

Abstract
High-grade gliomas continue to have dismal prognosis despite advances made in understanding the molecular genetics, signaling pathways, cytoskeletal dynamics, and the role of stem cells in gliomagenesis. Conventional treatment approaches, including surgery, radiotherapy, and cytotoxic chemotherapy, have been used with limited success. Therapeutic advances using molecular targeted therapy, immunotherapy, and others such as dietary treatments have not been able to halt tumor progression and disease-related death. High-grade gliomas (World Health Organization grades III/IV) are histologically characterized by cellular and nuclear atypia, neoangiogenesis, and necrosis. The expression of vascular endothelial growth factor, a molecular mediator, plays a key role in vascular proliferation and tumor survival. Targeting vascular endothelial growth factor has demonstrated promising results, with improved quality of life and progression-free survival. Bevacizumab, a humanized monoclonal antibody to vascular endothelial growth factor, is approved by the Food and Drug Administration as a single agent in recurrent glioblastoma and is associated with manageable toxicity. This review discusses the efficacy, practical aspects, and response assessment challenges with the use of bevacizumab in the treatment of high-grade gliomas.
Keywords: bevacizumab, antiangiogenesis, glioma, glioblastoma, vascular endothelial growth factor
Introduction
High-grade gliomas (HGGs) are lethal tumors with a poor prognosis. According to the World Health Organization (WHO) classification, HGGs include all grade III/IV primary brain tumors of astrocytic, oligodendroglial, and mixed cellular lineage. Glioblastoma (GBM) is a WHO grade IV astrocytoma that accounts for 50%–60% of adult astrocytomas and about 12%–15% of intracranial neoplasms.1,2 Anaplastic gliomas (AGs) (WHO grade III) include anaplastic astrocytoma, anaplastic oligoastrocytoma, and anaplastic oligodendroglial tumors.3 In this review, the term HGG is used to specifically discuss the role of bevacizumab (BEV) in WHO grade IV tumors or GBMs, with specific reference made to grade III tumors where necessary.
The European Organization for Research and Treatment of Cancer (EORTC)/National Cancer Institute of Canada trial incorporating temozolomide (TMZ) in GBM treatment resulted in a median survival benefit of 2.5 months (12.1–14.6 months) with increased survival in the number of patients treated at 2 years from 10% to 26%.4–6 This approach combining TMZ with radiation therapy is the Stupp protocol and consists of concomitant chemotherapy with TMZ at a dose of 75 mg/m2/day given 7 days a week, starting on the first day of radiotherapy to the last day, and not exceeding 49 days. Following a 4-week break, the patient receives adjuvant TMZ 5 days a week, every 28 days for up to six cycles.4 The survival benefit persists at 5 years,6 with a survival of 9.8% versus 1.9% for those who received combination therapy versus those receiving radiation alone. Hegi et al identified a distinct survival advantage in patients with methylated O6-methyl-guanine-DNA methyltransferase (MGMT) status receiving TMZ compared to those who received radiation alone, with a median survival of 21.7 months versus 15.3 months, respectively.7,8 The promoter methylation of MGMT achieves epigenetic silencing of MGMT which helps DNA repair, thus compromising its gene repair activity following treatment with an alkylating agent like TMZ. MGMT methylation has been established to be a prognostic factor, regardless of treatment modality. Hegi et al demonstrated the median overall survival (OS) among patients with promoter methylation of MGMT to be 18.2 months versus 12.2 months.9
Unfortunately, in spite of these advances almost all HGGs progress. Repeat surgery, alternative chemotherapeutic agents, and in certain cases reirradiation therapy may be considered in a select subset of patients.10 Historically, the response rates for patients with recurrent GBMs have been in the range of 5%–10%, with progression-free survival (PFS) at 6 months seen in only 10%–25% of these patients.11 BEV (Avastin®; Genentech, Inc, San Francisco, CA, USA), a monoclonal antibody against vascular endothelial growth factor (VEGF), was granted accelerated approval on May 5, 2009 by the US Food and Drug Administration as a single agent for the treatment of patients with recurrent GBMs that have failed initial therapy. Since then, it has been used in the treatment setting of both primary and recurrent GBMs. The clinical efficacy, safety, practical aspects, and future implications involving the use of BEV in patients with HGG (mainly GBMs) are reviewed here.
Targeting VEGF
Folkman described tumor angiogenesis in the early 1970s,12 with more recent studies confirming angiogenesis as a complex and dynamic process occurring during the growth of all solid tumors beyond 2–3 mm in size. Tumor growth is limited by the capacity of oxygen and nutrients to diffuse into the rapidly expanding core, necessitating the creation of new vascular networks (neoangiogenesis) to supply these various factors.13,14 Neoangiogenesis involves endothelial cell migration and proliferation, giving rise to new blood vessels. GBMs are one of the most vascular tumors known, thus making the factors associated with neoangiogenesis an attractive therapeutic target.15
VEGF is one of the most important factors regulating angiogenesis.16 There are five different isoforms of VEGF including VEGF-A, VEGF-B, VEGF-C, VEGF-D, and platelet-derived growth factor. Of these isoforms, VEGF-A is the cytokine most studied, being structurally related to platelet-derived growth factor and the cytokine that mainly promotes physiological and tumor angiogenesis.17 Abnormal angiogenesis mainly occurs in the tumor microenvironment, making VEGF a suitable target. The possible mechanisms of efficacy of an anti-VEGF approach include normalization of tumor vasculature and inhibition of new vessel formation, causing hypoxia and decreased nutrient availability and at the same time improving drug delivery by facilitating uniform distribution.18–20 However, enhanced drug delivery with the use of an anti-VEGF approach has been questioned recently. The interaction between GBM stem cells located in their perivascular niche and endothelial cells may also be disrupted by anti-VEGF agents, contributing to eventual cell death.21,22
In glioma models, preclinical data with the use of BEV demonstrated microvascular regression, normalization of mature blood vessels, and inhibition of new vessel growth.23 BEV blocks the action of VEGF on its receptor and prevents the migration and proliferation of endothelial cells, which in turn decreases tumor vascularization and results in hypoxia and cell death.24
BEV in HGG
GBM is characterized by neovascularization with endothelial cell proliferation, disruption of the blood–brain barrier, and abnormal contrast enhancement. Large amounts of VEGF are present in these areas of endothelial proliferation and in areas of necrosis that develop as the tumor rapidly enlarges.25,26 BEV is a humanized form of a mouse monoclonal antibody against human VEGF, which binds to and neutralizes mainly VEGF-A and its various isoforms including VEGF121/144/165/189/205.27–30 It has a half-life of 17–20 days.31 Clinical trials using BEV as a monotherapy as well as in combination with other cytotoxic chemotherapies has been reported in solid cancers such as metastatic colorectal cancers and lung cancer.32 Here, BEV is discussed in three settings: (1) recurrent HGGs, (2) BEV in the newly diagnosed GBM setting, and (3) BEV in the setting of WHO grade III gliomas, and subsequently the steroid sparing effect, toxicity, and challenges associated with the use of BEV.
BEV in the recurrent HGG setting
BEV was first used as an empirical treatment for recurrent GBMs by Stark-Vance in 2005.33 The results showed a dramatic radiological improvement as well as improvement in the patients’ neurological function. In their retrospective analysis of 29 patients treated with a combination of BEV and irinotecan, an overall response rate of 43% was observed, with one complete response, eight partial responses, and eleven patients with stable disease. There was one death that occurred secondary to intracranial hemorrhage and one due to gastrointestinal perforation.33
This initial efficacy of BEV led to a Phase II trial by Vredenburgh et al in a total of 35 patients with recurrent GBMs, using BEV and irinotecan (CPT-11), a topoisomerase-1 inhibitor.34 Their inclusion criteria were a minimum of 6 weeks following cranial surgery, a minimum of 4 weeks following radiation therapy and any other chemotherapy unless there was evidence of tumor progression on imaging, absolute neutrophil count of 1500, platelet count of more than 125,000, and hematocrit of 29. The exclusion criteria were evidence of hemorrhage on the baseline magnetic resonance imaging (MRI), prior treatment with BEV, or presence of a prior malignancy. They had two cohorts: an initial cohort of 23 patients using a dose of BEV at 10 mg/kg plus irinotecan every 2 weeks and a second cohort of twelve patients using a dose of BEV at 15 mg/kg every 21 days and irinotecan on days one, eight, 22, and 29. Their overall results in all 35 patients included a PFS at 6 months of 46% (95% confidence interval [CI] 32%–66%). A partial response was observed in 20 of 35 patients (57%; 95% CI 39%–74%), with the median OS in the study being 10.5 months.34 The adverse events included one central nervous system hemorrhage and four patients developed thromboembolic complications.
The subsequent study conducted by Friedman et al, an open-label, randomized Phase II study evaluated BEV with and without irinotecan in 167 recurrent GBM patients.35 Their inclusion criteria included progression of histologically confirmed GBMs at first or second relapse confirmed by MRI, initial treatment with radiation therapy and BEV, Karnofsky performance score ≥ 70, and adequate hematological, hepatic, and renal function. They excluded patients with history of hemorrhage, bleeding diathesis, coagulopathy, clinically significant cardiac disease, or recent treatment with anti-VEGF agents. Patients received BEV at a dose of 10 mg/kg alone or in combination with irinotecan at a dose of 340 mg/m2 or 125 mg/m2 (with or without concomitant enzyme-inducing antiepileptic drugs). A response rate of 37.8% and 28.2% in the BEV plus irinotecan (n = 82) and the BEV alone (n = 85) groups, respectively, and PFS at 6 months of 50.3% and 42.6% in the BEV plus irinotecan group and BEV alone groups, respectively, was seen in this study. Despite the higher response rate an OS of 8.7 months in the BEV plus irinotecan arm versus 9.2 months in the BEV alone arm was seen. The adverse events observed in the study included venous thromboembolism (BEV 3.6% and BEV plus irinotecan 8.9%), wound healing complications (BEV 2.4% and BEV plus irinotecan 1.3%), grade III gastrointestinal perforations (BEV plus irinotecan 2.5%), serious reversible posterior leukoencephalopathy syndrome (BEV plus irinotecan 1.3%), grade I intracranial hemorrhage (BEV 2.4%), and grade I, II, and IV intracranial hemorrhage (BEV plus irinotecan 3.8%). There were a total of three deaths attributed to adverse events: two patients in the BEV alone arm (one due to neutropenia and one to pulmonary thromboembolism) and one patient in the combined arm secondary to convulsions.35 In an update to this study submitted by Cloughesy et al, up to 16% of patients were alive at 30 months, with adverse events being consistent with those in the primary study.36
Another Phase II trial conducted by the National Cancer Institute used BEV monotherapy at a dose of 10 mg/kg every 14 days for a 28-day cycle. There were 48 pretreated recurrent GBM patients in the study, with PFS at 6 months being the primary endpoint of the study and MRI as well as positron emission tomography (PET) findings being the secondary endpoints. They found a median PFS of 16 weeks (95% CI 12–26 weeks), PFS at 6 months of 29% (95% CI 18%–48%), and a median OS of 31 weeks (95% CI 21–54 weeks).37
The US Food and Drug Administration granted an accelerated approval for BEV in patients with recurrent GBM in May 2009 following the results of the BEV Alone or in Combination With Irinotecan for Treatment of GMB Multiforme (BRAIN) study and the National Cancer Institute study.35,37,38 At the same time, the European Medicines Agency rejected the application for BEV registration in the treatment for recurrent GBM. They opined that a positive benefit–risk for BEV, either as a single agent or in combination with irinotecan, in the treatment of patients with recurrent GBMs had not been established because the evidence for efficacy was based on exploratory trial data. They did not consider the results in terms of objective response rate to be dramatic and the validity as a surrogate endpoint for clinical benefit had not been established. Additionally, the results presented in terms of OS and PFS were difficult to interpret owing to the lack of a randomized concurrent control.39Table 1 summarizes the various clinical trials and their results with the use of BEV in the recurrent HGG setting.
Table 1.
