

Abstract
Proteoglycans, key molecular effectors of cell surface and pericellular microenvironments, perform multiple functions in cancer and angiogenesis by virtue of their polyhedric nature and their ability to interact with both ligands and receptors that regulate neoplastic growth and neovascularization. Some proteoglycans such as perlecan, have pro- and anti-angiogenic activities, whereas other proteoglycans, such as syndecans and glypicans, can also directly affect cancer growth by modulating key signalling pathways. The bioactivity of these proteoglycans is further modulated by several classes of enzymes within the tumour microenvironment: (i) sheddases that cleave transmembrane or cell-associated syndecans and glypicans, (ii) various proteinases that cleave the protein core of pericellular proteoglycans and (iii) heparanases and endosulfatases which modify the structure and bioactivity of various heparan sulphate proteoglycans and their bound growth factors. In contrast, some of the small leucine-rich proteoglycans, such as decorin and lumican, act as tumour repressors by physically antagonizing receptor tyrosine kinases including the epidermal growth factor and the Met receptors or integrin receptors thereby evoking anti-survival and pro-apoptotic pathways. In this review we will critically assess the expanding repertoire of molecular interactions attributed to various proteoglycans and will discuss novel proteoglycan functions modulating cancer progression, invasion and metastasis and how these factors regulate the tumour microenvironment.
Keywords: perlecan, endorepellin, LG3, VEGF, syndecan, glypican, heparanase, small leucine-rich proteoglycans, decorin, lumican
Introduction
Proteoglycans were found to be involved in cancer biology over 50 years ago. Initially, pathologists noted that some carcinomas induce a desmoplastic hyperproliferative-like reaction within the host stroma and the connective tissue surrounding the cancer cells. Often this reaction stains metachromatically with cationic dyes such as alcian blue indicating a high glycosaminoglycan, and thus proteoglycan, content in the desmoplastic reaction. Given the fact that desmoplasia occurs in tumours with diverse histogenetic backgrounds [1], it seems plausible to consider this abnormal expression of proteoglycans a sort of general ‘reaction to invasion’ with agonistic and antagonistic elements embedded in the same stroma [2]. With the advancement of molecular and cellular biology, proteoglycans’ specific roles in cancer biology are being better delineated. For example, we now know that some heparan sulphate proteoglycans (HSPGs) act as pro-angiogenic factors by binding several growth factors and presenting them to their cognate receptors [3]. In contrast, C-terminal fragments of other proteoglycans counteract the angiogenic process by blocking the activity of several integrins necessary for endothelial cell movement [4–6]. The structural and functional characterization of cell surface heparan sulphate proteoglycans over the last two decades has led to an understanding of the broad impact these molecules have on cell behaviour [7]. Both the syndecan and glypican family have been implicated as regulators of cancer progression and in addition may serve as biomarkers for early detection or severity of disease [8–10]. Interestingly, the emerging data indicate that cell surface heparan sulphate proteoglycans may play diverse roles in cancer and act as either inhibitors or promoters of tumour progression depending on the type and stage of cancer. In addition, the function of these proteoglycans can be altered by two classes of enzymes within the tumour microenvironment, those that can release proteoglycans from the cell surface (sheddases) and those that can modify the structure of heparan sulphate chains (heparanases and endosulfatases) [11, 12]. Thus, the expression of these enzymes can act as a mechanism to dynamically control proteoglycan location and function and thus, tumour cell behaviour [9]. Another class of proteoglycans carrying primarily dermatan or chondroitin sulphate, the so-called small leucine-rich proteoglycans (SLRPs), act as tumour repressors by blocking the activity of receptor tyrosine kinases (RTKs) such as the epidermal growth factor receptor (EGFR) and Met [13]. In addition some of the SLRPs interact with integrins and Toll receptors and modulate adhesion and innate immunity [14, 15]. In this review we will discuss examples of both HSPGs and SLRPs to critically assess their roles in cancer biology, tumour microenvironment and angiogenesis.
Perlecan: a pro-angiogenic proteoglycan
Perlecan is a complex modular proteoglycan consisting of five domains with homology to growth factors, immunoglobulin and adhesion molecules [16–20] and encoded by a very large gene encompassing 95–97 exons [21]. Its biology is complicated by the large number of post-translational modifications including potential substitution with three heparan sulphate chains at the N-terminus, several O-linked oligosaccharides throughout the large protein core and the potential substitution with chondroitin sulphate [4, 22]. Its promoter region is quite complex [23] and is under positive and negative control by transforming growth factor (TGF)-β[24, 25] and interferon-γ[26], respectively. Perlecan is widely expressed [27] and targeted disruption of the perlecan gene Hspg2 causes embryonic lethality at about day 12. This is primarily caused by intra-pericardial haemorrhage likely due to the lack of perlecan causing failure of the basement membrane upon increased haemodynamic pressure concurrent with the enhanced cardiovascular circulation at that time [28]. The few surviving pups die very early from pulmonary insufficiency owed to severe cephalic and cartilage abnormalities [29]. It was soon realized, however, that about 75% of the Hspg2−/– mice developed transposition of the great vessels, aorta and pulmonary artery, as well as abnormal position of coronary arteries [30, 31], thereby implicating perlecan in cardiovascular development. These mouse results correlate well with the phenotype obtained by morpholino-mediated knockdown of zebrafish perlecan [32]. In addition to a severe muscular phenotype, the perlecan morphants show a markedly reduced angiogenic sprouting especially prominent in the intersegmental vessels which emerge from the dorsal aorta during development [32].
The initial report of perlecan involvement in cancer and angiogenesis described a 15-fold increase in perlecan mRNA in invasive melanomas which correlated with an abundant deposition of perlecan proetoglycan in the pericellular matrix of the tumour cells [33]. Concurrently it was discovered that perlecan promotes high-affinity binding of fibroblast growth factor (FGF)-2, an established pro-angiogenic factor [34], to cells lacking heparan sulphate and to soluble forms of FGF receptor [35]. Moreover, perlecan of both arterial and venous endothelial cells bind FGF-2 with a KD∼ 70 nM [36] and enhances FGF-2-mediated angiogenesis and its activity is as powerful as that obtained with heparin [35]. At the same time, in a study using PC3 prostate carcinoma tumour xenografts in nude mice, it was observed that perlecan of human origin is deposited in the basement membrane of the tumour vessels which, in theory, should be of mouse origin [18]. Thus, it was suggested that perlecan could serve as an initial scaffold upon which the murine endothelial cells would migrate and deposit an appropriate vascular basement membrane [18]. This concept has been subsequently validated by a study using mouse tumour xenografts in immunocompromised rats which showed that perlecan distributed around the tumour neovessels was of both host and tumour cell origin [37].
The central role of perlecan in angiogenesis, as a large multimodular heparan sulphate proteoglycan, has been confirmed by several independent studies using antisense RNA strategies in various tumour cells, both in vitro and in vivo [38]. For example, in both human colon carcinoma tumour xenografts and mouse melanoma tumour allografts, perlecan suppression caused substantial inhibition of tumour growth and angiogenesis [39]. Perlecan is required to inhibit thrombosis after deep vascular injury [40] and is essential to the strain-mediated effects on endothelial cell growth control [41]. Moreover, suppression of endogenous perlecan in mouse fibroblasts and human melanoma cells blocks both autocrine and paracrine functions of FGF-2 [42], further stressing a role for perlecan as co-receptor for FGFs. Knockdown of perlecan in melanoma cells has also been linked to a suppression of proliferation and invasion [43] and to a reduced responsiveness to both FGF-2 and vascular endothelial growth factor A (VEGFA) in human prostatic carcinoma cells [44]. In contrast, fibrosarcoma cells devoid of perlecan grow better, form more aggressive tumours and do not depend on FGF-2 for their proliferation, suggesting that cellular context is important [45]. We should also point out that perlecan, either via the protein core or the heparan sulphate chains, also binds platelet derived growth factors AA and BB [22, 46], FGF-1 [22], FGF-7 [47–49], FGF-9 [22], FGF-18 [50, 51], hepatocyte growth factor (HGF) [22] and FGF-binding protein [52] which could also modulate angiogenesis. Furthermore, perlecan binds progranulin [53] and extracellular matrix protein (ECM)1 [54], both of which have been involved in promoting angiogenesis [5], and is involved in regulating the Sonic Hedgehog pathway during development [55] and in prostate cancer [56, 57].
A central role for perlecan as a regulator of VEGFA/VEGFR2 axis has been recently reported in the zebrafish perlecan knockdown [58]. In this model system, the perlecan morphants show a surprising increase of VEGFA, which is also abnormally distributed in the embryos suggesting that perlecan is required for the proper localization of VEGFA. Moreover, a significant proportion of the perlecan morphants could be rescued by intra-embryonic injections of human VEGFA, suggesting that perlecan acts upstream of VEGFA/VEGFR2 axis [58]. This is interesting because the vascular abnormalities observed in the perlecan morphants are similar to those observed in VEGFA morphants [59], null mutants of VEGFR2 [60] or phospholypase Cγ1[61], a downstream effector of VEGFR2 [62, 63]. In human brain microvascular endothelial cells, VEGFA stimulates the synthesis of perlecan [64]. Notably, in human umbilical vein endothelial cells (HUVECs), addition of exogenous perlecan enhances VEGFA-induced phosphorylation of VEGFR2 [58] suggesting a positive feedback loop controlling VEGFR2 bioactivity. These findings further suggest that perlecan could be a key player in regulating not only the geographic distribution of various heparan sulphate-binding growth factors, but also the specific interaction with RTKs such as FGF receptors (FGFRs) and VEGFRs.
The pivotal role of perlecan heparan sulphate chains has been corroborated by transgenic animals lacking part of domain I where the heparan sulphate chains are attached [65]. These mice show impaired tumour growth and angiogenesis induced by FGF-2 [66]. In hepatoblastoma xenografts treated with a VEGF trap, consisting of soluble domains of both VEGFR1 and VEGFR2, there is an initial collapse of the tumour vasculature followed by a recovery. Strikingly, during the recovery phase there is a concurrent increase in both VEGFA and perlecan mRNA expression with abundant overlapping deposits of VEGFA protein and perlecan around the tumour vasculature [67]. Concomitantly, there is vascular expression of VEGFR2 and sustained VEFGR2 activation, suggesting that, like in the zebrafish model described above, perlecan might represent a potent reservoir of growth factors that can be presented to and utilized by VEGFR2 [67]. Indeed VEGF anchored to the extracellular matrix activates VEGFR2 in a different and more protracted way than soluble VEGF, in terms of receptor recruitment, phosphorylation kinetics and activation of downstream signalling [68]. This concept is further supported by the discovery that heparan sulphate proteoglycans in trans can potentiate VEGFR2 signalling and thus angiogenesis by evoking a sustained signal transduction due to heparan sulphate-dependent trapping of the active VEGFR2 signalling complex [69].
Thus, whole perlecan can be a pro-angiogenic factor by virtue of two main mechanisms. The first is by directly interacting with the VEGFR2 through its heparan sulphate chains as co-factors for VEGF and by binding to adjacent integrin receptors through the C-terminal domain V (see below) (Fig. 1). The second involves the activity of heparanase which would liberate the VEGF bound to the heparan sulphate chains located at the N-terminus (Figs 1 and 2). Indeed, heparan sulphate from perlecan has been shown to be sensitive to heparanase [70, 71], and during branching morphogenesis of the salivary gland, heparanase-mediated cleavage of perlecan heparan sulphate chains modulates FGF-10 bioactivity [72]. Of course, other heparan sulphate-binding growth factors would be equally presented to their cognate receptors by the enzymatic activity of heparanase [73], which generally cleaves the heparan sulphate chains into large fragments [12]. An additional mechanism involves the partial proteolytic cleavage of perlecan protein core mediated by various matrix metalloproteinases (MMPs) [70, 74], which together with heparanase, could generate soluble bioactive fragments [75].
fig 1.

Schematic representation of the dual activity of pelican. Perlecan can act as a powerful pro-angiogenic factor by either directly presenting VEGFA (and other heparan sulphate growth factors) to the VEGFR2, or indirectly following partial heparanase cleavage of the heparan sulphate side chains. Both can trigger VEGFR2 signalling with stimulation of migration, vascular permeability, survival and proliferation. Perlecan can also act as a powerful anti-angiogenic factor following cathepsin L or BMP-1/Tolloid-like protease cleavage of endorepellin and LG3, respectively. These C-terminal fragments bind with high affinity to the α2 I domain of the α2β1 integrin triggering a signal cascade that leads to disruption of endothelial cytoskeleton and endothelial cell motility. Endorepellin also activates the tyrosine phosphatase SHP-1 which is bound to the cytoplasmic domain of the α2β1 integrin. SHP-1 then dephosphorylates a number of RTKs including VEGFR2, thereby blocking endothelial cell migration, survival and proliferation.
fig 2.

Heparanase enhances gene expression and syndecan-1 (SDC1) shedding which together enhance tumour progression. Upon up-regulation of heparanase expression by the tumour cell, expression of syndecan-1, MMP-9, VEGF, HGF and receptor activator of nuclear factor κ-B ligand (RANKL) are elevated and released into the tumour microenvironment. MMP-9 acts as a ‘sheddase’ to increase release of syndecan-1 from the tumour cell surface (1). Shed syndecan-1 binds to VEGF (2) to promote angiogenesis and to HGF (3) to stimulate tumour growth. RANKL (4) activates differentiation of osteoclast precursors to accelerate bone turnover leading to release of factors that further drive tumour progression. Heparanase also cleaves the heparan sulphate chains of perlecan (5) thereby liberating additional VEGF that further stimulates angiogenesis.
Endorepellin, a C-terminal fragment of perlecan with anti-angiogenic activity
The discovery of endorepellin occurred by serendipity during a yeast two-hybrid screening utilizing the C-terminal domain V of perlecan as bait. Domain V of perlecan bound with high affinity to the non collagenous NC1 domain of collagen XVIII [76] which includes the well-established angiogenic inhibitor endostatin [77]. Soon, however, it was realized that perlecan domain V was as anti-angiogenic as endostatin and it was renamed endorepellin to signify its anti-endothelial nature [76]. Endorepellin binds with high affinity to the intercalated I domain, also known as von Willebrand/Integrin A domain, of the α2β1 integrin, a key receptor involved in angiogenesis [78–80] in both arterial and microvascular endothelial cells [81] as well as in platelets [82]. In endothelial cells, endorepellin evokes a signalling cascade that leads to disruption of the actin cytoskeleton thereby immobilizing endothelial cells and preventing them to establish capillary morphogenesis in either a collagen type I sandwich or Matrigel and in a number of angiogenic assays [5, 83, 84]. These functional studies correlate with the proteomic identification of at least five different proteins differentially regulated by exposing endothelial cells to recombinant endorepellin [85]. These proteins include β-actin, HSP60 and calreticulin (down-regulated), and vimentin and protein disulfide isomerase (up-regulated), all of which have been involved in angiogenesis. Notably, systemic delivery of recombinant human endorepellin to mice carrying two tumour xenografts, squamous cell carcinomas and lung carcinomas, caused a marked reduction of tumour growth, tumour metabolism and angiogenesis [86]. Further genetic experiments utilizing mice lacking the α2β1 integrin have revealed that this receptor is necessary for endorepellin-evoked anti-angiogenic activity insofar as microvascular endothelial cells isolated from the α2β1−/– integrin mice do not respond to exogenous endorepellin in contrast to α2β1+/+ mice [87]. Moreover, Lewis lung carcinoma allografts in the α2β1−/– mice fail to respond to systemic delivery of endorepellin, whereas α2β1+/+ mice did [87]. In support of a central role for the α2β1 integrin in perlecan-mediated pro- and anti-angiogenic activity are the recent results obtained with knockdown of the α2β1 integrin in zebrafish. In this case, reduced levels of α2β1 integrin evoke a vascular phenotype that is reminiscent of the perlecan morphants [88] with blunted intersegmental vessels which are largely non-functional by live video microscopy.
