

Abstract
Volume expansion and inotropic stimulation are used clinically to augment cardiac output during acute right ventricular (RV) pressure overload. We previously showed that a brief period of RV pressure overload causes RV free wall dysfunction that persists after normal loading conditions have been restored. However, the impact of volume expansion and inotropic stimulation on the severity of RV dysfunction after acute pressure overload is unknown. We hypothesized that the severity of RV dysfunction after RV pressure overload would be related to the level of RV free wall systolic stress during RV pressure overload, rather than to the specific interventions used to augment RV function. Chloralose-anesthetized, open-chest pigs were subjected to 1 h of RV pressure overload caused by pulmonary artery constriction, followed by 1 h of recovery after release of pulmonary artery constriction. A wide range of RV free wall systolic stress during RV pressure overload was achieved by either closing or opening the pericardium (to simulate volume expansion) and by administering or not administering dobutamine. The severity of RV free wall dysfunction 1 h after RV pressure overload was strongly and directly correlated with the values of two hemodynamic variables during RV pressure overload: RV free wall area at peak RV systolic pressure (determined by sonomicrometry) and peak RV systolic pressure, two of the major determinants of peak RV free wall systolic stress. Opening or closing the pericardium, and using or not using dobutamine during RV pressure overload, had no independent effects on the severity of RV dysfunction. The findings suggest that the goal of therapeutic intervention during RV pressure overload should be to achieve the required augmentation of cardiac output with the smallest possible increase in RV free wall systolic stress.
Keywords: pressure-volume relation, mechanics, pericardium, dobutamine, pig
WHEN FACED WITH an acute increase in afterload, the normal right ventricle (RV) is able to increase peak systolic pressure to ~60 mmHg before RV contractile failure and systemic hypotension ensue. Increases in RV afterload are normally accompanied by reciprocal decreases in ejection fraction in a relationship similar to that which has been well described in the left ventricle (LV; see Ref. 2). It had been assumed that the decrease in RV ejection fraction observed during increased RV pressure was reversible and that restoration of normal loading conditions would be sufficient for complete normalization of RV contractile function. However, we have demonstrated that, under open-chest, open-pericardium conditions, increases in RV systolic pressure not severe enough to cause RV ischemia or systemic hypotension may nonetheless cause persistent RV systolic dysfunction that becomes apparent after restoration of normal loading conditions (5).
Interventions such as volume expansion and inotropic stimulation are used clinically to augment RV function during acute RV pressure overload. However, the impact of such interventions on the severity of RV dysfunction persisting after relief of RV pressure overload is unknown.
We hypothesized that the severity of RV dysfunction after RV pressure overload would depend on the level of systolic stress in the RV free wall during RV pressure overload rather than on the specific intervention used to augment RV function. To test this hypothesis, we subjected open-chest pigs to acute RV pressure overload while opening or closing the pericardium (to simulate volume expansion or contraction) and administering or not administering dobutamine to vary the relationship among RV systolic pressure, RV dilatation, and RV free wall stress.
METHODS
This investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH publication no. 85–23, revised 1985). It was approved by the Animal Studies Subcommittee of the San Francisco Department of Veterans Affairs Medical Center.
Experimental Preparation
The experimental preparation was similar to that reported previously (5). Twenty-eight female Yorkshire-Landrace pigs weighing 30–42 kg were anesthetized with ketamine hydrochloride (20 mg/kg im) and α-chloralose (100 mg/kg iv; Sigma Chemical, St. Louis, MO); anesthesia was maintained with α-chloralose (30–50 mg·kg−1·h−1 iv). The pigs were wrapped in a circulating warm water blanket to prevent hypothermia. Normal saline solution was infused continuously at 150–250 ml/h iv. Pigs were intubated via tracheotomy and were ventilated with an air-O2 mixture using a pressure-cycled ventilator adjusted to maintain PCO2 between 35 and 45 mmHg, pH between 7.35 and 7.45, and PO2 >100 mmHg.
After exposure via a midline sternotomy, the pericardium was opened, and solid-state micromanometer catheters (Millar Instruments, Houston, TX) were introduced into the RV via an internal jugular vein and into the LV via a carotid artery (Fig. 1). Bipolar pacing wires were affixed to the left atrial appendage. A transit-time ultrasonic flow probe (Transonic Systems, Ithaca, NY) and an umbilical tape snare were placed around the main pulmonary artery, and a hydraulic occluder was placed around the inferior vena cava. Two orthogonal pairs of piezoelectric crystals were implanted in the mid-RV free wall for determination of segment shortening using a sonomicrometer (Triton Technology, San Diego, CA). The distance between each crystal pair ranged from 1 to 1.5 cm. The crystals were aligned parallel and transverse to the long axis of the RV outflow tract. An intramyocardial electrogram was recorded from the crystal leads and was used for timing of cardiac events. In experiments in which the pericardium was reapproximated, a flat latex balloon connected to a fluid-filled catheter was placed in the pericardial space to measure intrapericardial pressure.
