

Abstract
Peroxisome proliferator-activated receptor (PPAR)-γ modulates substrate metabolism and inflammatory responses. In experimental rats subjected to myocardial ischemia-reperfusion (I/R), thiazolidinedione PPAR-γ activators reduce infarct size and preserve left ventricular function. Troglitazone is the only PPAR-γ activator that has been shown to be protective in I/R in large animals. However, because troglitazone contains both α-tocopherol and thiazolidinedione moieties, whether PPAR-γ activation per se is protective in myocardial I/R in large animals remains uncertain. To address this question, 56 pigs were treated orally for 8 wk with troglitazone (75 mg·kg−1 ·day−1), rosiglitazone (3 mg·kg−1 ·day−1), or α-tocopherol (73 mg·kg−1 ·day−1, equimolar to troglitazone dose) or received no treatment. Pigs were then anesthetized and subjected to 90 min of low-flow regional myocardial ischemia and 90 min of reperfusion. Myocardial expression of PPAR-γ, determined by ribonuclease protection assay, increased with troglitazone and rosiglitazone compared with no treatment. Rosiglitazone had no significant effect on myocardial contractile function (Frank-Starling relations), substrate uptake, or expression of proinflammatory cytokines during I/R compared with untreated pigs. In contrast, preservation of myocardial contractile function and lactate uptake were greater and cytokine expression was attenuated in pigs treated with troglitazone or α-tocopherol compared with untreated pigs. Multivariate analysis indicated that presence of an α-tocopherol, but not a thiazolidinedione, moiety in the test compound was significantly related to greater contractile function and lactate uptake and lower cytokine expression during I/R. We conclude that PPAR-γ activation is not protective in a porcine model of myocardial I/R. Protective effects of troglitazone are attributable to its α-tocopherol moiety. These findings, in conjunction with prior rat studies, suggest interspecies differences in the response to PPAR-γ activation in the heart.
Keywords: nuclear receptor, thiazolidinedione, energy metabolism, cytokine, ventricular function
PEROXISOME PROLIFERATOR-ACTIVATED receptors (PPARs) are nuclear receptors involved in regulation of energy substrate metabolism and inflammatory responses. Thiazolidinedione PPAR-γ activators are used clinically to improve insulin sensitivity and glycemic control in patients with Type 2 diabetes (30). PPAR-γ is predominantly expressed in adipose tissue but is also expressed in myocardium (15). The function of PPAR-γ in the heart remains largely unknown; however, some evidence suggests that activation of myocardial PPAR-γ may have metabolic and anti-inflammatory effects.
Cardiac metabolic and anti-inflammatory effects of PPAR-γ have been demonstrated in rat and mouse models. PPAR-γ activation has been shown to increase myocardial expression of glucose transporters, promote carbohydrate substrate utilization in cardiomyocytes and intact hearts (1, 23, 34, 46), and attenuate proinflammatory cytokine expression in activated cardiomyocytes (51), experimental myocarditis (56), or chronic left ventricular (LV) failure due to myocardial infarction (45). Moreover, most (18, 23, 46, 53, 57), but not all (26), studies have shown that thiazolidinedione PPAR-γ activators reduce myocardial infarct size and enhance recovery of contractile function in intact rat hearts after ischemia and reperfusion. These findings are derived from studies in normal and insulin-resistant models.
Although the preponderance of evidence from rodent studies suggests cardioprotective effects of PPAR-γ activation in myocardial ischemia and reperfusion, effects in large animals are uncertain. This issue assumes greater importance in light of the fact that millions of patients with or at risk for ischemic heart disease are treated with thiazolidinediones, despite minimal knowledge of effects of these agents in the human heart. Key intermediate data concerning effects of PPAR-γ activation in large-animal models of myocardial ischemia and reperfusion are therefore needed.
We previously showed that pigs treated chronically with the prototypical thiazolidinedione drug troglitazone demonstrate improved LV function and enhanced carbohydrate utilization after an episode of acute ischemia and reperfusion (60). Another group demonstrated that troglitazone reduces infarct size in dogs (25). However, because the troglitazone molecule contains a thiazolidinedione moiety and an α-tocopherol moiety, it is uncertain whether beneficial effects of this agent are due to PPAR-γ activation by its thiazolidinedione moiety, antioxidant effects of its α-tocopherol moiety, or both.
In the present study, we tested the hypothesis that PPAR-γ activation promotes recovery of contractile function after myocardial ischemia and reperfusion in nondiabetic pigs, associated with enhanced myocardial carbohydrate metabolism and diminished expression of proinflammatory cytokines. Our approach was to compare responses to acute myocardial ischemia and reperfusion in pigs that received chronic pretreatment with troglitazone (thiazolidinedione with α-tocopherol moiety), rosiglitazone (thiazolidinedione without α-tocopherol moiety), or α-tocopherol (no thiazolidinedione moiety) with pigs that received no pretreatment.
METHODS
Treatment groups
The experimental protocol was approved by the responsible Institutional Animal Care and Use Committee. Fifty-six pigs of either gender (8 ± 1 kg body wt) were obtained from a breeding farm and fed a diet of dry chow containing 3% fat by weight (Growena, Purina Mills, St. Louis, MO). Four groups of pigs were studied. Twenty-four pigs served as controls and received no chronic drug treatment (untreated group). Twelve pigs were treated with troglitazone (Sankyo Pharmaceutical Research Institute, Tokyo, Japan; 75 mg·kg−1·day−1) for 8 wk (troglitazone group). Twelve pigs were treated with rosiglitazone (GlaxoSmithKline; 3 mg·kg−1·day−1) for 8 wk (rosiglitazone group). Pilot studies indicated that these doses of troglitazone and rosiglitazone result in trough plasma concentrations 10–20 times the EC50 of each agent for activation of PPAR-γ in vitro (41) and trough and peak plasma concentrations that bracket peak plasma concentrations achieved in clinical use of the agents (2, 32, 48). Eight pigs were treated with α-tocopherol (Leiner Health Products, Carson, CA; 73 mg·kg−1·day−1) for 8 wk(α-tocopherol group). This dose of α-tocopherol is equimolar to the dose of troglitazone administered to the troglitazone group.
Anesthesia, surgery, and instrumentation of the heart
After 8 wk of treatment, each pig underwent a terminal experiment of low-flow myocardial ischemia and reperfusion. The methods for anesthesia, surgery, instrumentation of the heart, and physiological monitoring have been described previously (60). After an overnight fast, pigs were sedated with ketamine HCl (25 mg/kg im), anesthetized with α-chloralose (100 mg/kg iv induction, 20–30 mg·kg−1·h−1 iv maintenance), and mechanically ventilated with oxygen-enriched air. Pigs were wrapped in a circulating warm-water blanket, which we have shown prevents significant hypothermia in this model (43). Arterial blood glucose was monitored every 15 min, and an intravenous infusion of 10% glucose was adjusted to maintain arterial blood glucose at ~4 mmol/l. Propranolol (1 mg/kg iv) and atropine (0.2 mg/kg iv) were administered to block autonomic reflexes that would otherwise influence measures of intrinsic myocardial function. Normal saline solution was administered at a rate of 150 ml/h iv.
Instrumentation of the heart is illustrated in Fig. 1. Micromanometer catheters were placed in the aortic arch and LV. A fluid-filled catheter was placed in the left atrium. Bipolar pacing electrodes were affixed to the left atrial appendage and used to maintain heart rate slightly greater than the spontaneous rate. An adjustable hydraulic occluder (In Vivo Metrics, Healdsburg, CA) and an ultrasonic flow probe (Transonic, Ithaca, NY) were placed around the proximal portion of the left anterior descending coronary artery (LAD) to produce and monitor the severity of regional myocardial ischemia. A catheter was placed in the anterior interventricular coronary vein to sample effluent blood from the ischemic region. Arrays of four ultrasonic crystals were implanted in the subendocardium of anterior and posterior LV free walls (ischemic and nonischemic regions, respectively). The crystals were connected to a digital sonomicrometer (Sonometrics, London, Ontario, Canada) to measure the instantaneous regional wall area subtended by each array.
Fig. 1.
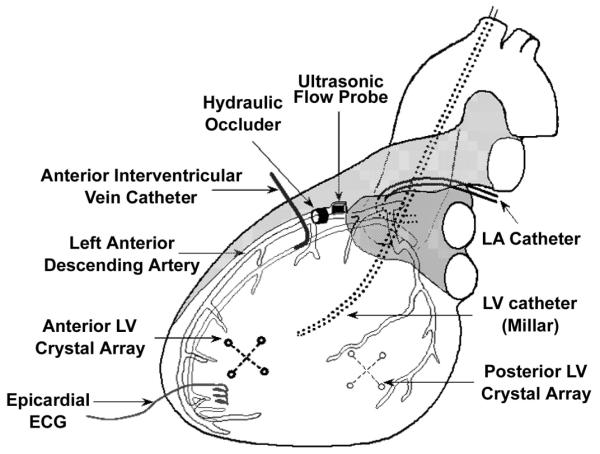
Instrumentation of the heart. LV, left ventricle; LA, left atrium.
