

Abstract
Neuroblastoma (NB), the most common extracranial solid tumor in childhood, is an extremely heterogeneous disease both biologically and clinically. Although significant progress has been made in identifying molecular and genetic markers for NB, this disease remains an enigmatic challenge. Since NB is thought to be an embryonal tumor that is derived from precursor cells of the peripheral (sympathetic) nervous system, understanding the development of normal sympathetic nervous system may highlight abnormal events that contribute to NB initiation. Therefore, this review focuses on the development of the peripheral trunk neural crest, the current understanding of how developmental factors may contribute to NB and on recent advances in the identification of important genetic lesions and signaling pathways involved in NB tumorigenesis and metastasis. Finally, we discuss how future advances in identification of molecular alterations in NB may lead to more effective, less toxic therapies, and improve the prognosis for NB patients.
1. Clinical and Biological Characteristics of Neuroblastoma (NB)
NB is the most common extracranial solid tumor in childhood, accounting for approximately 7–10% of pediatric cancers and 15% of all pediatric cancer deaths in patients less than 15 years old (Brodeur, 2003; Maris et al., 2007; Schor, 1999). NB is an extremely heterogeneous disease both biologically and clinically (Brodeur, 2003; Evans et al., 1971; Maris et al., 2007). NB is thought to be an embryonal tumor that is derived from precursor cells of the peripheral (sympathetic) nervous system (Brodeur, 2003; Grimmer and Weiss, 2006; Nakagawara and Ohira, 2004). The tumor can arise anywhere along the sympathetic chain but is most frequently in the adrenal medulla and paraspinal ganglia (Fig. 4.1; Johnsen et al., 2009; Nakagawara and Ohira, 2004).
Figure 4. 1.
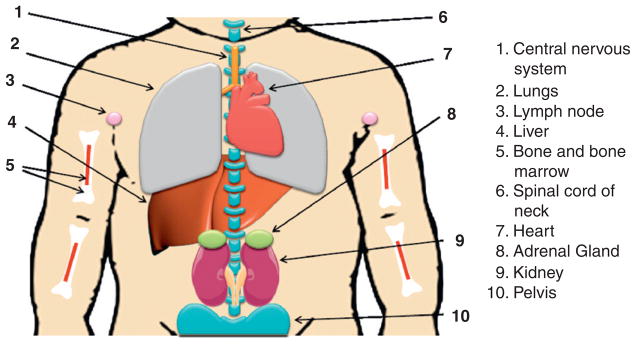
Neuroblastoma localization. Neuroblastoma primary tumors derive from precursor cells of the peripheral (sympathetic) nervous system and can arise anywhere along the sympathetic chain, most frequently in the adrenal gland (position 8 as shown). Neuroblastoma may also develop from spinal cord of neck (position 6) and pelvis (position 10). Neuroblastomas mainly metastasize to lymph nodes (position 3), liver (position 4), bone and bone marrow (position 5), and also spread to central nervous system (position 1) and lungs (position 2) in infants.
Historically, NB is clinically classified into five different stages (1–4 and 4S) according to the International Neuroblastoma Staging System (INSS; Brodeur, 2003; Maris et al., 2007; Schor, 1999; van Noesel and Versteeg, 2004). Early stage NB tumors (i.e., stages 1, 2) do not metastasize to bone or bone marrow and are treatable with chemotherapeutic drugs and irradiation. Advanced-stage NB tumors (stages 3 and 4) are highly metastatic and usually respond positively to initial treatment. However, they often become resistant to chemotherapy and irradiation. A fifth stage of NB tumors (stage 4S) undergoes spontaneous regression with minimum treatment or even without medical intervention.
Ninety percent of the children with this disease are diagnosed before the age of 5 years and in those patients older than 1 year, 75% of the patients present with stage 3 or 4 metastatic diseases (Brodeur, 2003; Maris et al., 2007). Metastatic disease remains a major clinical challenge in the treatment of NB since greater than 50% of all NB patients are diagnosed with metastatic diseases (Maris, 2005; Maris et al., 2007). In contrast, infants with this disease tend to be at lower stages (stage 1, 2, and 4s) and to have a better prognosis (Brodeur, 2003; Maris, 2005; Maris et al., 2007; van Noesel and Versteeg, 2004).
In addition to classification by stage, NB tumors are also classified into three risk groups (low, intermediate, and high risk) according to age, MYCN status, and histology (Table 4.1). A new system for tumor staging has recently been implemented by the International Neuroblastoma Risk Group Staging System (INRGSS; Cohn et al., 2009; Monclair et al., 2009). This system, which is based on a combination clinical and imaging data, classifies patients as L1 (localized disease without imaging risk factors), L2 (localized disease with imaging risk factors), M (metastatic tumors), and Ms (metastatic disease with metastasis only in skin, liver, and/or bone marrow; Cohn et al., 2009; Monclair et al., 2009). NB tumors are also graded in terms of histology using the International Pathology Classification System (INPC) which is based on the Shimada histology grading system (Shimada et al., 1999). This system distinguishes good and poor prognosis tumors based on the degree of differentiation, the Schwannian stromal content, the mitotic-karyorrhexis index (MKI), and the age at diagnosis (Shimada et al., 1999). Unfavorable tumors tend to include undifferentiated tumors with high MKI of any age, poorly differentiated tumors or intermediate MKI in patients older than 18 months, and differentiated tumor or low MKI in patients 5 years old or older. In contrast all other cases, including those with gang-lioneuroma or tumor with regions of ganglioneuroma intermixed, have a good prognosis.
Table 4. 1.
Neuroblastoma risk stratification based on age, MYCN status and histology
| INSS stage | Age | MYCN status | Histology | Risk group | 3-year survival rate |
|---|---|---|---|---|---|
| 1 | 0–21 years | Any | Any | Low | >90% |
| 2 | 0–21 years | Non-Amp | Any | Low | 70–90% in this stage |
| 1–21 years | Amp | Favorable | Low | ||
| 1–21 years | Amp | Unfavorable | High | ||
| 3 | <1 year | Non-Amp | Any | Intermediate | 30–50% in this stage |
| 1–21 years | Non-Amp | Favorable | Intermediate | ||
| 0–21 years | Amp | Any | High | ||
| 0–21 years | Non-Amp | Unfavorable | High | ||
| 4 | <1 year | Non-Amp | Any | Intermediate | <30% in this stage |
| 0–21 years | Amp | Any | High | ||
| 4S | <1 year | Non-Amp | Favorable | Low | 50–80% in this stage |
| <1 year | Non-Amp | Unfavorable | Intermediate | ||
| <1 year | Amp | Any | High |
INSS, International Neuroblastoma Staging System; Amp, amplified; Non-Amp, not amplified.
In the remainder of this review, we will discuss the development of the peripheral neural crest with a focus on how developmental factors may contribute to NB tumorigenesis and metastasis, and highlight the current understanding of other genetic changes related to NB and their importance in NB diagnosis and treatment.
2. Neural Development and NB
2.1. Neural crest contribution to sympathetic ganglia and adrenal gland
The majority of NB tumors appear to arise from neural crest-derived cells in the abdomen adjacent to the aorta in the region of the kidney or in the medullary region of the adrenal gland (Brodeur, 2003; Maris et al., 2007). Thus, NB is a sympaticoadrenal lineage neural crest-derived tumor. The neural crest arises from the dorsal region of the closing neural tube beneath the ectoderm (Le Dourin and Kalcheim, 1999). This transient population of cells produces multipotential progenitor cells that give rise to the peripheral nervous system, the enteric nervous system, pigment cells, Schwann cells, adrenal medullary cells, and cells of the craniofacial skeleton (Le Dourin and Kalcheim, 1999). This process is regulated by both extrinsic and intrinsic factors. The Hedgehog and Wnt signaling pathways are especially crucial for proper neural crest development (Dupin et al., 2007; Le Dourin and Kalcheim, 1999; Morales et al., 2005). Lineage studies in the developing embryo have shown that neural crest cells within the trunk region generate multiple neural crest derivatives such as melanocytes, Schwann cells, glia, and neurons of the dorsal root ganglia (Fontaine-Perus et al., 1982; Lallier and Bronner-Fraser, 1988; Teillet et al., 1987; Weston, 1963). A subset of these trunk crest cells, commonly referred to as the sympathoadrenal lineage, contributes to the sympathetic ganglia and medullary region of the adrenal gland (Anderson and Axel, 1986; Anderson et al., 1991). This lineage of cells is thought to be the origin of NB (Brodeur, 2003; Maris et al., 2007). However, given the fact that NB can develop anywhere along the sympathetic axis, it is likely that NB can also arise from earlier crest derivatives, before development of the sympathethoadreanal lineage but after the initial fate specification (Brodeur, 2003; Maris et al., 2007). This could contribute to the heterogeneous histology and pathology of NB.
Neural crest cells develop in response to extracellular signals (Bronner-Fraser, 1993). The signals that induce formation of the crest (BMP/Shh) appear to be similar along the dorsal/ventral axis of the embryo. In contrast, different factors appear to confer cell fate along the anterior/posterior axis of the embryo. Heterotopic crest cell transplantation studies indicate that the positional identity of the cells is based on their location during development, rather than the characteristics of cells in original locations (Bronner-Fraser et al., 1980; Ruffins et al., 1998). Although NB can arise anywhere along the developing sympathetic axis, the majority of cases arise in the abdomen (65%), frequently in the adrenal medulla, where the sympathoadrenal lineage is specified; while others are found in the paraspinal sympathetic ganglia in places such as the neck (5%), chest (20%), and pelvis (5%) (Maris et al., 2007). Therefore, it is reasonable to postulate that the positional identity of the cell along the anterior posterior axis of the embryo, and factors that specify this region, likely contribute to the oncogenic potential of the crest derivatives in this region. As such, developmental signals fating the proper development, migratory pathways, and regulated cell death will be examined here in the context of NB.