Endorepellin can be liberated by cathepsin L [89] and its terminal LG3 domain can be also specifically liberated by BMP1/Tolloid-like proteases [90] releasing an laminin-like globular domain (LG)3 domain that is biologically active [81, 91]. Perlecan-derived LG3 has been found in the amniotic fluid of women with premature rupture of foetal membranes [92], or mothers carrying Down Syndrome foetuses [93] and in the urine of patients with end-stage renal disease [94] and chronic allograft rejection [95]. Importantly, circulating levels of perlecan LG3 are decreased in women with breast cancer, suggesting that circulating LG3 titres could be a biomarker for breast cancer [96]. Interestingly, intact endorepellin has been found in the upper proliferative zone of cartilage foetal growth plate [97] suggesting that cartilage-derived endorepellin might be one of the key factors able to counteract invasion of blood vessels into cartilage. We note that the concept that certain tissues, such as cartilage, are not invaded by vascularized mesenchyme has been proposed nearly 40 years ago by Eisenstein et al. [98], implying that soluble factors from cartilage might prevent invasion by blood vessels.
Regarding the molecular mechanism of action of endorepellin, a recent study utilizing antibody arrays recognizing multiple tyrosine phosphorylated RTK has shown that endorepellin causes a global dephopsphorylation of several RTKs including VEGFR2 [99]. This is mediated by the Src homology-2 protein phosphatase-1 (SHP-1) which binds to the intracellular domain of α2 subunit of the α2β1 integrin and upon endorepellin engagement dephosphorylates several RTKS including VEGFR1, VEGFR2, EGFR, Tie2 and other receptors involved in angiogenesis, migration and growth control. Notably, endorepellin-mediated effects on SHP-1 require the α2β1 integrin insofar as human endothelial cells depleted in the receptor α2 subunit via siRNA-mediated strategies or endothelial cells derived from the α2β1−/– mice do not respond to endorepellin [99].
In summary, C-terminal fragments of perlecan, either endorepellin or its terminal LG3 module, can act as a powerful anti-angiogenic factor by interacting with the α2I domain of the α2β1 integrin. This unique interaction triggers a signal cascade that leads to disruption of endothelial cytoskeleton and endothelial cell motility (Fig. 1). Endorepellin also activates the tyrosine phosphatase SHP-1 which is bound to the cytoplasmic domain of the α2β1 integrin. SHP-1 then dephosphorylates a number of RTKs including VEGFR2, thereby blocking endothelial cell migration, survival and proliferation.
Syndecans in cancer biology
The syndecans are a family of transmembrane proteoglycans that bear predominantly heparan sulphate glycosaminoglycan chains [100, 101]. The core proteins consist of a short intracellular domain, a transmembrane domain that is highly conserved, and an ectodomain that is divergent in amino acid sequence among the four syndecan family members [100]. Syndecans are expressed ubiquitously throughout embryonic development and adulthood, although the individual syndecans exhibit differing patterns of expression. Importantly, the pattern of expression and/or the structure of heparan sulphate is altered in response to certain pathophysiological stimuli and this in turn can regulate tumour progression [7, 102]. As cellular components, syndecans can function both intracellularly and at the cell surface. In addition, via protease shedding that occurs at the cell surface, syndecans can be shed into the extracellular milieu where they can diffuse away from the cell, become part of the extracellular matrix or remain soluble and influence surrounding cells as well as cells distal to their original source. On the cell surface, the core protein and heparan sulphate chains of syndecans play a number of roles in promoting cell–cell and cell–ECM interactions and in regulating cell survival, adhesion and migration [103]. Syndecans also bind to numerous soluble molecules (e.g. cytokines, chemokines) via their heparan sulphate chains thereby facilitating downstream signalling events [104, 105]. Additionally, syndecans signal through their protein cores, largely by direct interaction with other molecules that mediate signalling cascades [68]. Thus, due to their diversity in localization and function, syndecans are well positioned to regulate tumorigenesis and tumour progression.
Syndecan-1 is highly expressed at the basolateral membrane of simple epithelial cells, where it promotes adhesion to the ECM and regulates cell shape by interacting with the actin cytoskeleton [100]. Transformed epithelial cells often lose expression of syndecan-1 and E-cadherin which together appear to be key steps in regulating epithelial to mesenchymal transition [106, 107]. This likely explains why loss of syndecan-1 expression is associated with accelerated tumour progression and reduced patient survival in some types of carcinomas [108, 109]. Conversely, in other tumour types such as breast carcinoma and haematological malignancies, syndecan-1 is often highly expressed and associated with increased tumour invasiveness and poor prognosis [110–114]. For example, in myeloma, expression of syndecan-1 regulates cell growth, adhesion and migration [9]. Myeloma cell lines expressing low levels of syndecan-1 grow poorly in vivo, whereas their counterparts expressing elevated levels of syndecan-1 grow very aggressively and form large, highly vascular tumours [115]. In addition to its expression by tumour cells, syndecan-1 expressed by stromal cells can also impact tumour behaviour. In breast cancer syndecan-1 is present in high levels in the stroma of some patients and stromal expression has been correlated with poor prognosis [116, 117]. Moreover, studies in mouse models have demonstrated that stromal syndecan-1 released into the tumour microenvironment enhances breast tumour growth and angiogenesis [118, 119].
In addition to regulating tumour growth and metastasis, studies with syndecan-1 null mice indicate that this proteoglycan may also be critical for initial steps leading to initiation of tumours. Crossing Wnt transgenic mice that normally have a high incidence of spontaneous breast tumour formation with syndecan-1 null mice results in a dramatic reduction in tumour formation [120]. Further studies probing the mechanism of syndecan-1 activity in tumorigenesis indicate that the proteoglycan may be required for the activation of tumour precursor cells that arise within the stem/progenitor cell compartment [121, 122].
Because heparan sulphate is known to have multiple roles in regulating tumour growth, much of the work on syndecans in cancer has focused on heparan sulphate. However, recent data indicate that a region within the extracellular portion of the core protein of syndecan-1 binds to and activates signalling of both αvβ3 and αvβ5 integrins present on mammary carcinoma cells [123]. This syndecan-1 facilitated activation of the integrins enhances the invasive phenotype of the carcinoma cells and a peptide mimic of the region of syndecan-1 responsible for this activation can block integrin activation and tumour growth in vivo. Also, it has been found that the cytoplasmic domain of syndecan-1 interacts with α6β4 integrin and regulates ErbB2 activation by the integrin [124]. Interestingly, although it has been known for some time that syndecan-1 promotes tumour cell binding to type I collagen and inhibits its invasion into collagen gels [125], a series of recent studies identify syndecan-1 as a co-receptor for α2β1 integrin-mediated adhesion to collagen [126, 127]. These studies reveal that even when the collagen receptor α2β1 integrin is present, suppression of syndecan-1 in carcinoma cells diminishes cell adhesion to collagen and enhances cell motility and invasion. Although the heparan sulphate chains were required for the effects of syndecan-1 on carcinoma cell behaviour, neither syndecan-2 nor syndecan-4 could substitute for syndecan-1 indicating that the core protein of syndecan-1 may participate in α2β1 integrin signalling. Also supporting this notion is the recent finding that shedding of syndecan-1 from the surface of injured lung epithelial cells weakens the affinity state of α2β1 integrin resulting in increased cell migration [128].
Syndecan-2 expression is up-regulated in a number of cancers where it plays an important role in mediating cell shape, adhesion and migration. Syndecan-2 functions in this manner by regulating organization of the actin cytoskeleton and activation of focal adhesion kinases through the interaction of its cytoplasmic domain with ezrin, radixin and moesin family members [129]. Co-dimerization of syndecan-2 with α5β1 integrin in Lewis lung carcinoma cells induces actin stress fibre formation [130]. Syndecan-2 overexpression in highly metastatic variants of Lewis lung carcinoma cells inhibited their metastasis to lung possibly via down-regulation of the activation of the pro-metastatic protease MMP-2 [131]. Similarly, overexpression of syndecan-2 by osteosarcoma cells reduces their migration and enhances their apoptosis in vitro as compared to controls expressing lower levels of syndecan-2 [132, 133]. However, in contrast, syndecan-2 can promote tumour progression of other cancer types. In colon cancer, syndecan-2 expression enhances cell adhesion, spreading and proliferation indicating that it is important in colon cancer survival and perhaps initial tumorigenesis and metastasis [134, 135]. These adhesion and migration functions of syndecan-2 appear to be, at least in part, due to its functioning as a co-receptor for α2β1 integrin because knockdown of the α2 integrin chain on HCT116 colon carcinoma cells diminished syndecan-2-induced cell migration [136]. Also, a recent report indicates that the cell migration promoted by syndecan-2 occurs through Tiam1-mediated activation of Rac [137].
In addition to its effects in tumour cells, syndecan-2 may also enhance angiogenesis. Syndecan-2 is highly expressed in the microvessels of mouse glioma tumours and when syndecan-2 expression is knocked down in brain microvascular endothelial cells both cell migration and in vitro capillary tube formation are inhibited [138]. Additional evidence of a role for syndecan-2 in angiogenesis has come from knockdown experiments in zebrafish where it appears that syndecan-2 may act as a co-receptor for VEGF [139].
Syndecan-4 is reportedly expressed at some level by most tumours and also within the tumour microenvironment. In contrast to syndecan-2 which enhances migration of tumour cells, syndecan-4 is similar to syndecan-1 in that it mediates tumour cell adhesion to extracellular matrix and inhibits cell invasion in some cancers. Syndecan-4 mediates adhesion and spreading of breast cancer cell lines [140] and is down-regulated on invasive colon carcinoma cells [135, 141]. Diminished expression of syndecan-4 in melanoma cells resulted in down-regulation of FGF-2 signalling leading to an increase in tumour cell motility and decreased adhesion to fibronectin [142]. In contrast, a different study reports that diminishing syndecan-4 expression caused a decrease in melanoma cell invasion by decreasing Wnt5A signalling [143]. This suggests that syndecan-4 can play differing roles in tumour invasive potential depending on the factor(s) that are present to regulate motile behaviour. Also, complexing of syndecan-4 with FGF-2/FGFR-1 promotes pro-angiogenic signalling events through the protein kinase Cα pathway [144]. Disruption of interactions between syndecan-4 and fibronectin by the extracellular matrix molecule tenascin-C decreases focal adhesions on tumour cells leading to increased tumour cell proliferation [145]. Thus, regulation of cell adhesion via syndecan-4 is critical in controlling both the migration and proliferation of some tumour types.
Glypicans and the control of cancer growth
Glypicans share similarities to the syndecans in that they are expressed in cell-, tissue- and development-specific patterns, interact with a multitude of extracellular matrix proteins, including chemokines, growth factors/morphogens and their receptors and regulate cell-signalling events during morphogenesis, adult physiology and carcinogenesis [146–148]. Glypicans differ from syndecans in their mode of interaction with the cell membrane which occurs via a glycosyl–phosphatidyl–inositol (GPI) anchor. The six-member family of glypicans is characterized by having protein cores of 60–70 kD and containing 14 conserved cysteine residues. All glypican C-terminal regions have attachment sites for heparan sulphate chains, as well as the signal sequence for the GPI anchor. In addition to their heparan sulphate chains, glypican core proteins can also mediate important cellular functions such as stimulating apoptosis and supporting Wnt signalling [149–151].
Within the context of human malignancies, glypican-3 has been studied most extensively within the glypican family members [146, 149, 152]. Glypican-3 expression and function correlate with tumour type and cell of origin and thus analysis of glypican-3 can aid in the diagnosis and prognosis of some cancers. In cancers of the breast [153], lung [154], ovary [155] or mesothelium [156], glypican-3 expression is often down-regulated during tumour progression. Down-regulation of glypican-3 could enhance growth of cells by indirectly causing an up-regulation of cell signalling through such mechanisms as competing with Patched, the receptor for Hedgehog, for binding to Hedgehog [157]. In contrast, in neoplasms originating from tissues that express glypican-3 only in the embryo, its expression reappears during malignant transformation as an oncofoetal protein [158, 159]. An example of this is hepatocellular carcinoma (HCC) where soluble glypican-3 is detected in serum of 40–53% of HCC patients, but is not present in serum of healthy individuals [160, 161]. In HCC, glypican-3 promotes Wnt signalling as well as oncogenesis via the IGF signalling pathway [162]. Interestingly, a recent study demonstrated that overexpression of soluble glypican-3 inhibits HCC growth at least in part by blocking Wnt signalling [163].
Studies have also revealed the potential of glypican-3 as an immunotherapeutic target in tumours that overexpress the proteoglycan. Preclinical studies have demonstrated that the antigenic peptide (amin0 acids 144–152 of glypican-3) induces peptide-reactive cytotoxic T lymphocytes that reduce tumour mass without inducing autoimmunity [164]. A protective effect was also achieved by the transfer of glypican-3 peptide pulsed dendritic cells or glypican-3-reactive cytotoxic T lymphocytes in a model of colon carcinoma [165].
Glypican-1 is reportedly up-regulated in pancreatic and breast cancers and is also frequently overexpressed in human gliomas, where it enhances FGF-2 signalling [166, 167]. Down-regulation of glypican-1 in the human pancreatic cancer cell line PANC-1 prolonged doubling times and decreased anchorage-independent growth in vitro and tumour growth, angiogenesis and metastasis in vivo [166, 168]. Release of glypican-1 from the surface of PANC-1 or COLO-357 pancreatic cell lines by treatment with phosphoinositide-specific phospholipase-C, which cleaves the GPI anchor of glypican-1, inhibited the mitogenic responses of these cells to FGF-2 and heparin binding EGF, two growth factors that are commonly overexpressed in pancreatic cancer [169]. Similarly, glypican-5 increases tumour proliferation in rhabdomyosarcomas by potentiating the action of FGF-2, HGF and Wnt1A [170].
Role of heparanase and proteoglycan remodelling in cancer
Enzymatic modification of heparan sulphate proteoglycans has emerged as an important mechanism of regulating proteoglycan function in cancer. These modifications consist primarily of three types: cleavage of the extracellular core protein by sheddases which can release soluble domains of the proteoglycans from the surface of cells, trimming of heparan sulphate chains by heparanase and removal of 6-O sulphates from heparan sulphate chains by extracellular sulfatases. These modifications to proteoglycan structure can dramatically impact the function of the molecule and lead to changes in tumour cell behaviour. The action of the sulfatases in cancer will not be considered here but are the subject of several recent reviews [171, 172].