Ischemia-reperfusion protocol
The protocol of low-flow myocardial ischemia and reperfusion has been described previously (60). After instrumentation of the heart, baseline measurements of hemodynamics, regional LV function, blood flow, and substrate uptake were obtained under steady-state conditions. Next, a microsyringe connected to the hydraulic occluder was used to produce regional low-flow myocardial ischemia by gradual constriction of the LAD, while coronary flow was continuously monitored with the flowmeter. LAD flow was reduced to 50% of baseline and maintained at this level (±1 ml/min) for 90 min by fine adjustment of the microsyringe. We and others previously showed that this duration and severity of ischemia in pigs result in profound myocardial stunning but not infarction (28, 42). At the end of 90 min of ischemia, a second set of measurements of hemodynamics, LV function, blood flow, and metabolism was made. The LAD occluder was then gradually released over 2–3 min to allow reperfusion. A final set of measurements was made after 90 min of reperfusion. Lidocaine (0.6 mg/kg iv) was administered every 30 min during ischemia and before reperfusion to prevent arrhythmias. Selected 10-s intervals of hemodynamic and sonomicrometry data were digitized at a frequency of 200 Hz and analyzed using custom software. After conclusion of the in vivo protocol and euthanasia, the heart was removed and myocardial samples were quickly excised from ischemic and nonischemic regions of the LV for analysis of regional myocardial blood flow and expression of PPAR-γ and cytokines.
Measurement of test compounds, regional myocardial blood flow, and substrate uptake
Plasma concentrations of troglitazone and rosiglitazone were measured by high-performance liquid chromatography (19, 33). Under each experimental condition, regional myocardial blood flow was measured in three transmural layers with fluorescent microspheres, as previously described (44). Paired blood samples were withdrawn simultaneously from arterial and anterior interventricular coronary vein catheters for measurement of substrate concentrations. Glucose and lactate concentrations were measured using an autoanalyzer (YSI, Yellow Springs, OH). Free fatty acid (FFA) concentration was measured using a spectrophotometric assay (Wako Diagnostics). Regional myocardial substrate uptake was calculated as the product of transmurally averaged myocardial blood flow and coronary arteriovenous concentration difference. Plasma insulin was measured by radioimmunoassay.
Assessment of LV systolic function
Hemodynamic data were digitized at 200 Hz and analyzed using custom software built on a LabView (National Instruments) platform. LV pressure vs. regional wall area loops were recorded in ischemic and nonischemic regions during brief suspension of mechanical ventilation.
Several measures of regional contractile function were calculated. Fractional systolic wall area reduction is a two-dimensional analog to segment shortening in one dimension or ejection fraction in three dimensions. It was calculated as the difference between end-diastolic and end-systolic wall area, divided by end-diastolic wall area. An index of regional external work was calculated as the area of LV pressure vs. wall area loop inscribed in one cardiac cycle. These measures, in conjunction with systemic hemodynamics, allowed comparison of baseline (preischemic) function among groups. However, because systolic wall area reduction and regional external work are sensitive to loading conditions, which change markedly during ischemia and reperfusion, a load-insensitive measure of systolic function was necessary under those conditions.
To obtain a measure of regional systolic function that is relatively insensitive to preload and afterload, preload-adjusted regional external work was calculated as follows (55): regional Frank-Starling relations were obtained under each experimental condition (baseline, ischemia, and reperfusion) by plotting regional external work against corresponding end-diastolic wall area for each cardiac cycle during 10 s of occlusion of the inferior vena cava. Preload-adjusted regional external work was computed from the Frank-Starling relation for each condition by setting preload (end-diastolic wall area) to its steady-state value under baseline conditions. Under each experimental condition in each pig, at least five measurements of preload-adjusted regional external work were made and averaged. It should be noted that preload-adjusted regional external work does not indicate actual work performed by the region under the prevailing loading conditions but provides a basis for comparing intrinsic myocardial function among pigs or groups of pigs at comparable loading conditions. In some cases, the calculated value of preload-adjusted regional external work was negative during ischemia or reperfusion. This does not necessarily imply that negative work was performed under the prevailing loading conditions; rather, it indicates that contractile dysfunction was so severe that positive work would not be performed if loading conditions were set back to baseline.
Myocardial expression of PPAR-γ
Myocardial homogenates were prepared from frozen specimens of subendocardium from the nonischemic and ischemic regions of five hearts from each of the four treatment groups. Tissue samples were obtained immediately after euthanasia. The 20 samples from ischemic-reperfused regions and the 20 samples from nonischemic regions were assayed on two gels, respectively. Expression of PPAR-γ mRNA was assessed by ribonuclease protection assay using a riboprobe derived from the published nucleotide sequence for porcine PPAR-γ1 (17). RNA was extracted using TRIzol reagent (Invitrogen). The PPAR-γ1 cDNA sequence was provided by Dr. Stefan Neuenschwander (Swiss Federal Institute of Technology, Zürich, Switzerland). Total RNA (40 μg) prepared from each homogenate was hybridized overnight with the PPAR-γ riboprobe at 42°C. Under these conditions, the riboprobe hybridizes with a 550-bp fragment of PPAR-γ1 (corresponding to nucleotides 12–562 of AJ006756) and with a 433-bp fragment of PPAR-γ2 (nucleotides 116–549 of AB097930). Digestion was carried out using 1:25 ribonuclease T1 (RPA III, Ambion), and the protected fragments were separated on a 6% denaturing polyacrylamide gel. PPAR-γ1 and PPAR-γ2 isoforms were quantified with a PhosphorImager and ImageQuant software (Amersham) and normalized to GAPDH, an internal control for RNA loading.
Expression of proinflammatory cytokines
Levels of mRNA and protein of IL-1β, IL-6, and IFN-γ were measured by ribonuclease protection assay and enzyme-linked immunosorbent assay in samples of subendocardium from ischemic-reperfused and nonischemic regions. Total RNA was hybridized overnight at 56°C with [α-32P]UTP-labeled antisense probes complementary to the porcine cytokine transcripts of interest (Pharmingen, San Diego, CA). Digestion was carried out with 1:100 dilution of ribonuclease A + T1 for 30 min at 37°C. Protected fragments were separated on 5% denaturing polyacrylamide gels. Each gel contained samples from the four treatment groups. The intensity of bands corresponding to IL-1β, IL-6, and IFN-γ mRNA was normalized to GAPDH. Cytokine protein expression was determined using commercially available kits containing antibodies to porcine IL-1β and IFN-γ (BioSource, Camarillo, CA) and IL-6 (R & D Systems, Minneapolis, MN). To determine whether ischemia and reperfusion in the anterior LV affected cytokine expression in the nonischemic posterior LV, three pigs were anesthetized and instrumented as described for the other experimental animals but were not subjected to ischemia. In these sham experiments, subendocardial tissue samples were obtained at a time corresponding to the end of reperfusion in ischemia-reperfusion experiments.
Effect of increasing arterial FFA concentration
Arterial FFA concentrations are considerably lower in pigs fed a low-fat diet than in humans, particularly under the conditions of β-adrenergic blockade in these experiments. To evaluate the effects of PPAR-γ activation on myocardial FFA utilization over a physiological range of arterial FFA concentrations, we performed additional experiments in six pigs from the untreated group and six pigs from the troglitazone group, in which arterial FFA concentration was increased by infusion of a 10% triglyceride emulsion (Liposyn, Abbott, Abbott Park, IL) and heparin. These pigs were surgically instrumented and subjected to the myocardial ischemia and reperfusion protocol described above. Liposyn was infused continuously at a rate of 50 ml/h, beginning 45 min before the onset of ischemia and continuing through reperfusion. Heparin was administered at the initiation of Liposyn infusion (15,000 IU iv), at 45 min of ischemia (5,000 IU iv), and at 45 min of reperfusion (5,000 IU iv). Myocardial substrate uptake was measured at baseline, at 45 min of preischemic treatment with Liposyn-heparin, at 90 min of ischemia, and at 90 min of reperfusion.