Trials with bevacizumab in recurrent high grade glioma (N ≥ 25)
| Study | Histology | Regimen | Response rate
|
PFS
|
OS
|
|||
|---|---|---|---|---|---|---|---|---|
| CR (%) | PR (%) | SD (%) | PFS-6 (%) | Mediana (months) | Mediana (months) | |||
| Single-agent bevacizumab | ||||||||
| Friedman 200935 | GB(N= 167) | BV (n = 85) | CR + PR: 28.2 | NA | 42.6 | 4.2 | 9.2 | |
| Kreisl 200937 | GB(N = 48) | BV → BV + irinotecan | Levin criteria: 71; MacDonald criteria: 35 | NA | 29 | 3.7 | 7.2 | |
| Chamberlain 2009116 | AA (N = 25) | BV | 0 | 64 | 8 | 60 | 7 | 9 |
| Raizer 2009117 | GB (n = 50); | BV | GB:0 | NA | NA | 25 | 2.7 | 6.4 |
| AG (n = II) | AG:0 | NA | NA | NA | NA | NA | ||
| All: 0 | 25 | 50.8 | 31 | 3.3 | 7.1 | |||
| Chamberlain 20I0118 | GB (N = 50) | BV | 0 | 42 | 0 | 42 | 1 | 8.5 |
| Combination of bevacizumab with chemotherapy | ||||||||
| Vredenburgh 200734,66 | GB(N = 35) | BV + irinotecan | CR + PR: 57 | 24 | 46 | 5.5 | 9.7 | |
| Vandenburgh 200734 | GB (n = 23); | BV + irinotecan | GB:4.3 | 56.5 | 34.4 | 30 | 4.6 | 9.23 |
| AG (n = 9) | AG:0 | 66.7 | 33.3 | 56 | 6.9 | NA | ||
| All: 3.1 | 59.4 | 34.4 | 38 | 5.3 | NA | |||
| Norden 200852 | GB(n = 33); | BV + CT | GB:NA | NA | NA | 42 | NA | NA |
| AG (n = 21); | AG:NA | NA | NA | 32 | NA | NA | ||
| MG NOS(n= 1) | All: 2.3 | 31.8 | 29.5 | 39 | 5.5 | 8.2 | ||
| Desjardins 2008119 | AA (n = 25); | BV + irinotecan | AA:NA | NA | NA | 52 | 6.5 | 15 |
| AO (n = 8) | AO:NA | NA | NA | 62 | 11.5 | 14.1 | ||
| All: 9.1 | 51.5 | 33.3 | 55 | 6.9 | 15 | |||
| Friedman 200935 | GB(N= 167) | BV + irinotecan (n = 82) | CR + PR: 37.8 | NA | 50.3 | 5.6 | 8.7 | |
| Gilbert 2009120 | GB (N = 57) | BV + irinotecan | NA | NA | NA | 37 | NA | NA |
| Nghiemphu 2009121 | GB(N= 123) | BV + CT (n = 44); | NA | NA | NA | BV + CT: 414.25 | 9.0 | |
| CT or other agent(s) [control] (n = 79) | Control: 18 1.82 | 6.1 | ||||||
| Narayana 2009122 | GB(n = 37); | BV + irinotecan or carboplatin | All: I3.2b | 60.4b | 20.7b | 44.3 | 5 | 9 |
| AG (n = 24) | ||||||||
| Zuniga 2009123 | GB(n = 37); | BV + irinotecan | GB: 5.4 | 62.2 | 16.2 | 63.7 | 7.6 | 11.5 |
| AG (n= 14) | AG: 14.3 | 64.3 | 14.3 | 78.6 | 13.4 | NA | ||
| All: CR + PR: 70.6 | 15.7 | NA | 9.5 | 13.4 | ||||
| Taillibert 2009124 | Oligodendroglial tumors, grade II/III (N = 25) | BV + irinotecan | 20 | 52 | 16 | 42 | 4.6 | NA |
| Quant 2009112 | GB(n = 35); | BV + CT | GB:0 | 23 | 57 | NA | NA | NA |
| AG (n= 15), MG NOS (n = 4) | AG:0 | 36 | 64 | NA | NA | NA | ||
| MG NOS: 0 | 50 | 25 | NA | NA | NA | |||
| All: 0 | 28 | 56 | 33 | 4.1 | NA | |||
| Hofer 2011125 | GB(n= 176); AG (n = 49) | BV alone (45);c | NA | NA | NA | NA | NA | GB:8.3 |
| BV + CT (180) | AG: 9.1 | |||||||
| All: 8.5 | ||||||||
Notes:
Median PFS and median OS that were reported in weeks were standardized to months using the following formula: weeks/52 × 12.
he response rates were calculated out of 53 evaluable patients.
No breakup was given for the two groups.
Abbreviations: AA, anaplastic astrocytoma; AG, anaplastic glioma; All: All patients with high grade glioma; AO, anaplastic oligodendroglioma; Be, bevacizumab; CR, complete response; CT, chemotherapy; GB, glioblastoma; MG NOS, malignant glioma not otherwise specified; NA, not available; OS, overall survival; PFS, progression-free survival; PFS-6, progression-free survival at 6 months; PR, partial response; SD, stable disease.
BEV in the newly diagnosed HGG setting
Following the results of the various trials in recurrent GBMs (Table 1), there has been considerable interest in evaluating the benefit of BEV in newly diagnosed GBMs. Narayana et al used BEV in conjunction with chemoradiation in 15 patients with newly diagnosed GBMs.40 BEV was given at a dose of 10 mg/kg every 14 days for a 28-day cycle with concurrent radiation therapy to a dose of 59.8 Gy (1.8 Gy/day) and TMZ at 75 mg/m2, which was then followed by BEV at 10 mg/kg every 14 days with TMZ at 150 mg/m2 for 12 months. Thirteen patients (86.6%) completed this course of therapy with radiographic responses seen in 13 of 14 patients (92.8%). There were six patients of the 15 (40%) who relapsed, three were at the primary site and three showed diffuse involvement. The median follow-up time in the study was 12 months, and the 1-year PFS and OS rates were 59.3% and 86.7%, respectively. The toxicity noted with upfront therapy in this series included four patients with grade II hematological and nonhematological toxicity, three patients with grade III/IV nonhematological toxicity, and three patients with grade III/IV hematological toxicity, with thrombocytopenia being the most common.
The drug was also evaluated as first-line therapy for newly diagnosed HGGs, with the first Phase II study of first-line BEV with TMZ and radiotherapy for treatment of HGGs reported by Lai et al.41 Their study, an open-label, prospective, single-arm Phase II trial combining BEV with radiation therapy and TMZ for newly diagnosed GBMs, enrolled 70 patients who received standard radiation therapy and concurrent TMZ biweekly (once every 2 weeks) followed by biweekly BEV with TMZ for 5 days in every 28-day cycle. Their study included all cases with a WHO histopathological diagnosis of GBM and gliosarcoma without any prior treatment, Karnofsky performance score ≥ 70, and no evidence of hematological, hepatic, or renal dysfunction. The standard exclusion criteria applied in their study and they did not take patients who only had a biopsy diagnosis without significant resection. They reported an OS of 19.6 months and PFS of 13.6 months, comparing favorably with the results from the Stupp (EORTC study) trial which had an OS of 14.6 months and PFS of 6.9 months.6 The toxicity was generally well tolerated in the study with four wound infections and two central nervous system hemorrhages, both being extraparenchymal. The overall toxicity pattern in their trial correlated well with the known toxicities of TMZ and BEV, without any evident supraadditive effects.
More recently, Narayana et al reported on a larger cohort of patients treated in a first-line setting, (adding biweekly BEV to standard EORTC protocol) reporting on the efficacy and toxicity of 51 patients.42 The efficacy was promising, with PFS at 6 months and 12 months noted as 85.1% and 51%, respectively, and 12-month and 24-month OS rates of 85.1% and 42.5%, respectively. There were ten patients (19.6) who experienced grade III/IV toxicity, but there were no treatment-related deaths. Of note, however, asymptomatic cranial bleeding was noted in five patients.
At present, there are two randomized Phase III trials evaluating the role of BEV in combination with TMZ and radiation therapy (both during the chemoradiation component and the adjuvant component of the EORTC/Stupp protocol) for untreated (newly diagnosed) GBMs. Radiation Therapy Oncology Group (RTOG) 0825 is a large Phase III trial targeting 720 patients and the other trial is the Effectiveness of Avastin in GBM (AVAGLIO) study targeting over 900 patients and sponsored by Hoffman-LaRoche (Basel, Switzerland). In November 2012 at the Society of Neuro-Oncology ASM, the AVAGLIO investigators presented the interim results of the AVAGLIO study which included the final PFS data and interim OS data. The result was positive for an improved PFS, with other positive outcomes in secondary endpoints such as quality of life. Specifically, the Investigator-Assessed PFS (Co-Primary Endpoint) had a 10.6 months median survival in the EORTC/Stupp protocol plus BEV arm compared to median survival of 6.2 months with the standard EORTC/Stupp protocol with a stratified hazard ratio of 0.64 (95% CI:0.55–0.74), P < 0.0001 (i.e. a 36% relative reduction in the risk of progression or death). This benefit applied to all subgroups but was not statistically significant in patients KPS ≥ 70; MGMT methylated patients, patients with biopsy only. However, these subgroups were very small (and there was a trend to benefit in the BEV group for each of these 3 groups). The central Independent Review Facility (IRF) Assessed PFS (secondary endpoint) demonstrated a median survival in the BEV group as 8.4 months compared with 4.3 months in the standard EORTC/Stupp group; again statistically significant with stratified hazard ratio of 0.61 (95% CI:0.53–0.71), P < 0.0001. The interim OS analysis demonstrated a small non-statistically significant benefit in the BEV plus EORTC/Stupp protocol group with a 1 year survival rate of 72% (68–76) compared with 66% (62–71) with P = 0.052; with 254 events in the EORTC/Stupp plus BEV arm compared with 263 events in the standard arm, with a stratified HR of 0.89 (0.75–1.07); P = 0.2135.
In the arm receiving up-front BEV there were statistically significant benefits in the five pre-specified domains of health-related quality of life (HRQoL) (secondary end-points), namely Global Health Status, physical functioning, social functioning, motor functioning, and communication deficit; with longer median duration that patients were stable/improved from baseline. The median duration those patients maintained a KPS ≥ 70 were 9 months in the BEV arm versus 6 months in the standard arm. In the BEV arm, 66% of patients who were on steroids at baseline discontinued their steroids compared with 47% on the standard arm. The time to steroid initiation for patients who had been off steroids at baseline was 12.3 months in the EORTC/Stupp plus BEV arm versus 3.7 months in the EORTC/Stupp arm with a stratified HR of 0.71 (95% CI:0.57–0.88), P = 0.0018. Overall, patients receiving BEV had a diminished steroid requirement.
There were no new serious adverse events or safety signals noted in the up-front setting. The patients on the EORTC/Stupp plus BEV arm were more likely to experience a serious adverse event (36.6% versus 25.7%) and were more likely to discontinue any treatment due to an adverse event (24.6% versus 13.2%). There was no significant increase in cerebral hemorrhage (2.6% versus 2.2% for all grades of cerebral bleed with 1.5% bleeds as ≥grade 3 versus 0.7%). There was more mucocutaneous bleeding in the BEV arm (26.7% versus 8.9%) but only 0.4% grade 3 or higher. There were slightly more wound-healing complications (3.7% versus 2.2%). There was an increase in arterial thromboembolic events (5% versus 1.6%) and a slightly lower incidence of venous thromboembolic events (7.8% all grades versus 9.6%). However, the incidence of hypertension was higher in the EORTC/Stupp protocol plus BEV arm, namely 37.5% versus 13.0% (with 10.3% versus 2.0% grade 3 or higher). Proteinuria was higher also (14% versus 4%). There was a slight excess of GI perforation (including GI fistula/abscess) at 1.7% versus 0.2%; with abscesses/fistulae elsewhere 0.6% versus 0.4%.
It is unclear how this data from AVAGLIO may impact on practice. As at this interim analysis the OS did not cross the threshold for significance; the final OS data is expected in 2013 and may impact on the ultimate FDA recommendation. Despite the benefits noted in PFS, HRQoL, and KPS it is difficult to anticipate the ultimate recommendations in European and other settings, particularly if there is no confirmatory OS benefit.43 Early data from published studies indicates the feasibility of using BEV in conjunction with chemoradiotherapy.40,41,44 Data from the RTOG and AVAGLIO trials with larger sample sizes will provide definitive evidence of the role of systemic antiangiogenic therapy. Issues of concern needing closer evaluation would include the optimal dose and timing to start treatment because wound healing problems may be potentiated when using combination therapy of BEV with radiotherapy/chemotherapy. Table 2 summarizes the various clinical trials and their results with the use of BEV in the newly diagnosed HGG setting.