It has become increasing clear that shedding of proteoglycans from the surface of cells plays an important role in regulating proteoglycan localization and function [11]. Shedding occurs through the activity of various MMPs which clip the core proteins of proteoglycans and release functional ectodomains [173]. These ectodomains can then diffuse into the microenvironment where they may impact other cells or they can become inserted into the insoluble extracellular matrix. In addition, they can diffuse into the serum where they can be detected and used as a marker for cancer. For example, elevated levels of soluble syndecan-1 have been detected in the serum of patients with Hodgkin’s lymphoma [174] and lung cancer [175, 176]. Syndecan-1 is also found in patients with multiple myeloma where high levels of circulating proteoglycan are associated with high tumour burden and a poor prognosis [113, 177]. Further analysis utilizing animal models of myeloma indicates that soluble syndecan-1 is not only a marker of poor prognosis by actually participates in driving the progression of this cancer by stimulating tumour growth, angiogenesis and metastasis [178]. In myeloma, shed syndecan-1 appears to be derived predominantly from the surface of the tumour cells themselves. However, in an animal model of breast cancer, syndecan-1 shed into the tumour microenvironment was found to originate predominantly from stromal cells adjacent to the tumour cells [119]. This shed syndecan-1 stimulates tumour cell proliferation via activation of FGF-2. A role for stromal cell derived syndecan-1 in breast cancer is also supported by the finding that syndecan-1 is highly expressed in the stroma of many breast cancer patients [116]. In addition to studies on syndecan-1, syndecan-2 was found to be highly expressed on endothelial cells in a murine glioma model and its inhibition leads to reduced tubulogenesis in vitro, consistent with its role in cell adhesion and migration [138]. In this same model, addition of exogenous MMPs or pro-angiogenic factors (e.g. VEGF, FGF-2) leads to increased syndecan-2 shedding and promotion of endothelial cell migration on Matrigel. Also, shedding of both syndecan-1 and glypican-1 may be important in the progression of pancreatic cancer [169, 179].
Interestingly, recent findings implicate heparanase as an important regulator of syndecan shedding. Heparanase is an endo-β-D-glucuronidase that cleaves heparan sulphate chains into fragments 5–7 kD in size. Through both enzymatic and non-enzymatic activities, heparanase enhances tumour progression by degrading the extracellular matrix, stimulating cell signalling and by up-regulating expression of genes that promote an aggressive tumour phenotype (reviewed in [180]). Heparanase is up-regulated in myeloma patients and high levels of heparanase expression correlate with enhanced angiogenesis and poor prognosis [181, 182]. These findings in part have recently been linked to the fact that heparanase enhances shedding of syndecan-1 from the surface of myeloma cells [182, 183]. Mechanistically, this occurs via heparanase stimulation of ERK signalling with a resulting increase in expression of MMP-9, a protease that acts as a sheddase for syndecan-1 [184]. Up-regulation of heparanase expression in myeloma cells also leads to enhanced expression of VEGF and HGF, two factors that bind to the shed syndecan-1 and form complexes that stimulate angiogenesis and tumour growth [185, 186] (Fig. 2). Thus, the dual effects of heparanase (i.e. stimulation of both syndecan-1 shedding and growth factor expression) together condition the tumour microenvironment thereby establishing an aggressive tumour phenotype. Additionally, heparanase within the tumour extracellular matrix may act to release heparin binding growth factors from matrix bound heparan sulphate proteoglycans such as perlecan (Fig. 2). These multifunctional activities of heparanase are consistent with is known roles in regulating tumour growth, angiogenesis and metastasis.
Decorin and growth control
Decorin is a prototype member of the SLRPs, an expanding family of about 18 homologous genes that encompass five distinct classes [14, 187, 188]. Decorin received its eponym because of its ability to ‘decorate’ collagen fibres [189] at specific sites [190] and to regulate fibrillogenesis both in vitro [191] and in vivo [192]. Decorin is also known to regulate growth factor bioactivity [193, 194]. The decorin-null animals are fertile and viable but show abnormal collagen architecture, most pronounced in the dermis, which causes a decrease tensile strength of the skin with a consequent skin fragility phenotype [192]. The initial observation linking decorin to growth control derives from studies using Chinese Hamster Ovary (CHO) cells which are unique because their growth is dependent on TGF-β secreted as an autocrine factor. These decorin-expressing CHO cells formed a more orderly monolayer and grew to a lower saturation density than control cells lacking decorin, and these changes correlated with the levels of decorin expression [195]. It was subsequently found that decorin secreted by the transgenic CHO cells bound with high affinity to TGF-β thereby blocking the growth-stimulatory activity of the growth factor [196]. The second observation linking decorin to cell growth emerged from studies focusing on the identification of differentially expressed genes in quiescence versus growing cells. These studies showed that the mRNA levels of decorin, one of the eight most up-regulated genes, increased several folds during quiescence [197]. In vitro transcriptional analysis indicated enhanced transcriptional activity in quiescent fibroblasts when compared to cells harvested in their logarithmic growth phase [198]. The third line of evidence for a direct involvement of decorin in growth control is based on the fact that de novo expression of decorin suppresses the malignant phenotype in a wide variety of transformed cells [199, 200] and the effects are mediated by decorin-evoked induction of the cyclin-dependent kinase inhibitor p21WAF1/CIP1 (p21) [201]. Induction of endogenous p21 has now been recorded in a number of experimental settings utilizing recombinant decorin, adenoviral gene delivery or stable transfection of decorin, both in normal and malignant cells [202–208]. It became apparent, however, that the growth of several of the tumour cells lines that responded to decorin did not depend on the presence of TGF-β or its downstream signalling like the original CHO cells mentioned above. A search for specific receptors for decorin revealed that decorin suppresses tumour cell growth by interacting with the epidermal growth factor receptor (EGFR) [209, 210]. Notably, the EGFR could be identified as an interacting partner for decorin using interaction cloning and the yeast two-hybrid system [211]. Decorin binds to a narrow region of the EGFR that is partially overlapping but distinct from the EGF-binding domain [211]. Decorin/EGFR interaction leads to a sustained down-regulation of the EGFR [212] and, in mammary carcinoma cells, decorin also interacts with ErbB4 and indirectly down-regulates ErbB2, presumably by down-regulating of ErbB2/ErbB4 heterodimers [213]. Decorin evokes protracted internalization of the EGFR via caveolar endocytosis rather than clathrin-mediated endocytosis as EGF usually does [214]. This leads to a profound physical down-regulation of the EGFR with consequent reduction of receptor number rather than affinity [212].
Biologically active decorin is likely to act as monomer in solution [215] and adenoviral gene delivery of decorin has been shown to inhibit in vivo tumorigenicity and metastatic spreading [205, 216–220]. Analogously, systemic delivery of decorin proteoglycan or decorin protein core inhibits tumour growth in various xenografts models, including squamous, breast and prostate carcinomas [208, 221, 222]. The in vivo effects of decorin, when tested, show a concomitant down-regulation of the EGFR in the tumour xenografts and induction of apoptosis, predominantly through activation of caspase-3 [221].
Collectively, these studies indicate that decorin is a component of the circuit that regulates growth of eukaryotic cells. The reported overexpression of decorin in the stroma of a variety of solid tumours [1, 223, 224] could indeed represent a protective mechanism, a sort of soluble tumour repressor, that could counteract in vivo tumour cell growth [225]. The recent report of reduced plasma decorin in oesophageal cancer patients suggests that plasma decorin could represent a novel biomarker for cancer recognition and prognosis [226].
Genetic evidence for a role for decorin in carcinogenesis
The human decorin gene is quite complex with two alternatively-spliced exons, potentially two distinct promoters [227–229] and its expression in colon cancer is regulated by methylation of its control regions [1, 230]. The original decorin null mice were generated in a mixed genetic background of 129/Sv and BL/Swiss [192] and did not develop spontaneous tumours. However, if decorin possesses anti-tumorigenic properties, lack of decorin would be permissive for tumorigenicity in a suitable background which is already predisposed to spontaneous tumour formation. To test this hypothesis, double-knockout mice were generated in which the genes encoding decorin and the tumour suppressor p53 were ablated [231]. Double-deficient mice showed a faster rate of tumour emergence, and succumbed very early with a mean survival age of ∼4 months versus 6 months for the p53 null mice. Moreover, the double-deficient mice presented with very aggressive thymic lymphomas positive for CD4 and CD8 markers [231]. Mice harbouring one decorin allele and no p53 gene developed the same spectrum of tumours as the double knockout mice but had a survival rate similar to that of singly p53 null mice. These results provide strong genetic evidence for a cooperative action of germ-line mutations between decorin, a secreted proteoglycan, and p53 an endogenous controller of cell growth. The absence of decorin is permissive for lymphomagenesis in an animal model predisposed to cancer and suggests that this cooperation may lead to a more aggressive phenotype perhaps by shortening tumour latency [231].
To investigate the potential role of decorin in oncogenesis and to eliminate the potential confounding effects of mixed genetic backgrounds, the original decorin null mice were backcrossed for 14 generations into C57BL/6 mice [232]. Notably, targeted inactivation of decorin was associated with the development of spontaneous intestinal tumours, mostly adenomas, in ∼30% of the mice studies. However, when mice were subjected for 36 weeks to a high risk western diet, high in fat and low in calcium and vitamin D, the animals developed malignant colon cancer that in some cases was invasive [232]. Moreover, it was observed that there was a concurrent disruption of intestinal epithelial cell maturation and concurrent up-regulation of β-catenin and down-regulation of p21 in the intestinal tumours [232], as well as the isolated epithelial cells.
Collectively, these two studies provide strong genetic support for a role of decorin in oncogenesis and suggest that pharmacological or dietary enhancement of decorin gene expression in the intestinal tract could be beneficial for prevention of colorectal cancer.
Mechanism of decorin action: suppression of β-catenin and Myc levels
The complex binding repertoire of decorin suggests that this proteoglycan might interact with other RTKs in addition to EGFR and ErbB4 [188]. Using an antibody array detecting 42 Tyrosine phosphorylated RTKs it was recently found that exogenous decorin binds to Met, the HGF receptor, and stimulates a process that leads to receptor down-regulation and shedding, both contributing to attenuation of Met signalling pathway [233]. Of note, human recombinant decorin proteoglycan and protein core induce a protracted suppression of intracellular β-catenin, a known downstream effector of Met, and concurrently inhibit cell growth. A recent study has shown that decorin blocks several biological activities mediated by the Met signalling axis, including cell scatter, evasion and migration. These effects were mediated by a profound down-regulation of non-canonical β-catenin levels [188]. In addition, Myc, a downstream target of β-catenin, was markedly down-regulated by decorin, whereas phosphorylation of Myc at Thr58 was markedly induced [234]. The latter is known to destabilize Myc and target it for proteasomal degradation [235]. We also discovered that systemic delivery of decorin using three distinct tumour xenograft models caused down-regulation of Met and a concurrent suppression of β-catenin and Myc levels [234].
It is well established that there is a positive feedback loop between HGF/Met axis and β-catenin in sustaining the growth of several malignant tumours [236–238]. In addition, HGF induces Wnt-independent nuclear translocation of β-catenin following Met/β-catenin dissociation in hepatocytes [239]. Under growing conditions, β-catenin and Myc are stabilized by activation of the Wnt signalling and various RTK pathways which are often hyperactive in cancer. Activation of Wnt and RTK pathways leads to phosphorylation of glycogen synthase kinase-3β (GSK-3β) which causes an inactivation of this important serine-threonine kinase and a concurrent release of active β-catenin and Myc (Fig. 3A). Both β-catenin and Myc translocate to the nucleus where they activate a large number of genes including Myc itself [240, 241], thereby establishing a positive feedback loop that favours growth and survival (Fig. 3A). Among the genes that are induced by Myc is activating enhancer-binding protein (AP)4, a transcriptional repressor of p21 [242], which also enhances the proliferative potential of transformed cells. Decorin, by neutralizing the activity of RTKs such as EGFR and Met, releases GSK-3β from being phosphorylated and thus inactivated. This leads to GSK-3β evoked phosphorylation of both β-catenin and Myc at specific sites that lead to recognition by ubiquitin ligases and degradation within the 26S proteasome (Fig. 3B). Notably, in the case of Myc, degradation through this pathway is initiated by phosphorylation at Ser62, which functions as a priming site for binding of GSK-3β which then phosphorylates Thr58, an activity facilitated by axin 1 [243]. Additional steps in this pathway include the removal of the phosphate from Ser62 by protein phosphatase 2A and the subsequent degradation of Myc. Thus, the synthesis of AP4, among other genes, would be stopped and consequently p21 would be released from Myc/AP4 repression. Collectively, the decorin-evoked suppression of RTK activity can provide a plausible mechanistic explanation for the decorin-evoked induction of endogenous p21 levels found in a variety of experimental conditions as discussed above. Overall the marked suppression of β-catenin by decorin appears to occur via a non-canonical pathway, i.e. insensitive to lithium chloride, an inhibitor of GSK-3β[233]. The concurrent induction of phosphorylated Thr58 in Myc and its down-regulation at the protein and transcriptional levels [234] indicate that this is also a consequence of decorin-evoked suppression of RTK activity.
fig 3.
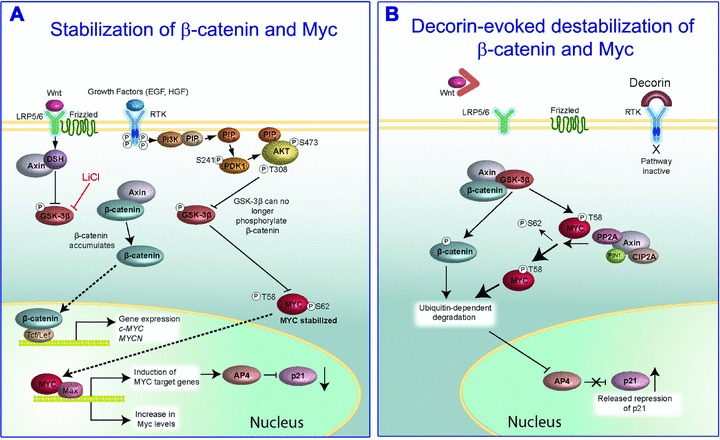
Schematic representation of decorin’s effects on β-catenin and Myc stabilization by directly down-regulating EGFR and Met. (A) Activation of both the Wnt and RTK pathways leads to stabilization of β-catenin and Myc proteins and their subsequent translocation into the nuclei where downstream transcription is activated. (B) When the Wnt pathway is blocked by absence or block of Wnt factors, or when RTK activity is attenuated by exogenous decorin, there is phosphorylation of β-catenin and Myc at specific residues. Especially important is phosphorylation at Threonine 58 (T58) of Myc, because this modification acts as priming for subsequent ubiquitination and 26S proteasomal degradation. For additional details see the text. Adapted in part from Albihn et al.