Statistical analysis
Values are means ± SE. Intergroup comparison of variables with a single measurement was performed by unpaired t-test (for 2 groups) or analysis of variance (for >2 groups). Within an individual group, changes in a variable from baseline to ischemia to reperfusion were assessed by a one-factor repeated-measures analysis of variance; if the F-test indicated a significant difference among conditions, Dunnett’s post hoc procedure was employed. Differences between groups in the response of a variable to ischemia and reperfusion were evaluated by a three-factor analysis of variance with repeated measures on one factor (experimental condition). The two other factors were binary grouping variables that described characteristics of the test compound in each group of pigs: one variable indicated presence or absence of a thiazolidinedione moiety (i.e., 1 for troglitazone and rosiglitazone, 0 for α-tocopherol and untreated), the other indicated presence or absence of an α-to-copherol moiety (i.e., 1 for troglitazone and α-tocopherol, 0 for rosiglitazone and untreated). In this analysis, the interaction terms between experimental condition and the grouping variables indicate which characteristic(s) of the test compounds influenced the response to ischemia and reperfusion. A significance level of 0.05 was specified for all analyses.
RESULTS
Growth, development, behavior, and weight gain were similar in each of the four groups of pigs. At the time of the terminal experiment, pigs weighed 28 ± 2 kg, with no significant differences among groups. No adverse effects of chronic treatment were discerned with any of the test compounds. Three pigs in the untreated group and one pig in the α-tocopherol group died of ventricular arrhythmia before completing the ischemia-reperfusion experiment. Although we previously observed an increase in lethal ventricular arrhythmias in pigs treated with troglitazone just before ischemia (58), there was no mortality in the chronic troglitazone or the chronic rosiglitazone group of the present study.
Plasma concentrations of test compounds
Plasma concentrations of test compounds are shown in Table 1. Trough and peak plasma concentrations of troglitazone and rosiglitazone bracketed the maximal plasma concentrations achieved in clinical use of these agents (2, 32, 48). Moreover, the relative plasma concentrations of troglitazone and rosiglitazone were commensurate with the relative affinities of these compounds for PPAR-γ: for both compounds, trough plasma concentrations were ~10 times higher than the EC50 for PPAR-γ activation in vitro (41).
Table 1.
Plasma concentrations of test compounds
| Experimental Plasma Concn, μM |
||||
|---|---|---|---|---|
| Compound | Trough | Peak | Cmax, μM | EC50, μM |
| Troglitazone | 3.6±1.3 | 18.1±6.8 | 6.3 | 0.36 |
| Rosiglitazone | 0.2±0.0 | 3.1±0.6 | 0.9 | 0.03 |
| α-Tocopherol | 6.8±1.0 | 13.6±2.2 | N/A | N/A |
Values are means ± SE. Concentrations were determined during chronic oral treatment with troglitazone (75 mg·kg−1·day−1, n = 12), rosiglitazone (3 mg·kg−1·day−1, n = 12), or α-tocopherol (73 mg·kg−1·day−1, n = 7). N/A, not applicable. Peak plasma concentrations were determined 2 h after treatment for troglitazone and rosiglitazone and 7 h after treatment for α-tocopherol. Cmax, published (2,32,48) peak plasma concentration measured in patients after administration of maximal clinical doses of troglitazone (600 mg) or rosiglitazone (8 mg). EC50, published (41) effective concentration for 50% maximal activation of periosome proliferation-activated receptor (PPAR)-γ in vitro. For troglitazone and rosiglitazone, experimental trough and peak plasma concentrations straddle clinical Cmax and trough concentrations are ≈10 times EC50.
Expression of PPAR-γ
Both isoforms of PPAR-γ were detected in porcine myocardium. The expression of PPAR-γ fulfills a necessary condition for specific actions of a PPAR-γ activator in the myocardium. Compared with untreated pigs, rosiglitazone increased expression of both isoforms of PPAR-γ in ischemic-reperfused and nonischemic regions (Fig. 2, Table 2). Troglitazone had a smaller effect on PPAR-γ expression that was significant only for PPAR-γ2. α-Tocopherol had no significant effect on PPAR-γ expression.
Fig. 2.

Myocardial expression of peroxisome proliferator-activated receptor (PPAR)-γ. Ribonuclease protection assay demonstrates expression of PPAR-γ1 and PPAR-γ2 mRNA in subendocardial samples from ischemic-reperfused region of 1 representative heart from each experimental group. Subcutaneous fat is a positive control. Quantitative analysis of group data is shown in Table 2.
Table 2.
Myocardial expression of PPAR-γ isoforms
| Ischemic-Reperfused Region |
NonIschemic Region |
|||
|---|---|---|---|---|
| Group | PPAR-γ1 | PPAR-γ2 | PPAR-γ1 | PPAR-γ2 |
| Untreated | 1.00±0.11 | 1.00±0.10 | 1.00±0.15 | 1.00±0.12 |
| Troglitazone | 1.25±0.16 | 1.73±0.09* | 1.23±0.14 | 1.75±0.06* |
| Rosiglitazone | 1.69±0.12* | 2.17±0.15* | 1.65±0.23 | 2.29±0.43* |
| α-Tocopherol | 0.98±0.05 | 1.00±0.07 | 1.47±0.15 | 1.28±0.08 |
Values are means ± SE; n = 5. Data were determined from ribonuclease protection assays. For each sample, bands corresponding to PPAR-γ1 and PPAR-γ2 were normalized to the band corresponding to GAPDH. Mean values of these ratios in the untreated group were assigned a value of 1.00, and values for the other groups are expressed as a fraction of the untreated group.
P < 0.05 vs. untreated.
Hemodynamics and myocardial blood flow
Baseline (preischemic) hemodynamics, myocardial blood flow, and systolic area reduction in the anterior LV were similar in each group (Table 3). Total LAD flow was higher in the rosiglitazone group than in the other groups. There are two likely explanations for this finding. 1) Hearts tended to be larger in the rosiglitazone group (mean 154 g) than in the other groups (mean 132–149 g). 2) Mean transmural blood flow (per gram myocardium, measured by microspheres) tended to be higher in the rosiglitazone group than in the other groups. This may have been related, in part, to slightly lower blood hemoglobin content in the rosiglitazone group (mean 11.0 g/dl) than in the other groups (12.0–12.3 g/dl, P = 0.07). Subendocardial blood flow did not differ among groups. This is important, because intergroup comparisons of regional contractile function and cytokine expression were made in the subendocardial layer. During ischemia, the relative reduction in LAD flow and the relative and absolute reductions in subendocardial blood flow were similar in each group.
Table 3.
Hemodynamics, myocardial blood flow, and LV systolic function
| Heart Rate, beats/min |
Mean Aortic Pressure, mmHg |
LV Systolic Pressure, mmHg |
LV End-Diastolic Pressure, mmHg |
LAD Coronary Flow, ml/min |
Anterior LV Mean Transmural Blood Flow, ml·g·min−1 |
Anterior LV Subendocardial Blood Flow, ml·g·min−1 |
Anterior LV Systolic Wall Area Reduction, % |
|
|---|---|---|---|---|---|---|---|---|
| Preischemia | ||||||||
| Untreated | 117±4 | 93±4 | 102±2 | 6±1 | 24±1 | 0.99±0.07 | 0.95±0.08 | 31±2 |
| Troglitazone | 117±3 | 93±6 | 108±3 | 8±1 | 22±2 | 1.10±0.19 | 1.11±0.22 | 30±3 |
| Rosiglitazone | 125±2 | 93±4 | 107±3 | 6±1 | 34±3* | 1.22±0.08 | 1.01±0.08 | 37±2 |
| α-Tocopherol | 126±2 | 94±7 | 95±3 | 4±1 | 23±3 | 1.03±0.12 | 1.14±0.16 | 34±3 |
| Ischemia | ||||||||
| Untreated | 116±4 | 84±3 | 95±1 | 12±2 | 12±1 | 0.40±0.04 | 0.20±0.02 | 1±2 |
| Troglitazone | 115±2 | 88±5 | 100±2 | 11±1 | 11±1 | 0.38±0.04 | 0.25±0.04 | 11±1* |
| Rosiglitazone | 124±1 | 92±2 | 108±2 | 11±1 | 18±2* | 0.61±0.08 | 0.21±0.05 | 1±1 |
| α-Tocopherol | 126±1 | 79±6 | 88±6 | 10±1 | 12±1 | 0.47±0.12 | 0.20±0.08 | 6±5 |
| Reperfusion | ||||||||
| Untreated | 116±4 | 86±3 | 94±2 | 12±2 | 25±2 | 1.00±0.17 | 0.86±0.22 | 2±2 |
| Troglitazone | 115±3 | 86±7 | 97±3 | 11±1 | 25±2 | 0.91±0.10 | 0.80±0.12 | 9±2* |
| Rosiglitazone | 124±2 | 81±7 | 98±3 | 10±1 | 33±4* | 1.02±0.13 | 0.81±0.11 | 2±1 |
| α-Tocopherol | 126±2 | 77±8 | 86±9 | 8±1 | 19±3 | 0.90±0.24 | 0.75±0.17 | 6±3 |
Values are means ± SE; n = 15 (untreated), n = 6 (troglitazone), n = 12 (rosiglitazone), and n = 7 (α-tocopherol). LV, left ventricle; LAD, left anterior descending coronary artery.