Fig. 1.
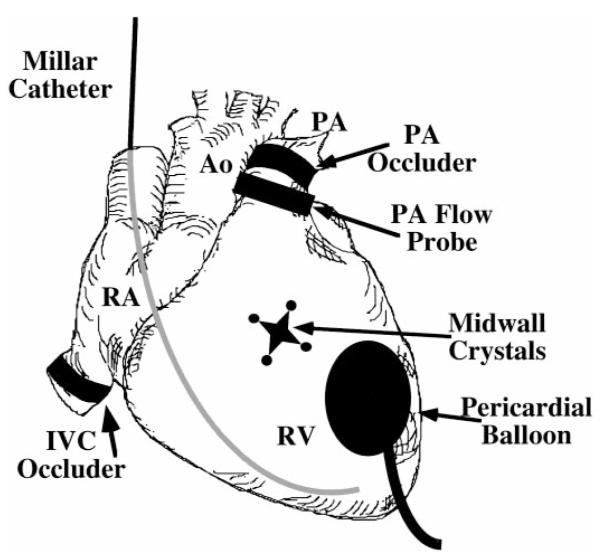
Instrumentation of the heart. RV, right ventricle; RA, right atrium; PA, pulmonary artery; Ao, aorta: IVC, inferior vena cava.
Experimental Protocol
Autonomic blockade was produced with atropine (0.2 mg/kg iv) and either propranolol (1.0 mg/kg iv) in pigs not receiving dobutamine or hexamethonium (7.5 mg/kg iv) in pigs receiving dobutamine. The heart was paced ~10 beats/min faster than the spontaneous heart rate (except during dobutamine infusion).
Baseline measurements of hemodynamics and RV function were made after verifying stability of the preparation for 30 min. The pulmonary artery was then gradually constricted over 15 min by using the umbilical tape snare to achieve the highest obtainable RV systolic pressure, while systemic arterial pressure was maintained near baseline by a titrated dose of phenylephrine (mean 175 μg/min iv). Pigs receiving dobutamine did not require or receive phenylephrine. After 1 h of pressure overload, the umbilical tape snare was released, and final measurements were obtained after a 1-h recovery period.
Four different conditions were employed during RV pressure overload using a 2 × 2 factorial design as follows: in 13 pigs, the pericardium was opened initially to permit instrumentation and baseline measurements, reapproximated snugly and left closed during RV pressure overload and the 1-h recovery period, and then reopened for final measurements; in 15 pigs, the pericardium was left open throughout the entire experiment; in 6 of the open pericardium and 6 of the closed pericardium pigs, dobutamine (6 μg·kg−1·min−1 iv) was infused during RV pressure overload. Measurements at baseline and at 1 h of recovery were obtained with the pericardium open and dobutamine off in all pigs.
Assessment of Regional and Global RV Function
Hemodynamic data were digitized at 200 Hz and recorded during brief suspensions of mechanical ventilation. To derive regional RV Frank-Starling relations under baseline and recovery conditions, three to six sets of data, spaced at intervals of at least 2 min, were recorded during 10-s occlusions of the inferior vena cava. Steady-state data (without inferior vena cava occlusion) were recorded under all three experimental conditions (baseline, RV pressure overload, and recovery).
RV free wall area
RV free wall area was defined as the instantaneous product of the orthogonal crystal pair separations, i.e., the area subtended by the four sonomicrometry crystals. Sonomicrometry measurements depend in part on the intercrystal distance, a source of experimental rather than physiological variability. Therefore, sonomicrometry measurements were normalized to their baseline values in each pig.
RV free wall systolic area reduction
RV free wall systolic area reduction (a 2-dimensional analog to segment shortening in 1 dimension or ejection fraction in 3 dimensions) was defined as the difference between end-diastolic and end-systolic RV free wall area, divided by end-diastolic RV free wall area. End diastole was defined as the point corresponding to the peak of the R wave of the intramyocardial electrogram. End systole was defined as the point of maximum negative RV dP/dt.
Regional external work
Regional external work was defined as the area of an RV pressure-wall area loop (i.e., the 2-dimensional plot of RV pressure vs. RV free wall area) obtained during a single cardiac cycle.