Lumican in cancer biology
Lumican is a keratan sulphate SLRP that participates in the acquisition of corneal transparency, hence its eponym, by regulating collagen fibril diameter and interfibrillar spacing [244, 245]. However, various forms of lumican often carrying polylactosamine chains, variants of undersulphated keratan sulphate, have been reported in various tissues outside of the cornea. Notably, lumican expression is increased in breast cancer, especially in the tumour stroma, and this correlates with higher tumour grade and lower oestrogen receptor levels [246]. More recently, lumican expression has been reported to be altered in various types of human malignancies, including melanomas [247, 248], osteosarcomas [249], pulmonary [250], pancreatic [251, 252] and colorectal carcinomas [253]. However, there are contrasting reports regarding the role of lumican in terms of potential biological function, i.e. pro- or anti-oncogenic role, and prognostic indicator. For example, in some studies lumican overexpression correlates with poor prognosis in advanced colorectal [253] and pancreatic [251] cancers. Similarly, in lung adenocarcinomas and squamous cell carcinomas lumican expression correlates with poor outcome [250]. It is not clear, however, whether the elevated expression of lumican in the tumour stroma would correlate with poor prognosis because of more invasive tumours or because lumican is indeed a biomarker for poor outcome. One possibility is that the poor prognosis might be related to the abundance of tumour stroma which is indicative of more advanced neoplasms. In contrast, lumican might play an opposite role in osteosarcomas where its expression correlates positively with differentiation and negatively with tumour progression [249]. In melanomas, stroma lumican levels decrease markedly with increasing malignancy as measured by vertical growth of the melanomas [247]. Moreover, B16F1 melanoma cells engineered to express lumican showed a marked reduction in the number and sizes of the pulmonary tumour nodules as compared to the wild-type cells [254, 255], suggesting that lumican might indeed be an anti-oncogenic agent. This concept is supported by a mechanistic study which has shown that de novo expression of lumican inhibits focus formation in cells transformed with the potent H-ras oncogene [256]. Moreover, colony formation in soft agar, a hallmark of malignant transformation, induced by the two viral oncogenes v-K-ras and v-src is suppressed in cells stably expressing lumican, without affecting cell proliferation [256]. These findings suggest that lumican might be able to selectively inhibit transformation by v-K-ras and v-src viral oncogenes. In support of these studies, cleavage of lumican by membrane type 1 metalloprotease (MT1-MMP) abrogates lumican-ability to suppress colony formation in soft agar [257]. Analogous to decorin, lumican-mediated cytostatic effects on cancer cells are associated with an induction of the cyclin-dependent kinase (CDK) inhibitor p21WAF1[257]. In murine embryonic fibroblasts induction of endogenous of p21WAF1 evoked by lumican follows a p53-dependent pathway and leads to suppression of cyclins A, D1 and E [258].
The precise molecular mechanisms of lumican’s mode of action, including receptors and signalling pathways, have not been completely delineated. However there are several emerging candidates for receptors and signalling pathways that could mediate lumican’s biological activity and most of these pathways are tissue specific. For example, lumican inhibits cell proliferation and aids apoptosis via Fas receptor (CD95)-Fas ligand [258], major regulators of programmed cell death. Moreover lumican regulates the induction of inflammatory cytokines and consequently wound healing by interacting with Fas–Fas ligand [259], it regulates neutrophil infiltration in experimental keratitis [260] and is required for neutrophil extravasation following corneal damage and inflammation [261]. Lumican adherent to the cell surface of neutrophils enhances their migration by interacting with β2-containing integrins [262]. In contrast, in melanoma cells lumican binds to β1-containing integrin receptors [263] and inhibits their migration by disrupting focal adhesion complexes [264]. The active site within the lumican protein core has been mapped to LRR9 and named lumcorin [265]. Several components of focal adhesion complexes, including vinculin and focal adhesion kinase, are modulated by lumican which leads to a disruption of the relationship between actin filaments and β1 integrin. Indeed, lumican protein core can directly bind the α2 I domain of the α2β1 integrin receptor on melanoma cells with relatively high affinity [266]. Thus, the reported anti-adhesive properties of lumican could be mediated by a specific interaction with the α2β1 integrin receptor, thereby providing a plausible explanation for the anti-invasive properties of this SLRP [264]. Finally, a recent study has shown that lumican can block not only melanoma invasion and metastasis, but it also can also induce tumour cell apoptosis and inhibit angiogenesis [255]. Collectively, these in vivo and in vitro studies suggest that lumican, in analogy to decorin, is an antagonist of cancer growth and possibly angiogenesis. These effects likely involve several receptors including integrins containing β1 and β2 subunits and other receptor complexes such as the Fas–Fas ligand complex involved in apoptosis.
Conclusions and perspectives
As a prototype basement membrane and cell-associated HSPG, perlecan, can exert pro- and anti-angiogenic activity. The concept of cryptic fragments of large protein cores with biological activity that differ from those of the parent molecule is now being accepted, insofar as other HSPGs such as collagen XVIII also contain similar bioactivities embedded at their C-terminus. Syndecans and glypicans can mediate an array of functions within tumours and have the ability to promote or inhibit the initiation and progression of cancer dependent on the structure, function and localization of the proteoglycan and on the tumour type and stage. The diversity inherent in these proteoglycans thus provides the potential for multiple layers of regulation of tumour behaviour. Although not discussed in this review, cell surface proteoglycans may represent viable targets for cancer therapy. Based on their functional attributes, therapeutic targets may include specific domains of proteoglycan core proteins and/or heparan sulphate chains. Additionally, the proteoglycan modifying enzymes, particularly the endosulfatases and heparanase, represent attractive therapeutic targets. Reports that heparan sulphate present within the nucleus can regulate gene transcriptional activity [267, 268] has led to speculation that proteoglycans may play direct roles in regulating transcription of genes that control tumour behaviour. This exciting possibility remains to be explored.
The SLRPs have emerged as powerful signalling molecules that control a wide variety of processes and could represent novel therapeutic modalities against cancer [188] as well as being targets themselves [269]. Deciphering the molecular mechanisms through which these stromal-derived proteoglycans affect cell cycle, survival and cancer growth could provide novel clues for a better understanding of how tumour cells modulate their environments.
Acknowledgments
We thank Angela McQuillan for her excellent work with the graphic designs. We also thank our numerous collaborators who have contributed to our work on proteoglycans during the past two decades. The original work in the authors’ laboratories was supported in part by National Institutes of Health grants CA39481, CA47282 and CA120975 (R.V.I.), a grant from the Mizutani Foundation (R.V.I.) and by National Institutes of Health grants CA135075 and CA055819 (R.D.S.). We apologize for the inability, due to space limitations, to reference all studies relevant to this review.
Conflicts of interest
The authors state that there are no conflicts of interest.
References
- 1.Adany R, Heimer R, Caterson B, et al. Altered expression of chondroitin sulfate proteoglycan in the stroma of human colon carcinoma. Hypomethylation of PG-40 gene correlates with increased PG-40 content and mRNA levels. J Biol Chem. 1990;265:11389–96. [PubMed] [Google Scholar]
- 2.Iozzo RV. Tumor stroma as a regulator of neoplastic behavior. Agonistic and antagonistic elements embedded in the same connective tissue. Lab Invest. 1995;73:157–60. [PubMed] [Google Scholar]
- 3.Iozzo RV. Heparan sulfate proteoglycans: intricate molecules with intriguing functions. J Clin Invest. 2001;108:165–7. doi: 10.1172/JCI13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iozzo RV. Basement membrane proteoglycans: from cellar to ceiling. Nature Rev Mol Cell Biol. 2005;6:646–56. doi: 10.1038/nrm1702. [DOI] [PubMed] [Google Scholar]
- 5.Iozzo RV, Zoeller JJ, Nyström A. Basement membrane proteoglycans: modulators par excellence of cancer growth and angiogenesis. Mol Cells. 2009;27:503–13. doi: 10.1007/s10059-009-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalluri R. Basement membranes: structure, assembly and role in tumor angiogenesis. Nature Rev Cancer. 2003;3:422–33. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 7.Sanderson RD. Heparan sulfate proteoglycans in invasion and metastasis. Semin Cell Dev Biol. 2001;12:89–98. doi: 10.1006/scdb.2000.0241. [DOI] [PubMed] [Google Scholar]
- 8.Sanderson RD, Yang Y, Suva LJ, et al. Heparan sulfate proteoglycans and heparanase – partners in osteolytic tumor growth and metastasis. Matrix Biol. 2004;23:341–52. doi: 10.1016/j.matbio.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Sanderson RD, Yang Y. Syndecan-1: a dynamic regulator of the myeloma microenvironment. Clin Exp Metastasis. 2008;25:149–59. doi: 10.1007/s10585-007-9125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filmus J, Selleck SB. Glypicans: proteoglycans with a surprise. J Clin Invest. 2001;108:497–501. doi: 10.1172/JCI13712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manon-Jensen T, Itoh Y, Couchman JR. Proteoglycans in health and disease: the multiple roles of syndecan shedding. FEBS J. 2010;277:3876–89. doi: 10.1111/j.1742-4658.2010.07798.x. [DOI] [PubMed] [Google Scholar]
- 12.Barash U, Cohen-Kaplan V, Dowek I, et al. Proteoglycans in health and disease: new concepts for heparanase function in tumor progression and metastasis. FEBS J. 2010;277:3890–903. doi: 10.1111/j.1742-4658.2010.07799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iozzo RV, Schaefer L. Proteoglycans in health and disease: novel regulatory signaling mechanisms evoked by the small leucine-rich proteoglycans. FEBS J. 2010;277:3864–75. doi: 10.1111/j.1742-4658.2010.07797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaefer L, Schaefer RM. Proteoglycans: from structural compounds to signaling molecules. Cell Tissue Res. 2010;339:237–46. doi: 10.1007/s00441-009-0821-y. [DOI] [PubMed] [Google Scholar]
- 15.Schaefer L. Extracellular matrix molecules: endogenous danger signals as new drug targets in kidney diseases. Curr Opin Pharmacol. 2010;10:185–90. doi: 10.1016/j.coph.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Noonan DM, Fulle A, Valente P, et al. The complete sequence of perlecan, a basement membrane heparan sulfate proteoglycan, reveals extensive similarity with laminin A chain, low density lipoprotein-receptor, and the neural cell adhesion molecule. J Biol Chem. 1991;266:22939–47. [PubMed] [Google Scholar]
- 17.Iozzo RV. Perlecan: a gem of a proteoglycan. Matrix Biol. 1994;14:203–8. doi: 10.1016/0945-053x(94)90183-x. [DOI] [PubMed] [Google Scholar]
- 18.Iozzo RV, Cohen IR, Grässel S, et al. The biology of perlecan: the multifaceted heparan sulphate proteoglycan of basement membranes and pericellular matrices. Biochem J. 1994;302:625–39. doi: 10.1042/bj3020625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knox SM, Whitelock JM. Perlecan: how does one molecule do so many things. Cell Mol Life Sci. 2006;63:2435–45. doi: 10.1007/s00018-006-6162-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farach-Carson MC, Carson DD. Perlecan – a multifunctional extracellular proteoglycan scaffold. Glycobiology. 2007;17:897–905. doi: 10.1093/glycob/cwm043. [DOI] [PubMed] [Google Scholar]
- 21.Cohen IR, Grässel S, Murdoch AD, et al. Structural characterization of the complete human perlecan gene and its promoter. Proc Natl Acad Sci USA. 1993;90:10404–8. doi: 10.1073/pnas.90.21.10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitelock JM, Iozzo RV. Heparan sulfate: a complex polymer charged with biological activity. Chem Rev. 2005;105:2745–64. doi: 10.1021/cr010213m. [DOI] [PubMed] [Google Scholar]
- 23.Iozzo RV, Danielson KG. Transcriptional and post-transcriptional control of proteoglycan gene expression. Progr Nucl Acids Res Mol Biol. 1999;62:19–53. doi: 10.1016/s0079-6603(08)60504-8. [DOI] [PubMed] [Google Scholar]
- 24.Dodge GR, Kovalszky I, Hassell JR, et al. Transforming growth factor β alters the expression of heparan sulfate proteoglycan in human colon carcinoma cells. J Biol Chem. 1990;265:18023–9. [PubMed] [Google Scholar]
- 25.Iozzo RV, Pillarisetti J, Sharma B, et al. Structural and functional characterization of the human perlecan gene promoter. Transcriptional activation by transforming factor-β via a nuclear factor 1-binding element. J Biol Chem. 1997;272:5219–28. doi: 10.1074/jbc.272.8.5219. [DOI] [PubMed] [Google Scholar]
- 26.Sharma B, Iozzo RV. Transcriptional silencing of perlecan gene expression by interferon-γ. J Biol Chem. 1998;273:4642–6. doi: 10.1074/jbc.273.8.4642. [DOI] [PubMed] [Google Scholar]