Table 2.
Trials with bevacizumab in newly diagnosed glioblastoma
| Study | Number of patients | Regimen | Response rate
|
PFS
|
OS
|
|||
|---|---|---|---|---|---|---|---|---|
| CR (%) | PR (%) | SD (%) | PFS-6 (%) | Mediana (months) | Mediana (months) | |||
| Upfront bevacizumab | ||||||||
| Chauffert 2011 (TEMAVIR)126 | 30b | BEV + irinotecan | NA | NA | NA | 57 | NA | NA |
| Hofland 2011127 | 31 | BEV + irinotecan | 0 | 19.4 | 22.6 | 55 | NA | 14.8 |
| 32 | BEV + TMZ | 0 | 31.2 | 18.7 | 56 | NA | 11.2 | |
| Lou 2011128 | 41 | BEV + TMZ | 0 | 25.8 | 61.3 | NA | 5.2 | 11.7 |
| 41 | BEV + TMZ + irinotecan | 0 | 41 | 44 | NA | 6.7 | 10.5 | |
| First-line bevacizumab | ||||||||
| Vredenburgh 2011129 | 75 | BEV + TMZ + irinotecan | NA | NA | NA | PFS12: 62.7 | 14.2 | 21.2 |
| Lai 201141 | 70 | BEV + TMZ | NA | NA | NA | NA | 13.6 | 19.6 |
| Omuro 2011130 | 40 | BEV + TMZ | 27c | 63 | 3 | NA | 11 | NA |
| Narayana 201242 | 51 | BEV + TMZ | NA | NA | NA | 85.1 | 13 | 23 |
Notes:
Median PFS and median OS that were reported in weeks were standardized to months using the following formula: weeks/52 × 12.
Results reported from an interim analysis 6 month after inclusion of the first 30 patients.
Response reported in 30 evaluable patients.
Abbreviations: BEV, bevacizumab; CR, complete response; CT, chemotherapy; NA, not available; PFS, progression-free survival; PFS6, 6-month progression-free survival; PR, partial response; SD, stable disease; TMZ, temozolomide.
BEV in the setting of WHO grade III gliomas
The median survival time for AG (anaplastic astrocytomas, anaplastic oligoastrocytomas, and anaplastic oligodendroglial tumors) is about 2–15 years, with pure oligodendroglial tumors having a better prognosis than astrocytic tumors. The anaplastic astrocytomas have a marginally better prognosis than GBMs. Without any prior existing standard recommendations to treat progression or recurrence in AGs, BEV as a monotherapy or in combination with irinotecan has been extensively used as a treatment option in patients with progressive AGs. Desjardin et al conducted a Phase II trial for 33 AG patients (25 anaplastic astrocytomas and eight anaplastic oligoastrocytomas) treated with BEV and irinotecan.45 PFS at 6 months and OS at 6 months was 55% and 79%, respectively. In addition, the steroid dosage was decreased in 67% of the patients and partial response was reported in 61% of patients. Vredenburgh et al published their results of a Phase II trial using BEV and irinotecan in the recurrent HGG setting, which included nine AG and 23 GBM patients.46 The median PFS was 23 weeks (20 weeks for the HGG group and 30 weeks for the AG group) with an overall radiologic response rate of 63% (six of nine in the AG group and 14 of 23 in the HGG group). The OS probability at 6-months was 72% with an overall PFS probability at 6 months of 38% (56% in the AG group versus 30% in the HGG group). There were two treatment-related deaths: one due to pulmonary thromboembolism and the other due to arterial ischemic stroke. Other toxicities observed in the study were thromboembolic complications in four patients and grade II proteinuria with resolution subsequent to discontinuing therapy in two patients, with no patient having any grade IV hematologic toxicity. At present in the absence of a high level of evidence, there is no definite role for BEV in the grade 3 setting.
BEV as a steroid-sparing agent
Steroids are used in the treatment of HGGs in order to reduce edema, restore blood–brain barrier function, and provide symptomatic improvement to the patient. The side effects of steroid use include weight gain, skin fragility, bleeding, infection, myopathy, lymphopenia, and hyperglycemia.10,47,48 Hyperglycemia in itself may be associated with poorer outcomes in these patients (OS 9.1 months versus 14.5 months in the better glycemic controlled group).49 The BRAIN study assessed corticosteroid use in recurrent GBMs treated with BEV. There was a sustained reduction in the use of corticosteroids in 30.2% and 46.5% and a complete reduction in 16.3% and 20.9% of patients on BEV and BEV plus irinotecan, respectively.50 Kreisl et al reported a reduction by an average of 59% in baseline corticosteroid use in 58% of their recurrent GBM patients while on BEV as a single therapy.37 Norden et al reported that 33% of their recurrent HGG patients had a reduction in the dose of corticosteroids used while on BEV.52 Although this may have an effect on the patient health-related quality of life, this has to be viewed with cautious optimism because of the inadequate data and variable results from the available studies. The true efficacy of this approach can only be answered by conducting well-designed and powered prospective clinical trials in a cohort of patients where a second surgery may not be feasible, ensuring that health-related quality of life endpoints are prioritized. Current literature, however, supports the corticosteroid-sparing effects of BEV, and clinicians should be able to individually assess patients and decide on dose reductions that can be tolerated without causing adverse events related to early corticosteroid withdrawal.51
BEV and thromboembolism
Patients with HGGs have a particularly increased risk of venous thromboembolic events.53 In a prospective study of 77 patients with HGG, the risk for venous thromboembolism at 12 months and 24 months was reported as 21% and 32%, respectively.54 Most literature acknowledges venous thromboembolism risk being 20%–30% during the course of malignant glioma, with the risk extending throughout the course of the disease.55,56 Venous thromboembolism remains one of the most common management problems in patients with HGG and anticoagulant medications are the generally accepted treatment for this complication.57 BEV, being an antiangiogenic agent, increases the risk of venous thromboembolism, and in addition there were initial concerns related to administering BEV therapy to patients on anticoagulant therapy; initially these patient were excluded from clinical trials, posing a challenge in the management of glioma patients receiving this drug since venous thromboembolism is so common.58
BEV and hemorrhagic complications
Another serious adverse effect associated with the use of BEV is hemorrhagic complications such as intracranial hemorrhage, epistaxis, and gastrointestinal bleeding.52,59 In a study of 21 patients on simultaneous treatment of BEV with anticoagulants, only one out of five patients who developed intracranial hemorrhage was found to have actual hemorrhage-related symptoms. Hence, these authors too did not consider BEV use a contraindication to therapeutic anticoagulation in HGG patients with venous thromboembolism.60 Norden et al assessed the risk and severity of hemorrhagic complications in a large cohort of 64 glioma patients receiving BEV with concurrent anticoagulation and compared them to 218 patients who did not receive anticoagulation. The results indicated that both the rate of intracranial hemorrhage of any grade and rate of serious hemorrhage (at least grade III) were significantly higher in the patients who received concurrent BEV and anticoagulation compared to those only on BEV (P = 0.02 and P = 0.03, respectively). However, the rate of serious intracranial hemorrhage remained within an acceptable range. Therefore, they supported the concurrent use of therapeutic anticoagulation and BEV in patients with symptomatic venous thromboembolism.61
Other toxicities related to BEV
Unique adverse events associated with the use of BEV make it essential to carefully select and monitor patients in neurooncology practice.62,63 There is an increased risk of bleeding and wound complications, with spontaneous opening of the craniotomy wound (ie, wound dehiscence) being reported as late as 2–6 months following therapy (<2% of patients).64 Other common systemic side effects are fatigue, hypertension, proteinuria, epistaxis, and rarely skin toxicity.29 Most of these toxicities appear to be the result of VEGF disruption in normal tissues.
Serious adverse events occur in <2% of patients using BEV and include gastrointestinal perforation, reversible posterior leukoencephalopathy, and cardiac failure. Life threatening intracranial hemorrhages were seen in <3% of patients and although the risk is low, a high degree of caution has to be maintained in order to avoid additional risk factors for bleeding.52,61,65 BEV is not contraindicated in patients on anticoagulants (as mentioned above) or enzyme-inducing antiepileptic drugs.34,66,67
Recommended dosing
The recommended dose of BEV as monotherapy is based on the previous reported trials (Tables 1 and 2). For recurrent HGGs, the recommended dose is 10 mg/kg bodyweight given intravenously once every 2 weeks (biweekly) until toxicity or progression occurs.10 An alternative would be 7.5–15 mg/kg bodyweight given intravenously once every 3 weeks and this could vary depending on the indication.68 The drug is initially administered intravenously over 90 minutes; if well tolerated, the second time is administered over 60 minutes, and then subsequent treatments are usually given as an infusion over 30 minutes. No standard recommendations for newly diagnosed HGGs exist and the data from the prospective RTOG 0825 and AVAGLIO studies will provide more information in this setting.
Patients on enzyme-inducing antiepileptic drugs and anticoagulants can remain on these drugs without potentiating any further significant increased risks.34,46 There is no evidence suggesting an increased risk of hemorrhage (intracranial or extracranial) with or without the concurrent use of BEV with anticoagulants.60 If any of the adverse events mentioned do occur, the drug should be discontinued. Hypertension and proteinuria should be carefully monitored during each visit and dosing interval. Hospital-based and clinical trial protocols should carefully take into account all the above factors in the care of patients on BEV. If the patient is to undergo any planned open surgical procedure, BEV should be stopped 4–6 weeks prior to any procedure, in view of its long half-life, and should not be recommenced for a minimum of 4 weeks postoperatively (and ensuring the wound is well healed).10
Imaging challenges and novel modalities to assess radiological response following BEV treatment
Imaging of tumor responses following radiation and chemotherapy is a valuable biomarker in the assessment of treatment response and OS.
Until recently, the most widely used criteria to assess response to various therapies in HGGs are the MacDonald criteria, introduced about 20 years ago. There are, however, limitations with the use of these criteria, but without any alternatives they have remained the standard to assess responses to therapies in HGGs. However, more recently newer criteria are being evaluated to assess response in HGGs and the various methods used are discussed in Table 3.
Table 3.
Imaging criteria to assess response to therapy
| Criteria | Definition | Advantages | Limitations |
|---|---|---|---|
| MacDonald’s criteria131 | • Use two-dimensional measurements of enhancing tumor (the product of the maximal cross-sectional enhancing diameters) on CT or MRI scans on a single axial section | • Easy to use | • Irregular shape of GBMs makes use of largest cross sectional area inaccurate |
| • Interobserver variations are minimal | |||
| • Objective method of assessment Enable comparison in clinical trials | • Non-enhancing portions of tumor and necrotic regions may not be taken into account or measured | ||
| • Takes into account steroid use | |||
| • Takes into account neurological status of patient | |||
| • Measurements from multiple lesions are summed | • Response evaluation based on finding a difference in the largest cross-sectional area of tumor on contrast-enhanced CT or MRI is difficult with antiangiogenic therapy. | ||
| Computer-aided volumetric methods87 | • Semi-automated tumor-segmentation software determines tumor volume | • Increased measurement accuracy | • Requires software validation and standardization |
| • Measures both the enhancing and non-enhancing components | |||
| • Images perimeters adjusted by a neuroradiologist | • Studies need to identify inter and intraobserver variability | ||
| • Program calculates an enhancing volume, a non-enhancing volume and total lesion volume in cubic centimeters. | • Can be used to measures volumetric changes on FLAIR/T2 Wl serially to assess response to therapy and identify early progression | ||
| • Used to assess response in clinical trials | |||
| • Measurement variation is low | |||
| Response Evaluation Criteria in Solid Tumors (RECIST version 1.1)80 | • Longest single linear enhancing diameter across a lesion in the axial plane | • Uniform, | • Rarely used in clinical trials for GBMs |
| • Simplified, and | • Not validated in any studies | ||
| • Repeat measurements with each study | • Conservative standard to determine response to therapy for solid tumors | • One dimensional measurements inaccurate for irregular tumor shapes in GBMs | |
| • Always using longest diameter, even if it varies from the original image | |||
| • Minimal lesion diameter 10 mm | • Bran tumor trials historically use 2-dimensional measurements | ||
| • Cystic or necrotic foci of tumor and leptomeningeal lesions are non-measurable | |||
| • Multiple lesions present—individual diameter measurements recorded separately and summed for response evaluation | |||
| Response Assessment in Neuro-oncology (RANO) criteria132 | • Based on two- dimensional tumor measurements | • Useful in measuring response to anti-angiogenic drugs | • Tumor-related edema or ischemia, radiation effect, demyelination and infection are difficult to discern on FLAIR/T2 W1 |
| • In presence of multiple lesions, a minimum of 2 and maximum of 5 can be taken into account | |||
| • Can measure changes in tumors that do not contrast enhance | |||
| • FLAIR/T2 Wl can be used as a surrogate for response/progression in non-enhancing tumors |
Abbreviations: FLAIR, fluid attenuated inversion recovery; T2 Wl, T2 weighted magnetic resonance images; GBM, glioblastoma; CT, computerized tomography scan; MRI, magnetic resonance imaging.