2.2. Neural crest migratory pathways
Neural crest-derived cells are highly migratory. Shortly after induction the crest cells undergo an epithelial to mesenchymal transition (EMT). This EMT transition results in acquisition of enhanced migratory abilities and decreased requirements for intercellular contact which allows the neural crest cells to leave the dorsal neural tube (Le Dourin and Kalcheim, 1999). They then migrate either between the dermatome and epidermis in a dorsolateral pathway or delaminate from the neural tube via a ventrolateral pathway (Le Dourin and Kalcheim, 1999). Importantly, a similar EMT transition may also play a role in NB metastasis as described below in Section 6.1.
Neural crest migration pathways are determined by signal from the mesoderm which develops prior to the arrival of the crest cells. Trunk region neural crest cells either migrate ventrolaterally and remain in the sclerotome form the doral root ganglia or continue migrating to a more ventral position to form sympathetic ganglia. N-myc, an oncogene which plays a role in aggressive NB, appears to be required for the migration, survival and/or differentiation of cells that migrate to the dorsal aorta since N-myc deficient mouse embryos have decreased numbers of mature cells in the both the dorsal root ganglia and sympathetic ganglia (Charron et al., 1992; Sawai et al., 1993; Stanton et al., 1992). Hedgehog, Wnt, and additional growth factors all play a role in inducing N-myc expression in the neural crest (Grimmer and Weiss, 2006). More detailed information on the role of N-myc in NB is present below in Sections 3.2.1 and 5.2.3.
The most complete studies on neural crest cell migration and development have been carried out in birds. These studies revealed that the avian trunk crest, which is located between somites 6 and the tail, gives rise to sympathetic neurons and that a subset of these (between somites 18 and 24) contribute to the adrenal medulla (Le Dourin and Kalcheim, 1999). The crest cells that contribute to the adrenal medulla, the site where the majority of NB tumors are found, follow the ventrolateral migratory pathway. Upon arrival to the proper target tissue, the cells then undergo final differentiation, and regulated cell death.
2.3. Sympathoadrenal lineage development
The sympathoadrenal lineage is thought to derive from a common progenitor that aggregates at the dorsal aorta after migrating from the crest utilizing the ventral pathway (Anderson, 1993). During the migration to the dorsal aorta, the crest cells encounter signals from the somites, the ventral neural tube, and the notochord. Local signals from the dorsal aorta, such as BMPS, specify the future differentiation of the crest cell as either a catecholaminergic/adrenal chromaffin cell or sympathetic neuron (Ernsberger et al., 2005; Howard et al., 2000; Reissmann et al., 1996; Schneider et al., 1999; Shah et al., 1996). The sympathoadrenal lineage is specified by a tightly regulated set of transcription factors. Trunk crest cells can first be identified by the expression of Sox10 (Betters et al., 2010; Huber et al., 2008). As the crest cells migrate along the ventral pathway, they are exposed to BMP signals (e.g., BMP2, 4, and 7) that induce the expression of Mash1, a helix-loop-helix transcription factor expressed throughout the autonomic progeny (Huber, 2006). Shortly thereafter Phox2b expression occurs in sympathoadrenal lineage cells. Phox2b is required for the maintenance of Mash1 and recent evidence has shown that temporal difference in expression of these two factors may separate the sympathoadrenal lineage into separate sympathetic and adrenal lineages earlier in development than thought (Huber, 2006). Mash 1 induces the expression of Phox2a which is required for the production of the biosynthetic enzymes, dopamine beta-hydroxylase (DBH) and tyrosine hydroxylase (TH) in noradrenergic cells. Phox2b also appears to be important in the development of NB since PHOX2b mutations have been found in a subset of familial NB patients (see Section 3.1.1). Shortly after the migrating cells reach the dorsal aorta, they begin to acquire their respective sympathetic and adrenal cell fates and undergo a secondary migration to the presumptive prevertebral ganglia, the adrenal medulla, and secondary sympathetic ganglia where they complete their differentiation (Fig. 4.2; Huber, 2006; Le Dourin and Kalcheim, 1999; Morales et al., 2005).
Figure 4. 2.

General schema of the development of chromaffin cells and sympathetic ganglia. Cells at the dorsal region of the neural tube undergo EMT (red population), delaminate from the neural tube (orange), and migrate ventrally to the aorta (green) where they are commonly referred to as the sympathoadrenal progenitors (blue and purple). From the aortic region, the cells then migrate to the developing adrenal gland (AP) to become chromaffin cells or differentiate to become sympathetic ganglia (SG). As cells begin to differentiate as sympathetic ganglia they upregulate neural markers while chromaffin cells upregulate proteins found in the adrenal gland. Recent studies suggest that the chromaffin cell and sympathetic ganglia may come from divergent lineages rather than a common sympathoadrenal lineage. The question marks between the migrating crest and the sympathoadrenal progenitors address this possibility. A more detailed discussion on the temporal expression of the transcription factors in the sym-pathoadreanal lineage (including developmental stages) can be found in (Howard et al., 2000) and details about the distinct chromaffin and sympathetic lineages can be found in (Ernsberger et al., 2005). Additional of neurotrophins and their receptors can be found in (Straub et al., 2007). Transcription Factors are shown in bold and factors implicated in neuroblastoma have been underlined. Abbreviations: NT, Neural Tube; NC, notochord; A, Aorta; SG, sympathetic ganglia; AP, adrenal primordial.
Although the complete mechanisms for proper chromaffin cell development are unknown, evidence indicates a highly intrinsic program. In animals that lack an adrenal cortex, chromaffin cells migrate to the suprarenal region, downregulate neuronal markers and contain large chromaffin granules. However, the generation of proper numbers of chromaffin cells and the expression of secretogranin II and PNMT requires intact glucocorticoid signaling. The human adrenal gland is remodeled throughout fetal development, infancy, and into adulthood in a process that is greatly affected by perturbations in levels of IGF II, FGF, and epidermal growth factor (EGF) levels. Importantly, as discussed below several of these growth factors are involved in cellular proliferation and signaling in NB (Section 4.2).
3. Genetic Lesions in NB
3.1. Familial genetic lesions
Hereditary NB is both rare and heterogeneous, accounting for less than 5% of all NBs (Maris et al., 2002). In addition to the known hereditary mutations that are described below, hereditary NB predisposition loci have been mapped to chromosomes 16p12–13 and 4p16 indicating other familiar predisposition mutations may exist, but no genes have been shown to be inactivated or mutated in these regions, to date (Maris et al., 2002; Perri et al., 2002).
3.1.1. Phox2b
Germline mutations in the paired-like homeobox 2B (PHOX2b) gene on chromosome 4p13 are the first predisposition mutations identified in NB (Mosse et al., 2004; Trochet et al., 2004). As mentioned earlier, Phox2b, as well as to MASH1, are expressed early in the developing sympathoadrenal progenitors (Alenina et al., 2006; Nakagawara, 2004; Nakagawara and Ohira, 2004). Shortly after expression of MASH1 and Phox2b in the sympathoadrenal lineage, Hand2, Phox2a, and GATA2/3 appear (Alenina et al., 2006; Nakagawara, 2004; Nakagawara and Ohira, 2004). Phox2b has also been shown to be essential for the expression of the glial family ligand tyrosine kinase coreceptor RET (REarranged during Transfection) and for the specification of noradrenergic fates, particularly the biosynthetic enzymes TH and DBH (Alenina et al., 2006; Nakagawara, 2004; Nakagawara and Ohira, 2004).
NB patients with PHOX2b mutations also have familial disorders of the neural crest such as Hirschsprung’s disease (HSCR) and congenital hypoventilation syndrome (CCHS; Mosse et al., 2004; Trochet et al., 2004). It is unclear that the mutations in PHOX2b found in familiar NB result in gain or loss of function, although many PHOX2b mutations stabilize the Phox2b protein and decrease or eliminate the ability of Phox2b to transactivate the DBH promoter (Raabe et al., 2008; Trochet et al., 2005). The findings that Phox2b is necessary for the differentiation of autonomic neurons and overexpression of Phox2b inhibits proliferation in neuron progenitors and cell lines suggests Phox2b is a tumor suppressor (Raabe et al., 2008; Trochet et al., 2005, 2009). However, the absence of tumors with loss of heterozygosity (LOH) or mutation in second allele suggests gain-of-function, dominant-negative effect, or haploinsufficiency (Benailly et al., 2003; Bourdeaut et al., 2005).
3.1.2. Anaplastic lymphoma kinase (ALK)
ALK is a member of receptor tyrosine kinases (RTK) and was first identified as a part of the fusion gene nucleophosmin (NMP)–ALK in anaplastic large cell lymphoma via chromosome translocation of t(2;5)(p23;q25) (Morris et al., 1994, 1997). ALK is thought to play a role in the normal development of the central and peripheral nervous system since ALK mRNA is expressed throughout the nervous system in mouse and rat, but is not present in normal hematopoietic cells (Degoutin et al., 2009; Hurley et al., 2006; Iwahara et al., 1997; Morris et al., 1997; Vernersson et al., 2006). More detailed studies in chick embryos have shown a similar pattern of ALK expression in the developing central nervous system in which ALK localizes primarily to the spinal motor neuron, the sympathetic ganglia, and the dorsal root ganglia. In mice, expression of ALK in the nervous system decreases after birth but is maintained at low levels in adults. Similar patterns of expression are observed in humans although additional ALK transcripts of differing size, most likely due to alternative splicing, have been observed in colon, prostrate, testis, small intestine, and brain of adults (Iwahara et al., 1997; Palmer et al., 2009).