- 27.Handler M, Yurchenco PD, Iozzo RV. Developmental expression of perlecan during murine embryogenesis. Dev Dyn. 1997;210:130–45. doi: 10.1002/(SICI)1097-0177(199710)210:2<130::AID-AJA6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 28.Costell M, Gustafsson E, Aszódi A, et al. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–22. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arikawa-Hirasawa E, Watanabe E, Takami H, et al. Perlecan is essential for cartilage and cephalic development. Nature Genet. 1999;23:354–8. doi: 10.1038/15537. [DOI] [PubMed] [Google Scholar]
- 30.Costell M, Carmona R, Gustafsson E, et al. Hyperplastic conotruncal endocardial cushions and transposition of great arteries in perlecan-null mice. Circ Res. 2002;91:158–64. doi: 10.1161/01.res.0000026056.81424.da. [DOI] [PubMed] [Google Scholar]
- 31.González-Iriarte M, Carmona R, Pérez-Pomares JM, et al. Development of the coronary arteries in a murine model of transposition of great arteries. J Mol Cell Cardio. 2003;35:795–802. doi: 10.1016/s0022-2828(03)00134-2. [DOI] [PubMed] [Google Scholar]
- 32.Zoeller JJ, McQuillan A, Whitelock J, et al. A central function for perlecan in skeletal muscle and cardiovascular development. J Cell Biol. 2008;181:381–94. doi: 10.1083/jcb.200708022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen IR, Murdoch AD, Naso MF, et al. Abnormal expression of perlecan proteoglycan in metastatic melanomas. Cancer Res. 1994;54:5771–4. [PubMed] [Google Scholar]
- 34.Nugent MA, Iozzo RV. Fibroblast growth factor-2. Int J Biochem Cell Biol. 2000;32:115–20. doi: 10.1016/s1357-2725(99)00123-5. [DOI] [PubMed] [Google Scholar]
- 35.Aviezer D, Hecht D, Safran M, et al. Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell. 1994;79:1005–13. doi: 10.1016/0092-8674(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 36.Whitelock JM, Graham LD, Melrose J, et al. Human perlecan immunopurified from different endothelial cell sources has different adhesive properties for vascular cells. Matrix Biol. 1999;18:163–78. doi: 10.1016/s0945-053x(99)00014-1. [DOI] [PubMed] [Google Scholar]
- 37.Jiang J, Multhaupt H, Chan E, et al. Essential contribution of tumor-derived perlecan to epidermal tumor growth and angiogenesis. J Histochem Cytochem. 2004;52:1575–90. doi: 10.1369/jhc.4A6353.2004. [DOI] [PubMed] [Google Scholar]
- 38.Iozzo RV, San Antonio JD. Heparan sulfate proteoglycans: heavy hitters in the angiogenesis arena. J Clin Invest. 2001;108:349–55. doi: 10.1172/JCI13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma B, Handler M, Eichstetter I, et al. Antisense targeting of perlecan blocks tumor growth and angiogenesis in vivo. J Clin Invest. 1998;102:1599–608. doi: 10.1172/JCI3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nugent MA, Nugent HM, Iozzo RV, et al. Perlecan is required to inhibit thrombosis after deep vascular injury and contributes to endothelial cell-mediated inhibition of intimal hyperplasia. Proc Natl Acad Sci USA. 2000;97:6722–7. doi: 10.1073/pnas.97.12.6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baker AB, Ettenson DS, Jonas M, et al. Endothelial cells provide feedback control for vascular remodeling through a mechanosensitive autocrine TGF-β signaling pathway. Circ Res. 2008;103:289–97. doi: 10.1161/CIRCRESAHA.108.179465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aviezer D, Iozzo RV, Noonan DM, et al. Suppression of autocrine and paracrine functions of basic fibroblast growth factor by stable expression of perlecan antisense cDNA. Mol Cell Biol. 1997;17:1938–46. doi: 10.1128/mcb.17.4.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adatia R, Albini A, Carlone S, et al. Suppression of invasive behavior of melanoma cells by stable expression of anti-sense perlecan cDNA. Ann Oncol. 1998;8:1257–61. doi: 10.1023/a:1008243115385. [DOI] [PubMed] [Google Scholar]
- 44.Savoré C, Zhang C, Muir C, et al. Perlecan knockdown in metastatic prostate cancer cells reduces heparin-binding growth factor responses in vitro and tumor growth in vivo. Clin Exp Metastasis. 2005;22:377–90. doi: 10.1007/s10585-005-2339-3. [DOI] [PubMed] [Google Scholar]
- 45.Mathiak M, Yenisey C, Grant DS, et al. A role for perlecan in the suppression of growth and invasion in fibrosarcoma cells. Cancer Res. 1997;57:2130–6. [PubMed] [Google Scholar]
- 46.Göhring W, Sasaki T, Heldin C-H, et al. Mapping of the binding of platelet-derived growth factor to distinct domains of the basement membrane proteins BM-40 and perlecan and distinction from the BM-40 collagen-binding epitope. Eur J Biochem. 1998;255:60–6. doi: 10.1046/j.1432-1327.1998.2550060.x. [DOI] [PubMed] [Google Scholar]
- 47.Mongiat M, Taylor K, Otto J, et al. The protein core of the proteoglycan perlecan binds specifically to fibroblast growth factor-7. J Biol Chem. 2000;275:7095–100. doi: 10.1074/jbc.275.10.7095. [DOI] [PubMed] [Google Scholar]
- 48.Ghiselli G, Eichstetter I, Iozzo RV. A role for the perlecan protein core in the activation of the keratinocyte growth factor receptor. Biochem J. 2001;359:153–63. doi: 10.1042/0264-6021:3590153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sher I, Zisman-Rozen S, Eliahu L, et al. Targeting perlecan in human keratinocytes reveals novel roles for perlecan in epidermal formation. J Biol Chem. 2006;281:5178–87. doi: 10.1074/jbc.M509500200. [DOI] [PubMed] [Google Scholar]
- 50.Smith SML, West LA, Hassell JR. The core protein of growth plate perlecan binds FGF-18 and alters its mitogenic effect on chondrocytes. Arch Biochem Biophys. 2007;468:244–51. doi: 10.1016/j.abb.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chuang CY, Lord MS, Melrose J, et al. Heparan sulfate-dependent signaling of fibroblast growth growth factor 18 by chondrocyte-derived perlecan. Biochemistry. 2010;49:5524–32. doi: 10.1021/bi1005199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mongiat M, Otto J, Oldershaw R, et al. Fibroblast growth factor-binding protein is a novel partner for perlecan protein core. J Biol Chem. 2001;276:10263–71. doi: 10.1074/jbc.M011493200. [DOI] [PubMed] [Google Scholar]
- 53.Gonzalez EM, Mongiat M, Slater SJ, et al. A novel interaction between perlecan protein core and progranulin: potential effects on tumor growth. J Biol Chem. 2003;278:38113–6. doi: 10.1074/jbc.C300310200. [DOI] [PubMed] [Google Scholar]
- 54.Mongiat M, Fu J, Oldershaw R, et al. Perlecan protein core interacts with extracellular matrix protein 1 (ECM1), a glycoprotein involved in bone formation and angiogenesis. J Biol Chem. 2003;278:17491–9. doi: 10.1074/jbc.M210529200. [DOI] [PubMed] [Google Scholar]
- 55.Park Y, Rangel C, Reynolds MM, et al. Drosophila perlecan modulates FGF and Hedgehog signals to activate neural stem cell division. Dev Biol. 2003;253:247–57. doi: 10.1016/s0012-1606(02)00019-2. [DOI] [PubMed] [Google Scholar]
- 56.Datta MW, Hernandez AM, Schlicht MJ, et al. Perlecan, a candidate gene for the CAPB locus, regulates prostate cancer cell growth via the Sonic Hedgehog pathway. Mol Cancer. 2006;5:9. doi: 10.1186/1476-4598-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Datta S, Pierce M, Datta MW. Perlecan signaling: helping hedgehog stimulate prostate cancer growth. Int J Biochem Cell Biol. 2006;38:1855–61. doi: 10.1016/j.biocel.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 58.Zoeller JJ, Whitelock J, Iozzo RV. Perlecan regulates developmental angiogenesis by modulating the VEGF-VEGFR2 axis. Matrix Biol. 2009;28:284–91. doi: 10.1016/j.matbio.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nasevicius A, Larson J, Ekker SC. Distinct requirements for zebrafish angiogenesis revealed by a VEGF-A morphant. Yeast. 2000;17:294–301. doi: 10.1002/1097-0061(200012)17:4<294::AID-YEA54>3.0.CO;2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Habeck H, Odenthal J, Walderich B, et al. Analysis of zebrafish VEGF receptor mutant reveals specific disruption of angiogenesis. Curr Biol. 2002;12:1405–12. doi: 10.1016/s0960-9822(02)01044-8. [DOI] [PubMed] [Google Scholar]
- 61.Lawson ND, Mugford JW, Diamond BA, et al. Phospholipase C gamma-1 is required downstream of vascular endothelial growth factor during arterial development. Genes Dev. 2003;17:1346–51. doi: 10.1101/gad.1072203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Olsson A-K, Dimberg A, Kreuger J, et al. VEGF receptor signalling – in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–71. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 63.Holmes K, Roberts OL, Thomas AM, et al. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signalling. 2007;19:2003–12. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 64.Kaji T, Yamamoto C, Oh-i M, et al. The vascular endothelial growth factor VEGF165 induces perlecan synthesis via VEGF receptor-2 in cultured human brain microvascular endothelial cells. Biochem Biophys Acta. 2006;1760:1465–74. doi: 10.1016/j.bbagen.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 65.Rossi M, Morita H, Sormunen R, et al. Heparan sulfate chains of perlecan are indispensable in the lens capsule but not in the kidney. EMBO J. 2003;22:236–45. doi: 10.1093/emboj/cdg019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou Z, Wang J, Cao R, et al. Impaired angiogenesis, delayed wound healing and retarded tumor growth in perlecan heparan sulfate-deficient mice. Cancer Res. 2004;64:4699–702. doi: 10.1158/0008-5472.CAN-04-0810. [DOI] [PubMed] [Google Scholar]
- 67.Kadenhe-Chiweshe A, Papa J, McCrudden KW, et al. Sustained VEGF blockade results in microenvironmental sequestration of VEGF by tumors and persistent VEGF receptor-2 activation. Mol Cancer Res. 2008;6:1–9. doi: 10.1158/1541-7786.MCR-07-0101. [DOI] [PubMed] [Google Scholar]
- 68.Lambaerts K, Wilcox-Adelman SA, Zimmermann P. The signaling mechanisms of syndecan heparan sulfate proteoglycans. Curr Opin Cell Biol. 2009;21:662–9. doi: 10.1016/j.ceb.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jakobsson L, Kreuger J, Holmborn K, et al. Heparan sulfate in trans potentiates VEGFR-mediated angiogenesis. Dev Cell. 2006;10:625–34. doi: 10.1016/j.devcel.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 70.Whitelock JM, Murdoch AD, Iozzo RV, et al. The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin and heparanases. J Biol Chem. 1996;271:10079–86. doi: 10.1074/jbc.271.17.10079. [DOI] [PubMed] [Google Scholar]
- 71.Reiland J, Sanderson RD, Waguespack M, et al. Heparanase degrades syndecan-1 and perlecan heparan sulfate: functional implications for tumor cell invasion. J Biol Chem. 2004;279:8047–55. doi: 10.1074/jbc.M304872200. [DOI] [PubMed] [Google Scholar]
- 72.Patel VN, Knox SM, Likar KM, et al. Heparanase cleavage of perlecan heparan sulfate modulates FGF10 activity during ex vivo submandibular gland branching morphogenesis. Development. 2007;134:4177–86. doi: 10.1242/dev.011171. [DOI] [PubMed] [Google Scholar]
- 73.Joyce JA, Freeman C, Meyer-Morse N, et al. A functional heparan sulfate mimetic implicates both heparanase and heparan sulfate in tumor angiogenesis and invasion in a mouse model of multistage cancer. Oncogene. 2005;24:4037–51. doi: 10.1038/sj.onc.1208602. [DOI] [PubMed] [Google Scholar]
- 74.D’Ortho M-P, Will H, Atkinson S, et al. Membrane-type matrix metalloproteinases 1 and 2 exhibit broad-spectrum proteolytic capacities comparable to many matrix metalloproteinases. Eur J Biochem. 1997;250:751–7. doi: 10.1111/j.1432-1033.1997.00751.x. [DOI] [PubMed] [Google Scholar]
- 75.Deryugina EI, Quigley JP. Pleiotropic roles of matrix metalloproteinases in tumor angiogenesis: contrasting, overlapping and compensatory functions. Biochim Biophys Acta – Mol Cell Res. 2010;1803:103–20. doi: 10.1016/j.bbamcr.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mongiat M, Sweeney S, San Antonio JD, et al. Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J Biol Chem. 2003;278:4238–49. doi: 10.1074/jbc.M210445200. [DOI] [PubMed] [Google Scholar]
- 77.O’Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–85. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 78.Senger DR, Claffey KP, Benes JE, et al. Angiogenesis promoted by vascular endothelial growth factor: regulation through α1β1 and α2β1 integrins. Proc Natl Acad Sci USA. 1997;94:13612–7. doi: 10.1073/pnas.94.25.13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Senger DR, Perruzzi CA, Streit M, et al. The α1β1 and α2β1 integrins provide critical support for vascular endothelial growth factor signaling, endothelial cell migration, and tumor angiogenesis. Am J Pathol. 2002;160:195–204. doi: 10.1016/s0002-9440(10)64363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sweeney SM, DiLullo G, Slater SJ, et al. Angiogenesis in collagen I requires α2β1 ligation of a GFP*GER sequence and possible p38 MAPK activation and focal adhesion disassembly. J Biol Chem. 2003;278:30516–24. doi: 10.1074/jbc.M304237200. [DOI] [PubMed] [Google Scholar]
- 81.Bix G, Fu J, Gonzalez E, Macro L, et al. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through the α2β1 integrin. J Cell Biol. 2004;166:97–109. doi: 10.1083/jcb.200401150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bix G, Iozzo RA, Woodall B, et al. Endorepellin, the C-terminal angiostatic module of perlecan, enhances collagen-platelet responses via the α2β1 integrin receptor. Blood. 2007;109:3745–8. doi: 10.1182/blood-2006-08-039925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bix G, Iozzo RV. Matrix revolutions: “tails” of basement-membrane components with angiostatic functions. Trends Cell Biol. 2005;15:52–60. doi: 10.1016/j.tcb.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 84.Bix G, Iozzo RV. Novel interactions of perlecan: unraveling perlecan’s role in angiogenesis. Microsc Res. 2008;71:339–48. doi: 10.1002/jemt.20562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zoeller JJ, Iozzo RV. Proteomic profiling of endorepellin angiostatic activity on human endothelial cells. Proteome Sci. 2008;6:7. doi: 10.1186/1477-5956-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bix G, Castello R, Burrows M, et al. Endorepellin in vivo: targeting the tumor vasculature and retarding cancer growth and metabolism. J Natl Cancer Inst. 2006;98:1634–46. doi: 10.1093/jnci/djj441. [DOI] [PubMed] [Google Scholar]