The abnormal vasculature in HGGs makes them permeable to contrast agents. Antiangiogenic drugs such as BEV decrease their permeability, leading to a reduction of contrast leakage into the tumor and thus altering the image characteristics on contrast-enhanced imaging. This makes it difficult to assess the actual radiologic response rate because if the MacDonald criteria are used, this can lead to a falsely high response rate (due to the decrease in contrast enhancement) or what is now referred to as a “pseudoresponse.” An example of the early radiological response with BEV can be seen in a 53-year-old patient with a progressive multifocal GBM treated with BEV His axial images reveal two adjacent posterior fossa lesions, one in the left cerebellar hemisphere and the other extending into the cerebellar peduncle and infiltrating the brainstem. Both the lesions have uniform contrast enhancement, along with ill-defined irregular margins and there is diffuse hyperintense involvement seen on the fluid attenuation inversion recovery images (Figure 1A and B). About 5 weeks following treatment with BEV, the contrast enhancement is significantly decreased along with the fluid attenuation inversion recovery changes (Figure 2 and B). Iwamoto et al reported the inadequate evaluation of disease status with contrast-enhanced MRI, with a nonenhancing tumor being a more significant indicator of recurrence with BEV-treated patients.69 The change in recurrence patterns of GBM was also reported by Norden et al, where a nonenhancing infiltrative tumor was seen to progress while contrast enhancing component regressed.52
Figure 1.
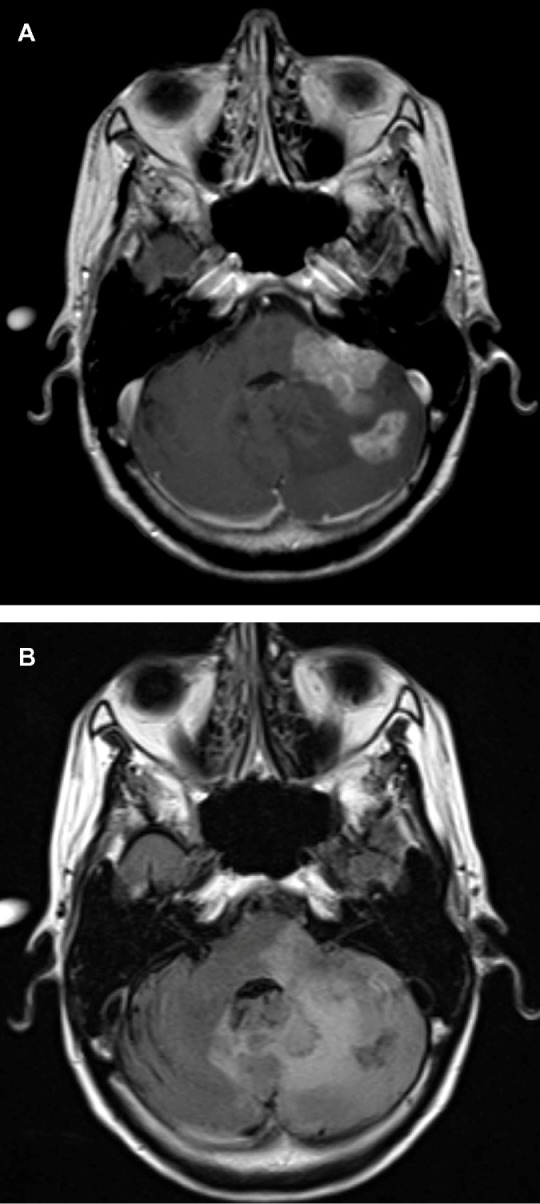
(A) Axial T1-weighted postgadolinium contrast-enhanced magnetic resonance image of a 53-year-old patient with a progressive multifocal glioblastoma revealing two lesions: one in the left cerebellar hemisphere and the other extending into the cerebellar peduncle and infiltrating the brainstem. Both the lesions have uniform contrast enhancement, along with ill-defined irregular margins. (B) Axial fluid attenuated inversion recovery images reveal a more diffuse hyperintense lesion infiltrating into the adjacent cerebellum and brainstem. The hyperintense signal crosses the midline vermis and involves the contralateral cerebellar hemisphere as well.
Figure 2.
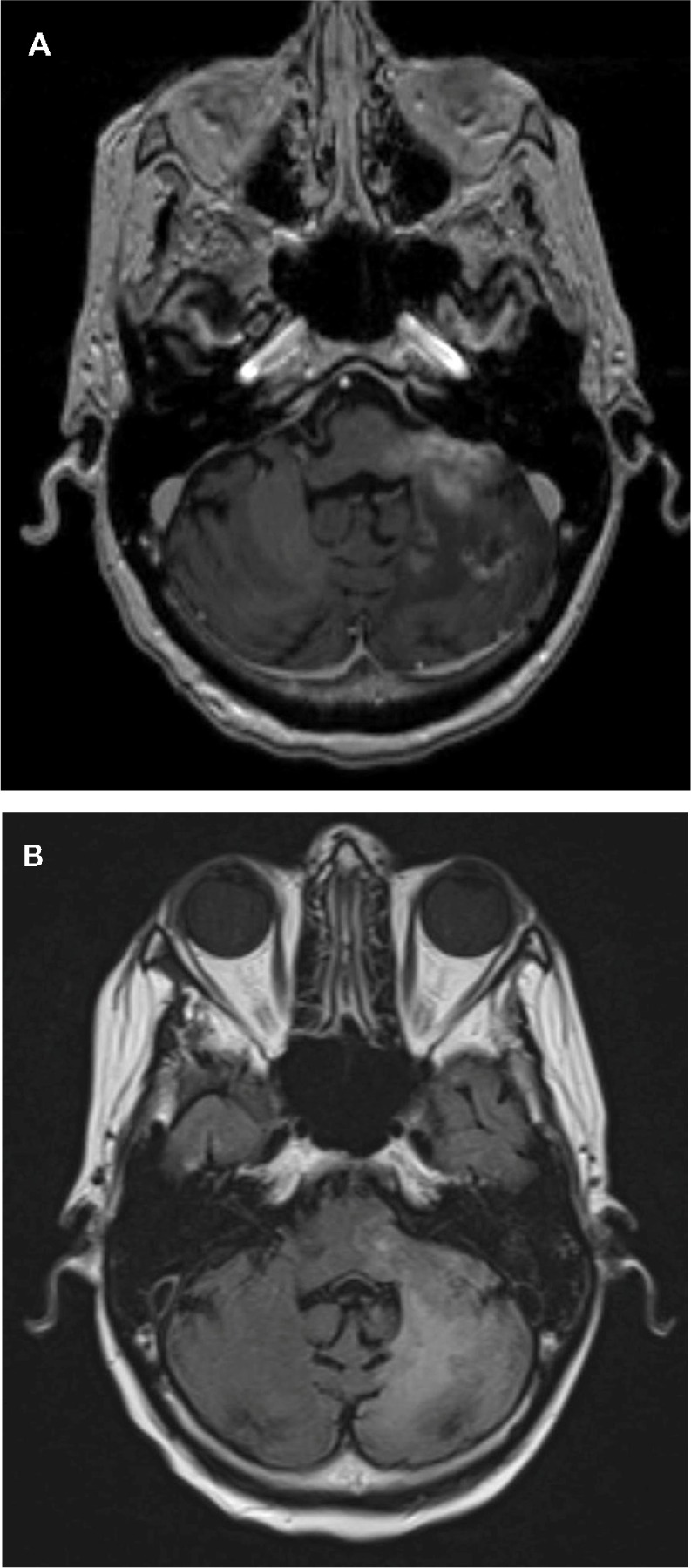
(A) An axial T1-weighted postgadolinium contrast-enhanced magnetic resonance image of the same patient after bevacizumab treatment reveals a significant reduction in the size and shape of the cerebellar/brainstem contrast enhancing tumor. There is also a decrease in the adjacent mass effect with opening up of the adjacent fourth ventricle. (B) An axial fluid attenuated inversion recovery image of the patient after bevacizumab treatment reveals a significant reduction in the hyperintensity of the cerebellar and brainstem involvement. This is associated with a decrease in the adjacent mass effect and opening up of the sulci and adjacent fourth ventricle.
A new reporting protocol, Response Assessment in Neurooncology (RANO), has been established by an international neurooncology and neuroradiology working group. The criteria continue to rely on two-dimensional measurements with a precise definition for measurable (> 10 mm diameter) and nonmeasurable disease (< 10 mm diameter).
The other issue that the RANO criteria aim to address is the issue of pseudoprogression, which has been recognized as a common phenomenon (in 20%–30% cases or up to 50% in some series). Pseudoprogression is defined as an increase of contrast enhancement and/or edema on MRI without true tumor progression, and classically occurs within 3 months of completion of chemoradiation (but can occur beyond this timeframe).70 Interestingly, pseudoprogression appears more commonly in patients with MGMT promoter methylation.71 In view of this phenomenon, RANO criteria distinguish between the MRIs within 12 weeks of chemoradiation and those beyond that timeframe.
Newer imaging modalities include dynamic susceptibility contrast MRI to measure relative cerebral blood volume, relative cerebral blood flow, and mean vessel density; dynamic contrast-enhanced MRI to measure vascular permeability; diffusion MRI to measure tissue density; magnetic resonance spectroscopy to measure tumor metabolites; and a variety of PET tracers to measure different physiologic tumor processes. These are described in detail in Table 4.
Table 4.