Full-length ALK protein is comprised of an extracellular region and an intracellular region containing a RTK domain, linked by a transmembrane (TM)-spanning segment, whereas the NMP–ALK fusion protein generated as a result of the t(2;5)(p23;125) translocation contains the N-terminal of NMP and C-terminal kinase domain of ALK. Translocation of the ALK gene is also found in other tumors, such as inflammatory myofibroblastic tumor (IMT), and nonsmall cell lung cancer (NSCLC), but not in NB (Palmer et al., 2009). Overexpression of wild-type ALK has also been observed in thyroid carcinoma, breast cancer, NB, melanoma, small cell lung carcinoma, glioblastoma, astrocytoma, retinoblastoma, Ewing sarcoma, and rhabdomyosarcomas NB (Cheng and Ott, 2010; Mosse et al., 2009; Palmer et al., 2009).
During 2008, at least five papers described ALK point mutations in 8–12% of all NB patient (both hereditary and sporadic) and some NB cell lines as well (Caren et al., 2008; Chen et al., 2008; George et al., 2008; Janoueix-Lerosey et al., 2008; Mosse et al., 2008). With one exception, all the point mutations identified to date occur in the kinase domain and result in the constitutive activation of ALK. Two of these activating ALK mutants were able to transform NIH3T3 fibroblasts and induce tumor formation in nude mice (Chen et al., 2008). In addition, knockdown of ALK or small molecular ALK inhibitors could reduce cell proliferation and induce apoptosis (George et al., 2008; Janoueix-Lerosey et al., 2008). Amplification of the ALK gene and/or overexpression of the ALK protein is seen in as many as 77% of all NB tumors (Passoni et al., 2009) suggesting that overexpression of the ALK protein may also contribute to NB.
The downstream effects of ALK in NB need to be defined. Current data suggest that ALK may function through the Shc and MAP kinase pathways (Motegi et al., 2004; Osajima-Hakomori et al., 2005; Souttou et al., 2001). More recent studies also suggest that activation of ALK enhances RAP1 activity via interaction with C3G, a Crk-binding protein and Crk-like protein (CRKL), and that this complex contributes to NB tumor cell growth and neurite outgrowth (Schonherr et al., 2010).
3.2. Chromosome gain and oncogene activation
Many genetic abnormalities have been identified in nonfamilial NB tumors, including amplification of the MYCN proto-oncogene (25–33% of patients) and consistent areas of chromosomal deletion and rearrangement that result in loss of 1p36 (25–35%), 11q23 (35–45%), and 14q23 (16–27%), as well as unbalanced gain of 17q22 (~50%) (Table 4.2; Brodeur, 2003; Maris et al., 2007; Schor, 1999). In contrast, known tumor suppressor genes (TSGs) such as p16INK4a, pRb, p53, and p14ARF are not frequently deleted or mutated in NB, although the nuclear localization of the p16INK4a and p53 proteins has been reported to be altered in some tumor cell lines (Brodeur, 2003; Maris et al., 2007; Schor, 1999; Teitz et al., 2001; van Noesel and Versteeg, 2004). Many of these abnormalities are powerful prognostic markers and are highly related to clinical outcome. For example, amplification of MYCN in NB patients is correlated with chromosome 1p36 LOH. NB tumors which harbor 1p36 LOH and MYCN amplification are usually advanced-stage (stages 3 and 4) aggressive tumors that are frequently metastatic and generally respond poorly to chemotherapy/irradiation (Brodeur, 2003; Maris et al., 2007). In the recent years, clinical trials are increasingly based on the tumor genetic characteristics.
Table 4. 2.
Frequent chromosomal region abnormalities in neuroblastoma
| Chromosomal region | Status | Frequency | Relation with MYCN amplification | Involved genes | Clinical group |
|---|---|---|---|---|---|
| 1p36 | Loss | 25–35% | Correlation | CHD5, miR-34a, KIF1Bβ | Unfavorable |
| 11q23 | Loss | 35–45% | Inversed correlation | TSLC1/IGSF4 | Unfavorable |
| 14q23 | Loss | 16–27% | Inversed correlation | All groups | |
| 17q22 | Gain | ~50% | Correlation | Survivin, NM23A | Unfavorable |
3.2.1. Amplification of MYCN and the 2p24 locus
In 1983, Schwab et al. found that a novel myc homolog gene was amplified in several NB cell lines and one NB tumor (Schwab et al., 1983). Later, several papers termed this gene as MYCN based on homology to c-myc and expression pattern in the developing nervous system, and identified its location at chromosome 2p24 (Kohl et al., 1983; Schwab et al., 1984). Additional studies have shown that N-myc protein is a nuclear phospho-protein that is a member of the myc family of helix-loop-helix transcription factors (Pelengaris et al., 2002). Amplification of the MYCN gene in patient tumors ranges from 10-fold to more than 500-fold, although the majority of tumors exhibit 50- to 100-fold MYCN gene amplification levels. The amplified DNA typically contains a large region of chromosome 2 ranging from 100 kb to 1 Mb which includes the entire MYCN gene and varying amounts of adjacent DNA. Although other genes may be coamplified with MYCN, MYCN is only consistent amplified gene from this region (Reiter and Brodeur, 1996, 1998).
MYCN amplification is rarely observed on chromosome 2p24 in primary tumors but is found to be at homogeneously staining regions (HSRs) on different chromosomes or, more frequently, as double minutes (DMs; which are small fragments of extrachromosomal DNA; Emanuel et al., 1985; Schwab et al., 1984). During cell culture, the amplification unit frequently integrates into chromosomes to become HSRs. The reason for the differences in the location of the amplicon in primary tumors and cultured cells remains unclear.
Amplification of MYCN is highly associated with aggressive NB tumors and poor outcome. Although the entire role of MYCN in NB is still being uncovered, amplification of the MYCN gene is usually accompanied by overexpression of the N-myc protein. Studies on N-myc regulation suggest that the transcription factor and signaling pathways responsible for the upregulation of N-myc are dependent on cell type (Hurlin, 2005). These factors include IL-7 and Pax-5, NF-κB in pre-B cells, and insulin-like growth factors I and II (IGFI and IGFII) in NB cells (Strieder and Lutz, 2003). In contrast, N-myc transcription is repressed by retinoic acid (RA) in association with E2F binding, nerve growth factor (NGF) binding to TrkA receptor, the iron chelator deferoxamine mesylate, and transforming growth factor-beta 1 (TGF-β1; Strieder and Lutz, 2003; Wada et al., 1992).
Myc proteins form heterodimers with the Max protein. These heterodimers bind to E-box elements (CACGTG) to activate transcription. However, Myc–Max dimers can also associate with other transcription factors such as Miz-1 and Smad and bind to Inr (initiator) element to repress transcription. Max can also form homodimers or heterodimers with Mad to compete or suppress Myc–Max binding (Pelengaris et al., 2002; Thompson, 1998; Fig. 4.3). The targets of Myc–Max are involved in various cellular processes, including cell growth, proliferation, loss of differentiation, and apoptosis (Adhikary and Eilers, 2005; Pelengaris et al., 2002; Thompson, 1998), and include proteins such as MASH1 and important molecules in the normal development of sympathocoadrenal lineage cells, such as the multidrug resistance protein 1 (MRP1), α-prothymosin, telomerase, Id2, MCM7; leukemia inhibitory factor, activin A, Pax-3, and MDM2 (Breit and Schwab, 1989; Haber et al., 1999; Harris et al., 2002; Hatzi et al., 2002; Lasorella et al., 2002; Mac et al., 2000; Pelengaris et al., 2002; Shohet et al., 2002; Slack et al., 2005). Many other putative N-myc targets with E-boxes in or near the promoter have also been identified although studies are still ongoing to determine which E-boxes actually bind Myc. Overexpression of N-myc is also reported to influence the expression of IL-6, NDRG1, MHC class I genes, and integrins (Chambery and Mohseni-Zadeh, 1999; Lutz et al., 1996; Mac et al., 2000), although the mechanisms responsible for these effects are unknown.
Figure 4. 3.

The structure of N-myc protein and transcription regulation by N-myc. (A) The structure of N-myc protein. The N-terminal transactivation domain (TAD) contains two conserved Myc box I and II (MBI and MBII), which are essential for DNA binding. The C-terminal domain (CTD) harbors basic region (BR), helix-loop-helix (HLH) motif, and leucine zipper (LZ) for dimerization with Max. There is a nuclear localization signal (NLS) before CTD. (B) The model for the transcription regulation by N-myc. Myc–Max heterodimer may bind to E-box element (CACGTG) to activate transcription, however, Myc–Max dimer can associate with other transcription factors such as Miz-1, Smad, and bind to Inr (initiator, weak consensus) element to repress transcription. Max can also form homodimers or heterodimers with Mad to compete or suppress Myc–Max binding to E-box.
The transgenic mouse model demonstrates that MYCN overexpression is an initial event in NB tumorigenesis. In this model, overexpression of the human MYCN is driven by the rat TH promoter, which is expressed in migrating cells of the neural crest early in development (Banerjee et al., 1992), causes the formation of NB tumors in transgenic mice (Weiss et al., 1997). These tumors recapitulate most of the histological and pathological aspects of the human disease, including tumor localization, positive staining for neuronal markers, and gains and losses of chromosomes in regions syntenic with those observed in human NB (Weiss et al., 1997). However, other factors are also likely to be involved in the early stages of tumor formation since amplification of the MYCN oncogene occurs in only about one-third of NBs. In addition, the tumors in these transgenic mice rarely exhibit significant metastasis despite the presence of high levels of N-myc protein suggesting that other genetic alterations and/or epigenetic changes are needed for tumor formation and metastasis. These and other studies suggest that N-myc regulates neural progenitor cell proliferation, nuclear size and differentiation (Knoepfler et al., 2002). Importantly, studies using chick/quail chimera reveal that overexpression of N-myc in the early neural crest induces premature ventral migration of neural crest cells and promotes the differentiation of these cells (Wakamatsu et al., 1997). In addition, other studies have shown that high level N-myc mRNA or protein expression in NB cells accelerate cell cycle progression (Lutz et al., 1996), and that overexpression of N-myc in postmitotic sympathetic neurons causes quiescent cells to reenter the cell cycle and enhances the survival of these cells upon NGF withdrawal (Wartiovaara et al., 2002).