To compare RV regional external work at recovery with the value determined at baseline under conditions of matched preload, RV regional Frank-Starling relations were used to calculate preload-adjusted regional external work (5). Briefly, the area of each RV free wall area-pressure loop obtained during a dynamic inferior vena cava occlusion (i.e., regional external work) was plotted against its corresponding end-diastolic RV free wall area, and the slope and intercept of the resulting Frank-Starling relation were determined by linear regression. RV regional external work was then calculated from the regional Frank-Starling relation slope and intercept in each pig, with preload (end-diastolic RV free wall area) set to its baseline, steady-state value. Figure 2 indicates how preload-adjusted regional external work is determined from regional Frank-Starling relations.
Fig. 2.
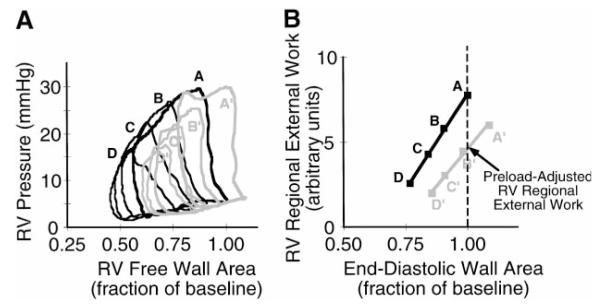
A: representative pressure-wall area loops obtained by brief occlusion of the inferior vena cava under baseline conditions (black lines, loops A–D) and at 1 h of recovery (gray lines, loops A’–D’). Only four loops are shown under each condition for clarity. Loops A (baseline) and A’ (recovery) were obtained under steady-state conditions before inferior vena cava occlusion. Note that loop A’ (recovery) has greater end-diastolic wall area and nearly identical end-diastolic pressure compared with loop A (baseline), but loop area (i.e., external work) is 24% lower and RV free wall systolic area reduction is 37% lower at 1 h of recovery compared with baseline. B: regional Frank-Starling relation derived from the data in A, demonstrating a downward and rightward shift in the relation at 1 h of recovery. The definition of preload-adjusted regional external work is shown (see text).
Global RV stroke work
Global RV stroke work was defined as the integral of the instantaneous product of pulmonary artery flow (from the transit-time flow probe) and RV pressure over a single cardiac cycle.
Echocardiography
RV short axis echocardiographic views at the midwall were obtained using a GE RT6800 echocardiography system by placing a 5-MHz biplane transesophogeal echocardiography probe directly on the epicardial surface of the posterolateral LV free wall and imaging through the heart. Views were standardized by adjusting the image to obtain a circular LV cross section and imaging the RV just distal to the tips of the tricuspid valve leaflets during systole; this plane includes the area of RV free wall in which the sonomicrometry crystals were placed. Echocardiographic frames corresponding to peak RV systolic pressure (timed from the simultaneous electrocardiogram recording) were digitized and analyzed using NIH ObjectImage 1.62 software. For each cross-sectional view, points on the epicardial surface of the RV free wall at the anterior and posterior junctions with the interventricular septum and midway between these points were identified and used to calculate the short axis radius of curvature of the RV free wall.
Statistical Analysis
Measured hemodynamic variables at baseline, during pressure overload, and at recovery were compared using Student’s t-test and the Bonferroni correction for multiple comparisons. Derived indexes of RV contractile function were normalized to baseline and tested for significance using one-sample t-tests. In addition, measured variables, including peak RV systolic pressure, RV end-diastolic pressure, end-diastolic RV free wall area, RV free wall area at peak RV systolic pressure, RV short axis radius of curvature, and dummy variables representing the presence or absence of inotropic stimulation or pericardial restraint during pressure overload, were entered into a forward stepwise linear regression model (F to enter = 4.000, F to remove = 3.996, where F is the F-test statistic). Those variables that predicted the severity of RV dysfunction at recovery (as measured by the fraction of baseline RV preload-adjusted regional external work) with P < 0.05 were considered to be significant. All data are reported as means ± SE.
RESULTS
Table 1 shows steady-state hemodynamic data under each experimental condition for each of the four treatment groups. Pulmonary artery constriction simultaneously increased peak RV systolic pressure and decreased RV free wall systolic area reduction, as anticipated. Pigs receiving dobutamine maintained somewhat higher RV free wall systolic area reduction and cardiac output despite greater peak RV systolic pressure compared with pigs not receiving dobutamine during pressure overload. Opening the pericardium during RV pressure overload permitted the RV to dilate and RV free wall area to expand compared with baseline, especially in the absence of dobutamine. Closing the pericardium during pressure overload limited both RV free wall area at end diastole and RV free wall area at peak RV systolic pressure to no more than their baseline values. The RV short axis radius of curvature did not change significantly from baseline (2.5 ± 0.04 cm) to RV pressure overload (2.4 ± 0.04 cm) in any group. At 1 h of recovery, RV systolic and diastolic pressures did not differ significantly from baseline, but RV free wall systolic area reduction, global RV stroke work, and cardiac output were all depressed compared with baseline.