P < 0.05 vs. other groups.
Regional LV systolic function
At baseline before ischemia, regional LV systolic function in the anterior LV was similar in all groups as assessed by fractional systolic area reduction (Table 3) and by regional external work (Fig. 3). We used a load-insensitive measure of contractile function, preload-adjusted regional external work, to assess effects of test compounds on contractile function during ischemia and reperfusion. During these conditions, preload-adjusted regional external work remained greater in pigs treated with troglitazone or α-tocopherol (i.e., groups treated with test compounds containing an α-tocopherol moiety) than in pigs treated with rosiglitazone or untreated pigs (i.e., groups not treated with a test compound containing an α-tocopherol moiety; Fig. 3). In multivariate analysis, presence of an α-tocopherol moiety in the test compound was associated with significantly greater preload-adjusted external work during ischemia and reperfusion (P < 0.01); conversely, presence of a thiazolidinedione moiety in the test compound had no independent effect on preload-adjusted external work during ischemia and reperfusion (P = 0.85).
Fig. 3.
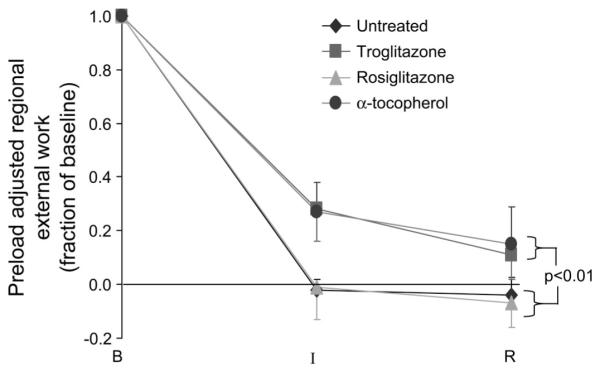
Systolic function in the ischemic region. Preload-adjusted regional external work (see text) was utilized as a load-insensitive measure of regional systolic function. Under baseline (preischemic) conditions, regional external work index did not differ among groups (25 ± 2, 26 ± 4, 26 ± 1, and 25 ± 5 mmHg/cm2 in untreated, troglitazone, rosiglitazone, and α-tocopherol groups, respectively). Therefore, data during ischemia and reperfusion are expressed as a fraction of baseline. Although each group exhibited contractile dysfunction during ischemia and reperfusion, dysfunction was less severe among troglitazone (n = 6) and α-tocopherol (n = 7) groups than among rosiglitazone (n = 12) and untreated (n = 15) groups. B, baseline; I, after 90 min of low-flow ischemia; R, after 90 min of reperfusion.
Effects of treatment on expression of proinflammatory cytokines
Having established a functional benefit of treatment with troglitazone or α-tocopherol, but not rosiglitazone, we sought to determine whether these findings might be attributable to differences in inflammatory responses and/or energy substrate metabolism. Figure 4 shows an example of ribonuclease protection assays for IL-1β, IL-6, and IFN-γ, and Fig. 5 shows group data for cytokine mRNA and protein expression. In untreated pigs, ischemia and reperfusion caused significant increases in expression of IL-1β, IL-6, and IFN-γ mRNA and protein compared with nonischemic regions of the same hearts. Treatment with troglitazone or α-tocopherol, but not rosiglitazone, reduced expression of cytokine mRNA and cytokine protein in ischemic-reperfused myocardium compared with the ischemic-reperfused myocardium from untreated pigs. In multivariate analysis, presence of an α-tocopherol moiety in the test compound, but not presence of a thiazolidinedione moiety, was significantly associated with reduced expression of cytokine mRNA (P < 0.01) and protein (P < 0.05). Cytokine protein content in hearts of sham pigs (not subjected to ischemia-reperfusion) did not differ significantly from content in nonischemic regions of hearts subjected to regional ischemia and reperfusion (data not shown).
Fig. 4.
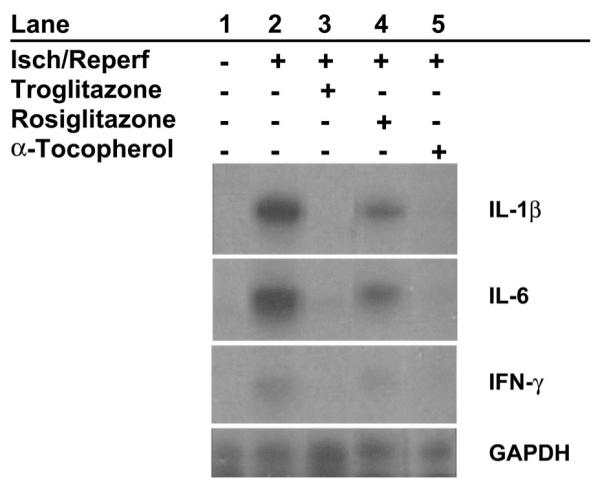
Myocardial cytokine mRNA expression. Representative ribonuclease protection assays show expression of IL-1β, IL-6, and IFN-γ in subendocardial tissue. Lane 1, minimal expression in nonischemic myocardium; lane 2, stimulation of expression of all 3 cytokines by 90 min of low-flow ischemia (Isch) and 90 min of reperfusion (Reperf); lanes 3–5, attenuation of cytokine expression in ischemic-reperfused myocardium by pretreatment with troglitazone and α-tocopherol, but not rosiglitazone.
Fig. 5.

Myocardial cytokine mRNA and protein expression. Pooled data are shown for pigs pretreated with troglitazone (n = 6), rosiglitazone (n = 12), or α-tocopherol (n = 6) and untreated pigs (n = 7). Top: cytokine mRNA expression by ribonuclease protection assay, normalized to expression of GAPDH in each heart. Data are medians ± interquartile range. Bottom: cytokine protein expression by enzyme-linked immunosorbent assay. Values are means ± SE. U, untreated; T, troglitazone; R, rosiglitazone; E, α-tocopherol; ND, not detected. *P < 0.05 vs. nonischemic. ‡P < 0.05 vs. untreated.
Effects of treatment on myocardial substrate utilization
Table 4 shows plasma insulin, arterial substrate concentrations, and myocardial uptake of glucose, lactate, and FFA under baseline (nonischemic) conditions, at 90 min of low-flow ischemia, and after 90 min of reperfusion. Under baseline conditions, there were no significant differences between groups in any of these variables. It is noteworthy that arterial FFA levels are very low in this model, as in other studies in pigs fed a low-fat diet (7, 12) or after β-adrenergic blockade; therefore, lactate and glucose are the predominant myocardial substrates.
Table 4.
Arterial substrate concentrations and myocardial substrate uptake
| Plasma Insulin, μU/ml |
Arterial Glucose, mmol/l |
Glucose Uptake, μmol·g−1·min−1 |
Arterial Lactate, mmol/l |
Lactate Uptake, μmol·g−1·min−1 |
Arterial FFA, mmol/l |
FFA Uptake, μmol·g−1·min−1 |
|
|---|---|---|---|---|---|---|---|
| Preischemia | |||||||
| Untreated | 4±1 | 4.0±0.2 | 0.19±0.04 | 1.2±0.2 | 0.63±0.11 | 0.16±0.04 | 0.02±0.02 |
| Troglitazone | 2±1 | 3.8±0.2 | 0.15±0.04 | 1.3±0.1 | 0.87±0.10‡ | 0.12±0.03 | 0.02±0.02 |
| Rosiglitazone | 3±1 | 4.2±0.1 | 0.17±0.04 | 1.0±0.1 | 0.66±0.07 | 0.10±0.04 | 0.00±0.01 |
| α-Tocopherol | 3±0 | 4.0±0.0 | 0.22±0.09 | 0.8±0.1 | 0.52±0.09‡ | 0.16±0.05 | 0.00±0.03 |
| Ischemia | |||||||
| Untreated | 4±1 | 4.1±0.2 | 0.33±0.05 | 1.0±0.1 | −0.28±0.05 | 0.09±0.03 | 0.01±0.01 |
| Troglitazone | 4±2 | 4.2±0.4 | 0.25±0.07 | 1.6±0.1* | −0.14±0.04‡ | 0.08±0.02 | 0.00±0.00 |
| Rosiglitazone | 4±1 | 4.3±0.1 | 0.42±0.08 | 1.1±0.1 | −0.26±0.10 | 0.06±0.01 | 0.01±0.01 |
| α-Tocopherol | 4±1 | 4.1±0.1 | 0.26±0.11 | 1.1±0.1 | −0.10±0.11‡ | 0.06±0.02 | 0.00±0.01 |
| Reperfusion | |||||||
| Untreated | 5±1 | 3.9±0.1 | 0.23±0.04 | 0.9±0.4 | 0.10±0.05 | 0.10±0.02 | 0.02±0.01 |
| Troglitazone | 6±2 | 3.7±0.2 | 0.21±0.02 | 1.3±0.2 | 0.39±0.03‡ | 0.05±0.01 | 0.01±0.00 |
| Rosiglitazone | 5±1 | 4.3±0.1 | 0.31±0.11 | 1.1±0.1 | 0.25±0.05 | 0.05±0.02 | 0.00±0.01 |
| α-Tocopherol | 3±0 | 4.0±0.1 | 0.19±0.06 | 1.2±0.1 | 0.38±0.18‡ | 0.07±0.03 | 0.03±0.02 |
Values are means ± SE; n = 15 (untreated), n = 6 (troglitazone), n = 12 (rosiglitazone), and n = 7 (α-tocopherol). FFA, free fatty acid.
