ther. 1988;244:1067–1080. [PubMed] [Google Scholar]
- Dodt HU, Eder M, Schierloh A, Zieglgansberger W. Infrared-guided laser stimulation of neurons in brain slices. Sci STKE. 2002;2002:PL2. doi: 10.1126/stke.2002.120.pl2. [DOI] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geffen LB, Jessell TM, Cuello AC, Iversen LL. Release of dopamine from dendrites in rat substantia nigra. Nature. 1976;260:258–260. doi: 10.1038/260258a0. [DOI] [PubMed] [Google Scholar]
- Gysling K, Wang RY. Morphine-induced activation of A10 dopamine neurons in the rat. Brain Res. 1983;277:119–127. doi: 10.1016/0006-8993(83)90913-7. [DOI] [PubMed] [Google Scholar]
- Heijna MH, Padt M, Hogenboom F, Portoghese PS, Mulder AH, Schoffelmeer AN. Opioid receptor-mediated inhibition of dopamine and acetylcholine release from slices of rat nucleus accumbens, olfactory tubercle and frontal cortex. Eur J Pharmacol. 1990;181:267–278. doi: 10.1016/0014-2999(90)90088-n. [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO, Fields HL. Kappa opioid receptor activation in the nucleus accumbens inhibits glutamate and GABA release through different mechanisms. J Neurophysiol. 2003;89:2389–2395. doi: 10.1152/jn.01115.2002. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992a;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992b;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P. Effect of acute and daily neurotensin and enkephalin treatments on extracellular dopamine in the nucleus accumbens. J Neurosci. 1990;10:2940–2949. doi: 10.1523/JNEUROSCI.10-09-02940.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P. A comparison of axonal and somatodendritic dopamine release using in vivo dialysis. J Neurochem. 1991;56:961–967. doi: 10.1111/j.1471-4159.1991.tb02015.x. [DOI] [PubMed] [Google Scholar]
- Liss B, Haeckel O, Wildmann J, Miki T, Seino S, Roeper J. K-ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat Neurosci. 2005;8:1742–1751. doi: 10.1038/nn1570. [DOI] [PubMed] [Google Scholar]
- Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:6629–6636. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Kappa-opioid agonists directly inhibit midbrain dopaminergic neurons. J Neurosci. 2003;23:9981–9986. doi: 10.1523/JNEUROSCI.23-31-09981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, Fields HL. Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc Natl Acad Sci USA. 2006;103:2938–2942. doi: 10.1073/pnas.0511159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhoff H, Neu A, Liss B, Roeper J. I(h) channels contribute to the different functional properties of identified dopaminergic subpopulations in the midbrain. J Neurosci. 2002;22:1290–1302. doi: 10.1523/JNEUROSCI.22-04-01290.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirenberg MJ, Chan J, Liu Y, Edwards RH, Pickel VM. Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. J Neurosci. 1996;16:4135–4145. doi: 10.1523/JNEUROSCI.16-13-04135.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirenberg MJ, Chan J, Vaughan RA, Uhl GR, Kuhar MJ, Pickel VM. Immunogold localization of the dopamine transporter: an ultrastructural study of the rat ventral tegmental area. J Neurosci. 1997;17:5255–5262. doi: 10.1523/JNEUROSCI.17-14-05255.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AG, LePiane FG. Reinforcing effects of morphine microinjection into the ventral tegmental area. Pharmacol Biochem Behav. 1980;12:965–968. doi: 10.1016/0091-3057(80)90460-8. [DOI] [PubMed] [Google Scholar]
- Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. Pharmacological characterization of the cloned kappa-, delta-, and muopioid receptors. Mol Pharmacol. 1994;45:330–334. [PubMed] [Google Scholar]
- Schoffelmeer AN, Hogenboom F, Mulder AH. Kappa1- and kappa2-opioid receptors mediating presynaptic inhibition of dopamine and acetylcholine release in rat neostriatum. Br J Pharmacol. 1997;122:520–524. doi: 10.1038/sj.bjp.0701394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Loughlin SE, Leslie FM. kappa-Opioid inhibition of [3H]dopamine release from rat ventral mesencephalic dissociated cell cultures. Mol Pharmacol. 1992;42:575–583. [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS. The effects of opioid peptides on dopamine release in the nucleus accumbens: an in vivo microdialysis study. J Neurochem. 1990;55:1734–1740. doi: 10.1111/j.1471-4159.1990.tb04963.x. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Weiss F. The dopamine hypothesis of reward: past and current status. Trends Neurosci. 1999;22:521–527. doi: 10.1016/s0166-2236(99)01447-2. [DOI] [PubMed] [Google Scholar]
- Spangler R, Unterwald EM, Kreek MJ. “Binge” cocaine administration induces a sustained increase of prodynorphin mRNA in rat caudate-putamen. Brain Res Mol Brain Res. 1993;19:323–327. doi: 10.1016/0169-328x(93)90133-a. [DOI] [PubMed] [Google Scholar]
- Svingos AL, Colago EE, Pickel VM. Cellular sites for dynorphin activation of kappa-opioid receptors in the rat nucleus accumbens shell. J Neurosci. 1999;19:1804–1813. doi: 10.1523/JNEUROSCI.19-05-01804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW. The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull. 1982;9:321–353. doi: 10.1016/0361-9230(82)90145-9. [DOI] [PubMed] [Google Scholar]
- Thompson AC, Zapata A, Justice JB, Jr, Vaughan RA, Sharpe LG, Shippenberg TS. Kappa-opioid receptor activation modifies dopamine uptake in the nucleus accumbens and opposes the effects of cocaine. J Neurosci. 2000;20:9333–9340. doi: 10.1523/JNEUROSCI.20-24-09333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchan J, Przewlocka B, Lason W, Przewlocki R. Effects of repeated psychostimulant administration on the prodynorphin system activity and kappa opioid receptor density in the rat brain. Neuroscience. 1998;85:1051–1059. doi: 10.1016/s0306-4522(97)00639-8. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA, Christie MJ. How opioids inhibit GABA-mediated neurotransmission. Nature. 1997;390:611–614. doi: 10.1038/37610. [DOI] [PubMed] [Google Scholar]
- Werling LL, Frattali A, Portoghese PS, Takemori AE, Cox BM. Kappa receptor regulation of dopamine release from striatum and cortex of rats and guinea pigs. J Pharmacol Exp Ther. 1988;246:282–286. [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- You ZB, Herrera-Marschitz M, Terenius L. Modulation of neurotransmitter release in the basal ganglia of the rat brain by dynorphin peptides. J Pharmacol Exp Ther. 1999;290:1307–1315. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.