- 87.Woodall BP, Nyström A, Iozzo RA, et al. Integrin α2β1 is the required receptor for endorepellin angiostatic activity. J Biol Chem. 2008;283:2335–43. doi: 10.1074/jbc.M708364200. [DOI] [PubMed] [Google Scholar]
- 88.San Antonio JD, Zoeller JJ, Habursky K, et al. A key role for the integrin α2β1 in experimental and developmental angiogenesis. Am J Pathol. 2009;175:1138–347. doi: 10.2353/ajpath.2009.090234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cailhier J-F, Sirois I, Raymond M-A, et al. Caspase-3 activation triggers extracellular release of cathepsin L and endorepellin proteolysis. J Biol Chem. 2008;283:27220–9. doi: 10.1074/jbc.M801164200. [DOI] [PubMed] [Google Scholar]
- 90.Gonzalez EM, Reed CC, Bix G, et al. BMP-1/Tolloid-like metalloproteases process endorepellin, the angiostatic C-terminal fragment of perlecan. J Biol Chem. 2005;280:7080–7. doi: 10.1074/jbc.M409841200. [DOI] [PubMed] [Google Scholar]
- 91.Laplante P, Raymond M-A, Labelle A, et al. Perlecan proteolysis induces α2β1 integrin and src-family kinases dependent anti-apoptotic pathway in fibroblasts in the absence of focal adhesion kinase activation. J Biol Chem. 2006;281:30383–92. doi: 10.1074/jbc.M606412200. [DOI] [PubMed] [Google Scholar]
- 92.Thadikkaran L, Crettaz D, Siegenthaler MA, et al. The role of proteomics in the assessment of premature rupture of fetal membranes. Clin Chim Acta. 2005;360:27–36. doi: 10.1016/j.cccn.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 93.Tsangaris GT, Karamessinis P, Kolialexi A, et al. Proteomic analysis of amniotic fluid in pregnancies with Down syndrome. Proteomics. 2006;6:4410–9. doi: 10.1002/pmic.200600085. [DOI] [PubMed] [Google Scholar]
- 94.Oda O, Shinzato T, Ohbayashi K, et al. Purification and characterization of perlecan fragment in urine of end-stage renal failure patients. Clin Chim Acta. 1996;255:119–32. doi: 10.1016/0009-8981(96)06395-4. [DOI] [PubMed] [Google Scholar]
- 95.O’Riordan E, Orlova TN, Mendelev N, et al. Urinary proteomic analysis of chronic renal allograft nephropathy. Proteomics Clin Appl. 2008;2:1025–35. doi: 10.1002/prca.200780137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chang JW, Kang U-B, Kim DH, et al. Identification of circulating endorepellin LG3 fragment: potential use as a serological biomarker for breast cancer. Proteomics Clin Appl. 2008;2:23–32. doi: 10.1002/prca.200780049. [DOI] [PubMed] [Google Scholar]
- 97.West L, Govindraj P, Koob TJ, et al. Changes in perlecan during chondrocyte differentiation in the fetal bovine rib growth plate. J Orthop Res. 2006;24:1317–26. doi: 10.1002/jor.20160. [DOI] [PubMed] [Google Scholar]
- 98.Eisenstein R, Sorgente N, Soble LW, et al. Resistance of certain tissue to invasion. Penetrability of explanted tissues by vascularized mesenchyme. Am J Pathol. 1973;73:765–74. [PMC free article] [PubMed] [Google Scholar]
- 99.Nyström A, Shaik ZP, Gullberg D, et al. Role of tyrosine phosphatase SHP-1 in the mechanism of endorepellin angiostatic activity. Blood. 2009;114:4897–906. doi: 10.1182/blood-2009-02-207134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bernfield M, Kokenyesi R, Kato M, et al. Biology of the syndecans: a family of transmembrane heparan sulfate proteoblycans. Annu Rev Cell Biol. 1992;8:365–93. doi: 10.1146/annurev.cb.08.110192.002053. [DOI] [PubMed] [Google Scholar]
- 101.Couchman JR. Syndecans: proteoglycan regulators of cell-surface microdomains. Nat Rev Mol Cell Biol. 2003;4:926–37. doi: 10.1038/nrm1257. [DOI] [PubMed] [Google Scholar]
- 102.Fears CY, Woods A. The role of syndecans in disease and wound healing. Matrix Biol. 2006;25:443–56. doi: 10.1016/j.matbio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 103.Couchman JR, Chen L, Woods A. Syndecans and cell adhesion. Int Rev Cytol. 2001;207:113–50. doi: 10.1016/s0074-7696(01)07004-8. [DOI] [PubMed] [Google Scholar]
- 104.Tkachenko E, Rhodes JM, Simons M. Syndecans: new kids on the signaling block. Circ Res. 2005;96:488–500. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- 105.Perrimon N, Bernfield M. Cellular functions of proteoglycans-an overview. Cell Develop Biol. 2001;12:65–7. doi: 10.1006/scdb.2000.0237. [DOI] [PubMed] [Google Scholar]
- 106.Kato M, Saunders S, Nguyen H, et al. Loss of cell surface syndecan-1 causes epithelial to transform into anchorage-independent mesenchyme-like cells. Mol Biol Cell. 1995;6:559–76. doi: 10.1091/mbc.6.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun D, Mcalmon KR, Davies JA, et al. Simultaneous loss of expression of syndecan-1 and E-cadherin in the embryonic palate during epithelial-mesenchymal transformation. Int J Dev Biol. 1998;42:733–6. [PubMed] [Google Scholar]
- 108.Nackaerts K, Verbeken E, Deneffe G, et al. Heparan sulfate proteoglycan expression in human lung-cancer cells. Int J Cancer. 1997;74:335–45. doi: 10.1002/(sici)1097-0215(19970620)74:3<335::aid-ijc18>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 109.Pulkkinen J, Penttinen M, Jalkanen M, et al. Syndecan-1: a new prognostic marker in laryngeal cancer. Acta Otolaryngol. 1997;117:312–5. doi: 10.3109/00016489709117794. [DOI] [PubMed] [Google Scholar]
- 110.Conejo JR, Kleeff J, Koliopanos A, et al. Syndecan-1 expression is up-regulated in pancreatic but not in other gastrointestinal cancers. Int J Cancer. 2000;88:12–20. doi: 10.1002/1097-0215(20001001)88:1<12::aid-ijc3>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 111.Davies EJ, Backhall FH, Shanks JH, et al. Distribution and clinical significance of heparan sulfate proteoblycans in ovarian cancer. Clin Cancer Res. 2004;10:5178–86. doi: 10.1158/1078-0432.CCR-03-0103. [DOI] [PubMed] [Google Scholar]
- 112.Barbareschi M, Maisonneuve P, Aldovini D, et al. High syndecan-1 expression in breast carcinoma is related to an aggressive phenotype and to poorer prognosis. Cancer. 2003;98:474–83. doi: 10.1002/cncr.11515. [DOI] [PubMed] [Google Scholar]
- 113.Seidel C, Sundan A, Hjorth M, et al. Serum syndecan-1: a new independent prognostic marker in multiple myeloma. Blood. 2000;95:388–92. [PubMed] [Google Scholar]
- 114.Sebbestyén A, Berczi L, Mihalik R, et al. Syndecan-1 (CD138) expression in human non-Hodgkin lymphomas. Brit J Haematol. 1999;104:412–9. doi: 10.1046/j.1365-2141.1999.01211.x. [DOI] [PubMed] [Google Scholar]
- 115.Khotskaya YB, Dai Y, Ritchie JP, et al. Syndecan-1 is required for robust growth, vascularization, and metastasis of myeloma tumors in vivo. J Biol Chem. 2009;284:26085–95. doi: 10.1074/jbc.M109.018473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Stanley MJ, Stanley MW, Sanderson RD, et al. Syndecan-1 expression is induced in the stroma of infiltrating breast carcinoma. Am J Clin Pathol. 1999;112:377–83. doi: 10.1093/ajcp/112.3.377. [DOI] [PubMed] [Google Scholar]
- 117.Tsanou E, Ioachim E, Briasoulis E, et al. Clinicopathological study of the expression of syndecan-1 in invasive breast carcinomas. Correlation with extracellular matrix components. J Exp Clin Cancer Res. 2004;23:641–50. [PubMed] [Google Scholar]
- 118.Maeda T, Desouky J, Friedl A. Syndecan-1 expression by stromal fiborblasts promotes breast carcinoma growth in vivo and stimulates tumor angiogenesis. Oncogene. 2006;25:1408–12. doi: 10.1038/sj.onc.1209168. [DOI] [PubMed] [Google Scholar]
- 119.Su G, Blaine SA, Qiao D, et al. Shedding of syndecan-1 by stromal fibroblasts stimulates human breast cancer cell proliferation via FGF2 activation. J Biol Chem. 2007;282:14906–15. doi: 10.1074/jbc.M611739200. [DOI] [PubMed] [Google Scholar]
- 120.Alexander CM, Reichsman F, Hinkes MT, et al. Syndecan-1 is required for Wnt-1-induced mammary tumorigenesis in mice. Nat Genet. 2000;25:329–32. doi: 10.1038/77108. [DOI] [PubMed] [Google Scholar]
- 121.Liu BY, McDermott SP, Khwaja SS, et al. The transforming activity of Wnt effectors correlates with their ability to induce the accumulation of mammary progenitor cells. Proc Natl Acad Sci USA. 2004;101:4158–63. doi: 10.1073/pnas.0400699101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McDermott SP, Ranheim EA, Leatherberry VS, et al. Juvenile syndecan-1 null mice are protected from carcinogen-induced tumor development. Oncogene. 2007;26:1407–16. doi: 10.1038/sj.onc.1209930. [DOI] [PubMed] [Google Scholar]
- 123.Beauvais DM, Ell BJ, McWhorter AR, et al. Syndecan-1 regulates αvβ3 and αvβ5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J Exp Med. 2009;206:691–705. doi: 10.1084/jem.20081278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang H, Leavitt L, Ramaswamy R, et al. Interaction of syndecan and α6β4 integrin cytoplasmic domains. Regulation of ErbB2-mediated integrin activation. J Biol Chem. 2010;285:13569–79. doi: 10.1074/jbc.M110.102137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liebersbach BF, Sanderson RD. Expression of syndecan-1inhibits cell invasion into type I collagen. J Biol Chem. 1994;269:20013–9. [PubMed] [Google Scholar]
- 126.Ishikawa T, Kramer RH. Sdc1 negatively modulates carcinoma cell motility and invastion. Exp Cell Res. 2010;316:951–65. doi: 10.1016/j.yexcr.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vuoriluoto K, Jokinen J, Kallio K, et al. Syndecan-1 supports integrin α2β1 – mediated adhesion to collagen. Exp Cell Res. 2008;314:3369–81. doi: 10.1016/j.yexcr.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 128.Chen P, Abacherli LE, Nadler ST, et al. MMP7 shedding of syndecan-1 facilitates re-epithelialization by affecting α2β1 integrin activation. PLoS ONE. 2009;4:e6565. doi: 10.1371/journal.pone.0006565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Granés F, Ureña JM, Rocamora N, et al. Ezrin links syndecan-2 to the cytoskeleton. J Cell Sci. 2000;113:1267–76. doi: 10.1242/jcs.113.7.1267. [DOI] [PubMed] [Google Scholar]
- 130.Munesue S, Kusano Y, Oguri K, et al. The role of syndecan-2 in regulation of actin-cytoskeletal organization of Lewis lung carcinoma-derived metastatic clones. Biochem J. 2002;363:201–9. doi: 10.1042/0264-6021:3630201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Munesue S, Yoshitomi Y, Kusano Y, et al. A novel function of syndecan-2, suppression of matrix metalloprotease-2 activation, which causes suppression of metastasis. J Biol Chem. 2007;282:28164–74. doi: 10.1074/jbc.M609812200. [DOI] [PubMed] [Google Scholar]
- 132.Modrowski D, Orosco A, Thévenard J, et al. Syndecan-2 overexpression induces osteosarcoma cell apoptosis: implication of syndecan-2 cytoplasmic domain and JNK signaling. Bone. 2005;37:180–9. doi: 10.1016/j.bone.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 133.Orosco A, Fromigué O, Bazille C, et al. Syndecan-2 affects the basal and chemotheray-induced apoptosis in osteosarcoma. Cancer Res. 2007;67:3708–15. doi: 10.1158/0008-5472.CAN-06-4164. [DOI] [PubMed] [Google Scholar]
- 134.Contreras HR, Fabre M, Granés F, et al. Syndecan-2 expression in colorectal cancer-derived HT-29 M6 epithelial cells induces a migratory phenotype. Biochem Biophys Res Commun. 2001;286:742–51. doi: 10.1006/bbrc.2001.5459. [DOI] [PubMed] [Google Scholar]
- 135.Park H, Kim Y, Lim Y, et al. Syndecan-2 mediates adhesion and proliferation of colon carcinoma cells. J Biol Chem. 2002;277:29730–6. doi: 10.1074/jbc.M202435200. [DOI] [PubMed] [Google Scholar]
- 136.Choi S, Kim Y, Park H, et al. Syndecan-2 overexpression regulates adhesion and migration through cooperation with integrin α2. Biochem Biophys Res Commun. 2009;384:231–5. doi: 10.1016/j.bbrc.2009.04.093. [DOI] [PubMed] [Google Scholar]
- 137.Choi Y, Kim H, Chung H, et al. Syndecan-2 regulates cell migration in colon cancer cells through Tiam1-mediated rac activation. Biochem Biophys Res Commun. 2010;391:921–5. doi: 10.1016/j.bbrc.2009.11.165. [DOI] [PubMed] [Google Scholar]
- 138.Fears CY, Gladson CL, Woods A. Syndecan-2 is expressed in the microvasculature of gliomas and regulates angiogenic processes in microvascular endothelial cells. J Biol Chem. 2006;281:14533–6. doi: 10.1074/jbc.C600075200. [DOI] [PubMed] [Google Scholar]
- 139.Chen E, Hermanson S, Ekker SC. Syndecan-2 is essential for angiogenic sprouting during zebrafish development. Blood. 2004;103:1710–9. doi: 10.1182/blood-2003-06-1783. [DOI] [PubMed] [Google Scholar]
- 140.Beauvais DLM, Rapraeger AC. Syndecan-1-mediated cell spreading requires signalinb by αvβ3 integrins in human breast carcinoma cells. Exp Cell Res. 2003;286:219–32. doi: 10.1016/s0014-4827(03)00126-5. [DOI] [PubMed] [Google Scholar]
- 141.Jayson GC, Yives C, Paraskeva C, et al. Coordinated modulation of the fibroblast growth factor dual receptor mechanism during transformation from human colon adenoma to carcinoma. Int J Cancer. 1999;82:298–304. doi: 10.1002/(sici)1097-0215(19990719)82:2<298::aid-ijc23>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 142.Chalkiadaki G, Nikitovic D, Berdiaki A, et al. Fibroblast growth factor-2 modulates melanoma adhesion and migration through a syndecan-4-dependent mechanism. Int J Biochem Cell Biol. 2009;41:1323–31. doi: 10.1016/j.biocel.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 143.O’Connell MP, Fiori JL, Kershner EK, et al. Heparan sulfate proteoglycan modulation of Wnt5A signal transduction in metastatic melanoma cells. J Biol Chem. 2009;284:28704–12. doi: 10.1074/jbc.M109.028498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mundhenke C, Meyer K, Drew S, et al. Heparan sulfate proteoglycans as regulators of fibroblast growth factor-2 receptor binding in breast carcinomas. Am J Pathol. 2002;160:185–94. doi: 10.1016/S0002-9440(10)64362-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Huang W, Chiquet-Ehrismann R, Moyano JV, et al. Interference of Tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res. 2001;61:8586–94. [PubMed] [Google Scholar]