Advanced imaging modalities for gliomas
| Imaging modality | Technique | Advantages | Limitations |
|---|---|---|---|
| Contrast MRI133 | Standard T1 W1 with and without contrast; T2 W1 and FLAIR studies | • Simple | • Limited to depicting morphological abnormalities |
| • Standardized, easy to use | • Non-specific | ||
| • Early identification of edema | • Cannot assess underlying metabolic or functional integrity | ||
| • FLAIR sequences sensitive to subtle changesin soft tissue and hence possible early detectionof tumor changes and identifying leptomeningeal tumor spread | • Cannot differentiate progression from pseudoprogression | ||
| • Not useful in follow-up of HGGs treated with anti-angiogenic agents | |||
| MR perfusion | • Dynamic susceptibility contrast imaging (DSC) measures rCBV, rCBF, and MVD | • Prognostic value for standard treatment (chemotherapy and radiation)134 | • Accurate perfusion imaging requires significant expertise to correct for BBB leakage effects |
| • Uses MRI uses rapid echo-planar measurement of T2-weghted changes after injecting the bolus of contrast-like gadolinium | • Predictive value for response to anti-angiogenic therapy135 | • There is a critical need to standardize this technique to permit comparison across centers | |
| • Differentiating true progression from pseudoprogression136 | • Prone to susceptibility artifact and geometric distortion especially at bone-brain-air interface | ||
| • Signal drop by passage of contrast calculates rCBV and rCBF | |||
| • Comparison of spin and gradient echo DSC imagesblood vessel diameter measured Dynamic contrast-enhanced imaging (DCE) -Sorensen et al have calculated a ’vascular permeability index’ by combining the value of K-trans (rate at which contrast material moves from vasculature into the extracellular space), micro-vessel volume, and circulating collagen IV after a day of anti-VEGF therapy to predict OS and PFS137 | • Prognostic value for standard treatment (chemotherapy and radiation)138 | • Requires standardization and expertise across various centers | |
| • Susceptibility artifact remains high as in DSC imaging | |||
| • Predictive value for response to antiangiogenic therapy137 | |||
| DWI | Measures the diffusivity of water within brain tissue and can provide apparent diffusion coefficient maps (ADC) which correlates well with cell density and proliferation.135Edema increases interstitial fluid, thereby increasing ADC whereas dense structures have lower ADC values.140 | Help measure non-enhancing tumor and does not require the administration of contrast dye Greater the density of the structure impeding water mobility, the lower is the ADC value and hence is a noninvasive marker of cellularity. ADC values can be potentially used to determine response of tumorsto BEV, since ADCs are influenced by cellularity, necrosis and edema.141The rate of change of ADC maps within regions of T2 abnormality has been shown to be an early predictor of progression, time to progression and OS subsequent to treatment with BEV.142 | Not sensitive or specific Requires further evaluation and standardization Extreme susceptibility caused by para/ferromagnetic materials such as blood products, calcium or metal |
| MR spectroscopy | Allows for the characterization of the chemical makeup of a particular region of interest143 | • May be able to differentiate true progression from pseudoprogression | • Initial studies, small amounts of data |
| • Due to small voxel size, the entire tumor volume not evaluated missing important areas of tumor | |||
| • Undergoing evaluation as a possible biomarker to assess response to radiation therapy | |||
| • No characteristic correlates with tumor malignancy, | |||
| • Guiding surgical biopsy site | • Nonspecific findings are not uncommon | ||
| • Glioma grading | • Data processing is complex | ||
| PET | Coupling radioactive tracers to molecules that are taken up by metabolically active cells, PET scans can detect and localize the presence of tumors and other cells with increased metabolism. | Can be used as an early biomarker of GBM progression Early differentiation of progression from pseudoprogression | Not available at all centers Requires expertise and training to read scans Cost |
Abbreviations: MRI, magnetic resonance imaging; BEV, bevacizumab; rCBV, relative cerebral blood volume; rCBF, relative cerebral blood flow; MVD, mean vessel diameter; DWI, diffusion weighted images; ADC, apparent diffusion coefficient; DTI, diffusion tensor imaging; PET, positron emission tomography
Dynamic susceptibility contrast MRI uses rapid echo-planar measurement of T2-weighted changes after injecting the bolus of contrast-like gadolinium. The signal drop by passage of contrast agent through the tissue is used to calculate the relative cerebral blood volume and relative cerebral blood flow. By comparing spin and gradient echo dynamic susceptibility contrast images, blood vessel diameter can be measured.72 Dynamic contrast-enhanced MRI has been used to predict clinical outcome by combining it with biomarkers.
Glioma proliferation is marked by indices such as Ki-67 which strongly correlates with fluorothymidine (FLT; 3′-18F-fluoro-3′-deoxy-L-thymidine) analog uptake.73,74 FLT is a promising tracer used in PET imaging and acts as a marker for tumor cell proliferation and is presently used mostly in a research setting. Increased FLT uptake correlates with DNA synthesis and tumor proliferation. In the presence of antiangiogenic therapy where standard imaging is not reliable, FLT PET scans may be able to more accurately assess glioma progression as shown by Chen et al.75 18F-fluoromisonidazole PET predicts hypoxia and increased uptake is associated with poorer survival and a more aggressive tumor.76
Assessing optimal clinical trial endpoints for BEV therapy
Optimal endpoints for identifying the efficacy of antiangiogenic drugs in HGGs remain a challenge because of the use of multiple agents with different mechanisms of action and variable affects on imaging.77 Clinical trials designed to assess new treatments in HGGs use a standardized approach.78 There are three phases in trials. Phase I studies primarily assess toxicity of the drug as its endpoint and secondarily characterize its pharmacokinetics. Phase II studies use radiographic response rates and clinical status evaluations, which often include PFS at 6 and 12 months as the primary endpoint. Radiographic criteria used in Phase II studies assume that measurement of radiographic response is a valid surrogate for a response to the drug. Due to the random nature of timing in imaging, PFS using clinical criteria such as the patients’ neurological status and/or a decline in Karnofsky performance score by >20 is a better indicator to evaluate the drug as compared to disease progression on imaging studies.79 PFS is dependent on radiological findings, which may be confounded by factors such as pseudoresponse and pseudoprogression as well as the inability to objectively quantify radiologic progression. Phase III studies generally use OS as the endpoint, comparing it to whichever agent or modality is the best available standard therapy (often referred to as the “gold standard”). OS, however, has its limitations, as factors related to disease biology may produce variable outcomes; studies need longer assessment times; the impact of salvage (or crossover) therapy can impact on eventual OS figures; and studies require a greater sample size (making them considerably more expensive).
Response Evaluation Criteria in Solid Tumors (RECIST) criteria, introduced in 2000 and recently updated to RECIST 1.1 in 2009, using one- and two-dimensional imaging have been used in the assessment of systemic malignancies.80,81 Studies comparing RECIST criteria with two-dimensional, three-dimensional, and volumetric measurements in gliomas have shown good concordance but have not been used to validate prospective trials.82,83 Conventionally the response of treatment in HGGs is assessed using contrast-enhanced MRI and computed tomography scans and the MacDonald criteria.
The limitations of the MacDonald criteria, as already alluded to above, include variability between observer measurements, inability to assess the noncontrast enhancing component of the tumor, irregular-shaped tumors which are difficult to measure, no guidance in the assessment of multifocal tumors, and difficulty measuring enhancing lesions in the wall of cystic or surgical cavities.84–87 The responses are defined as complete response, partial response, stable disease, and progressive disease and use contrast-enhancement for categorizing a response. Contrast enhancement is nonspecific and a number of factors can effect this including the variability in contrast injections, use and dose of corticosteroids at the time of imaging, postsurgical changes, inflammatory changes, infarction, seizures, and the effects of radiation therapy.88–91 The modified MacDonald criteria also include a category of minor response that has been described earlier in this section.
With the evolution of various therapeutic modalities, the increasing use of concurrent chemoradiation therapy, and newer antiangiogenic therapies affecting the permeability of the blood–brain barrier, it has become more difficult to objectively assess response using the MacDonald criteria. Due to the unreliability and limitations of conventional imaging in assessing treatment outcome, the RANO Working Party has proposed updated criteria to more accurately monitor the efficacy of anti-VEGF therapies. In patients on VEGF therapy, to determine tumor response, the RANO criteria take into account measurable and nonmeasurable disease, changes taking place on patients’ imaging in the first 12 weeks after radiation therapy versus the first 12 weeks of immunotherapy treatment, strict criteria for eligibility and progression in clinical trials, and inclusion of nonenhancing disease with the T2-weighted and fluid attenuation inversion recovery sequence images. Newer imaging modalities will also be taken into account, such as physiological imaging and diffusion imaging with restricted diffusion indicating tumor progression, and will be used to assess clinical trial endpoints.92–94
Other criteria which could be used to supplement the primary endpoints and provide meaningful insight and data into tumor response and progression include neurocognitive function, neurological status, corticosteroid use, and health-related quality of life. All these secondary endpoints could provide important information to incorporate into the design of trials.
Mechanisms of resistance to BEV
Although a number of strategies have been utilized in the treatment of these tumors, most treatments eventually fail, with tumor progression and disease-related death as the ultimate outcome. In some patients, treatment failure occurs without any initial response to the drug. Two questions need to be critically evaluated. (1) Why do some patients respond whereas others do not? (2) For those who initially respond, what factors lead to therapy failure, with the tumor subsequently progressing extremely rapidly?
HGGs appear to develop adaptive responses to the therapies, leading to resistance to these treatments and the loss of a drug-induced response. Clinical and preclinical studies hypothesize some sort of fusion between endothelial cells and malignant cells enabling them to acquire genotypic alterations, such as aneuploidy and abnormal chromosomal architecture.95,96 Spontaneous genetic mutations within the endothelial cells may lead to altered targets, alternative pathways, and resistance to agents inhibiting neoangiogenesis (Figure 3).
Figure 3.

Mechanisms of resistance to BEV appear to be of two major types.
Notes: The first being mechanisms that develop in ECs. ECs can possess primary resistance to VEGF therapies (BEV) and not have any initial response to treatment; this may be a reason for unresponsiveness in certain patients. An alternative mechanism of EC resistance is acquired secondary to fusion with malignant cells followed by altered genetic characteristics such as chromosomal aberrations and aneuploidy, making them resistant to antiangiogenic agents. They may thus become activated via alternative pathways via ligand-independent mechanisms and by recruiting factors other than VEGF (eg, FGF and SDF-α). An alternative pathway of resistance lies with the malignant glioma cells themselves and could involve increased invasiveness, upregulation of altered p53 variants, and recruitment of pericytes, which are proangiogenic and release factors such as Ang-1. Additionally, it could involve the release of factors such as PlGF, which recruit myeloid cells. Myeloid cells produce various cytokines such as IL6 and IL8, which are proangiogenic and resistant to various antiangiogenic therapies.
Abbreviations: Ang-1, angiopoietin-1; BEV, bevacizumab; CED, cediranib; EC, endothelial cell; FGF, fibroblast growth factor; IL, interleukin; p53, protein-53; PlGF, placental-derived growth factor; SDF-α, stromal-derived factor-α; VEGF, vascular endothelial growth factor.
Endothelial cells may become activated via ligand-independent mechanisms, and in those cases targeting receptors may be a less effective method of inhibiting angiogenic signal pathways. Experimental spontaneous mouse tumor models have shown VEGF receptor-2 blockade initially disrupts angiogenesis but the late-stage tumors become resistant, inducing other hypoxia-related factors including fibroblast growth factor and angiopoietins (angiogenic rescue).97,98 These models demonstrate a blockade of single angiogenic factors causes a switch, with a changeover to alternate pathways. Though we would have expected decreased levels of VEGF in the context of VEGF inhibitors, a paradoxical response involving increased measured levels of VEGF has sometimes been seen in tumors when anti-VEGF drugs are used. Other mechanisms of developing resistance include cluster of differentiation-11b+/Gr1+ myeloid cells infiltrating the tumor and producing cytokines interleukin-6 and interleukin-8 to induce tumor angiogenesis.99 Cytotoxic chemotherapy using myeloid suppression may improve therapeutic benefit when combined with anti-VEGF therapy and is the strategy used in combinatorial drug regimens. Tumor stem cells also contribute via “selection of the fittest” cells, which are resistant to antiangiogenic agents used.
Other proposed mechanisms by which they circumvent antiangiogenic actions include upregulating alternative proangiogenic pathways (basic fibroblast growth factor and platelet-derived growth factor), conversion from angiogenesis to the cooption of adjacent vasculature from the normal parenchyma resulting in a vasculocentric growth pattern, an increase in glioma invasiveness by upregulation of proinvasive molecules, selective overgrowth of protein-53 mutated glioma cells resistant to the effects of hypoxia, the recruitment of pericytes stabilizing the tumor vasculature, and expressing endothelial survival factors such as angiopoietin-1 along with colonization by bone marrow proangiogenic precursors.97,100–106 Another contributor to the anti-VEGF ligand effects is the production of placental-derived growth factor, a pleiotropic cytokine that stimulates endothelial cell growth, migration, and survival. This factor also attracts bone marrow-derived precursors, which are resistant to the effects of antiangiogenic therapy.107,108
The explanation for the primary intrinsic resistance to antiangiogenic therapies could possibly lie in the fact that patients harboring certain tumor types have a preexisting predilection to use proangiogenic pathways alternate to the VEGF pathways and appear unresponsive both radiologically and clinically.109 BEV has also been reported to induce a more invasive tumor phenotype by expressing insulin-like growth factor binding protein-2 and matrix metalloprotease-2 in GBMs.110
Horizon scanning post BEV
Withdrawal of BEV in cases of progressive disease, or following significant drug-related toxicity, can result in a rapid regrowth or rebound phenomenon on imaging with a clinical decline and median OS of only 47.5 days.111 Various reports indicate the need to gradually taper off BEV or to continue it along with other alternate chemotherapy regimens. Quant et al retrospectively analyzed their data for patients who progressed on BEV and reported a poor median PFS of 37.5 days and PFS at 6 months of 2%.112
Torcuator et al looked into the role of salvage irradiation once progressive disease has occurred on BEV.113 The median PFS was 2.6 months versus 1.9 months in the group of patients who underwent salvage irradiation with concurrent BEV combined with a different chemotherapy as compared to having no radiation and combining BEV with a different chemotherapy. Reardon et al in a recent meta-analysis of five recurrent GBM trials concluded that outcome following BEV failure is poor and continuing BEV improves OS compared to any other presently available therapies.114
In the absence of any other therapies which can produce comparable results to BEV, limited data exist as to the best therapy following progressive disease. Prospective randomized studies evaluating the role of repeating radiation therapy needs to be further evaluated in the setting of new combination chemotherapy. Cilengitide, an antiintegrin inhibitor, is being evaluated in combination with BEV as a first-line treatment for recurrent disease as well as in the setting of disease progression on BEV at Duke University (Durham, NC, USA).