The high level expression of N-myc in NB is also consistent with the hypothesis that NB arises during development. N-myc is normally expressed at the beginning of the preimplantation stage of development. As the embryo develops, N-myc expression is observed in the central nervous system and neural crest. By embryonic day 9.5, relatively high levels of N-myc are observed in the fetal brain, kidney, and in the neural crest and early stage migrating neural crest cells (Zimmerman et al., 1986). During later stages of neural crest migration N-myc expression is only observed in cells undergoing neuronal migration. Even in these cells, N-myc expression is gradually downregulated as cells differentiate and become quiescent (Lee et al., 1984; Zimmerman et al., 1986). These data are consistent with studies using N-myc knockout mice that demonstrate that the loss of N-myc results in embryonic death at day 10.5 of gestation due to defects in the nervous system, limb, heart, liver, lung, gut, mesonephros, and genital ridge (Sawai et al., 1991).
3.2.2. Gain of 17q
Gain of chromosome 17q was first identified by G-banded cytogenetic analyses in early 1980s (Gilbert et al., 1984). However, researchers paid little attention to these observations since their interests focused on the genetic abnormalities of MYCN amplification and 1p LOH at that time. In the middle 1990s, NB scientists realized the importance of 17q abnormalities since FISH technology indicated that translocation of this chromosome arm occurs in about half of the NB primary tumors. The translocation results in unbalanced gain of one to three copies of 17q (Brodeur, 2003; Maris et al., 2007; Schor, 1999). Although the breakpoint of 17q varies, the frequent gain of regions from 17q22 suggests that increased dosage of one or more genes from this region may confer a selective survival advantage for NB tumor cells. It is estimated that as much as 20 Mb of the 17q chromosome fragment, which could include more than 200 genes, can be translocated in NB tumors. Therefore, it is difficult to identify the genes responsible for the selective advantages (Brodeur, 2003; Maris et al., 2007; Schor, 1999). Several genes in this area are considered good candidate oncogenes or tumor suppressors based on correlations between expression levels and unbalanced gain of 17q. These include survivin, NM23A, and PPM1D (Godfried et al., 2002; Islam et al., 2000; Saito-Ohara et al., 2003). Notably, survivin is a member of apoptosis inhibiting protein family and is frequently overexpressed in many tumor types, including NB where expression has been correlated with late stage disease and poor prognosis; whereas NM23A is metastasis-related gene and PPM1D a phosphatase that suppresses stress induced apoptosis (Almgren et al., 2004; Bown et al., 1999).
Unbalanced gain of 17q correlates with other chromosomal deletions. The most frequent deletion site is the short arm of chromosome 1, followed by 11q. At least 30 translocation sites on 20 different chromosomes have been identified in various patient samples and cell lines (Bown et al., 1999; Lastowska et al., 1997; Meddeb et al., 1996). Nevertheless, NB tumors harboring unbalanced gain of 17q exhibit a more aggressive phenotype and a poorer prognosis than those without this abnormality.
3.2.3. Amplification and chromosome gain of other loci
In addition to the amplification of MYCN gene, several other regions of gene amplifications have been identified in small groups of NB cases. These include amplification of the MDM2 gene at 12q13, the DDX1 gene at 2p24, the MYCL gene at 1p32, and unidentified DNA from chromosome 2p22 and 2p13 (Corvi et al., 1995a, b; Jinbo et al., 1989; Van et al., 1995). The MDM2 gene was initially found to be amplified in three NB cell lines and one primary tumor (Corvi et al., 1995a). Like the MYCN gene amplification, the MDM2 amplification unit first developed within DMs and then integrates into a different chromosome to form HSRs (Corvi et al., 1995b). The DDX1 gene, which encodes a RNA helicase, was found to be coamplified with MYCN in 4/6 NB cell lines and 6/16 tumors with MYCN amplification; however, DDX1 amplification was not found without MYCN amplification (George et al., 1996). One paper also indicated that MYCL gene is coamplified with MYCN in NB cell lines. MYCL, another member of myc gene family, is frequently overexpressed in small cell lung carcinoma (Jinbo et al., 1989). In addition to gain of 17q, other chromosome gains have been identified on 1q, 4q, 5q, 6p, 7q, 18q using comparative genomic hybridization (CGH) methodology, although their biological and clinical significance remain unclear (Hirai et al., 1999; Lastowska et al., 1997; Meltzer et al., 1996; Takita et al., 2000; Vandesompele et al., 1998).
3.3. Chromosome loss and tumor supressor genes (TSGs)
In addition to mutation, gene amplification and increased chromosome copy number, NB tumors also experience loss of genetic material and deletion of putative TSGs.
3.3.1. LOH of chromosome 1p and CHD5, miR-34, KIF1Bβ
Loss of the short arm of chromosome 1 occurs in about 25–35% NB tumors. 1p LOH is correlated with amplification of MYCN in NB patients. As mentioned above, loss of 1p correlates with and may be a result of unbalanced gain of 17q, however, the exact mechanism that is responsible for these two events is not clear. The importance of 1p LOH is highlighted by studies showing that transferring chromosome 1p material into human NB cells in vitro led to differentiation and suppression of tumorigenicity (Bader et al., 1991). In search of potential TSGs that reside in this region, extensive efforts have been made to identify the smallest region of overlap (SRO) that would include the TSG candidate. These studies delineate the 1p36.1–36.3 as the SRO. Several candidate genes, such as p73, reside in this area. However, further studies failed to demonstrate a correlation between p73 loss and NB development (Ichimiya et al., 1999).
Whereas patients with 1p36 abnormalities without MYCN amplification have been identified, the reverse situation virtually never occurs suggesting either that 1p36 LOH provides a permissive environment for MYCN amplification or that tumors with these two associated genetic defects have a high degree of genomic instability (Brodeur, 2003). NB tumors with 1p36 LOH and MYCN amplification are usually aggressive tumors that are frequently metastatic and generally resistant to chemotherapy/irradiation. Although the chromosomal regions described above are known to be important in NB, the TSGs that reside within these regions have not been definitively identified.
Recent studies have identified three new putative tumor suppressors on chromosome 1p36: the chromodomain helicase DNA-binding domain 5 (CHD5), microRNA-34a (mir-34a), and the kinesin superfamily protein 1B beta (KIF1Bβ; Bagchi et al., 2007; Munirajan et al., 2008; Welch et al., 2007). All three of these proteins have affects on cell growth. For example, Bagchi et al. demonstrated that the effects of CHD5 on cell growth were dependent on p53 and that CDH5 positively regulates p53 via effects on p19ARF expression. Thus, overexpression of CHD5 results in enhanced apoptosis and senescence, increased p53 and p19ARF levels, and sequestration of MDM2, the negative regulator of p53, by p19ARF. Conversely, cells lacking CHD5 exhibit decreased p16 and p19ARF expression. This decrease in p19ARF was mirrored by a decrease in p53 levels and enhanced cellular proliferation. Thus, CHD5 appears to function as a tumor suppressor that controls proliferation, apoptosis, and senescence via effects on the p19ARF/p53 pathway. These effects are most likely due to changes in the accessibility of the p16/p19ARF gene locus resulting from the chromatin remodeling function of CHD5 (Bagchi et al., 2007).
In addition to CHD5, Chen et al. found that mir-34a was expressed at very low levels in unfavorable primary tumors and NB cell lines. This group further showed that introduction of this microRNA (miRNA) into cell lines resulted in decreased cell proliferation and caspase-dependent apoptosis. They also found that mir-34a directly targeted the E2F3 mRNA and repressed its expression (Chen and Stallings, 2007; Welch et al., 2007). E2F3 is a transcription factor that induces the expression of many genes with roles in cellular proliferation.
Finally, overexpression of KIF1Bβ induced cell death while decreased KIF1Bβ levels correlated with cell proliferation and enhanced tumor formation in nude mice, suggesting that KIF1Bβ is also a potential TSG candidate (Munirajan et al., 2008). Kaelin’s group also found that KIF1Bβ is a downstream target of prolyl hydroxylase EglN3 and induced apoptosis in neuronal progenitor cells or NB cells when NGF is limited. In addition, they identified missense mutations of KIF1Bβ in inherited NBs and pheochromocytomas (Schlisio et al., 2008), supporting the hypothesis that KIF1Bβ is a potential TSG candidate.
3.3.2. Loss of 11q and TSLC1
Loss of the long arm of chromosome 11 has been identified in 35–45% NB primary tumors with a single copy MYCN gene. Two large patient studies analyzed 295 NB primary tumors. These studies found loss of 11q in 44% cases, and common regions of LOH located at 11q23, suggesting there are TSGs residing in this area (Guo et al., 1999; Maris and Matthay, 1999). Loss of 11q correlated with adverse clinical features including late stage disease, older age of disease onset and unfavorable histology, although it is strikingly inversely correlated with MYNC amplification and 1p loss. Therefore, 11q loss is a useful and important marker in determining the clinical prognosis for those advanced-stage tumors without MYCN amplification. Transfer of chromosome 11 induced differentiation in NB cell lines supporting the importance of loss of 11q in tumorigenesis (Bader et al., 1991).