Table 1.
Hemodynamics
| Measurement | Baseline | Pressure Overload |
One-Hour Recovery |
|---|---|---|---|
| Pericardium open without dobutamine (n=9) | |||
| Heart rate, beats/min | 128±1 | 128±1 | 128±1 |
| LV systolic pressure, mmHg | 98±5 | 103±5 | 97±4 |
| RV systolic pressure, mmHg | 30±1 | 56±1* | 31±2 |
| RV end-diastolic pressure, mmHg | 5±1 | 8±1* | 6±1 |
| RV free wall area at end diastole, fraction of baseline | 1.0±0 | 1.17±0.04* | 1.10±0.04* |
| RV free wall area at peak RV systolic pressure, fraction of baseline | 1.0±0 | 1.24±0.05* | 1.20±0.06* |
| RV free wall systolic area reduction, fraction of baseline | 0.36±0.03 | 0.17±0.04* | 0.23±0.04* |
| Global RV stroke work, J | 0.12±0.01 | 0.13±0.01 | 0.10±0.01 |
| Cardiac output, l/min | 4.5±0.3 | 2.6±0.1* | 3.3±0.2* |
| Pericardium closed without dobutamine (n=7) | |||
| Heart rate, beats/min | 129±1 | 128±1 | 128±1 |
| LV systolic pressure, mmHg | 88±3 | 96±3* | 93±6 |
| RV systolic pressure, mmHg | 29±2 | 55±2* | 34±2 |
| RV end-diastolic pressure, mmHg | 5±1 | 9±1* | 6±1 |
| RV free wall area at end diastole, fraction of baseline | 1.0±0 | 0.85±0.02* | 0.96±0.02 |
| RV free wall area at peak RV systolic pressure, fraction of baseline | 1.0±0 | 0.94±0.02 | 0.94±0.04 |
| RV free wall systolic area reduction, fraction of baseline | 0.34±0.03 | 0.19±0.04* | 0.28±0.02 |
| Global RV stroke work, J | 0.12±0.01 | 0.11±0.01 | 0.08±0.01 |
| Cardiac output, l/min | 4.4±0.2 | 1.9±0.2* | 2.4±0.3* |
| Pericardium open with dobutamine (n=6) | |||
| Heart rate, beats/min | 128±2 | 153±7 | 128±2 |
| LV systolic pressure, mmHg | 88±4 | 91±7 | 79±7 |
| RV systolic pressure, mmHg | 33±1 | 63±2* | 31±2 |
| RV end-diastolic pressure, mmHg | 5±1 | 8±1 | 8±1 |
| RV free wall area at end diastole, fraction of baseline | 1.0±0 | 1.05±0.07 | 1.08±0.06 |
| RV free wall area at peak RV systolic pressure, fraction of baseline | 1.0±0 | 1.05±0.11 | 1.17±0.08 |
| RV free wall systolic area reduction, fraction of baseline | 0.39±0.02 | 0.34±0.06* | 0.21±0.04* |
| Global RV stroke work, J | 0.15±0.01 | 0.17±0.018 | 0.10±0.02 |
| Cardiac output, l/min | 5.1±0.3 | 3.6±0.1* | 3.3±0.4* |
| Pericardium closed with dobutamine (n=6) | |||
| Heart rate, beats/min | 132±2 | 167±5 | 132±3 |
| LV systolic pressure, mmHg | 89±4 | 92±4 | 80±3 |
| RV systolic pressure, mmHg | 31±2 | 69±2* | 34±2 |
| RV end-diastolic pressure, mmHg | 5±1 | 9±1* | 7±1 |
| RV free wall area at end diastole, fraction of baseline | 1.0±0 | 0.89±0.05 | 1.06±0.03 |
| RV free wall area at peak RV systolic pressure, fraction of baseline | 1.0±0 | 0.90±0.07 | 1.06±0.05 |
| RV free wall systolic area reduction, fraction of baseline | 0.34±0.02 | 0.38±0.04* | 0.26±0.02 |
| Global RV stroke work, J | 0.12±0.01 | 0.16±0.02* | 0.11±0.01 |
| Cardiac output, l/min | 4.4±0.5 | 3.8±0.3* | 3.4±0.2 |
Values are means ± SE; n, no. of pigs. LV, left ventricle; RV, right ventricle.