Partnering BEV with other agents both old and new in a prospective randomized manner will hopefully provide more answers and come to the forefront where no treatments are presently available. An Australian study (Carboplatin and BEV in Recurrent GBM Multiforme [CABARET]) is comparing BEV alone to BEV plus carboplatin and will be evaluating the predictive role of an early MRI (at 4 weeks). The second part of the study will also randomize patients who have progressed to further chemotherapy with or without ongoing BEV. Neurocognitive endpoints are also incorporated into the study design.115
The ongoing RTOG 0825 and the AVAGLIO studies which are nearing completion will provide more information on the feasibility, efficacy, dosing, and toxicity of BEV as an upfront therapy in HGGs. Until there are more data, BEV will likely remain a cornerstone of therapy in the progressive/recurrent HGG disease setting for the neurooncologist in the US (with its use in Europe and other countries limited by registration and financial constraints).
The limited response seen with available regimens of antiangiogenic therapy necessitates continuing investigations that will determine the best use of BEV for newly diagnosed or recurrent tumors. The results from these Phase III trials will establish the optimum combinations with radiation, cytotoxic chemotherapy, and other newer targeted therapies. As the understanding of mechanisms of resistance to antiangiogenic therapies are further elucidated, better selection of patients will be crucial to improve outcomes for treating malignant gliomas.
Conclusion
As BEV becomes more widely used in neurooncology, the results of these initial studies remain encouraging. However, malignant gliomas eventually progress due to their resistance to current drug therapies. Targeting GBM stem cells, development of drugs that block the invasion of glioma cells, and combinations of these approaches remain ongoing challenges for physicians treating this devastating disease. The future availability of new guidelines and imaging techniques for assessment of endpoints in various types of neurooncology trials are being made by an international working group with an effort to optimize the evaluation of these experimental agents on HGGs. Antiangiogenic therapies play an important role in the treatment pathway of these extremely malignant tumors, and future clinical trials will address a number of unanswered questions.
Footnotes
Disclosure
Dr Elizabeth J Hovey is a member of the ROCHE Glioma Advisory Board in Australia. Manmeet Ahluwalia has served on the board of Genentech. The authors report no other conflicts of interest in this work.
References
- 1.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008 Jul 31;359(5):492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Wrensch M, Minn Y, Chew T, Bondy M, Berger MS. Epidemiology of primary brain tumors: current concepts and review of the literature. Neuro Oncol. 2002 Oct;4(4):278–299. doi: 10.1093/neuonc/4.4.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giglio P, Villano JL. Newly diagnosed high-grade gliomas. Curr Treat Options Neurol. 2010 Jul;12(4):309–320. doi: 10.1007/s11940-010-0077-6. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005 Mar 10;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Stupp R, Hegi ME, Gilbert MR, Chakravarti A. Chemoradiotherapy in malignant glioma: standard of care and future directions. J Clin Oncol. 2007 Sep 10;25(26):4127–4136. doi: 10.1200/JCO.2007.11.8554. [DOI] [PubMed] [Google Scholar]
- 6.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009 May;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 7.Hegi ME, Diserens AC, Godard S, et al. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004 Mar 15;10(6):1871–1874. doi: 10.1158/1078-0432.ccr-03-0384. [DOI] [PubMed] [Google Scholar]
- 8.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005 Mar 10;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 9.Hegi ME, Liu L, Herman JG, et al. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol. 2008 Sep 1;26(25):4189–4199. doi: 10.1200/JCO.2007.11.5964. [DOI] [PubMed] [Google Scholar]
- 10.Chamberlain MC. Emerging clinical principles on the use of bevacizumab for the treatment of malignant gliomas. Cancer. [DOI] [PubMed]
- 11.Wong ET, Hess KR, Gleason MJ, et al. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol. 1999 Aug;17(8):2572–2578. doi: 10.1200/JCO.1999.17.8.2572. [DOI] [PubMed] [Google Scholar]
- 12.Folkman J. Tumor angiogensis: role in regulation of tumor growth. Symp Soc Dev Biol. 1974;30(0):43–52. [PubMed] [Google Scholar]
- 13.Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990 Jan 3;82(1):4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 14.Geiger TR, Peeper DS. Metastasis mechanisms. Biochimica et biophysica acta. 2009 Dec;1796(2):293–308. doi: 10.1016/j.bbcan.2009.07.006. [Research Support, Non-U.S. Gov’t Review] [DOI] [PubMed] [Google Scholar]
- 15.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 Jan 7;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 16.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nature reviews Cancer. [Review] 2003 Jun;3(6):401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 17.Parikh AA, Ellis LM. The vascular endothelial growth factor family and its receptors. Hematology/oncology clinics of North America. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, PHS. Review. 2004 Oct;18(5):951–971. doi: 10.1016/j.hoc.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007 Apr;6(4):273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 19.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005 Jan 7;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 20.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006 Jan;3(1):24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 21.Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007 Jan;11(1):69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 22.Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006 Aug 15;66(16):7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 23.Siemann DW, Shi W. Targeting the tumor blood vessel network to enhance the efficacy of radiation therapy. Seminars in radiation oncology. [Research Support, U.S. Gov’t, P.H.S. Review] 2003 Jan;13(1):53–61. doi: 10.1053/srao.2003.50005. [DOI] [PubMed] [Google Scholar]
- 24.Miletic H, Niclou SP, Johansson M, Bjerkvig R. Anti-VEGF therapies for malignant glioma: treatment effects and escape mechanisms. Expert Opin Ther Targets. 2009 Apr;13(4):455–468. doi: 10.1517/14728220902806444. [DOI] [PubMed] [Google Scholar]
- 25.Lamszus K, Heese O, Westphal M. Angiogenesis-related growth factors in brain tumors. Cancer treatment and research. [Review] 2004;117:169–190. doi: 10.1007/978-1-4419-8871-3_12. [DOI] [PubMed] [Google Scholar]
- 26.Tuettenberg J, Friedel C, Vajkoczy P. Angiogenesis in malignant glioma–a target for antitumor therapy? Critical reviews in oncology/hematology. [Review] 2006 Sep;59(3):181–193. doi: 10.1016/j.critrevonc.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 27.Presta LG, Chen H, O’Connor SJ, Chisholm V, Meng YG, Krummen L, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer research. 1997 Oct 15;57(20):4593–4599. [PubMed] [Google Scholar]
- 28.Zondor SD, Medina PJ. Bevacizumab: an angiogenesis inhibitor with efficacy in colorectal and other malignancies. The Annals of pharmacotherapy. [Review] 2004 Jul-Aug;38(7–8):1258–1264. doi: 10.1345/aph.1D470. [DOI] [PubMed] [Google Scholar]
- 29.Dietrich J, Norden AD, Wen PY. Emerging antiangiogenic treatments for gliomas—efficacy and safety issues. Curr Opin Neurol. 2008 Dec;21(6):736–744. doi: 10.1097/WCO.0b013e3283131370. [DOI] [PubMed] [Google Scholar]
- 30.Cardones AR, Banez LL. VEGF inhibitors in cancer therapy. Current pharmaceutical design. [Review] 2006;12(3):387–394. doi: 10.2174/138161206775201910. [DOI] [PubMed] [Google Scholar]
- 31.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nature reviews Drug discovery. [Review] 2004 May;3(5):391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 32.Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochemical and biophysical research communications. [Review] 2005 Jul 29;333(2):328–335. doi: 10.1016/j.bbrc.2005.05.132. [DOI] [PubMed] [Google Scholar]
- 33.Stark-Vance V. Bevacizumab and CPT-11 in the treatment of relapsed malignant glioma [abstract] Neuro Oncology. 2005;7:369. [Abstract] [Google Scholar]
- 34.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007 Oct 20;25(30):4722–4729. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
- 35.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009 Oct 1;27(28):4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 36.Cloughesy T, JJV B, Day A, Das HS, Friedman and for the BRAIN Investigators Updated safety and survival of patients with relapsed glioblastoma treated with bevacizumab in the BRAIN study. Journal of Clinical Oncology. 2010;28(15) 2010 ASCO Annual Meeting Proceedings (PostMeeting Edition). [Abstract] [Google Scholar]
- 37.Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009 Feb 10;27(5):740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: Bevacizumab (Avastin (R)) as treatment of recurrent glioblastoma multiforme. The Oncologist. 2009;14(11):1131. doi: 10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- 39.Balana C, Etxaniz O, Buges C, Martinez A. Approval denied by the European Medicines Agency (EMA) for bevacizumab in the treatment of high-grade glioma recurrence: a good idea or a grave error? Clin Transl Oncol. [Letter] 2011 Mar;13(3):209–210. doi: 10.1007/s12094-011-0642-9. [DOI] [PubMed] [Google Scholar]
- 40.Narayana A, Golfinos JG, Fischer I, et al. Feasibility of using bevacizumab with radiation therapy and temozolomide in newly diagnosed high-grade glioma. Int J Radiat Oncol Biol Phys. 2008 Oct 1;72(2):383–389. doi: 10.1016/j.ijrobp.2008.05.062. [DOI] [PubMed] [Google Scholar]
- 41.Lai A, Tran A, Nghiemphu PL, et al. Phase II study of bevacizumab plus temozolomide during and after radiation therapy for patients with newly diagnosed glioblastoma multiforme. Journal of clinical oncology: Official journal of the American Society of Clinical Oncology. 2011;29(2):142–148. doi: 10.1200/JCO.2010.30.2729. [Clinical Trial, Phase II Multicenter Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Narayana A, Gruber D, Kunnakkat S, et al. A clinical trial of bevacizumab, temozolomide, and radiation for newly diagnosed glioblastoma. Journal of neurosurgery. [Clinical Trial] 2012;116(2):341–345. doi: 10.3171/2011.9.JNS11656. [DOI] [PubMed] [Google Scholar]
- 43.Chinot O, Wick W, Mason W, et al. OT3: Phase III trial of Bevacizumab added to standard radiotherapy and temozolomide for newly-diagnosed glioblastoma: mature progression-free survival and preliminary overall survival results in AVAGLIO. Neuro Oncology. [Abstract] 2012;14(6):vi101–vi105. [Google Scholar]
- 44.Lai A, Filka E, McGibbon B, Nghiemphu PL, Graham C, Yong WH, et al. Phase II pilot study of bevacizumab in combination with temozolomide and regional radiation therapy for up-front treatment of patients with newly diagnosed glioblastoma multiforme: interim analysis of safety and tolerability. Int J Radiat Oncol Biol Phys. 2008;71(5):1372–1380. doi: 10.1016/j.ijrobp.2007.11.068. [DOI] [PubMed] [Google Scholar]
- 45.Desjardins A, Reardon DA, Herndon JE, 2nd, Marcello J, Quinn JA, Rich JN, et al. Bevacizumab plus irinotecan in recurrent WHO grade 3 malignant gliomas. Clinical cancer research: an official journal of the American Association for Cancer Research. Clinical Trial, Phase II, Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. 2008;14(21):7068–7073. doi: 10.1158/1078-0432.CCR-08-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, Dowell JM, Reardon DA, Quinn JA, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;1513(4):1253–1259. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 47.Wick W, Kuker W. Brain edema in neurooncology: radiological assessment and management. Onkologie. [Review] 2004;27(3):261–266. doi: 10.1159/000077976. [DOI] [PubMed] [Google Scholar]