P < 0.05 vs. other groups.
P < 0.05 vs. untreated and rosiglitazone groups over the course of the experiment.
In all groups, myocardial glucose uptake increased during ischemia, consistent with an increase in anaerobic glycolysis, whereas FFA uptake decreased, consistent with a decline in oxidative metabolism. Myocardial lactate uptake became negative in all groups, indicating net myocardial lactate release. However, lactate release during ischemia was lower in the troglitazone and α-tocopherol groups than in the rosiglitazone and untreated groups (Fig. 6). At 90 min of reperfusion, myocardial glucose uptake declined slightly from levels during ischemia and net lactate uptake returned to positive values in all groups. However, net lactate uptake after reperfusion was greater in the troglitazone and α-tocopherol groups than in the rosiglitazone and untreated groups. In multivariate analysis, presence of an α-tocopherol, but not a thiazolidinedione, moiety in the test compound was significantly associated with greater myocardial lactate uptake during ischemia and reperfusion (P < 0.05). Greater net lactate uptake generally reflects greater oxidative metabolism of carbohydrate substrates as a result of greater uptake and oxidation of exogenous lactate and diminished release of endogenous lactate from nonaerobic metabolism of glucose. There were no differences among groups in myocardial FFA uptake, which was low because of the low prevailing arterial FFA concentrations.
Fig. 6.
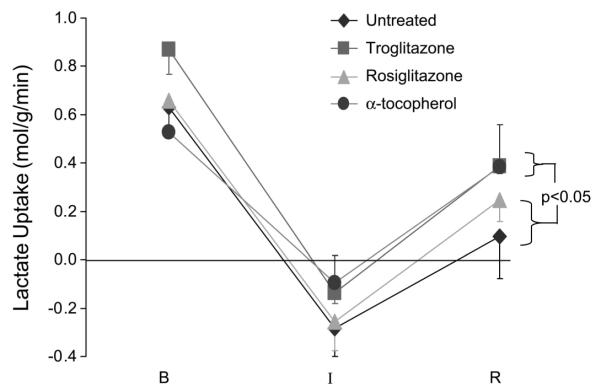
Myocardial lactate uptake. Myocardial lactate uptake becomes negative (net release) during ischemia and recovers partially with reperfusion. Over the course of the experiment, lactate uptake was greater in troglitazone and α-tocopherol groups (test compounds with an α-tocopherol moiety) than in rosiglitazone and untreated groups (no α-tocopherol moiety). P value indicates an intergroup difference over the course of the experiment.
Effect of increasing arterial FFA concentration
Because thiazolidinedione treatment had no discernible effect on myocardial FFA uptake at low arterial FFA concentrations, additional experiments were undertaken to determine whether thiazolidinedione treatment affects myocardial FFA uptake at higher arterial FFA concentrations comparable to those measured in human subjects. In subsets of pigs in the troglitazone and untreated groups, arterial FFA concentration was increased by infusion of the 10% triglyceride emulsion (Liposyn) and heparin. Troglitazone treatment had no effect on myocardial FFA uptake over a wide range of arterial FFA concentrations at baseline (preischemia), ischemia, or reperfusion (Fig. 7).
Fig. 7.
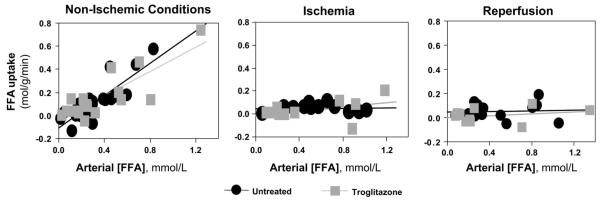
Effect of chronic troglitazone treatment on myocardial free fatty acid (FFA) uptake. Myocardial FFA uptake is plotted as a function of arterial FFA concentration ([FFA]) under nonischemic, ischemic, and reperfusion conditions for 14 untreated pigs and 12 troglitazone-pretreated pigs. In 6 pigs from each group, arterial FFA concentrations were increased by infusion of triglyceride emulsion (10% Liposyn) and heparin. Lines indicate linear regression of data. Chronic troglitazone treatment had no significant effect on myocardial FFA uptake under any condition.
DISCUSSION
The present study indicates that chronic activation of PPAR-γ is not protective in a model of myocardial ischemia and reperfusion in normal, nondiabetic pigs. This model results in severe myocardial contractile dysfunction, increased expression of proinflammatory cytokines (mRNA and protein), and metabolic impairment. Each abnormality was ameliorated by pretreatment with troglitazone (a thiazolidinedione that contains an α-tocopherol moiety) or with α-tocopherol itself. In contrast, pretreatment with rosiglitazone, a thiazolidinedione that lacks an α-tocopherol moiety, had no beneficial effect on any measured variable, even though rosiglitazone increased myocardial expression of PPAR-γ. Multivariate analysis indicated that presence of an α-tocopherol, but not a thiazolidinedione, moiety in the test compound protected against ischemia-reperfusion injury. These findings stand in contrast to the predominant finding of myocardial protection with rosiglitazone or pioglitazone (which also lacks an α-tocopherol moiety) in prior rat studies of ischemia and reperfusion (18, 23, 45, 46, 53, 57).
In these prior rat studies, rosiglitazone or pioglitazone reduced myocardial infarct size, preserved postischemic myocardial function, enhanced myocardial glucose utilization, and limited the activation of inflammatory signaling pathways. The cardioprotective effects of rosiglitazone or pioglitazone in rats were observed in isolated hearts and in situ, with acute or chronic pretreatment before ischemia, with no-flow or low-flow ischemia, with brief or prolonged periods of reperfusion, and in normal, insulin-resistant, or diabetic animals. Three of these studies (46, 53, 57) utilized the same dose of rosiglitazone as the present study, and the fourth (23) used a perfusate concentration of rosiglitazone similar to the plasma concentration in the present study. Species differences in PPAR action have been clearly described in other tissues (6) and likely account for the widely disparate findings in prior rat vs. present porcine studies of myocardial ischemia-reperfusion injury. The present findings highlight the need for realistic, large-animal studies as an intermediate step toward defining effects of PPAR-γ activation in the human heart.
The oxidative stress associated with myocardial ischemia and reperfusion increases the expression of proinflammatory mediators, including several cytokines. The proinflammatory cytokines include those expressed in myocardium (e.g., IL-1β, IL-6, and TNF-α) and those expressed by infiltrating inflammatory cells (e.g., IFN-γ). These mediators may act as myocardial depressants (4, 13, 14, 24, 29, 36-38, 50). In the present study, the cytokine concentrations measured in the ischemic-reperfused region of untreated hearts are within the range of concentrations shown to depress myocardial function in isolated perfused hearts or cardiomyocytes (4, 13, 14, 29, 36). Therefore, it is likely that a causal relation exists between cytokine expression and contractile dysfunction in this model. In a variety of in vitro and in vivo systems using rats or mice, including isolated cardiomyocytes exposed to lipopolysaccharide, lung or intestine subjected to ischemia-reperfusion injury, or experimental myocarditis, PPAR-γ activation has been shown to exert anti-inflammatory effects, including attenuation of the expression of proinflammatory cytokines (3, 9, 21, 31, 35, 39, 51, 56). By the same token, antioxidants, in general, and α-tocopherol, in particular, have been shown to abrogate the activation of transcription factors and attenuate the expression of IL-1β and IL-6 in vitro and in vivo (10, 47, 54) and to mitigate myocardial ischemia-reperfusion injury in some, but not all, studies (11). Troglitazone is an effective antioxidant in a variety of experimental systems and has been shown to reduce transmyocardial release of superoxide anions after ischemia and reperfusion (25). Thus, on the basis of prior studies, it was plausible that any of the test compounds employed in the present experiments could have abrogated inflammatory responses after ischemia and reperfusion. However, our data indicate that the presence of an α-tocopherol moiety in the test compound, but not the presence of a thiazolidinedione moiety, diminished proinflammatory cytokine expression (mRNA and protein content).