P < 0.05 vs. baseline.
Figure 2A shows representative RV pressure-wall area loops obtained by brief occlusion of the inferior vena cava under baseline conditions and at 1 h of recovery. Note that the loop areas, which are an index of regional external work, are smaller for any given end-diastolic RV free wall area at 1 h of recovery compared with baseline. Figure 2B shows the regional Frank-Starling relations derived from the data in Fig. 2A. The downward and rightward shift in the relation at 1 h of recovery compared with baseline indicates persistent contractile dysfunction of the RV free wall.
Table 2 summarizes the derived indexes of systolic function at baseline and 1 h of recovery. (Inferior vena cava occlusion was not performed, and regional Frank-Starling relations therefore were not determined during pressure overload because these relations are not linear, and therefore not interpretable, under these conditions.) In the closed-pericardium/no-dobutamine group, there were no significant changes from baseline to recovery in regional Frank-Starling slope and intercept or preload-adjusted regional RV external work; peak RV systolic pressure and RV free wall area during RV pressure overload were lower in these pigs than in the other three groups. In the other three groups, RV preload-adjusted regional external work at 1 h of recovery was persistently and significantly reduced compared with baseline even though peak RV systolic pressure had returned to baseline. In these three groups, there were also trends to decreased regional Frank-Starling relation slope and increased intercept, consistent with a decline in contractility. Although these changes were not all statistically significant, this may be because neither one of these measurements alone is as sensitive to changes in contractility as preload-adjusted regional external work, which incorporates both of these variables.
Table 2.
Derived indexes of contractile function at 1 h of recovery
| Regional Frank-Starling Relation Slope |
Regional Frank-Starling Relation Intercept |
RV Preload- Adjusted Regional External Work |
|
|---|---|---|---|
| Pericardium open, without dobuta- mine (n=9) |
0.82±0.08* | 1.17±0.05* | 0.55±0.13* |
| Pericardium closed, without dobuta- mine (n=7) |
1.05±0.07 | 0.99±0.02 | 1.11±0.10† |
| Pericardium open, with dobutamine (n=6) |
0.70±0.13 | 1.19±0.08 | 0.53±0.15* |
| Pericardium closed, with dobutamine (n=6) |
0.74±0.08* | 1.04±0.02 | 0.67±0.09* |
Values are means ± SE (fraction of baseline); n, no. of pigs.
P < 0.05 vs. baseline.
P < 0.05 vs. other groups.
All variables measured during RV pressure overload were entered into a stepwise multiple linear regression to determine the significant predictors of RV dysfunction at recovery. Of the variables measured, only RV free wall area at peak RV pressure and peak RV systolic pressure during pressure overload were significant independent predictors of the severity of RV dysfunction at recovery, with the former a stronger predictor than the latter. These two variables together accounted for 83% of the variability in the severity of RV dysfunction (i.e., r2 = 0.83, P < .0001). Note that closing or opening the pericardium and using or not using dobutamine were not independent predictors of RV dysfunction at recovery. Figure 3 shows the univariate dependence of RV dysfunction after pressure overload on peak RV systolic pressure (A) and RV free wall area at peak RV systolic pressure (B) during RV pressure overload.
Fig. 3.
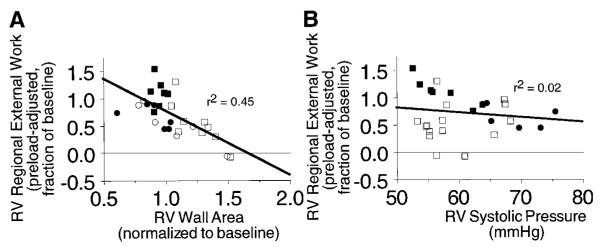
Correlation of recovery of RV preload-adjusted regional external work 1 h after release of pressure overload with RV systolic pressure (A) or normalized RV free wall area at peak RV pressure (B) during acute pressure overload. ○, Open pericardium, no dobutamine; □, open pericardium + dobutamine; ●, closed pericardium, no dobutamine; ■, closed pericardium + dobutamine.
To permit a graphical representation of the multivariate dependence of RV dysfunction on peak RV systolic pressure and RV free wall area, we used the above linear regression model to define a dysfunction predictor index as
where [RVSP]RVPO is peak RV systolic pressure during RV pressure overload (mmHg), [RVWA]RVPO and [RVWA]baseline are instantaneous RV free wall area at peak RV systolic pressure during RV pressure overload and baseline, and the constants a and b, which are the first-order coefficients determined from the linear regression model, are 0.045 and 1.9, respectively. Note that the dysfunction predictor index is not equivalent to wall stress, which cannot be determined precisely in the RV. Instead, it is a linear combination of the two measured hemodynamic variables that together best predict the severity of RV dysfunction after acute pressure overload. Figure 4 graphically shows the relation between the severity of RV dysfunction at recovery and the dysfunction predictor index.