- 48.Chang SM, Parney IF, Huang W, et al. Patterns of care for adults with newly diagnosed malignant glioma. JAMA. 2005;293(5):557–564. doi: 10.1001/jama.293.5.557. [DOI] [PubMed] [Google Scholar]
- 49.Derr RL, Ye X, Islas MU, Desideri S, Saudek CD, Grossman SA. Association between hyperglycemia and survival in patients with newly diagnosed glioblastoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27(7):1082–1086. doi: 10.1200/JCO.2008.19.1098. [Research Support, N.I.H., Extramural] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. Clinical Trial, Phase II Multicenter Study, Randomized Controlled Trial. Research Support, Non-U.S. Govt. 2009;27(28):4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 51.Vredenburgh JJ, Cloughesy T, Samant M, et al. Corticosteroid use in patients with glioblastoma at first or second relapse treated with bevacizumab in the BRAIN study. The Oncologist. 2010;15(12):1329–1334. doi: 10.1634/theoncologist.2010-0105. [Clinical Trial, Phase II Multicenter Study Randomized Controlled Trial] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Norden AD, Young GS, Setayesh K, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70(10):779–787. doi: 10.1212/01.wnl.0000304121.57857.38. [DOI] [PubMed] [Google Scholar]
- 53.Semrad TJ, O’Donnell R, Wun T, et al. Epidemiology of venous thromboembolism in 9489 patients with malignant glioma. J Neurosurg. 2007 Apr;106(4):601–608. doi: 10.3171/jns.2007.106.4.601. [DOI] [PubMed] [Google Scholar]
- 54.Brandes AA, Scelzi E, Salmistraro G, Ermani M, Carollo C, Berti F, et al. Incidence of risk of thromboembolism during treatment high-grade gliomas: a prospective study. Eur J Cancer. 1997;33(10):1592–1596. doi: 10.1016/s0959-8049(97)00167-6. [DOI] [PubMed] [Google Scholar]
- 55.Jenkins EO, Schiff D, Mackman N, Key NS. Venous thromboembolism in malignant gliomas. J Thromb Haemost. 2010 Feb;8(2):221–217. doi: 10.1111/j.1538-7836.2009.03690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marras LC, Geerts WH, Perry JR. The risk of venous thromboembolism is increased throughout the course of malignant glioma: an evidence-based review. Cancer. 2000;89(3):640–646. doi: 10.1002/1097-0142(20000801)89:3<640::aid-cncr20>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 57.Drappatz J, Schiff D, Kesari S, Norden AD, Wen PY. Medical management of brain tumor patients. Neurol Clin. 2007 Nov;25(4):1035–1071. doi: 10.1016/j.ncl.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 58.Nalluri SR, Chu D, Keresztes R, Zhu X, Wu S. Risk of venous thromboembolism with the angiogenesis inhibitor bevacizumab in cancer patients: a meta-analysis. JAMA. 2008 Nov 19;300(19):2277–2285. doi: 10.1001/jama.2008.656. [DOI] [PubMed] [Google Scholar]
- 59.Gordon MS, Cunningham D. Managing patients treated with bevacizumab combination therapy. Oncology. 2005;69( Suppl 3):25–33. doi: 10.1159/000088481. [DOI] [PubMed] [Google Scholar]
- 60.Nghiemphu PL, Green RM, Pope WB, Lai A, Cloughesy TF. Safety of anticoagulation use and bevacizumab in patients with glioma. Neuro Oncol. 2008 Jun;10(3):355–360. doi: 10.1215/15228517-2008-009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Norden AD, Bartolomeo J, Tanaka S, et al. Safety of concurrent bevacizumab therapy and anticoagulation in glioma patients. J Neurooncol. 2011 Jun 26; doi: 10.1007/s11060-011-0642-1. [DOI] [PubMed] [Google Scholar]
- 62.Verheul HM, Pinedo HM. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat Rev Cancer. 2007;7(6):475–485. doi: 10.1038/nrc2152. [DOI] [PubMed] [Google Scholar]
- 63.Eskens FA, Verweij J. The clinical toxicity profile of vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor (VEGFR) targeting angiogenesis inhibitors; a review. Eur J Cancer. 2006;42(18):3127–3139. doi: 10.1016/j.ejca.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 64.Chamberlain MC. Bevacizumab plus irinotecan in recurrent glioblastoma. J Clin Oncol. 2008 Feb 20;26(6):1012–1013. doi: 10.1200/JCO.2007.15.1605. author reply 3. [DOI] [PubMed] [Google Scholar]
- 65.Norden AD, Drappatz J, Ciampa AS, et al. Colon perforation during antiangiogenic therapy for malignant glioma. Neuro Oncol. 2009;11(1):929–925. doi: 10.1215/15228517-2008-071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clinical cancer research: an official journal of the American Association for Cancer Research. Clinical Trial, Phase II Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. 2007 Feb 15;13(4):1253–1259. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 67.Chamberlain MC. Bevacizumab and irinotecan for recurrent oligodendroglial tumors. Neurology. 74(2):181. doi: 10.1212/WNL.0b013e3181c775e3. [DOI] [PubMed] [Google Scholar]
- 68.Specenier P. Bevacizumab in glioblastoma multiforme. Expert review of anticancer therapy. [Review] 2012;12(1):9–18. doi: 10.1586/era.11.179. [DOI] [PubMed] [Google Scholar]
- 69.Iwamoto FM, Abrey LE, Beal K, et al. Patterns of relapse and prognosis after bevacizumab failure in recurrent glioblastoma. Neurology. 2009 Oct 13;73(15):1200–1206. doi: 10.1212/WNL.0b013e3181bc0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brandsma D, van den Bent MJ. Pseudoprogression and pseudoresponse in the treatment of gliomas. Curr Opin Neurol. 2009;22(6):633–638. doi: 10.1097/WCO.0b013e328332363e. [DOI] [PubMed] [Google Scholar]
- 71.Brandes AA, Franceschi E, Tosoni A, et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J Clin Oncol. 2008;26(13):2192–2197. doi: 10.1200/JCO.2007.14.8163. [DOI] [PubMed] [Google Scholar]
- 72.Batchelor TT, Sorensen AG, di Tomaso E, et al. AZD2171, a panVEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11(1):83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ullrich R, Backes H, Li H, et al. Glioma proliferation as assessed by 3’-fluoro-3’-deoxy-L-thymidine positron emission tomography in patients with newly diagnosed high-grade glioma. Clinical cancer research: an official journal of the American Association for Cancer Research. Research Support, Non-U.S. Gov’t. 2008;14(7):2049–2055. doi: 10.1158/1078-0432.CCR-07-1553. [DOI] [PubMed] [Google Scholar]
- 74.Chen W, Cloughesy T, Kamdar N, et al. Imaging proliferation in brain tumors with 18F-FLT PET: comparison with 18F-FDG. J Nucl Med. 2005;46(6):945–952. [PubMed] [Google Scholar]
- 75.Chen W, Delaloye S, Silverman DH, et al. Predicting treatment response of malignant gliomas to bevacizumab and irinotecan by imaging proliferation with [18F] fuorothymidine positron emission tomography: a pilot study. J Clin Oncol. 2007;25(30):4714–4721. doi: 10.1200/JCO.2006.10.5825. [DOI] [PubMed] [Google Scholar]
- 76.Szeto MD, Chakraborty G, Hadley J, et al. Quantitative metrics of net proliferation and invasion link biological aggressiveness assessed by MRI with hypoxia assessed by FMISO-PET in newly diagnosed glioblastomas. Cancer research. 2009;1569(10):4502–4509. doi: 10.1158/0008-5472.CAN-08-3884. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reardon DA, Galanis E, DeGroot JF, et al. Clinical trial end points for high-grade glioma: the evolving landscape. Neuro-oncology. [Review] 2011;13(3):353–361. doi: 10.1093/neuonc/noq203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Batchelor T, Stanley K, Andersen J. Clinical trials in neuro-oncology. Current opinion in neurology. [Review] 2001;14(6):689–694. doi: 10.1097/00019052-200112000-00003. [DOI] [PubMed] [Google Scholar]
- 79.Prados MD, Seiferheld W, Sandler HM, Buckner JC, Phillips T, Schultz C, et al. Phase III randomized study of radiotherapy plus procarbazine, lomustine, and vincristine with or without BUdR for treatment of anaplastic astrocytoma: final report of RTOG 9404. Int J Radiat Oncol Biol Phys. 2004 Mar 15;58(4):1147–1152. doi: 10.1016/j.ijrobp.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 80.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. Journal of the National Cancer Institute. [Guideline Practice Guideline] 2000;92(3):205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 81.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) European Journal of Cancer. [Practice Guideline] 2009 Jan;45(2):228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 82.Warren KE, Patronas N, Aikin AA, Albert PS, Balis FM. Comparison of one-, two-, and three-dimensional measurements of childhood brain tumors. Journal of the National Cancer Institute. [Comparative Study Evaluation Studies] 2001;93(18):1401–1405. doi: 10.1093/jnci/93.18.1401. [DOI] [PubMed] [Google Scholar]
- 83.Galanis E, Buckner JC, Maurer MJ, et al. Validation of neuroradiologic response assessment in gliomas: measurement by RECIST, two-dimensional, computer-assisted tumor area, and computer-assisted tumor volume methods. Neuro-oncology. 2006;(2):156–165. doi: 10.1215/15228517-2005-005. [Comparative Study Research Support, N.I.H., Extramural Validation Studies] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28(11):1963–1972. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 85.Sorensen AG, Batchelor TT, Wen PY, Zhang WT, Jain RK. Response criteria for glioma. Nat Clin Pract Oncol. 2008;5(11):634–644. doi: 10.1038/ncponc1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van den Bent MJ, Vogelbaum MA, Wen PY, Macdonald DR, Chang SM. End point assessment in gliomas: novel treatments limit usefulness of classical Macdonald’s Criteria. J Clin Oncol. 2009;27(18):2905–2908. doi: 10.1200/JCO.2009.22.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Henson JW, Ulmer S, Harris GJ. Brain tumor imaging in clinical trials. AJNR Am J Neuroradiol. 2008;29(3):419–424. doi: 10.3174/ajnr.A0963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cairncross JG, Macdonald DR, Pexman JH, Ives FJ. Steroid-induced CT changes in patients with recurrent malignant glioma. Neurology. 1988;38(5):724–726. doi: 10.1212/wnl.38.5.724. [DOI] [PubMed] [Google Scholar]
- 89.Cairncross JG, Pexman JH, Rathbone MP, DelMaestro RF. Postoperative contrast enhancement in patients with brain tumor. Ann Neurol. 1985;17(6):570–572. doi: 10.1002/ana.410170607. [DOI] [PubMed] [Google Scholar]
- 90.Ulmer S, Braga TA, Barker FG, 2nd, Lev MH, Gonzalez RG, Henson JW. Clinical and radiographic features of peritumoral infarction following resection of glioblastoma. Neurology. 2006;1467(9):1668–1670. doi: 10.1212/01.wnl.0000242894.21705.3c. [DOI] [PubMed] [Google Scholar]
- 91.Kumar AJ, Leeds NE, Fuller GN, et al. Malignant gliomas: MR imaging spectrum of radiation therapy- and chemotherapy-induced necrosis of the brain after treatment. Radiology. 2000;217(2):377–384. doi: 10.1148/radiology.217.2.r00nv36377. [DOI] [PubMed] [Google Scholar]
- 92.Hylton N. Dynamic contrast-enhanced magnetic resonance imaging as an imaging biomarker. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. Review. 2006;24(20):3293–3298. doi: 10.1200/JCO.2006.06.8080. [DOI] [PubMed] [Google Scholar]
- 93.Barrett T, Brechbiel M, Bernardo M, Choyke PL. MRI of tumor angiogenesis. Journal of magnetic resonance imaging: JMRI. [Research Support, N.I.H., Intramural Review] 2007;26(2):235–249. doi: 10.1002/jmri.20991. [DOI] [PubMed] [Google Scholar]
- 94.Popperl G, Kreth FW, Herms J, et al. Analysis of 18F-FET PET for grading of recurrent gliomas: is evaluation of uptake kinetics superior to standard methods? Journal of nuclear medicine: official publication, Society of Nuclear Medicine. [Controlled Clinical Trial Research Support, Non-U.S. Gov’t] 2006;47(3):393–403. [PubMed] [Google Scholar]
- 95.Hida K, Hida Y, Amin DN, et al. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer research. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.] 2004;64(22):8249–8255. doi: 10.1158/0008-5472.CAN-04-1567. [DOI] [PubMed] [Google Scholar]