One putative tumor suppressor, the IGSF4 (immunoglobulin superfamily 4) gene, was first localized to the common 11q23 LOH region in 1999 (Gomyo et al., 1999). This gene which is also known as TSLC1/CADM1 (Tumor suppressor in lung cancer 1/cell adhesion molecule 1), is considered as a potential TSG for lung cancers. A recent CGH study which examined 236 primary tumor samples found TSLC1 LOH locus in 35% tumors. Importantly, the level of TSLC1 expression correlated with tumor stage, histological classification, MYCN and TrkA expression levels. Reduced expression of TSLC1 was found in unfavorable tumors. Further, introduction of TSLC1 decreased cell proliferation in NB cell lines (Ando et al., 2008). These results indicated that TSLC1 is a good NB tumor suppressor candidate. Interestingly, a recent study indicates that expression of both KIF1Bβ and TSLC1 is controlled by the polycomb protein Bmi1, whose expression is regulated by N-myc (Ochiai et al., 2010).
3.3.3. LOH of 14q
Loss of the long arm of chromosome 14 is also commonly found in NB primary tumors (~16–27% of the patients). LOH on chromosome 14q was first identified in 1989 using a polymorphic DNA marker which detected allelic deletion at specific 14q23 loci (Suzuki et al., 1989). LOH analysis of 14q in a large number of primary tumors using 11 polymorphic DNA markers found 14q LOH in 83 of 372 tumors (22%) (Thompson et al., 2001). 14q LOH was highly correlated with 11q loss and had an inverse relationship with 1p loss and MYCN amplification (Thompson et al., 2001). However, LOH for 14q was present in tumors from all clinical stages, suggesting this abnormality may be a universal early event during tumor development.
In addition, to the genetic changes described above, there have also been reports of LOH and/or allelic imbalance at chromosome arms 2q, 3p, 4p, 9p, and 19q (Caron, 1996; Ejeskar et al., 1998; Marshall et al., 1997; Mora et al., 2001; Takita et al., 2001), however, the significance of these genetic changes is not clear.
4. The Role of Neurotrophins and Growth Factors in the Development of the Sympathetic Nervous System and in NB
As the neural crest cells migrate to the aorta but prior to reaching the adrenal medulla they begin to express TH which in turn controls the expression of other enzymes needed for catecholamine biosynthesis as described above in Section 2.3. Since NB appears to arise from cells that are transformed at various times during this migration, the majority of NB tumors secrete catecholamines. Indeed, the presence of high levels of cate-cholamines in patient urine samples is used as one of the diagnostic criteria for the disease (LaBrosse et al., 1976). Based on this data, Sawada began to screen the urine of 6-month-old infants for increased catecholamine metabolites from 1984 and found that the incidence of in situ NB was much higher than the number of sporadic cases that had been observed previously (Sawada, 1992). These data agree with a previous hypothesis of Beckwith and Perrin who postulated that during the development of sympathetic neurons the incidence of in situ NB is higher than the incidence of sporadic cases (Beckwith and Perrin, 1963). Most of these in situ NBs spontaneously regress as the child ages (Brodeur, 2003; Maris, 2005; van Noesel and Versteeg, 2004), suggesting they are resolved using normal developmental programs. Developmental studies and studies from knockout mice suggest the TrkA is crucial for the development of many sympathetic lineage cell types (Brodeur et al., 2009). This is consistent with data indicating the NGF is required for the differentiation and survival of many sympathetic lineage cells (Nakagawara, 2004; Nakagawara and Ohira, 2004). In addition to NGF and TRKs several other growth factors also play roles in the development of the sympathoadrenal lineage cells. These include EGF which is expressed in neural crest cells and is thought to contribute to the formation of neuron and melanocytes at later point during neural crest migration (Erickson and Turley, 1987), vascular endothelial growth factor (VEGF) which is expressed by surrounding cells (McLennan et al., 2010), and IGFI and IGFII which is expressed in the neural crest, the dorsal root, the sympathetic ganglia, and the adrenal medulla (Coppola et al., 2009; D’Ercole et al., 1996).
4.1. Neurotrophin receptors
Although the steps in the transformation of sympathetic neuroblasts to NB cells is not clear, extensive evidence suggests that neurotrophin receptors are involved in NB tumorigenesis and in development of the nervous system and sympaticoadrenal lineage cells. The neurotrophin receptors TrkA, TrkB, and TrkC, are encoded by the NTRK1, 2, 3 genes, respectively. Upon binding to their ligands, NGF, brain-derived neurotrophic factor (BDNF), neurotro-phin-3, respectively, these receptors regulate proliferation, survival, and differentiation in normal neuronal cells (Brodeur et al., 2009; Maris and Matthay, 1999; Maris et al., 2007; Straub et al., 2007). These receptors all associate with p75, a low affinity receptor that may enhance the binding of ligand to the Trk proteins or alter the function of the Trk receptors (Brodeur et al., 2009; Maris and Matthay, 1999; Maris et al., 2007; Straub et al., 2007). High levels of TrkA, in association with very low/no N-myc expression, are detected in favorable NB tumors which often spontaneous regress (Nakagawara, 1993; Nakagawara et al., 1992). These favorable NB tumors cells usually express a small amount of NGF as do some of the surrounding cells (Nakagawara, 1993). Cells that express the most NGF are thought to undergo differentiation, while those that express less NGF undergo apoptosis (Nakagawara, 1993). TrkA expression is dramatically decreased in MYCN amplified NB tumors (Nakagawara et al., 1992). Thus, the TrkA/NGF pathway could play an important role in determining the ability of these favorable NB tumors to differentiate or to regress in response to the microenvironment. While in general TrkA expression is correlated with a good prognosis, a novel TrkA splice variant has been found in advanced-stage tumors with adverse biological features (Tacconelli et al., 2004). This TrkA isoform is constitutively active and promotes cell survival and angiogenesis independently of NGF expression. NGF signaling may also be linked to IGFII expression since a study in which SHSY5Y cells were transfected with TrkA found increased expression of IGFII in response to NGF binding to the transfected receptor (Kim et al., 1999).
In contrast, TrkB is preferentially expressed in clinically unfavorable NB tumors and expression of TrkB strongly correlates with MYCN amplification (Nakagawara et al., 1994). The TrkB ligand, BDNF, is also highly expressed in these tumors. Coexpression of ligand and receptor may form an autocrine loop to enhance survival, metastasis, and drug resistance (Douma et al., 2004; Nakagawara, 1994). Interestingly, a truncated form of TrkB which lacks tyrosine kinase activity is expressed in some favorable NB tumors (Ho et al., 2002).
Finally, TrkC is commonly expressed in favorable NB tumors. These tumors also express very limited amount of the TrkC ligand neurotrophin-3 and coexpress TrkA (Svensson et al., 1997; Yamashiro et al., 1996).
4.2. Other growth factors and growth factor receptors
In addition to NGF and BDNF several other growth factors also play roles in the development of the sympathoadrenal lineage cells and NB tumorigenesis. These include EGF, VEGF, and insulin-like IGFI and IGFII. EGF receptor 1 (EGFR1) expression is found on both primary NB tumors and tumor-derived cell lines (Ho et al., 2005). Binding of EGF to EGFR1 causes receptor autophosphorylation and increases proliferation via effects on the MAPK and PI3K/AKT pathways (Henson and Gibson, 2006). Exogenous VEGF also stimulates the PI3K/AKT pathway and increases expression of survivin, an antiapoptotic gene in NB cells (Beierle et al., 2005). Endocrine-derived VEGF has also been shown to play a role in the proliferation and differentiation of neural crest cells during development and to promote NB cell growth. IGF1 receptors (IGF1Rs) are expressed in the majority of NB primary tumors (Martin et al., 1992). This receptor binds both IGFI and IGFII suggesting that these growth factors are important for NB tumorigenesis. Expression of IGF1R activates the PI3K/AKT and MAPK pathways and enhances cellular proliferation, cell survival, migration, and invasion, and induces chemotherapeutic resistance and reduced response to other apoptotic stimuli (Valentinis and Baserga, 2001). IGFII has been reported to be upregulated upon TRKA activation and downregulated upon TRKA overexpression suggesting potential feedback loops between these proteins (Kim et al., 1999). In addition, IGF1R is transcriptionally activated by N-myc and in turn high IGF1R levels induce N-myc protein and mRNA expression suggesting the presence of an amplification loop that enhances the ability of these proteins to promote tumorigenesis (Chambery and Mohseni-Zadeh, 1999). Inhibition of IGF1R signaling has also been shown to increase N-myc phosphorylation by GSK-3β which inactivates N-myc and enhances N-myc turnover resulting in decreased cell growth both in culture and in mice model systems (Coulter et al., 2009). IGF1R has also been shown to enhance NB metastasis to bone most likely due to its ability to enhance migration and invasion and to the presence of IGF ligand in bone marrow (van Golen et al., 2006). In addition, IGF increases cellular survival under hypoxic conditions via increased expression of hypoxia-inducing factor 1α (HIF1α) and VEGF (Treins et al., 2005).
5. Programmed Cell Death (PCD; Apoptosis) in Development of NB
5.1. The role of cell death in development
Another important process during development of the peripheral nervous system is PCD, also known as apoptosis. This process is used during development to eliminate redundant cells, control cell number, and for remodeling and repair. Cell death also occurs in the developing peripheral nervous system in response to loss of essential growth factors and cytokines (De Zio et al., 2005). Neural crest development therefore is a balance between proliferation, cell death, migration, and differentiation. Errors in any of these processes may leave the cell more prone to transformation and potentially increase tumorigenesis. This is especially true of pediatric cancers, such as NB, since these cancers develop during normal development. Indeed, stage 4S NB spontaneously regresses with little to no intervention (Blaschke et al., 1998; De Zio et al., 2005; Johnsen et al., 2009). Tumors of this stage are present in younger children and the prognosis is good. It is unknown why these tumors suddenly die or cease to grow, although it is thought that apoptosis is likely to be involved in the disappearance of these tumors (Brodeur, 2003; Maris et al., 2007; Schor, 1999). Several hypotheses have been suggested to explain the regression of these tumors: (1) these tumors are dependent on growth factors or other proteins that are present in low levels and that once a tumor reaches a certain size the factors are depleted and the tumor undergoes apoptosis, (2) as the child develops, the tumor is recognized by the immune system and destroyed, and/or (3) that the cells become responsive to the environmental cues and developmental regulatory apoptotic pathways as they mature which triggers apoptosis. Though any of these possibilities may be the case, there is sparse literature investigating apoptotic pathways in the developing trunk crest as well as the peripheral nervous system. Although it is known that crest cells from rhombomeres 3 and 5 in the chicken and mammals undergo PCD (Kulesa et al., 2004; Morales et al., 2005) and that expression of Snail in the early migratory crest cells protects the migratory crest cells form apoptosis (Vega et al., 2004), a more detailed investigation regarding specific cell death programs in crest cells contributing to the developing sympathoadrenal lineage requires more characterization.