Fig. 4.
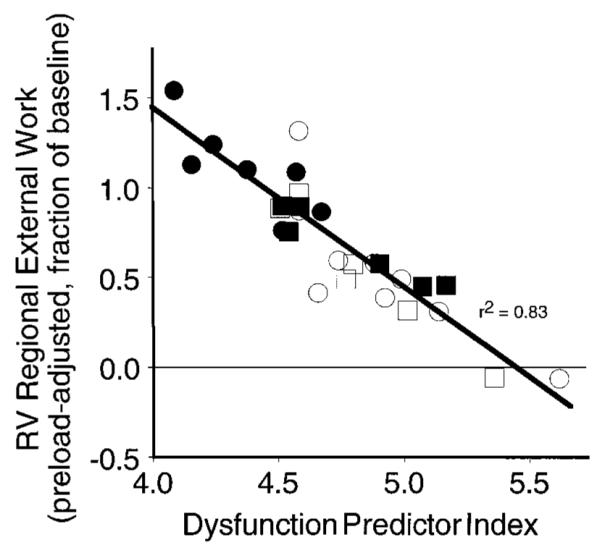
Correlation of recovery of RV preload-adjusted regional external work with the dysfunction predictor index (see text for explanation). There was no independent effect on recovery of function due to opening or closing the pericardium or using or not using dobutamine. ○, Open pericardium, no dobutamine; □, open pericardium + dobutamine; ●, closed pericardium, no dobutamine; ■, closed pericardium + dobutamine.
DISCUSSION
We found that the severity of RV dysfunction persisting at recovery after relief of RV pressure overload was closely related to two variables measured during RV pressure overload: RV free wall area at peak RV systolic pressure and peak RV systolic pressure. Neither pericardial restraint nor inotropic stimulation with dobutamine during RV pressure overload had independent effects on the severity of RV dysfunction after restoration of baseline loading conditions. Instead, pericardial restraint during RV pressure overload attenuated RV contractile dysfunction at recovery by reducing RV free wall area during pressure overload, whereas inotropic stimulation by dobutamine during RV pressure overload exerted a variable effect by increasing RV systolic pressure and decreasing RV free wall area at peak RV systolic pressure.
Because of the complex shape of the RV, wall stress cannot be determined exactly. However, we can identify several variables that affect wall stress and as a result would be anticipated to contribute most significantly to RV dysfunction. First, according to the Laplace relation, wall stress (σ) in a thin-walled container is proportional to the intracavitary pressure (P) and the local radius of curvature (ROC) and is inversely proportional to the wall thickness (h)
Assuming that the volume of myocardium subtended by the sonomicrometry crystals is constant over time, instantaneous wall thickness (h) is inversely proportional to instantaneous wall area (WA)
Therefore
Thus our hypothesis that the severity of persistent RV free wall dysfunction after RV pressure overload is related to the level of peak RV free wall stress during RV pressure overload predicts that the severity of RV dysfunction after recovery from RV pressure overload will be directly related to RV free wall area at peak intracavitary pressure and to peak RV systolic pressure, as confirmed in this study. Although we had anticipated that the radius of curvature at peak intracavitary pressure would also be a significant predictor of RV dysfunction after RV pressure overload, this was not the case because there was no significant change in radius of curvature from baseline to RV pressure overload. However, our ability to identify the radius of curvature as an additional independent predictor of RV dysfunction after RV pressure overload may have been limited by variability in our determination of the radius of curvature or because we determined the radius of curvature in only one axis.
Only a few other investigators have studied changes in ventricular function consequent to acute volume or pressure overload. All of these prior studies were performed in the LV, and the results were not concordant. Pagliaro et al. (10) and Downing et al. (3) both reported that ventricular contractility declined after acute volume overload. The observed decline in contractility was transient in the former study but was persistent in the latter. However, in both of these studies, diastolic dilatation was severe (160% of baseline in the study by Downing et al. and >230% of baseline in the study by Pagliaro et al.). Lew (7) observed a transient increase in contractility after more modest volume loading in dogs (mean 12% increase in end-diastolic segment length). LeWinter et al. (8) also observed a transient increase in contractility in dogs subjected to simultaneous volume and pressure overload. In our study, diastolic dilatation was minimal, with mean diastolic RV free wall area during RV pressure overload unchanged from the baseline value. Our results in the RV may have differed from previous results in the LV because of a relatively greater increase in wall stress in the thin-walled RV.