The oxidative stress associated with myocardial ischemia and reperfusion also impairs mitochondrial function and oxidative energy substrate metabolism, resulting in reduced flux through the tricarboxylic acid cycle (20). In particular, a metabolic signature of viable reperfused (stunned) myocardium is impaired oxidation of carbohydrate substrates (i.e., glucose and lactate) with a shift toward nonoxidative metabolism of glucose, despite ample oxygen availability (27). This metabolic abnormality is characterized by reduced myocardial lactate uptake relative to glucose uptake. Net myocardial lactate uptake is the difference between uptake of exogenous lactate and release of lactate formed in the myocardium from anaerobic metabolism of glucose. These processes occur simultaneously. With impaired carbohydrate oxidation, exogenous lactate uptake is reduced, while lactate production from glucose is increased, resulting in low net lactate uptake. As oxidative carbohydrate metabolism is restored, exogenous lactate uptake increases and glucose metabolism to lactate decreases, resulting in increased net lactate uptake. In the present experiments, greater myocardial lactate uptake after ischemia and reperfusion was observed in the troglitazone and α-tocopherol groups than in the untreated and rosiglitazone groups, without intergroup differences in glucose uptake. These observations are consistent with improved carbohydrate oxidation in the troglitazone and α-tocopherol groups compared with the untreated and rosiglitazone groups. Reduced carbohydrate oxidation in reperfused myocardium has been linked to impairment of pyruvate dehydrogenase (PDH) activity by proinflammatory cytokines (52, 58, 59) and may contribute to myocardial calcium overload and contractile dysfunction. Conversely, restoration of carbohydrate oxidation in reperfused myocardium by PDH activators is associated with improved contractile function (16). PPAR-γ activation may result in enhanced PDH activity by attenuating expression of proinflammatory cytokines and/or by downregulating expression of the principal inhibitor of PDH, PDH kinase (22). In fact, thiazolidinediones have been shown to normalize depressed PDH activity in skeletal muscle (22, 49) and to increase myocardial glucose utilization after ischemia and reperfusion in Zucker obese rats (46). Similarly, antioxidant agents may preserve oxidative metabolism of carbohydrates in the face of oxidant stress (40). Therefore, on the basis of prior studies, an improvement in oxidative carbohydrate metabolism might have been expected with any of the test compounds employed in the present study. However, multivariate analysis again indicated that presence of an α-tocopherol, but not a thiazolidinedione, moiety in the test compound was related to increased myocardial lactate uptake after ischemia and reperfusion compared with untreated pigs.
In insulin-resistant rodents and patients, thiazolidinediones improve insulin sensitivity, in large part, through reduction of circulating FFA concentrations. In the present experiments, we did not observe an effect of troglitazone or rosiglitazone on plasma FFA levels or myocardial FFA uptake. The absence of such effects in this model may be related to the fact that circulating FFA levels are very low in lean swine fed a low-fat chow diet and with lipolysis inhibited by β-blockade during the terminal experiment. However, even when FFA levels were augmented by infusion of triglyceride emulsion and heparin, we did not see an effect of troglitazone on myocardial FFA uptake. These findings suggest that, in normal swine, chronic thiazolidinedione treatment has no discernible effect on myocardial FFA utilization.
The absence of any detected beneficial effects of treatment with rosiglitazone, a PPAR-γ activator without intrinsic antioxidant properties, in contrast to the benefit of treatment with troglitazone, a PPAR-γ activator with antioxidant properties, suggests that PPAR-γ activation per se is not protective in this model of ischemia-reperfusion injury and that antioxidant effects most likely account for the benefits of treatment with troglitazone or α-tocopherol. Alternatively, it is possible that the different effects of rosiglitazone and troglitazone reflect different patterns of gene regulation by these compounds. For example, in a study that compared effects of rosiglitazone and troglitazone in a variety of cell culture lines, as many genes were regulated discordantly by these compounds as were regulated concordantly (5). Such differences in transcriptional regulation may be due to different affinities for PPAR coactivator and corepressor complexes. As another example, troglitazone, but not rosiglitazone, has been shown to inhibit sodium-proton exchange in endothelial cells (8); an analogous effect of troglitazone in myocardium could be protective in ischemia and reperfusion.
Several limitations of the present study must be recognized. First, we evaluated only one dose of each test compound; therefore, our data do not provide dose-response relations. We chose doses of troglitazone and rosiglitazone that resulted in clinically relevant plasma concentrations and that would beexpected to produce similar activation of PPAR-γ on the basis of the relative affinities of each compound for PPAR-γ. We chose a dose of α-tocopherol that was equimolar to the dose of troglitazone. Second, our examination of inflammatory mediators was selective, rather than comprehensive. We examined the effects of treatment on expression of three proinflammatory cytokines (IL-1β, IL-6, and IFN-γ) that are believed to play important roles as myocardial depressants and contributors to myocardial ischemia-reperfusion injury. Using two commercial kits, we were not able to obtain reproducible measurements of myocardial TNF-α concentrations. We did not examine effects on treatment on other signaling pathways that may be affected by PPAR-γ activation, such as nuclear factor-κB, activator protein 1, or MAP kinases, nor did we measure myocardial high-energy phosphates. Finally, we evaluated the effects of the test compounds on responses to a single duration and severity of ischemia in open-chest anesthetized, nondiabetic pigs. It is possible that the divergent results of prior studies in rats and the present study in pigs are related to the severity of myocardial injury imposed: In each of the prior rat studies (18, 23, 46, 53, 57), ischemia and reperfusion resulted in myocardial infarction; in contrast, our porcine model of low-flow ischemia and reperfusion results in myocardial stunning without significant infarction (28, 42). We cannot exclude the possibility that PPAR-γ activation would have had different effects in pigs subjected to greater or lesser severity of ischemia, in conscious chronically instrumented pigs, or in pigs with insulin resistance or overt diabetes. These questions require further investigation.
ACKNOWLEDGMENTS
The authors thank Drs. Takashi Izumi and Hidekuni Takahashi (Sankyo Research Institute, Tokyo, Japan) for measurement of plasma troglitazone concentration, Drs. Robin Buckingham and Anne-Marie Muxlow, Sonia Puri, Kate Hutton, and Stephen White (GlaxoSmithKline, The Frythe, UK) for measurement of plasma rosiglitazone concentrations, and Teri Armstrong and staff of the Denver Veterans Affair Medical Center animal care facility.
GRANTS This work was supported by National Heart, Lung, and Blood Institute Grants HL-49944 (to G. G. Schwartz) and HL-68606 (to C. Greyson) and the Medical Research Service of the Department of Veterans Affairs.