5.2. The role of apoptosis genes in NB
As mentioned above, apoptosis or PCD in multicellular organisms is a tightly regulated process required for normal growth, development, and cellular specialization (Danial and Korsmeyer, 2004; Hengartner, 2000). Defective expression of proteins and aberrant function of constituents in the apoptotic cascade have been implicated in oncogenesis, tumor progression, and treatment resistance (Fesik, 2005; Kaufmann and Vaux, 2003; Reed and Tomaselli, 2000). There are two major apoptotic pathways in mammalian cells: the death receptor (or extrinsic) pathway and the mitochondrial (or intrinsic) pathway. Caspases (cysteine proteases) are the central components of the apoptotic machinery. At least 14 distinct caspases have been identified in mammals and are grouped into three categories (nonapoptotic, initiators, and effectors; Danial and Korsmeyer, 2004; Shi, 2004). Importantly for this review, alterations in caspase-8 expression have been observed in NB.
5.2.1. Caspase-8
Human caspase-8 (also known as FLICE) is encoded by the CASP8 gene which is located on chromosome 2q33–2q34 (Grenet et al., 1999). Caspase-8 exists as a monomer in the cell and requires dimerization or oligomerization for its activation (Boatright and Salvesen, 2003; Salvesen and Abrams, 2004). Subsequent cleavage events, although not essential for activity, further stabilize the activated protein (Boatright and Salvesen, 2003; Boatright et al., 2003; Salvesen and Abrams, 2004). Activated caspase-8 is an important initiator caspase in the death receptor-mediated apoptotic pathway. The death receptor pathway is triggered by members of the death receptor superfamily (FasR, TNFRI, DR5, etc.; Hengartner, 2000). Binding of the ligand (e.g., FasL) to its receptor (FasR) induces the formation of the death-inducing signaling complex (DISC) containing death receptors, adaptor proteins and procaspase-8 (Danial and Korsmeyer, 2004; Hengartner, 2000). Procaspase-8 then dimerizes, resulting in activation and cleavage (Danial and Korsmeyer, 2004; Hengartner, 2000; Shi, 2004). The active enzyme subsequently cleaves downstream effector caspases, resulting in their activation and leading to apoptosis (Danial and Korsmeyer, 2004; Hengartner, 2000; Shi, 2004). Active caspase-8 can also cleave Bid, which is a Bcl2 family member. Cleaved Bid, termed t-Bid, translocates to the mitochondria and promotes cytochrome c release, thereby activating the mitochondrial pathway (Danial and Korsmeyer, 2004; Hengartner, 2000; Shi, 2004).
Our laboratory first found that caspase-8 is deleted or more commonly, silenced in most NB cell lines and patient tumor samples. We further identified a region within the CASP8 gene which is methylated (Teitz et al., 2000). This finding has subsequently been verified by several groups (Fulda and Debatin, 2006; Fulda et al., 2001; Teitz et al., 2000; Yang et al., 2007). Several additional genes with roles in tumorigenesis are also hyper-methylated in NB including the PCDHB gene family, BLU, TSP-1, RASSFIA, TIG1, HIN-1, DcR1, DcR2, DcR4, etc. (Abe et al., 2005; Astuti et al., 2001; van Noesel et al., 2002; Yang et al., 2003, 2007). Among these, CASP8, PCDHB, BLU, DcR2, and HIN-1 are found to be associated with high-risk factors and poor outcome in NB patients (Abe et al., 2007; Yang et al., 2007). Methylation of this region of the CASP8 gene has been correlated with decreased caspase-8 protein expression in many (Fulda et al., 2001; Hopkins-Donaldson et al., 2000; Teitz et al., 2000), but not all studies (Banelli et al., 2002; van Noesel et al., 2002). However, even in studies where methylation has not been correlated with expression, decreased caspase-8 protein levels are observed in 50–70% of NB patient tumors. Since our initial discovery in NB, loss of caspase-8 expression has been found in many other tumors, including peripheral neuroectodermal tumors, medulloblastoma, glioblastoma multiforme, rhabdomyosarcoma, retinoblastoma, small cell lung carcinoma, and Wilms tumors (Gonzalez-Gomez et al., 2004; Harada et al., 2002; Hopkins-Donaldson et al., 2003). Caspase-8 mutations in adult gastric and colorectal tumors have also been reported (Kim et al., 2003; Soung et al., 2005). Finally, loss of caspase-8 expression has been correlated with poor prognosis in medulloblastoma (Pingoud-Meier et al., 2003) and with relapse and aggressive metastatic disease in glioblastoma multiforme (Martinez et al., 2007) A role for caspase-8 in tumorigenesis is also supported by recent studies demonstrating increased transformation of caspase-8 null SV40 immortalized mouse embryo fibroblasts (Krelin et al., 2008). In addition, we have demonstrated that the loss of caspase-8 expression has biological significance in NB cell metastasis (Lahti et al., 2006; Stupack et al., 2006; Teitz et al., 2006). Loss of caspase-8 increases metastasis by blocking integrin-mediated death, a caspase-8-dependent process (Lahti et al., 2006; Stupack et al., 2006; Teitz et al., 2006). The role of caspase-8 in metastasis will be discussed in more detail in the Section 6.2 describing proteins that play a role in NB metastasis.
Multiple groups have shown that caspase-8 deficient NB cells are resistant to death receptor signals and some chemotherapeutic drugs. Importantly, these defects can be corrected by reexpression of caspase-8 via demethylating agents, IFN-γ treatment and caspase-8 expressing retroviral vectors. However, in a few instances secondary defects in death receptor proteins and/or signaling pathway or increased expression of FLIP prevented restoration of the extrinsic cell death pathway (Fulda et al., 2001; Johnsen et al., 2004; Muhlethaler-Mottet et al., 2003; Teitz et al., 2000; Tekautz et al., 2006; Yang et al., 2003).
5.2.2. Bcl-2 family
Another group of proteins that play a role in NB tumorigenesis and metastasis are the Bcl-2 family members. This family of proteins plays a central role in the intrinsic apoptotic pathway. There are more than 20 genes of Bcl-2 family identified in mammals, including antiapoptotic members (Bcl-2, Bcl-XL, Bcl-W, Bcl-G, Bfl1, Mcl-1) and proapoptotic members (Bcl-XS, Bcl-B, Bax, Bak, Bad, Bid, Bik, Bok, Bim, Puma, Noxa, Nix, Nip3, Hrk, Mtd). They usually regulate apoptosis by controlling mitochondrial outer membrane permeabilization (MOMP) to promote or prevent the release of cytochrome c (Green and Kroemer, 2004; Johnsen et al., 2009; Reed, 2006).
The antiapoptotic members, such as Bcl-2, Bcl-XL, Mcl-1, are highly expressed in neural progenitor cells, indicating their protection role in neuron development. Some proapoptotic regulators of the intrinsic pathway, such as Bid and caspase-9, are expressed preferentially in favorable tumors, whereas antiapoptotic regulators such as Mcl-1 were expressed at high levels in unfavorable tumors (Abel et al., 2005), suggesting an imbalance between anti- and proapoptotic factors in NB tumors with favorable or unfavorable biology. Bcl-2 is highly expressed in the majority of NB cell lines and primary tumors and inversely correlates with the degree of cell differentiation (Ramani and Lu, 1994). Although conflicting data exists about the correlation between Bcl-2 levels and MYCN amplification or poor prognosis (Castle et al., 1993; Ikegaki et al., 1995; Mejia et al., 1998), high Bcl-2 level is considered as an important reason for chemoresistance and transfection of Bcl-2 or Bcl-XL into NB cells conferred drug resistance (Dole et al., 1994; Dole et al., 1995).
5.2.3. Regulation of apoptosis-related genes by MYCN
Although the Myc gene was originally identified as an oncogene, it is involved in various cellular processes, including cell growth, proliferation, loss of differentiation, and apoptosis (Adhikary and Eilers, 2005; Pelengaris et al., 2002; Thompson, 1998). N-myc has been found to sensitize NB cells to death receptor induced apoptosis in the absence of cytokines, growth factors or other conditions of stress (Cui et al., 2005; Fulda et al., 1999; Lutz et al., 1998). Cell death in response to MYCN results in caspase-8 cleavage which can be blocked using the caspase inhibitor zVAD-fmk (Cui et al., 2005; Fulda et al., 1999). Upregulation of p53, a direct target of MYCN, is an important mechanism of sensitization of the cell to apoptosis by N-myc (Chen et al., 2010). These data suggest that resistance of NB cells with MYCN amplification to chemotherapy requires additional dysfunction in apoptotic pathways, such as silencing of caspase-8 or inhibiting the p53 pathway. Currently, there are conflicting data on a possible direct relationship between N-myc and caspase-8. Genome wide studies looking for E-box binding sites have identified caspase-8 as a target of both N-myc and c-myc (Fernandez et al., 2003; Perini et al., 2005). Perini and coworkers further demonstrated that the E-box sites in the caspase-8 promoter are methylated in NB preventing binding of N-myc/Max heterodimers. Furthermore, when these NB cells were treated with the demethylating agent 5-azacytidine, N-myc/Max binding was restored and caspase-8 expression was increased (Perini et al., 2005). In contrast to this data which suggests a direct relationship, Fulda and coworkers failed to observe any changes in caspase-8 expression upon overexpression of N-myc or after downregula-tion of N-myc with antisense RNA; however, since these authors did not examine the methylation state of the E-box sequences in caspase-8, this study is somewhat difficult to interpret (Fulda et al., 1999). In addition to data suggesting a possible direct relationship between N-myc and caspase-8, several studies have reported that myc expression promotes activation or priming of the mitochondrial pathway which increases the sensitivity of cells to death receptor signaling. Once the pathway is activated, caspase-8 cleavage of Bid provides a further amplification loop (Klefstrom et al., 2002; Nieminen et al., 2007). Additionally others have shown that the death receptor DR5 contains two E-box sequences that bind N-myc resulting in increased expression of this receptor and further augmentation of the extrinsic pathway (Cui et al., 2005). Some proapoptotic Bcl-2 family members such as Bax and puma are transactivated by MYC, which play an important role in switching p53 downstream effects from G1 arrest to apoptosis (Seoane et al., 2002).