This study was designed to elucidate hemodynamic factors that contribute to RV dysfunction after acute pressure overload. The potential cellular or structural mechanisms of persistent RV dysfunction after RV systolic pressure overload are uncertain. We previously showed that regional myocardial blood flow does not decrease and regional myocardial lactate release does not increase during RV pressure overload in this model, suggesting that ischemia was unlikely to explain the RV dysfunction (5). Alterations in excitation-contraction coupling or in the function of myocardial contractile proteins could have been induced by RV pressure overload. For example, acute pressure overload has been shown to activate mitogen-activated protein kinases in mouse hearts (6). Because such protein kinase activation is implicated in other conditions causing myocardial dysfunction (13), it is conceivable that it could lead to ventricular dysfunction in this model as well. Alternatively, it is possible that disruption of extracellular myocardial structural elements, such as collagen, or intracellular structural proteins could have occurred consequent to the elevated systolic wall stress. Alterations in both types of elements have been implicated in LV dysfunction in conditions such as ischemia and cardiomyopathy (1, 9, 14). Moreover, disruption of the extracellular collagen matrix has been demonstrated in isolated rat hearts subjected to elevated intracavitary pressure (4).
Limitations
Although both RV preload-adjusted regional external work and global stroke work were depressed after RV pressure overload, there was no significant relation between these two variables. This is not surprising, since global RV stroke work is dependent on global RV preload (end-diastolic volume) and interventricular interaction as well as intrinsic contractility of the RV free wall. We did not determine either global RV volume or the extent of interventricular interaction in this study; therefore, we could not adjust for these effects.
Absolute quantification of wall stress in the RV free wall is extremely difficult with the use of currently available techniques because of heterogeneities in wall thickness and geometry. Moreover, because global function of the RV is a result of the combined contributions of the morphologically distinct conus and sinus regions (11) and the LV septum (12), a single global measurement of RV wall stress would not be physiologically meaningful. Instead, we measured several factors that contribute to wall stress over a small area of the RV free wall. We did not attempt to determine the radius of curvature in the RV long axis because of marked variability in the echocardiographic measurement. We therefore cannot exclude the possibility that incorporating an estimate of radius of curvature in the longitudinal direction would have affected our results. In addition, we chose a central portion of the RV free wall for our analysis; therefore, we cannot exclude the possibility that other regions of the RV behave differently. However, we did not identify differences in the response of the conus and sinus regions to acute RV pressure overload in a previous study (5).
Wall stress is most closely related to transmural RV pressure rather than to absolute intraventricular pressure. Therefore, we could have overestimated the effective transmural RV pressure in pigs undergoing pericardial restraint because of elevated intrapericardial pressure. In every experiment in which the pericardium was closed, we measured intrapericardial pressure with a flat, latex balloon attached to a fluid-filled catheter. Uncertainty in synchronizing the timing of measurements made with a fluid-filled system with those made with the intracavitary micromanometer made determination of instantaneous transmural RV pressure uncertain. However, when we repeated our analysis by subtracting either intrapericardial pressure at the time of peak RV systolic pressure or mean intrapericardial pressure from peak RV systolic pressure to obtain an estimate of RV transmural pressure, our findings were not significantly altered from the preceding analysis. Indeed, we had anticipated that pericardial restraint would have at most a small effect on effective peak RV systolic wall stress, because total cardiac volume within the pericardial space is diminishing at the time of peak RV systolic pressure.
It was not possible to determine directly from this study whether elevated RV free wall stress during acute RV pressure overload was a cause or merely a consequence of the RV free wall dysfunction, since RV free wall dysfunction might itself lead to RV systolic dilatation and hence to increased RV free wall area. However, peak RV systolic pressure and RV free wall area at the time of peak RV systolic pressure were more strongly correlated with the severity of RV dysfunction when those variables were measured at the beginning of the hour of RV pressure overload than when those values were measured at the end of the hour. Thus it seems more likely that elevated wall stress was the cause of subsequent dysfunction than vice versa.
Implications
Our findings show that, for any given RV systolic pressure, systolic RV dilatation during acute pressure overload significantly exacerbates RV contractile dysfunction that persists after recovery from acute pressure overload. In the management of patients with hemodynamic compromise due to acute pulmonary hypertension, consideration of these effects suggests a potential advantage to earlier use of inotropic therapy, compared with volume loading, when necessary to augment cardiac output.