REFERENCES
- 1.Bähr M, Spelleken M, Bock M, von Holtey M, Kiehn R, Eckel J. Acute and chronic effects of troglitazone on isolated rat ventricular cardiocytes. Diabetologia. 1996;39:766–774. doi: 10.1007/s001250050509. [DOI] [PubMed] [Google Scholar]
- 2.Barman Balfour JA, Plosker GL. Rosiglitazone. Drugs. 1999;57:921–930. doi: 10.2165/00003495-199957060-00007. [DOI] [PubMed] [Google Scholar]
- 3.Cabrero A, Laguna JC, Vazquez M. Peroxisome proliferator-activated receptors and the control of inflammation. Curr Drug Targets Inflamm Allergy. 2002;1:243–248. doi: 10.2174/1568010023344616. [DOI] [PubMed] [Google Scholar]
- 4.Cain BS, Meldrum DR, Dinarello CA, Meng X, Joo KS, Banerjee A, Harken AH. Tumor necrosis factor-α and interleukin-1β synergistically depress human myocardial function. Crit Care Med. 1999;27:1309–1318. doi: 10.1097/00003246-199907000-00018. [DOI] [PubMed] [Google Scholar]
- 5.Camp HS, Li O, Wise SC, Hong YH, Frankowski CL, Shen X, Vanbogelen R, Leff T. Differential activation of peroxisome proliferator-activated receptor-γ by troglitazone and rosiglitazone. Diabetes. 2000;49:539–547. doi: 10.2337/diabetes.49.4.539. [DOI] [PubMed] [Google Scholar]
- 6.Choudhury AI, Chahal S, Bell AR, Tomlinson SR, Roberts RA, Salter AM, Bell DR. Species differences in peroxisome proliferation: mechanisms and relevance. Mutat Res. 2000;448:201–212. doi: 10.1016/s0027-5107(99)00237-7. [DOI] [PubMed] [Google Scholar]
- 7.Cleale RM, Ingling JM, Search DJ, Hadcock JR, Pausch MH. Effects of α2-adrenoceptor antagonists on metabolic processes of swine. I. Effects on nonesterified fatty acid and plasma urea nitrogen concentrations in jugularly catheterized pigs. J Anim Sci. 1998;76:1838–1848. doi: 10.2527/1998.7671838x. [DOI] [PubMed] [Google Scholar]
- 8.De Dios ST, Hannan KM, Dilley RJ, Hill MA, Little PJ. Troglitazone, but not rosiglitazone, inhibits Na/H exchange activity and proliferation of microvascular endothelial cells. J Diabetes Complications. 2001;15:120–127. doi: 10.1016/s1056-8727(01)00141-6. [DOI] [PubMed] [Google Scholar]
- 9.Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol. 2001;169:453–459. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- 10.Devaraj S, Li D, Jialal I. The effects of α-tocopherol supplementation on monocyte function. Decreased lipid oxidation, interleukin 1β secretion, and monocyte adhesion to endothelium. J Clin Invest. 1996;98:756–763. doi: 10.1172/JCI118848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dhalla NS, Elmoselhi AB, Hata T, Makino N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc Res. 2000;47:446–456. doi: 10.1016/s0008-6363(00)00078-x. [DOI] [PubMed] [Google Scholar]
- 12.Diersen-Schade DA, Richard M, Beitz DC, Jacobson NL. Effects of beef, soy and conventional diets on body composition and plasma lipids of young pigs fed restricted or liberal amounts of diet. J Nutr. 1985;115:1016–1024. doi: 10.1093/jn/115.8.1016. [DOI] [PubMed] [Google Scholar]
- 13.Ferdinandy P, Danial H, Ambrus I, Rothery RA, Schulz R. Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circ Res. 2000;87:241–247. doi: 10.1161/01.res.87.3.241. [DOI] [PubMed] [Google Scholar]
- 14.Gao CQ, Sawicki G, Suarez-Pinzon WL, Csont T, Wozniak M, Ferdinandy P, Schulz R. Matrix metalloproteinase-2 mediates cytokine-induced myocardial contractile dysfunction. Cardiovasc Res. 2003;57:426–433. doi: 10.1016/s0008-6363(02)00719-8. [DOI] [PubMed] [Google Scholar]
- 15.Gilde AJ, Van Bilsen M. Peroxisome proliferator-activated receptors (PPARs): regulators of gene expression in heart and skeletal muscle. Acta Physiol Scand. 2003;178:425–434. doi: 10.1046/j.1365-201X.2003.01161.x. [DOI] [PubMed] [Google Scholar]
- 16.Griffin JL, White LT, Lewandowski ED. Substrate-dependent proton load and recovery of stunned hearts during pyruvate dehydrogenase stimulation. Am J Physiol Heart Circ Physiol. 2000;279:H361–H367. doi: 10.1152/ajpheart.2000.279.1.H361. [DOI] [PubMed] [Google Scholar]
- 17.Grindflek E, Sundvold H, Klungland H, Lien S. Characterisation of porcine peroxisome proliferator-activated receptors γ1 and γ2: detection of breed and age differences in gene expression. Biochem Biophys Res Commun. 1998;249:713–718. doi: 10.1006/bbrc.1998.9212. [DOI] [PubMed] [Google Scholar]
- 18.Ito H, Nakano A, Kinoshita M, Matsumori A. Pioglitazone, a peroxisome proliferator-activated receptor-γ agonist, attenuates myocardial ischemia/reperfusion injury in a rat model. Lab Invest. 2003;83:1715–1721. doi: 10.1097/01.lab.0000106724.29121.da. [DOI] [PubMed] [Google Scholar]
- 19.Izumi T, Enomoto S, Hosiyama K, Sasahara K, Shibukawa A, Nakagawa T, Sugiyama Y. Prediction of the human pharmacokinetics of troglitazone, a new and extensively metabolized antidiabetic agent, after oral administration with an animal scale-up approach. J Pharmacol Exp Ther. 1996;277:1630–1641. [PubMed] [Google Scholar]
- 20.Janero DH, Hreniuk D. Suppression of TCA cycle activity in the cardiac muscle cell by hydroperoxide-induced oxidant stress. Am J Physiol Cell Physiol. 1996;270:C1735–C1742. doi: 10.1152/ajpcell.1996.270.6.C1735. [DOI] [PubMed] [Google Scholar]
- 21.Jiang C, Ting AT, Seed B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 22.Jove M, Salla J, Planavila A, Cabrero A, Michalik L, Wahli W, Laguna JC, Vazquez-Carrera M. Impaired expression of NADH dehydrogenase subunit 1 and PPAR γ coactivator-1 in skeletal muscle of ZDF rats: restoration by troglitazone. J Lipid Res. 2004;45:113–123. doi: 10.1194/jlr.M300208-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Khandoudi N, Delerive P, Berrebi-Bertrand I, Buckingham RE, Staels B, Bril A. Rosiglitazone, a peroxisome proliferator-activated receptor-γ, inhibits the Jun NH2-terminal kinase/activating protein 1 pathway and protects the heart from ischemia/reperfusion injury. Diabetes. 2002;51:1507–1514. doi: 10.2337/diabetes.51.5.1507. [DOI] [PubMed] [Google Scholar]
- 24.Kukielka GL, Smith EW, Manning AM, Youker KA, Michael LH, Entman ML. Induction of interleukin-6 synthesis in the myocardium: potential role in postreperfusion inflammatory injury. Circulation. 1995;92:1866–1875. doi: 10.1161/01.cir.92.7.1866. [DOI] [PubMed] [Google Scholar]
- 25.Lee TM, Chou TF. Troglitazone administration limits infarct size by reduced phosphorylation of canine myocardial connexin43 proteins. Am J Physiol Heart Circ Physiol. 2003;285:H1650–H1659. doi: 10.1152/ajpheart.00407.2002. [DOI] [PubMed] [Google Scholar]
- 26.Lygate CA, Hulbert K, Monfared M, Cole MA, Clarke K, Neubauer S. The PPAR-γ activator rosiglitazone does not alter remodeling but increases mortality in rats post-myocardial infarction. Cardiovasc Res. 2003;58:632–637. doi: 10.1016/s0008-6363(03)00289-x. [DOI] [PubMed] [Google Scholar]
- 27.Lopaschuk GD, Stanley WC. Glucose metabolism in the ischemic heart. Circulation. 1997;95:313–315. doi: 10.1161/01.cir.95.2.313. [DOI] [PubMed] [Google Scholar]
- 28.Lu L, Xu Y, Greyson CR, Ursell PC, Schwartz GG. Nonelastic deformation of myocardium in low-flow ischemia and reperfusion: ultrastructure-function relations. J Mol Cell Cardiol. 1999;31:1157–1169. doi: 10.1006/jmcc.1999.0948. [DOI] [PubMed] [Google Scholar]
- 29.Maass DL, White J, Horton JW. IL-1β and IL-6 act synergistically with TNF-α to alter cardiac contractile function after burn trauma. Shock. 2002;18:360–366. doi: 10.1097/00024382-200210000-00012. [DOI] [PubMed] [Google Scholar]
- 30.Martens FM, Visseren FL, Lemay J, de Koning EJ, Rabelik TJ. Metabolic and additional vascular effects of thiazolidinediones. Drugs. 2002;62:1463–1480. doi: 10.2165/00003495-200262100-00004. [DOI] [PubMed] [Google Scholar]