- 146.Filmus J, Capurro M, Rast J. Glypicans. Genome Biol. 2008;9:224. doi: 10.1186/gb-2008-9-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Häcker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6:530–41. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- 148.Lin X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 2004;131:6009–21. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- 149.Capurro M, Xiang YY, Lobe C, et al. Glypican-3 promotes the growth of hepatocellular carcinoma by stimulating canonical Wnt signaling. Cancer Res. 2005;65:6245–54. doi: 10.1158/0008-5472.CAN-04-4244. [DOI] [PubMed] [Google Scholar]
- 150.De Cat B, Muyldermans S-Y, Coomans C, et al. Processing by proprotein convertases is required for glypican-3 modulation of cell survival, Wnt signaling, and gastrulation movements. J Cell Biol. 2003;163:625–35. doi: 10.1083/jcb.200302152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gonzales AD, Kaya M, Shi W, et al. OCI-5/GPC3, a glypican encoded by a gene that is mutated in the Simpson-Golabi-Behmel overgrowth syndrome, induces apoptosis in a cell line-specific manner. J Cell Biol. 1998;141:1407–14. doi: 10.1083/jcb.141.6.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Filmus J. Glypicans in growth control and cancer. Glycobiology. 2001;11:19–23R. doi: 10.1093/glycob/11.3.19r. [DOI] [PubMed] [Google Scholar]
- 153.Xiang YY, Ladeda V, Filmus J. Glypican-3 expression is silenced in human breast cancer. Oncogene. 2001;20:7408–12. doi: 10.1038/sj.onc.1204925. [DOI] [PubMed] [Google Scholar]
- 154.Kim H, Xu G-L, Borczuk AC, et al. The heparan sulfate proteoglycan GPC3 is a potential lung tumor suppressor. Am J Resp Cell Mol Biol. 2003;29:694–701. doi: 10.1165/rcmb.2003-0061OC. [DOI] [PubMed] [Google Scholar]
- 155.Lin H, Huber R, Schlessinger D, et al. Frequent silencing of the GPC3 gene in ovarian cancer cell lines. Cancer Res. 1999;59:807–10. [PubMed] [Google Scholar]
- 156.Murthy SS, Shen T, De Rienzo A, et al. Expression of GPC3, an X-linked recessive overgrowth gene, is silenced in malignant mesothelioma. Oncogene. 2000;19:410–6. doi: 10.1038/sj.onc.1203322. [DOI] [PubMed] [Google Scholar]
- 157.Capurro M, Xu P, Shi W, et al. Glypican-3 inhibits Hedgehog signaling during develpment by competing with Patched for Hedgehog binding. Dev Cell. 2008;14:700–11. doi: 10.1016/j.devcel.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 158.Ota S, Hishinuma M, Yamauchi N, et al. Oncofetal protein glypican-3 in testicular germ-cell tumor. Virchows Arch. 2006;449:308–14. doi: 10.1007/s00428-006-0238-x. [DOI] [PubMed] [Google Scholar]
- 159.Yamauchi N, Watanabe A, Hishinuma M, et al. The glypican 3 oncofetal protein is a promising diagnostic marker for hepatocellular carcinoma. Modern Pathol. 2005;18:1591–8. doi: 10.1038/modpathol.3800436. [DOI] [PubMed] [Google Scholar]
- 160.Capurro M, Wanless I, Sherman M, et al. Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology. 2003;125:89–97. doi: 10.1016/s0016-5085(03)00689-9. [DOI] [PubMed] [Google Scholar]
- 161.Nakatsura T, Yoshitake Y, Senju S, et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem Biophys Res Commun. 2003;306:16–25. doi: 10.1016/s0006-291x(03)00908-2. [DOI] [PubMed] [Google Scholar]
- 162.Cheng W, Tseng C-J, Lin TTC, et al. Glypican-3-mediated oncogenesis involves the Insulin-like growth factor-signaling pathway. Carcinogenesis. 2008;29:1319–26. doi: 10.1093/carcin/bgn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zittermann SI, Capurro M, Shi W, et al. Soluble glypican 3 inhibits the growth of hepatocellular carcinoma in vitro and in vivo. Int J Cancer. 2010;126:1291–301. doi: 10.1002/ijc.24941. [DOI] [PubMed] [Google Scholar]
- 164.Komori H, Nakatsura T, Senju S, et al. Identification of HLA-A2- or HLA-A24-restricted CTL epitopes possibly useful for glypican-3-specific immunotherapy of hepatocellular carcinoma. Clin Cancer Res. 2006;12:2689–97. doi: 10.1158/1078-0432.CCR-05-2267. [DOI] [PubMed] [Google Scholar]
- 165.Nakatsura T, Komori H, Kubo T, et al. Mouse homologue of a novel human oncofetal antigen, glypican-3, evokes T-cell-mediated tumor rejection without autoimmune reactions in mice. Clin Cancer Res. 2004;10:8630–40. doi: 10.1158/1078-0432.CCR-04-1177. [DOI] [PubMed] [Google Scholar]
- 166.Kleef J, Wildi S, Kumbasar A, et al. Stable transfection of a glypican-1 antisense construct decreases tumorigenicity in PANC-1 pancreatic carcinoma cells. Pancreas. 1999;19:281–8. doi: 10.1097/00006676-199910000-00009. [DOI] [PubMed] [Google Scholar]
- 167.Su G, Meyer K, Nandini CQ, et al. Glypican-1 is frequently overexpressed in human gliomas and enhances FGF-2 signaling in glioma cells. Am J Pathol. 2006;168:2014–26. doi: 10.2353/ajpath.2006.050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Aikawa T, Whipple CA, Lopez ME, et al. Glypican-1 modulates the angiogenic and metastatic potential of human and mouse cancer cells. J Clin Inv. 2008;118:89–99. doi: 10.1172/JCI32412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kleeff J, Ishiwata T, Kumbasar A, et al. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J Clin Invest. 1998;102:1662–73. doi: 10.1172/JCI4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Williamson D, Selfe J, Gordon T, et al. Role for amplification and expression of Glypican-5 in rhabdomyosarcoma. Cancer Res. 2007;67:57–65. doi: 10.1158/0008-5472.CAN-06-1650. [DOI] [PubMed] [Google Scholar]
- 171.Lai JP, Sandhu DS, Shire AM, et al. The tumor suppressor function of human sulfatase 1 (SULF1) in carcinogenesis. J Gastrointest Cancer. 2008;39:149–58. doi: 10.1007/s12029-009-9058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rosen SD, Hassan L-A. Sulf-2: an extracellular modulator of cell signaling and a cancer target candidate. Exp Opin Therap Targets. 2010;14:935–49. doi: 10.1517/14728222.2010.504718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hayashida K, Bartlett AH, Chen Y, et al. Molecular and cellular mechanisms of ectodomain shedding. Anatomic Rec. 2010;293:925–37. doi: 10.1002/ar.20757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Vassilakopoulos TP, Kyrtsonis MC, Papadogiannis A, et al. Serum levels of soluble syndecan-1 in Hodgkin’s lymphoma. AntiCancer Res. 2005;25:4743–6. [PubMed] [Google Scholar]
- 175.Anttonen A, Leppä S, Ruotsalainen T, et al. Pretreatment serum syndecan-1 levels and outcome in small cell lung cancer patients treated with platinum-based chemotherapy. Lung Caner. 2003;41:171–7. doi: 10.1016/s0169-5002(03)00196-x. [DOI] [PubMed] [Google Scholar]
- 176.Joensuu H, Anttonen A, Eriksson M, et al. Soluble Syndecan-1 and serum basic fibroblast growth factor are new prognostic factors in lung cancer. Cancer Res. 2002;62:5210–7. [PubMed] [Google Scholar]
- 177.Dhodapkar MV, Kelly T, Theus A, et al. Elevated levels of shed syndecan-1 correlate with tumour mass and decreased matrix metalloproteinase-9 activity in the serum of patients with multiple myeloma. Brit J Haematol. 1997;99:368–71. doi: 10.1046/j.1365-2141.1997.3893203.x. [DOI] [PubMed] [Google Scholar]
- 178.Yang Y, Yaccoby S, Liu W, et al. Soluble syndecan-1 promotes growth of myeloma tumors in vivo. Blood. 2002;100:610–7. doi: 10.1182/blood.v100.2.610. [DOI] [PubMed] [Google Scholar]
- 179.Ding K, Lopez-Burks M, Sánchez-Duran JA, et al. Growth factor-induced shedding of syndecan-1 confers glypican-1 dependence on mitogenic responses of cancer cells. J Cell Biol. 2005;171:729–38. doi: 10.1083/jcb.200508010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Fux L, Ilan N, Sanderson RD, et al. Heparanase: busy at the cell surface. Trends Biochem Sci. 2009;34:511–9. doi: 10.1016/j.tibs.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kelly T, Miao H-Q, Yang Y, et al. High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Res. 2003;63:8749–56. [PubMed] [Google Scholar]
- 182.Mahtouk K, Hose D, Raynaud P, et al. Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood. 2007;109:4914–23. doi: 10.1182/blood-2006-08-043232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yang Y, MacLeod V, Miao H-Q, et al. Heparanase enhances syndecan-1 shedding. A novel mechanism for stimulation of tumor growth and metastasis. J Biol Chem. 2007;282:13326–33. doi: 10.1074/jbc.M611259200. [DOI] [PubMed] [Google Scholar]
- 184.Purushothaman A, Chen L, Yang Y, et al. Heparanase stimulation of protease espression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem. 2008;283:32628–36. doi: 10.1074/jbc.M806266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Purushothaman A, Uyama T, Kobayashi F, et al. Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood. 2010;115:2449–57. doi: 10.1182/blood-2009-07-234757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Derksen PWB, Keehnen RMJ, Evers LM, et al. Cell surface proteoglycan syndecan-1 mediates hepatocyte growth factor binding and promotes Met signaling in multiple myeloma. Blood. 2002;99:1405–10. doi: 10.1182/blood.v99.4.1405. [DOI] [PubMed] [Google Scholar]
- 187.Weber IT, Harrison RW, Iozzo RV. Model structure of decorin and implications for collagen fibrillogenesis. J Biol Chem. 1996;271:31767–70. doi: 10.1074/jbc.271.50.31767. [DOI] [PubMed] [Google Scholar]
- 188.Iozzo RV, Goldoni S, Berendsen A. Small leucine-rich proteoglycans. In: Mecham RP, et al., editors. Extracellular matrix: an overview. Springer; 2011. In press. [Google Scholar]
- 189.Ruoslahti E, Yamaguchi Y. Proteoglycans as modulators of growth factor activities. Cell. 1991;64:867–9. doi: 10.1016/0092-8674(91)90308-l. [DOI] [PubMed] [Google Scholar]
- 190.Keene DR, San Antonio JD, Mayne R, et al. Decorin binds near the C terminus of type I collagen. J Biol Chem. 2000;275:21801–4. doi: 10.1074/jbc.C000278200. [DOI] [PubMed] [Google Scholar]
- 191.Vogel KG, Paulsson M, Heinegård D. Specific inhibition of type I and type II collagen fibrillogenesis by the small proteoglycan of tendon. Biochem J. 1984;223:587–97. doi: 10.1042/bj2230587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Danielson KG, Baribault H, Holmes DF, et al. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol. 1997;136:729–43. doi: 10.1083/jcb.136.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Iozzo RV. The family of the small leucine-rich proteoglycans: key regulators of matrix assembly and cellular growth. Crit Rev Biochem Mol Biol. 1997;32:141–74. doi: 10.3109/10409239709108551. [DOI] [PubMed] [Google Scholar]
- 194.Iozzo RV. The biology of the small leucine-rich proteoglycans. Functional network of interactive proteins. J Biol Chem. 1999;274:18843–6. doi: 10.1074/jbc.274.27.18843. [DOI] [PubMed] [Google Scholar]
- 195.Yamaguchi Y, Ruoslahti E. Expression of human proteoglycan in Chinese hamster ovary cells inhibits cell proliferation. Nature. 1988;336:244–6. doi: 10.1038/336244a0. [DOI] [PubMed] [Google Scholar]
- 196.Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor-β by the proteoglycan decorin. Nature. 1990;346:281–4. doi: 10.1038/346281a0. [DOI] [PubMed] [Google Scholar]
- 197.Coppock DL, Kopman C, Scandalis S, et al. Preferential gene expression in quiescent human lung fibroblasts. Cell Growth & Differ. 1993;4:483–93. [PubMed] [Google Scholar]
- 198.Mauviel A, Santra M, Chen YQ, et al. Transcriptional regulation of decorin gene expression. Induction by quiescence and repression by tumor necrosis factor-α. J Biol Chem. 1995;270:11692–700. doi: 10.1074/jbc.270.19.11692. [DOI] [PubMed] [Google Scholar]
- 199.Santra M, Skorski T, Calabretta B, et al. De novo decorin gene expression suppresses the malignant phenotype in human colon cancer cells. Proc Natl Acad Sci USA. 1995;92:7016–20. doi: 10.1073/pnas.92.15.7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Santra M, Mann DM, Mercer EW, et al. Ectopic expression of decorin protein core causes a generalized growth suppression in neoplastic cells of various histogenetic origin and requires endogenous p21, an inhibitor of cyclin-dependent kinases. J Clin Invest. 1997;100:149–57. doi: 10.1172/JCI119507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.De Luca A, Santra M, Baldi A, et al. Decorin-induced growth suppression is associated with upregulation of p21, an inhibitor of cyclin-dependent kinases. J Biol Chem. 1996;271:18961–5. doi: 10.1074/jbc.271.31.18961. [DOI] [PubMed] [Google Scholar]
- 202.Ständer M, Naumann U, Wick W, et al. Transforming growth factor-β and p-21: multiple molecular targets of decorin-mediated suppression of neoplastic growth. Cell Tissue Res. 1999;296:221–7. doi: 10.1007/s004410051283. [DOI] [PubMed] [Google Scholar]
- 203.Xaus J, Comalada M, Cardó M, et al. Decorin inhibits macrophage colony-stimulating factor proliferation of macrophages and enhances cell survival through induction of p27Kip1 and p21Waf1. Blood. 2001;98:2124–33. doi: 10.1182/blood.v98.7.2124. [DOI] [PubMed] [Google Scholar]
- 204.Schönherr E, Levkau B, Schaefer L, et al. Decorin-mediated signal transduction in endothelial cells. Involvement of Akt/protein kinase B in up-regulation of p21WAF1/CIP1 but not p27KIP1. J Biol Chem. 2001;276:40687–92. doi: 10.1074/jbc.M105426200. [DOI] [PubMed] [Google Scholar]
- 205.Tralhão JG, Schaefer L, Micegova M, et al. in vivo selective and distant killing of cancer cells using adenovirus-mediated decorin gene transfer. FASEB J. 2003;17:464–6. doi: 10.1096/fj.02-0534fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Wu H, Wang S, Xue A, et al. Overexpression of decorin induces apoptosis and cell growth arrest in cultured rat mesangial cells in vitro. Nephrology. 2008;13:607–15. doi: 10.1111/j.1440-1797.2008.00961.x. [DOI] [PubMed] [Google Scholar]
- 207.Li X, Pennisi A, Yaccoby S. Role of decorin in the antimyeloma effects of osteoblasts. Blood. 2008;112:159–68. doi: 10.1182/blood-2007-11-124164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hu Y, Sun H, Owens RT, et al. Decorin suppresses prostate tumor growth through inhibition of epidermal growth factor and androgen receptor pathways. Neoplasia. 2009;11:1042–53. doi: 10.1593/neo.09760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Moscatello DK, Santra M, Mann DM, et al. Decorin suppresses tumor cell growth by activating the epidermal growth factor receptor. J Clin Investig. 1998;101:406–12. doi: 10.1172/JCI846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Iozzo RV, Moscatello D, McQuillan DJ, et al. Decorin is a biological ligand for the epidermal growth factor receptor. J Biol Chem. 1999;274:4489–92. doi: 10.1074/jbc.274.8.4489. [DOI] [PubMed] [Google Scholar]