Although p53 is a transcriptional target of N-myc, N-myc also inhibits its function through increased levels of p53 negative regulators, such as MDM2 and TWIST. MDM2, the essential inhibitor of p53, is a direct target of N-myc and inhibition of N-myc results in decreased MDM2, stabilized p53, and apoptosis. TWIST, a transcription factor of bHLH family with antiapoptotic activity, is frequently overexpressed in MYCN amplified NB (Valsesia-Wittmann et al., 2004). Furthermore, TWIST overexpression inhibits ARF/p53 preventing apoptosis in response to N-myc overexpression (Valsesia-Wittmann et al., 2004).
6. The Role of Epithelial to Mesenchymal Transition (EMT) in Development and Metastasis
6.1. EMT in development
Although greater than 50% of all NB patients present with metastatic disease, little is known of the process and the mechanism. Microarray mRNA expression analysis studies comparing highly metastatic human NBs (stage 4) to nonmetastatic human tumors (stage 1 and 2) have provided information on some of the proteins that are involved in NB metastasis (Scaruffi et al., 2005). Of note, transcripts encoding proteins related to the developmental program of the epithelial to mesenchymal transition or EMT, are expressed at higher levels in metastatic human NB (Scaruffi et al., 2005). As mentioned above, early in development, as the neural folds join to form the neural tube, the epidermal ectoderm expresses BMP signals and the floorplate of the neural tube produces Shh. These gradients interact and result in the formation of the neural crest at the dorsal aspect of the neural tube, between the epidermal ectoderm and the dorsal neural tube. The epithelial-like cells of the former neural folds undergo the EMT program. EMT is characterized by: (1) lose of epithelial morphology, (2) downregulation of junctional complexes (E-cadherin, cytokeratin, occludins, and claudins), (3) upregulation of intracellular migratory proteins (RhoB), (4) increased expression of matrix modulators (collagenase, matrilysin, urokinase, heparanase, matrix metalloproteinases—MMP), and (5) upregulation of matrix recognition molecules (N-cadherin; Thiery et al., 2009). Metastatic cells exhibit many of these features and evidence is accumulating that several regulators of EMT are misregulated in NB (Shimono et al., 2000; Valsesia-Wittmann et al., 2004; Vitali et al., 2008).
The transcription factors Snail, Twist, and SIP1/ZEB2 are considered to be master regulators of the EMT transition. Snail is induced primarily by TGFB signaling and directly represses E-cadherin by binding of the E-boxes in the E-cadherin promoter (Kang and Massague, 2004). The repression of E-cadherin is a hallmark of the induction of the EMT program, allowing for the motility of the crest cells. Similarly Twist, a basic helix-loop-helix transcription factor developmentally regulated via the NF-κB pathway, represses E-cadherin expression via the E-boxes within the E-cadherin promoter, although this interaction has not been shown to be direct (Sosic and Olson, 2003). Snail also induces the expression of ZEB factors (inhibitors of E-cadherin expression) which are involved in an EMT regulation loop with miRNA-200 family (Bracken et al., 2008). The miRNA-200 family inhibits the expression of the ZEB family that relieves repression of E-cadherin (Bracken et al., 2008).
6.2. Metastasis-related genes
Although greater than 50% of all NB patients present with metastatic disease, little is known of the process and the mechanism. The data from our group and collaborators first indicated that loss of caspase-8 increases metastasis by blocking integrin-mediated death. Loss of caspase-8 facilitates survival in foreign environments both during development and during metastasis. This data may also explain the observation that at least 50% of NB patients present with metastatic disease (Lahti et al., 2006; Stupack et al., 2006; Teitz et al., 2006).
Another protein that is involved in NB metastasis is CD44, a cell surface glycoprotein that plays a role in cell adhesion and metastasis. CD44 is highly expressed in colon tumors where it affects tumor invasion, however, CD44 expression variable in NB tumors (Shtivelman and Bishop, 1991; Tanabe et al., 1993). There are conflicting data about the correlation between CD44 expression and MYCN amplification, however, high expression of CD44 always correlates with favorable tumors and with the presence of more differentiated cells (Combaret et al., 1996; Munchar et al., 2003). The observation that CD44 is only expressed in favorable tumors is consistent with its functions as a metastasis inhibitor (Munchar et al., 2003; Shtivelman and Bishop, 1991).
The Nm23A protein (nucleoside diphosphate kinase A) encoded by NM23-H1 gene mapped to 17q22 locus is highly expressed in unfavorable NB tumors. A point mutation (S120G) was identified in a subset of NB tumors with elevated NM23A expression (Chang et al., 1996). In addition, NM23A promotes NB metastasis in the xenograft NB animal model and S120G mutation reduced cell adhesion and increased cell migration (Almgren et al., 2004). In contrast, NM23A expression is very low in melanoma, breast, colon tumors and NM23A appears to serve as metastasis suppressor in these tumors (Bown et al., 1999). The reason for the opposite expression pattern may be due to the different regulatory mechanisms in specific tissues (Okabe-Kado et al., 2005).
The matrix metalloproteinases (MMPs), such as MMP-2 and MMP-9, are highly expressed in advanced-stage NB tumors. MMP-2 and MMP-9 promote cell invasion and metastasis by degrading extracellular matrix including type IV, V, VII, and X collagens as well as fibronectin. However, there is no correlation between these MMPs and MYCN status (Ribatti et al., 2004; Sugiura et al., 1998).
Twist1, which is originally identified as a key inducer of mesoderm formation in Drosophila, is found to play an essential role during metastasis in many types of tumors. First evidence of Twist1 involvement in metastasis came from human breast cancers. High levels of Twist1 are associated with invasive carcinoma and loss of EMT. Suppression of Twist1 inhibits the ability of cancer cells to metastasize, while overexpression of Twist1 increases cell motility and decreases cell adhesion (Yang et al., 2004). Further study demonstrated Twist1 may promote metastasis through direct induction of microRNA-10b, which inhibits homeobox D10, resulting in upregulation of a well-characterized prometastatic gene, RHOC (Ma et al., 2007). Later studies found that Twist1 also promotes migration and invasion in bladder and prostate cancer, hepatocellular carcinoma and colorectal cancer (Matsuo et al., 2009; McConkey et al., 2009; Valdes-Mora and Gomez Test, 2009). Twist1 is also overexpressed in N-myc amplified NB, where it inhibits N-myc induced apoptosis by inhibiting the p53 pathway in part via downregulation of p19ARF (Valsesia-Wittmann et al., 2004).
7. The Role of miRNA in Development and NB
miRNAs are endogenous small noncoding RNAs of ~22 nucleotides in length that negatively regulate gene expression by mRNA cleavage or translational repression of the target mRNA (Bartel, 2004). miRNAs play an important role in regulating most cellular processes, and contribute to the process of tumorigenesis and metastasis (Zhang et al., 2010). miRNA expression profiles have been correlated with prognosis, differentiation, and apoptosis in NB tumors (Chen and Stallings, 2007), suggesting that miRNAs could function as TSGs or oncogenes in NB. As mentioned above, miR-34a, which is located at 1p36, is a good TSG candidate (Welch et al., 2007). Mir-34a is a direct target of p53 and knockdown of mir-34a reduced p53-dependent apoptosis (He et al., 2007). In addition, one study found that N-myc is a direct target of mir-34a (Wei et al., 2008), although this data is contrary to a previous study (Welch et al., 2007). The discrepancy between these two studies may reflect the slightly different systems that were used for these two studies.
Recent data showed that seven miRNAs including miR-17 to -92 cluster were induced by N-myc in vitro, and their high expression correlates with MYCN amplification in tumors (Schulte et al., 2008), suggesting this cluster is a potential oncogene in NB. This cluster, which is transcribed as a polycis-tronic transcript containing miR-17, -18, -19, -20, and -92, was first found to be a target of c-myc, a potential oncogene in B-cell lymphoma (He et al., 2005). The miR-17 to -92 cluster is also overexpressed in lung cancers where it has been shown to enhance cell proliferation (Hayashita et al., 2005). miRNAs also mediate tumor metastasis, for example, miR-10b was shown to initiate tumor invasion and metastasis in breast cancer (Ma et al., 2007).