- 31.Marx N, Kehrle B, Kohlhammer K, Grub M, Koenig W, Hombach V, Libby P, Plutzky J. PPAR activators as anti-inflammatory mediators in human T lymphocytes: implications for atherosclerosis and transplantation-associated arteriosclerosis. Circ Res. 2002;90:703–710. doi: 10.1161/01.res.0000014225.20727.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller AK, Inglis AM, Culkin KT, Jorkasky DK, Freed MI. The effect of acarbose on the pharmacokinetics of rosiglitazone. Eur J Clin Pharmacol. 2001;57:105–109. doi: 10.1007/s002280100275. [DOI] [PubMed] [Google Scholar]
- 33.Muxlow AM, Fowles S, Russell P. Automated high performance liquid chromatography method for the determination of rosiglitazone in human plasma. J Chromatogr B Biomed Appl. 2001;752:77–84. doi: 10.1016/s0378-4347(00)00519-3. [DOI] [PubMed] [Google Scholar]
- 34.Oakes ND, Kennedy CJ, Jenkins AB, Laybutt DR, Chisholm DJ, Kraegen EW. A new antidiabetic agent, BRL 49653, reduces lipid availability and improves insulin action and glucoregulation in the rat. Diabetes. 1994;43:1203–1210. doi: 10.2337/diab.43.10.1203. [DOI] [PubMed] [Google Scholar]
- 35.Okada M, Yan SF, Pinsky DJ. Peroxisome proliferator-activated receptor-γ (PPAR-γ) activation suppresses ischemic induction of Egr-1 and its inflammatory gene targets. FASEB J. 2002;16:1861–1868. doi: 10.1096/fj.02-0503com. [DOI] [PubMed] [Google Scholar]
- 36.Panas D, Khadour FH, Szabo C, Schulz R. Proinflammatory cytokines depress cardiac efficiency by a nitric oxide-dependent mechanism. Am J Physiol Heart Circ Physiol. 1998;275:H1016–H1023. doi: 10.1152/ajpheart.1998.275.3.H1016. [DOI] [PubMed] [Google Scholar]
- 37.Paulus WJ. Cytokines and heart failure. Heart Fail Monit. 2000;1:50–56. [PubMed] [Google Scholar]
- 38.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1β. Proc Natl Acad Sci USA. 2001;98:2871–2876. doi: 10.1073/pnas.041611398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ricote M, Li AC, Wilson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 40.Sadek HA, Nulton-Persson AC, Szweda PA, Szweda LI. Cardiac ischemia/reperfusion, aging, and redox-dependent alterations in mitochondrial function. Arch Biochem Biophys. 2003;420:201–208. doi: 10.1016/j.abb.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 41.Sakamoto J, Kimura H, Moriyama S, Odaka H, Momose Y, Sugiyama Y, Sawada H. Activation of human peroxisome proliferator-activated receptor (PPAR) subtypes by pioglitazone. Biochem Biophys Res Commun. 2000;278:704–711. doi: 10.1006/bbrc.2000.3868. [DOI] [PubMed] [Google Scholar]
- 42.Schulz R, Guth BD, Pieper K, Martin C, Heusch G. Recruitment of an inotropic reserve in moderately ischemic myocardium at the expense of metabolic recovery. Circ Res. 1992;70:1282–1295. doi: 10.1161/01.res.70.6.1282. [DOI] [PubMed] [Google Scholar]
- 43.Schwartz GG, Schaefer S, Trocha SD, Garcia J, Steinman S, Massie BM, Weiner MW. Effect of supranormal coronary blood flow on energy metabolism and systolic function of porcine left ventricle. Cardiovasc Res. 1992;26:1001–1006. doi: 10.1093/cvr/26.10.1001. [DOI] [PubMed] [Google Scholar]
- 44.Schwartz GG, Xu Y, Greyson CR, Cohen J, Lu L. Low-dose inotropic stimulation during left ventricular ischaemia does not worsen post-ischaemic dysfunction. Cardiovasc Res. 1996;32:1024–1037. doi: 10.1016/s0008-6363(96)00150-2. [DOI] [PubMed] [Google Scholar]
- 45.Shiomi T, Tsutsui H, Hayashidani S, Suematsu N, Ikeuchi M, Wen J, Ishibashi M, Kubota T, Egashira K, Takeshita A. Pioglitazone, a peroxisome proliferator-activated receptor-γ agonist, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2002;106:3126–3132. doi: 10.1161/01.cir.0000039346.31538.2c. [DOI] [PubMed] [Google Scholar]
- 46.Sidell RJ, Cole MA, Draper NJ, Desrois M, Buckingham RE, Clarke K. Thiazolidinedione treatment normalizes insulin resistance and ischemic injury in the Zucker fatty rat heart. Diabetes. 2002;51:1110–1117. doi: 10.2337/diabetes.51.4.1110. [DOI] [PubMed] [Google Scholar]
- 47.Slim R, Toborek M, Robertson LW, Hennig B. Antioxidant protection against PCB-mediated endothelial cell activation. Toxicol Sci. 1999;52:232–239. doi: 10.1093/toxsci/52.2.232. [DOI] [PubMed] [Google Scholar]
- 48.Spencer CM, Markham A. Troglitazone. Drugs. 1997;54:89–101. doi: 10.2165/00003495-199754010-00010. [DOI] [PubMed] [Google Scholar]
- 49.Sreenan S, Keck S, Fuller T, Cockburn B, Burant CF. Effects of troglitazone on substrate storage and utilization in insulin-resistant rats. Am J Physiol Endocrinol Metab. 1999;276:E1119–E1129. doi: 10.1152/ajpendo.1999.276.6.E1119. [DOI] [PubMed] [Google Scholar]
- 50.Stein B, Frank P, Schmitz W, Scholz H, Thoenes M. Endotoxin and cytokines induce direct cardiodepressive effects in mammalian cardiomyocytes via induction of nitric oxide synthase. J Mol Cell Cardiol. 1996;28:1631–1639. doi: 10.1006/jmcc.1996.0153. [DOI] [PubMed] [Google Scholar]
- 51.Takano H, Nagai T, Asakawa M, Toyozaki T, Oka T, Komuro I, Saito T, Masuda Y. Peroxisome proliferator-activated receptor activators inhibit lipopolysaccharide-induced tumor necrosis factor-α expression in neonatal rat myocytes. Circ Res. 2000;87:596–602. doi: 10.1161/01.res.87.7.596. [DOI] [PubMed] [Google Scholar]
- 52.Tridgett EE, Yu YM, Zhong S, Burini R, Okusawa S, Gelfand JA, Dinarello CA, Young VR, Burke JF. Role of interleukin 1 and tumor necrosis factor on energy metabolism in rabbits. Am J Physiol Endocrinol Metab. 1988;255:E760–E768. doi: 10.1152/ajpendo.1988.255.6.E760. [DOI] [PubMed] [Google Scholar]
- 53.Wayman NS, Hattori Y, McDonald MC, Mota-Filipe H, Cuzzocrea S, Pisano B, Chatterjee PK, Thiemermann C. Ligands of the peroxisome proliferator-activated receptors (PPAR-γ and PPAR-α) reduce myocardial infarct size. FASEB J. 2002;16:1027–1040. doi: 10.1096/fj.01-0793com. [DOI] [PubMed] [Google Scholar]
- 54.Webel DM, Mahan DC, Johnson RW, Baker DH. Pretreatment of young pigs with vitamin E attenuates the elevation of plasma interleukin-6 and cortisol caused by a challenge dose of lipopolysaccharide. J Nutr. 1998;128:1657–1660. doi: 10.1093/jn/128.10.1657. [DOI] [PubMed] [Google Scholar]
- 55.Xu Y, Lu L, Lee J, Gen M, Kinugawa K, Long CS, Schwartz GG. Deleterious effects of acute treatment with a peroxisome proliferatoractivated receptor-γ activator in myocardial ischemia and reperfusion in pigs. Diabetes. 2003;52:1187–1194. doi: 10.2337/diabetes.52.5.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yuan Z, Liu Y, Liu Y, Zhang J, Kishimoto C, Wang Y, Ma A, Liu Z. Peroxisome proliferation-activated receptor-γ ligands ameliorate experimental autoimmune myocarditis. Cardiovasc Res. 2003;59:685–694. doi: 10.1016/s0008-6363(03)00457-7. [DOI] [PubMed] [Google Scholar]
- 57.Yue T, Chen J, Bao W, Narayan PK, Bril A, Jiang W, Lysko PG, Gu JL, Boyce R, Zimmerman DM, Hart TK, Buckingham RE, Ohlstein EH. In vivo myocardial protection from ischemia/reperfusion injury by the peroxisome proliferator-activated receptor-γ agonist rosiglitazone. Circulation. 2001;104:2588–2594. doi: 10.1161/hc4601.099403. [DOI] [PubMed] [Google Scholar]
- 58.Zell R, Geck P, Werdan K, Boekstegers P. TNF-α and IL-1α inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: evidence for primary impairment of mitochondrial function. Mol Cell Biochem. 1997;177:61–67. doi: 10.1023/a:1006896832582. [DOI] [PubMed] [Google Scholar]
- 59.Zentella A, Manogue K, Cerami A. Cachectin/TNF-mediated lactate production in cultured myocytes is linked to activation of a futile substrate cycle. Cytokine. 1993;5:436–447. doi: 10.1016/1043-4666(93)90033-2. [DOI] [PubMed] [Google Scholar]
- 60.Zhu P, Lu L, Xu Y, Schwartz GG. Troglitazone improves recovery of left ventricular function after regional ischemia in pigs. Circulation. 2000;101:1165–1171. doi: 10.1161/01.cir.101.10.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]