- 211.Santra M, Reed CC, Iozzo RV. Decorin binds to a narrow region of the epidermal growth factor (EGF) receptor, partially overlapping with but distinct from the EGF-binding epitope. J Biol Chem. 2002;277:35671–81. doi: 10.1074/jbc.M205317200. [DOI] [PubMed] [Google Scholar]
- 212.Csordás G, Santra M, Reed CC, et al. Sustained down-regulation of the epidermal growth factor receptor by decorin. A mechanism for controlling tumor growth in vivo. J Biol Chem. 2000;275:32879–87. doi: 10.1074/jbc.M005609200. [DOI] [PubMed] [Google Scholar]
- 213.Santra M, Eichstetter I, Iozzo RV. An anti-oncogenic role for decorin: downregulation of ErbB2 leads to growth suppression and cytodifferentiation of mammary carcinoma cells. J Biol Chem. 2000;275:35153–61. doi: 10.1074/jbc.M006821200. [DOI] [PubMed] [Google Scholar]
- 214.Zhu J-X, Goldoni S, Bix G, et al. Decorin evokes protracted internalization and degradation of the EGF receptor via caveolar endocytosis. J Biol Chem. 2005;280:32468–79. doi: 10.1074/jbc.M503833200. [DOI] [PubMed] [Google Scholar]
- 215.Goldoni S, Owens RT, McQuillan DJ, et al. Biologically active decorin is a monomer in solution. J Biol Chem. 2004;279:6606–12. doi: 10.1074/jbc.M310342200. [DOI] [PubMed] [Google Scholar]
- 216.Reed CC, Gauldie J, Iozzo RV. Suppression of tumorigenicity by adenovirus-mediated gene transfer of decorin. Oncogene. 2002;21:3688–95. doi: 10.1038/sj.onc.1205470. [DOI] [PubMed] [Google Scholar]
- 217.Ständer M, Naumann U, Dumitrescu L, et al. Decorin gene transfer-mediated suppression of TGF-β synthesis abrogates experimental malignant glioma growth in vivo. Gene Therapy. 1998;5:1187–94. doi: 10.1038/sj.gt.3300709. [DOI] [PubMed] [Google Scholar]
- 218.Biglari A, Bataille D, Naumann U, et al. Effects of ectopic decorin in modulating intracranial glioma progression in vivo, in a rat syngeneic model. Cancer Gene Therapy. 2004;11:721–32. doi: 10.1038/sj.cgt.7700783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Reed CC, Waterhouse A, Kirby S, et al. Decorin prevents metastatic spreading of breast cancer. Oncogene. 2005;24:1104–10. doi: 10.1038/sj.onc.1208329. [DOI] [PubMed] [Google Scholar]
- 220.Araki K, Wakabayashi H, Shintani K, et al. Decorin suppresses bone metastasis in a breast cancer cell line. Oncology. 2009;77:92–9. doi: 10.1159/000228253. [DOI] [PubMed] [Google Scholar]
- 221.Seidler DG, Goldoni S, Agnew C, et al. Decorin protein core inhibits in vivo cancer growth and metabolism by hindering epidermal growth factor receptor function and triggering apoptosis via caspase-3 activation. J Biol Chem. 2006;281:26408–18. doi: 10.1074/jbc.M602853200. [DOI] [PubMed] [Google Scholar]
- 222.Goldoni S, Seidler DG, Heath J, et al. An anti-metastatic role for decorin in breast cancer. Am J Pathol. 2008;173:844–55. doi: 10.2353/ajpath.2008.080275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Iozzo RV, Wight TN. Isolation and characterization of proteoglycans synthesized by human colon and colon carcinoma. J Biol Chem. 1982;257:11135–44. [PubMed] [Google Scholar]
- 224.Goldoni S, Iozzo RV. Tumor microenvironment: modulation by decorin and related molecules harboring leucine-rich tandem motifs. Int J Cancer. 2008;123:2473–9. doi: 10.1002/ijc.23930. [DOI] [PubMed] [Google Scholar]
- 225.Iozzo RV. Proteoglycans and neoplasia. Cancer Metastasis Rev. 1988;7:39–50. doi: 10.1007/BF00048277. [DOI] [PubMed] [Google Scholar]
- 226.Wu I-C, Wu D-C, Huang C-C, et al. Plasma decorin predicts the presence of esophageal squamous cell carcinoma. Int J Cancer. 2010;127:2138–46. doi: 10.1002/ijc.25239. [DOI] [PubMed] [Google Scholar]
- 227.Danielson KG, Fazzio A, Cohen I, et al. The human decorin gene: intron-exon organization, discovery of two alternatively spliced exons in the 5’ untranslated region, and mapping of the gene to chromosome 12q23. Genomics. 1993;15:146–60. doi: 10.1006/geno.1993.1022. [DOI] [PubMed] [Google Scholar]
- 228.Santra M, Danielson KG, Iozzo RV. Structural and functional characterization of the human decorin gene promoter. J Biol Chem. 1994;269:579–87. [PubMed] [Google Scholar]
- 229.Mauviel A, Korang K, Santra M, et al. Identification of a bimodal regulatory element encompassing a canonical AP-1 binding site in the proximal promoter region of the human decorin gene. J Biol Chem. 1996;271:24824–9. doi: 10.1074/jbc.271.40.24824. [DOI] [PubMed] [Google Scholar]
- 230.Adany R, Iozzo RV. Hypomethylation of the decorin proteoglycan gene in human colon cancer. Biochem J. 1991;276:301–6. doi: 10.1042/bj2760301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Iozzo RV, Chakrani F, Perrotti D, et al. Cooperative action of germline mutations in decorin and p53 accelerates lymphoma tumorigenesis. Proc Natl Acad Sci USA. 1999;96:3092–7. doi: 10.1073/pnas.96.6.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Standker L, Schrader M, Kanse SM, et al. Isolation and characterization of the circulating form of human endostatin. FEBS Lett. 1997;420:129–33. doi: 10.1016/s0014-5793(97)01503-2. [DOI] [PubMed] [Google Scholar]
- 233.Goldoni S, Humphries A, Nyström A, et al. Decorin is a novel antagonistic ligand of the Met receptor. J Cell Biol. 2009;185:743–54. doi: 10.1083/jcb.200901129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Buraschi S, Pal N, Tyler-Rubinstein N, et al. Decorin antagonizes Met receptor activity and downregulates β-catenin and Myc levels. J Biol Chem. 2010;285:42075–85. doi: 10.1074/jbc.M110.172841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sears RC. The life cycle of c-Myc. From synthesis to degradation. Cell Cycle. 2004;3:1133–7. [PubMed] [Google Scholar]
- 236.Herynk MH, Tsan R, Radinsky R, et al. Activation of c-Met in colorectal carcinoma cells leads to constitutive association of tyrosine-phopshorylated β-catenin. Clin Exp Metastasis. 2003;20:291–300. doi: 10.1023/a:1024024218529. [DOI] [PubMed] [Google Scholar]
- 237.Rasola A, Fassetta M, De Bacco F, et al. A positive feedback loop between hepatocyte growth factor receptor and β-catenin sustains colorectal cancer cell invasive growth. Oncogene. 2007;26:1078–87. doi: 10.1038/sj.onc.1209859. [DOI] [PubMed] [Google Scholar]
- 238.Li Y, Guessous F, Johnson EB, et al. Functional and molecular interactions between the HGF/c-Met pathway and c-Myc in large-cell medulloblastoma. Lab Invest. 2008;88:98–111. doi: 10.1038/labinvest.3700702. [DOI] [PubMed] [Google Scholar]
- 239.Seo D-W, Li H, Qu C-K, et al. Shp-1 mediates the antiproliferative activity of tissue inhibitor of metalloproteinase-2 in human microvascular endothelial cells. J Biol Chem. 2006;281:3711–21. doi: 10.1074/jbc.M509932200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Moore D, Kohn AD, De Ferrari GV, et al. Wnt and β-catenin signaling: diseases and therapies. Nat Rev Genet. 2004;5:689–99. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 241.Albihn A, Johnsen JI, Henriksson MA. MYC in oncogenesis and as a target for cancer therapies. Adv Cancer Res. 2010;107:163–224. doi: 10.1016/S0065-230X(10)07006-5. [DOI] [PubMed] [Google Scholar]
- 242.Jung P, Menssen A, Mayr D, et al. AP4 encodes a c-Myc-inducible reperessor of p21. Proc Natl Acad Sci USA. 2008;105:15046–51. doi: 10.1073/pnas.0801773105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Arnold HK, Zhang X, Daniel CJ, et al. The Axin1 scaffold protein promotes formation of a degradation complex for c-Myc. EMBO J. 2010;28:500–12. doi: 10.1038/emboj.2008.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Chakravarti S, Magnuson T, Lass JH, et al. Lumican regulates collagen fibril assembly: skin fragility and corneal opacity in the absence of lumican. J Cell Biol. 1998;141:1277–86. doi: 10.1083/jcb.141.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Chakravarti S. Functions of lumican and fibromodulin: lessons from knockout mice. Glycoconj J. 2003;19:287–93. doi: 10.1023/A:1025348417078. [DOI] [PubMed] [Google Scholar]
- 246.Leygue E, Snell L, Dotzlaw H, et al. Expression of lumican in human breast carcinoma. Cancer Res. 1999;58:1348–52. [PubMed] [Google Scholar]
- 247.Brézillon S, Venteo L, Ramont L, et al. Expression of lumican, a small leucine-rich proteoglycan with antitumour activity, in human malignant melanoma. Clin Exp Derm. 2007;32:405–16. doi: 10.1111/j.1365-2230.2007.02437.x. [DOI] [PubMed] [Google Scholar]
- 248.Sifaki M, Assouti M, Nikitovic D, et al. Lumican, a small leucine-rich proteoglycan substituted with keratan sulfate chains is expressed and secreted by human melanoma cells and not normal melanocytes. IUBMB Life. 2008;58:606–10. doi: 10.1080/15216540600951605. [DOI] [PubMed] [Google Scholar]
- 249.Nikitovic D, Berdiaki A, Zafiropoulos A, et al. Lumican expression is positively correlated with the differentiation and negatively with the growth of human osteosarcoma cells. FEBS J. 2008;275:350–61. doi: 10.1111/j.1742-4658.2007.06205.x. [DOI] [PubMed] [Google Scholar]
- 250.Matsuda Y, Yamamoto T, Kudo M, et al. Expression and roles of lumican in lung adenocarcinoma and squamous cell carcinoma. Int J Oncol. 2008;33:1177–85. [PubMed] [Google Scholar]
- 251.Ishiwata T, Cho K, Kawahara K, et al. Role of lumican in cancer cells and adjacent stromal tissues in human pancreatic cancer. Onc Reports. 2007;18:537–43. [PubMed] [Google Scholar]
- 252.Lu YP, Ishiwata T, Asano G. Lumican expression in alpha cells of islets in pancreas and pancreatic cancer cells. J Pathol. 2002;196:324–30. doi: 10.1002/path.1037. [DOI] [PubMed] [Google Scholar]
- 253.Li X, Carthew RW. A microRNA mediates EGF receptor signaling and promotes photoreceptor differentiation in the Drosophila eye. Cell. 2005;123:1267–77. doi: 10.1016/j.cell.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 254.Vuillermoz B, Khoruzhenko A, D’Onofrio MF, et al. The small leucine-rich proteoglycan lumican inhibits melanoma progression. Exp Cell Res. 2004;296:294–306. doi: 10.1016/j.yexcr.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 255.Brézillon S, Zeltz C, Schneider L, et al. Lumican Inhibits B16F1 melanoma cell lung metastasis. J Physiol Pharmacol. 2009;60:15–22. [PubMed] [Google Scholar]
- 256.Yoshioka N, Inoue H, Nakanishi K, et al. Isolation of transformation suppressor genes by cDNA substraction: lumican suppresses transformation induced by v-src and v-K-ras. J Virol. 2000;74:1008–13. doi: 10.1128/jvi.74.2.1008-1013.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Li Y, Aoki T, Mori Y, et al. Cleavage of lumican by membrane-type matrix metalloprotease-1 abrogates this proteoglycan-mediated suppression of tumor cell colony formation in soft agar. Cancer Res. 2004;64:7058–64. doi: 10.1158/0008-5472.CAN-04-1038. [DOI] [PubMed] [Google Scholar]
- 258.Vij N, Roberts L, Joyce S, et al. Lumican suppresses cell proliferation and aids Fas-Fas ligand mediated apoptosis: implications in the cornea. Exp Eye Res. 2004;78:957–71. doi: 10.1016/j.exer.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 259.Vij N, Roberts L, Joyce S, et al. Lumican regulates corneal inflammatory responses by modulating Fas-Fas ligand signaling. Invest Ophthalmol Vis Sci. 2005;46:88–95. doi: 10.1167/iovs.04-0833. [DOI] [PubMed] [Google Scholar]
- 260.Carlson EC, Lin M, Liu C-Y, et al. Keratocan and lumican regulate neutrophil infiltration and corneal clarity in lipopolysaccharide-induced keratitis by direct interaction with CXCL1. J Biol Chem. 2007;282:33502–9. doi: 10.1074/jbc.M705823200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Hayashi Y, Call MK, Chikama T-I, et al. Lumican is required for neutrophil extravasation following corneal injury and wound healing. J Cell Sci. 2010;123:2987–95. doi: 10.1242/jcs.068221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Lee S, Bowrin K, Hamad AR, et al. Extracellular matrix lumican deposited on the surface of neutrophils promotes migration by binding to β2 integrin. J Biol Chem. 2009;284:23662–9. doi: 10.1074/jbc.M109.026229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.D’Onofrio M-F, Brézillon S, Baranek T, et al. Identification of β1 integrin as mediator of melanoma cell adhesion to lumican. Biochem Biophys Res Commun. 2008;365:266–72. doi: 10.1016/j.bbrc.2007.10.155. [DOI] [PubMed] [Google Scholar]
- 264.Brézillon S, Radwanska A, Zeltz C, et al. Lumican core protein inhibits melanoma cell migration via alterations of focal adhesion compleses. Cancer Lett. 2009;283:92–100. doi: 10.1016/j.canlet.2009.03.032. [DOI] [PubMed] [Google Scholar]
- 265.Zeltz C, Brézillon S, Perreau C, et al. Lumcorin: a leucine-rich repeat 9-derived peptide from human lumican inhibiting melanoma cell migration. FEBS Lett. 2009;583:3027–32. doi: 10.1016/j.febslet.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 266.Zeltz C, Brézillon S, Käpylä J, et al. Lumican inhibits cell migration through α2β1 integrin. Exp Cell Res. 2010;316:2922–31. doi: 10.1016/j.yexcr.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 267.Buczek-Thomas JA, Hsia E, Rich CB, et al. Inhibition of histone acetyltransferase by glycosaminoglycans. J Cell Biochem. 2008;105:108–20. doi: 10.1002/jcb.21803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Kovalszky I, Dudás J, Oláh-Nagy J, et al. Inhibition of DNA topoisomerase I activity by heparan sulfate and modulation by basic fibroblast growth factor. Mol Cell Biochem. 1998;183:11–23. doi: 10.1023/a:1006898920637. [DOI] [PubMed] [Google Scholar]
- 269.Theocharis AD, Tzanakakis G, Karamanos NK. Proteoglycans in health and disease: novel roles for proteoglycan in malignancy and their pharmacological targeting. FEBS J. 2010;277:3904–23. doi: 10.1111/j.1742-4658.2010.07800.x. [DOI] [PubMed] [Google Scholar]