Acknowledgments
This work was supported in part by National Heart, Lung, and Blood Institute Grants HL-3475 (to C. Greyson) and HL-49944 (to G. G. Schwartz) and by the Department of Veterans Affairs Medical Research Service (to G. G. Schwartz).
REFERENCES
- 1.Charney RH, Takahashi S, Zhao M, Sonnenblick EH, Eng C. Collagen loss in the stunned myocardium. Circulation. 1992;85:1483–1490. doi: 10.1161/01.cir.85.4.1483. [DOI] [PubMed] [Google Scholar]
- 2.Dell’Italia LJ, Walsh RA. Application of a time varying elastance model to right ventricular performance in man. Cardiovasc Res. 1988;22:864–874. doi: 10.1093/cvr/22.12.864. [DOI] [PubMed] [Google Scholar]
- 3.Downing SW, Savage EB, Streicher JS, Bogen DK, Tyson GS, Edmunds LJ. The stretched ventricle. Myocardial creep and contractile dysfunction after acute nonischemic ventricular distention. J Thorac Cardiovasc Surg. 1992;104:996–1005. [PubMed] [Google Scholar]
- 4.Factor SM, Flomenbaum M, Zhao MJ, Eng C, Robinson TF. The effects of acutely increased ventricular cavity pressure on intrinsic myocardial connective tissue. J Am Coll Cardiol. 1988;12:1582–1589. doi: 10.1016/s0735-1097(88)80029-9. [DOI] [PubMed] [Google Scholar]
- 5.Greyson C, Xu Y, Cohen J, Schwartz GG. Right ventricular dysfunction persists following brief right ventricular pressure overload. Cardiovasc Res. 1997;34:281–288. doi: 10.1016/s0008-6363(97)00038-2. [DOI] [PubMed] [Google Scholar]
- 6.Harada K, Komuro I, Zou Y, Kudoh S, Kijima K, Matsubara H, Sugaya T, Murakami K, Yazaki Y. Acute pressure overload could induce hypertrophic responses in the heart of angiotensin II type 1a knockout mice. Circ Res. 1998;82:779–785. doi: 10.1161/01.res.82.7.779. [DOI] [PubMed] [Google Scholar]
- 7.Lew WY. Time-dependent increase in left ventricular contractility following acute volume loading in the dog. Circ Res. 1988;63:635–647. doi: 10.1161/01.res.63.3.635. [DOI] [PubMed] [Google Scholar]
- 8.LeWinter MM, Engler R, Pavelec RS. Time-dependent shifts of the left ventricular diastolic filling relationship in conscious dogs. Circ Res. 1979;45:641–653. doi: 10.1161/01.res.45.5.641. [DOI] [PubMed] [Google Scholar]
- 9.Morano I, Hadicke K, Grom S, Koch A, Schwinger RH, Bohm M, Bartel S, Erdmann E, Krause EG. Titin, myosin light chains and C-protein in the developing and failing human heart. J Mol Cell Cardiol. 1994;26:361–368. doi: 10.1006/jmcc.1994.1045. [DOI] [PubMed] [Google Scholar]
- 10.Pagliaro P, Gattullo D, Linden RJ, Losano G, Westerhof N. Systolic coronary flow impediment in the dog: role of ventricular pressure and contractility. Exp Physiol. 1998;83:821–831. doi: 10.1113/expphysiol.1998.sp004162. [DOI] [PubMed] [Google Scholar]
- 11.Raines RA, LeWinter MM, Covell JW. Regional shortening patterns in canine right ventricle. Am J Physiol. 1976;231:1395–1400. doi: 10.1152/ajplegacy.1976.231.5.1395. [DOI] [PubMed] [Google Scholar]
- 12.Santamore WP, Dell’Italia LJ. Ventricular interdependence: significant left ventricular contributions to right ventricular systolic function. Prog Cardiovasc Dis. 1998;40:289–308. doi: 10.1016/s0033-0620(98)80049-2. [DOI] [PubMed] [Google Scholar]
- 13.Sugden PH, Clerk A. “Stress-responsive” mitogen-activated protein kinases (c-Jun N-terminal kinases and p38 mitogen-activated protein kinases) in the myocardium. Circ Res. 1998;83:345–352. doi: 10.1161/01.res.83.4.345. [DOI] [PubMed] [Google Scholar]
- 14.Zhao MJ, Zhang H, Robinson TF, Factor SM, Sonnenblick EH, Eng C. Profound structural alterations of the extracellular collagen matrix in postischemic dysfunctional (“stunned”) but viable myocardium. J Am Coll Cardiol. 1987;10:1322–1334. doi: 10.1016/s0735-1097(87)80137-7. [DOI] [PubMed] [Google Scholar]