- 96.Cao Y, Zhong W, Sun Y. Improvement of antiangiogenic cancer therapy by understanding the mechanisms of angiogenic factor interplay and drug resistance. Seminars in cancer biology. [Research Support, Non-U.S. Gov’t Review] 2009;19(5):338–343. doi: 10.1016/j.semcancer.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 97.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.] 2005;8(4):299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 98.Chamberlain MC. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2009;72(8):772–773. doi: 10.1212/01.wnl.0000339387.03225.0a. author reply 3–4. [DOI] [PubMed] [Google Scholar]
- 99.Shojaei F, Wu X, Malik AK, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nature biotechnology. 2007;25(8):911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 100.Chamberlain MC, Raizer J. Antiangiogenic therapy for high-grade gliomas. CNS Neurol Disord Drug Targets. 2009;8(3):184–194. doi: 10.2174/187152709788680706. [DOI] [PubMed] [Google Scholar]
- 101.Paez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lamszus K, Kunkel P, Westphal M. Invasion as limitation to anti-angiogenic glioma therapy. Acta neurochirurgica Supplement. [Review] 2003;88:169–177. doi: 10.1007/978-3-7091-6090-9_23. [DOI] [PubMed] [Google Scholar]
- 103.Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. Review. 2005;23(10):2411–2422. doi: 10.1200/JCO.2005.03.089. [DOI] [PubMed] [Google Scholar]
- 104.Nakada M, Nakada S, Demuth T, Tran NL, Hoelzinger DB, Berens ME. Molecular targets of glioma invasion. Cellular and molecular life sciences: CMLS. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review] 2007;64(4):458–478. doi: 10.1007/s00018-007-6342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rubenstein JL, Kim J, Ozawa T, et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.] 2000;2(4):306–314. doi: 10.1038/sj.neo.7900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rao JS. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer. 2003;3(7):489–501. doi: 10.1038/nrc1121. [DOI] [PubMed] [Google Scholar]
- 107.Jain RK, Xu L. alphaPlGF: a new kid on the antiangiogenesis block. Cell. 2007;131(3):443–445. doi: 10.1016/j.cell.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 108.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;(8):592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brastianos PK, Batchelor TT. Vascular endothelial growth factor inhibitors in malignant gliomas. Targeted oncology. [Review] 2010;5(3):167–174. doi: 10.1007/s11523-010-0158-1. [DOI] [PubMed] [Google Scholar]
- 110.de Groot JF, Fuller G, Kumar AJ, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010;12(3):233–242. doi: 10.1093/neuonc/nop027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zuniga RM, Torcuator R, Jain R, et al. Rebound tumour progression after the cessation of bevacizumab therapy in patients with recurrent high-grade glioma. J Neurooncol. 2010;99(2):237–242. doi: 10.1007/s11060-010-0121-0. [DOI] [PubMed] [Google Scholar]
- 112.Quant EC, Norden AD, Drappatz J, Muzikansky A, Doherty L, Lafrankie D, et al. Role of a second chemotherapy in recurrent malignant glioma patients who progress on bevacizumab. Neuro Oncol. 2009;11(5):550–555. doi: 10.1215/15228517-2009-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Torcuator RG, Thind R, Patel M, et al. The role of salvage reirradiation for malignant gliomas that progress on bevacizumab. J Neurooncol. 2010;97(3):401–407. doi: 10.1007/s11060-009-0034-y. [DOI] [PubMed] [Google Scholar]
- 114.Reardon DA, Herndon JE, 2nd, Peters KB, et al. Bevacizumab continuation beyond initial bevacizumab progression among recurrent glioblastoma patients. British journal of cancer. 2012;107(9):1481–1487. doi: 10.1038/bjc.2012.415. 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kathryn M. Field LC, Helen Wheeler, et al. Abstracts from the 16th Annual Scientific Meeting of the Society for Neuro-Oncology in Conjunction with the AANS/CNS Section on Tumors, November 17–20, 2011. Orange County, California: Lot-14. A randomized phase ii study of carboplatin and bevacizumab in recurrent glioblastoma multiforme (cabaret study) Neuro-Oncology. 2011;13(3):iii85–iii91. [Abstract] [Google Scholar]
- 116.Chamberlain MC, Johnston S. Salvage chemotherapy with bevacizumab for recurrent alkylator-refractory anaplastic astrocytoma. J Neurooncol. 2009;91(3):359–367. doi: 10.1007/s11060-008-9722-2. [DOI] [PubMed] [Google Scholar]
- 117.Raizer JJ, Grimm S, Chamberlain MC, et al. A phase 2 trial of single-agent bevacizumab given in an every-3-week schedule for patients with recurrent high-grade gliomas. Cancer. 2010;116(22):5297–5305. doi: 10.1002/cncr.25462. [DOI] [PubMed] [Google Scholar]
- 118.Chamberlain MC, Johnston SK. Salvage therapy with single agent bevacizumab for recurrent glioblastoma. Journal of neuro-oncology. [Clinical Trial] 2010;96(2):259–269. doi: 10.1007/s11060-009-9957-6. [DOI] [PubMed] [Google Scholar]
- 119.Desjardins A, Reardon DA, Herndon JE, 2nd, et al. Bevacizumab plus irinotecan in recurrent WHO grade 3 malignant gliomas. Clin Cancer Res. 2008;14(21):7068–7073. doi: 10.1158/1078-0432.CCR-08-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gilbert M, Wang M, Aldape K, Sorensen A, Mikkelson T, Bokstein F. RTOG 0625: a randomized Phase II trial of bevacizumab with either irinotecan (CPT) or dose-dense temozolomide (TMZ) in recurrent glioblastoma (GBM) Neuro Oncol. 2010;12 doi: 10.1007/s11060-016-2288-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nghiemphu PL, Liu W, Lee Y, et al. Bevacizumab and chemotherapy for recurrent glioblastoma: a single-institution experience. Neurology. 2009;72(14):1217–1222. doi: 10.1212/01.wnl.0000345668.03039.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Narayana A, Kelly P, Golfinos J, et al. Antiangiogenic therapy using bevacizumab in recurrent high-grade glioma: impact on local control and patient survival. J Neurosurg. 2009;110(1):173–180. doi: 10.3171/2008.4.17492. [DOI] [PubMed] [Google Scholar]
- 123.Zuniga RM, Torcuator R, Jain R, et al. Efficacy, safety and patterns of response and recurrence in patients with recurrent high-grade gliomas treated with bevacizumab plus irinotecan. J Neurooncol. 2009;91(3):329–336. doi: 10.1007/s11060-008-9718-y. [DOI] [PubMed] [Google Scholar]
- 124.Taillibert S, Vincent LA, Granger B, et al. Bevacizumab and irinotecan for recurrent oligodendroglial tumors. Neurology. 2009;72(18):1601–1606. doi: 10.1212/WNL.0b013e3181a413be. [DOI] [PubMed] [Google Scholar]
- 125.Hofer S, Elandt K, Greil R, et al. Clinical outcome with bevacizumab in patients with recurrent high-grade glioma treated outside clinical trials. Acta oncologica. [Evaluation Studies Multicenter Study Research Support, Non-U.S. Gov’t] 2011;50(5):630–635. doi: 10.3109/0284186X.2011.572913. [DOI] [PubMed] [Google Scholar]
- 126.Chauffert B, Feuvret L, Bonnetain F, et al. Randomized multicenter phase II trial of irinotecan and bevacizumab as neoadjuvant and adjuvant to temozolomide-based chemoradiation versus chemoradiation for unresectable glioblastoma: Interim results of the TEMAVIR study from the ANOCEF group. ASCO Meeting Abstracts. 2011;29(15):2029. [Google Scholar]
- 127.Hofand KFHS, Sørensen M, Schultz H, et al. Randomized phase ii study of neoadjuvant bevacizumab and irinotecan versus bevacizumab and temozolomide followed by concomitant chemoradiotherapy in newly diagnosed primary glioblastoma multiforme. Neuro-Oncology. [Abstract] 2011;13(3):iii51. doi: 10.3109/0284186X.2013.879607. [DOI] [PubMed] [Google Scholar]
- 128.Lou E, Reardon DA, Peters K, et al. Upfront bevacizumab with temozolomide or with temozolomide and irinotecan for unresectable or multifocal glioblastoma. J Clin Oncol. 2011;29 (suppl; abstr 2055) [Google Scholar]
- 129.Vredenburgh JJ, Desjardins A, Reardon DA, Peters KB, Herndon JE, 2nd, Marcello J, et al. The addition of bevacizumab to standard radiation therapy and temozolomide followed by bevacizumab, temozolomide, and irinotecan for newly diagnosed glioblastoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17(12):4119–4124. doi: 10.1158/1078-0432.CCR-11-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Omuro AMP, Beal K, Karimi S, Correa D, Chan TA, DeAngelis LM, et al. Phase II study of bevacizumab (BEV), temozolomide (TMZ), and hypofractionated stereotactic radiotherapy (HFSRT) for newly diagnosed glioblastoma (GBM) J Clin Oncol. 2011;29(15):2028. [Google Scholar]
- 131.Macdonald DR, Cascino TL, Schold SC, Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990 Jul;8(7):1277–1280. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 132.Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(11):1963–1772. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 133.Cha S. Neuroimaging in neuro-oncology. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. [Review] 2009 Jul;6(3):465–477. doi: 10.1016/j.nurt.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Law M, Young RJ, Babb JS, Peccerelli N, Chheang S, Gruber ML, et al. Gliomas: predicting time to progression or survival with cerebral blood volume measurements at dynamic susceptibility-weighted contrast-enhanced perfusion MR imaging. Radiology. [Research Support, N.I.H., Extramural] 2008;247(2):490–498. doi: 10.1148/radiol.2472070898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Akella NS, Twieg DB, Mikkelsen T, et al. Assessment of brain tumor angiogenesis inhibitors using perfusion magnetic resonance imaging: quality and analysis results of a phase I trial. Journal of magnetic resonance imaging : JMRI. Clinical Trial Clinical Trial, Phase I Multicenter Study Research Support, U.S. Gov’t, P.H.S. 2004;20(6):913–922. doi: 10.1002/jmri.20202. [DOI] [PubMed] [Google Scholar]
- 136.Barajas RF, Jr, Chang JS, Segal MR, et al. Differentiation of recurrent glioblastoma multiforme from radiation necrosis after external beam radiation therapy with dynamic susceptibility-weighted contrast-enhanced perfusion MR imaging. Radiology. 2009;253(2):486–496. doi: 10.1148/radiol.2532090007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sorensen AG, Batchelor TT, Zhang WT, Chen PJ, Yeo P, Wang M, et al. A “vascular normalization index” as potential mechanistic biomarker to predict survival after a single dose of cediranib in recurrent glioblastoma patients. Cancer Res. 2009;69(13):5296–5300. doi: 10.1158/0008-5472.CAN-09-0814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cao Y, Tsien CI, Nagesh V, et al. Survival prediction in high-grade gliomas by MRI perfusion before and during early stage of RT [corrected] International journal of radiation oncology, biology, physics. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] 2006;64(3):876–885. doi: 10.1016/j.ijrobp.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 139.Gerstner ER, Batchelor TT. Imaging and response criteria in gliomas. Current opinion in oncology. [Research Support, Non-U.S. Gov’t Review] 2010;22(6):598–603. doi: 10.1097/CCO.0b013e32833de96e. [DOI] [PubMed] [Google Scholar]
- 140.Pope WB, Lai A, Mehta R, Kim HJ, Qiao J, Young JR, et al. Apparent diffusion coefficient histogram analysis stratifes progression-free survival in newly diagnosed bevacizumab-treated glioblastoma. AJNR. [Research Support, Non-U.S. Gov’t] 2011;32(5):882–889. doi: 10.3174/ajnr.A2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ahluwalia MS, Wen PY. Antiangiogenic therapy for patients with glioblastoma: current challenges in imaging and future directions. Expert review of anticancer therapy. 2011;11(5):653–656. doi: 10.1586/era.11.35. [Editorial] [DOI] [PubMed] [Google Scholar]
- 142.Ellingson BM, Malkin MG, Rand SD, et al. Volumetric analysis of functional diffusion maps is a predictive imaging biomarker for cytotoxic and anti-angiogenic treatments in malignant gliomas. Journal of neuro-oncology. 2011;102(1):95–103. doi: 10.1007/s11060-010-0293-7. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Weybright P, Sundgren PC, Maly P, et al. Differentiation between brain tumor recurrence and radiation injury using MR spectroscopy. AJR. 2005;185(6):1471–1476. doi: 10.2214/AJR.04.0933. [DOI] [PubMed] [Google Scholar]