One very recent paper compared miRNA expression patterns between primary and metastatic NB tumors and found significant changes of 54 miRNAs in metastatic samples, among which 35 miRNAs were upregulated and 19 miRNAs were downregulated (Guo et al., 2010). Some miRNAs, such as miR-10b, miR-29a/b, miR-335, which are known to promote metastasis were upregulated in metastatic NB tumors. In contrast, miR-7, miR-338-3p, and the let-7 family were the three of the top 10 downregulated miRNAs in the metastatic group. These miRNAs have been shown to play antimetastatic roles in other tumors (Zhang et al., 2010). The authors also analyzed the predicted miRNA targets of these miRNAs and found that many of these targets are related to metastasis. For instance, both caspase-8 and integrin beta1 are the predicted targets of miR-29a and miR-29b, miRNAs that promote metastasis in breast cancer (Gebeshuber et al., 2009). However, the actual function of these identified miRNAs in metastasis needs to be investigated.
8. Other Important Genes in NB
8.1. Telomerase
Telomerase is a specialized ribonucleoprotein polymerase that synthesizes the TTAGGG telomeric repeats found at the end of chromosomes to maintain the length of the telomere. This enzyme is expressed in germ line cells but not in the majority of somatic cells. Thus, telomeres in somatic cells undergo progressive shortening and eventually lose the ability to protect chromosome ends, resulting in cell senescence and/or death. Increased telomerase expression, which results in unlimited cell replication and repression of cell senescence, is found in many tumors (Bodnar et al., 1998; Hahn et al., 1999). Telomerase activity was detected in most NBs, but not in ganglioneuromas or normal adrenal tissue (Hiyama et al., 1997). In addition, high telomerase activity usually correlated with MYCN amplification and poor outcome, suggesting telomerase could be a prognostic marker for poor survival (Hiyama et al., 1997; Ohali et al., 2006; Reynolds et al., 1997).
8.2. MDR1 and MRP gene family
Most NBs exhibit a strong initial response to chemotherapy followed by the appearance of multidrug resistance (MDR) in more than half of cases with remission/remaining tumor (Keshelava et al., 2001; Tweddle et al., 2003; Xue et al., 2007). Although p53 seems to have an important role, the MDR1 gene (multidrug resistance gene 1) and the MRP (multidrug transporter MDR-associated protein) gene family also contribute to this resistance. MDR1 expression was found to be increased after treatment of NB tumors (Bourhis et al., 1989; Chan et al., 1991). In one report, MDR1 expression was inversely correlated with MYCN amplification and poor outcome (Chan et al., 1991), while other groups did not find this correlation (Dhooge et al., 1997; Kutlik et al., 2002). Likewise, some studies found that expression of MRP gene family members such as MRP1, MRP4, strongly correlated with MYCN expression and chemoresistance in NBs (Haber et al., 2006; Norris et al., 1996, 2005), while other studies failed to find a correlation (De Cremoux et al., 2007; Goto et al., 2000).
8.3. GD2 and Bmi-1
Several other genes are also abnormally expressed in NB tumors such as GD2 and Bmi-1. The ganglioside GD2 is a glycolipid that is most commonly expressed in the majority of NB tumors, thus representing a good diagnostic marker and therapeutic target. Indeed recent studies using an anti-GD2 antibodies in association with GMC-SF and IL2 and RA results in a significant increase in event free survival (2-year estimates of 66% ±5% vs. 46% ±5%), and preliminary data suggest in overall survival (86% ±4% vs. 75% ±5% at 2 years p = 0.0223) (Yu et al., 2009). Bmi-1, a polycomb ring finger gene, was found to be strongly expressed in primary NB tumors in 2006 and very recent data indicated that it is a MYCN target gene that promotes tumorigenesis through inhibition of two potential TSGs (KIF1Bβ and TSCL1) in NB cells (Nowak et al., 2006; Ochiai et al., 2010).
9. Clinical Treatment Overview
Current treatment for NB consists of surgery, chemotherapy, radiation, and biotherapy. The clinical strategy usually depends on a patient’s risk stratification (Table 4.1). For examples, exposure to chemotherapy is generally limited for low risk group patients, whereas, high-risk group patients are treated with multiagent chemotherapy to reduce the overall burden of the disease before the surgical removal of the primary tumor (Haase et al., 1999; Park et al., 2008).
9.1. Low risk group and intermediate risk group
Low risk group encompasses diseases at stages 1, 2, and 4S with favorable characteristics (Brodeur, 2003; Castleberry et al., 1997; Matthay, 1995). Most low risk group patients at stages 1 or 2 are successfully treated with surgery alone and complete resection of the tumor is the goal. The chances of these tumors recurring or progressing to advanced-stage NB are very low and chemotherapy is reserved for those patients with recurrences. NBs at stage 4S without MYCN amplification almost always spontaneously regresses and these patients can be safely observed without any treatment. The survival rate of patients of this group is greater than 95% (Alvarado et al., 2000; Park et al., 2008; Simon et al., 2004).
Intermediate risk group patients include stage 3 patients of any age with favorable features, stage 4 infants with favorable features, and 4S patients without MYCN amplification and with unfavorable histology (Brodeur, 2003; Castleberry et al., 1997; Matthay, 1995). For intermediate risk group patients, surgery, and moderate dose of multiagent chemotherapy are the basic therapeutic strategies. Sometimes radiation is also used to remove the residual tumors. Aggressive chemotherapy is utilized for patients who respond poorly to initial treatment or experience recurrence. Current treatments for intermediate risk group patients have a cure rate about 70–90% (Matthay et al., 1998; Park et al., 2008; Schmidt et al., 2000).
9.2. High-risk group patients
High-risk group consist of patients with unfavorable biological features at stages 2, 3, 4, and those with MYCN amplification at stage 4S (Brodeur, 2003; Castleberry et al., 1997; Matthay, 1995). The standard clinical strategy for this group is comprised of 4 steps: initial induction chemotherapy, local control, consolidation, and biology therapy (Haase et al., 1999; Park et al., 2008). Initial induction chemotherapy consists of combinations of chemo-therapeutic agents such as cisplatin, etoposide, doxorubicin, cyclophosphamide, topectan/ironotecan, and vincristine. Following the completion of induction therapy, local control is achieved by aggressive surgical resection of the primary tumor followed by external beam radiation. After that, consolidation is provided by high-dose chemotherapy and autologous hematopoietic stem cell rescue, using stem cells that are prepared during the induction therapy. The purpose of consolidation therapy is to eliminate any remaining tumors. Consolidation therapy typically employs agents such as carboplatin, etoposide, and melphalan. Sometime focal radiotherapy is also applied to the primary tumor sites. After recovery from the consolidation, patients receive biological therapy to eradicate minimal residual disease because relapse is a frequent occurrence after autologous transplantation in this group of patients. The most common used biological agent is cis-retinoic acid (cis-RA), which is a noncytotoxic differentiation inducer. The use of monoclonal antibodies against tumor-specific antigens, such as GD2, provides an alternative and promising therapy to eliminate minimal residual tumor cells as described above (Matthay et al., 1999; Pearson et al., 2008; Zage et al., 2008).
Even with these complicated and aggressive treatments, the overall cure rate for high-risk patients is only about 30% during the last two decades (Matthay et al., 1999; Pearson et al., 2008; Zage et al., 2008). Therefore, the development of new agents and methods to more effectively treat these patients is underway. These therapeutic approaches include immunetherapy aimed at NB-specific antigens, targeted delivery of radioactive molecules to NB, or new chemotherapy such as retinoids for inducing differentiation, tyrosine kinase inhibitors targeted at ALK, demethylating agents and HDAC inhibitors for inducing reexpression of caspase-8 or other epigenetically silenced genes (Kelleher and McDermott, 2010; Schor, 2009; Wagner and Danks, 2009; Witt et al., 2009).
10. Conclusion
The investigation and identification of genomic abnormalities and gene expression changes has improved the understanding of the molecular basis of biological and clinical characteristics of NBs (summary in Tables 4.2 and 4.3). Although great progress has been made in recent 20 years, much work needs to be done to identify tumor-specific targets for therapy. Also, deeper understanding of the development of normal sympathetic nervous system will help us find the important abnormal events that initiate NB development. In conclusion, more precise identification of molecular alterations should allow more effective and less toxic therapies with improved cure rate.
Table 4. 3.
Summary of genetic changes in neuroblastoma
| Gene symbol | Gene name | Chromosomal locus | Gene function | Gene alterations | References |
|---|---|---|---|---|---|
| ALK | Anaplastic lymphoma receptor tyrosine kinase | 2p23 | Receptor tyrosine kinase | Mutation/amplification | Caren et al. (2008), Chen et al. (2008), George et al. (2008), Janoueix-Lerosey et al. (2008), Mosse et al. (2008), Passoni et al. (2009) |
| Bcl2 | B-cell CLL/lymphoma 2 | 18q21.3 | Apoptosis suppression | High expression | Abel et al. (2005), Ramani and Lu (1994), Castle et al. (1993), Ikegaki et al. (1995), Mejia et al. (1998) |
| Bmi1 | BMI1 polycomb ring finger oncogene | 10p11.23 | Oncogene | Overexpression | Ochiai et al. (2010), Nowak et al. (2006) |
| Casp8 | Caspase 8 | 2q33–q34 | Apoptosis/metastasis | Silence/deletion | Teitz et al. (2000, 2006), Yang et al. (2007), Fulda et al. (2001), Fulda and Debatin (2006), Lahti et al. (2006), Stupack et al. (2006) |
| CD44 | CD44 molecule | 11p13 | Integrin/antimetastasis | Expression in favorable tumors | Munchar et al. (2003), Combaret et al. (1996) |
| CHD5 | Chromodomain helicase DNA-binding protein 5 | 1p36.31 | Helicase/tumor suppressor | Deletion/low expression | Bagchi et al. (2007) |
| DDX1 | ATP-dependent RNA helicase DDX1 | 2p24 | RNA helicase/Oncogene | Amplification | George et al. (1996) |
| B4GALNT1 | GM2/GD2 synthase | 12q13.3 | Ganglioside | High expression | Yu et al. (2009) |
| KIF1b | Kinesin family member 1beta | 1p36.2 | Kinesin/tumor suppressor | Deletion/low expression | Munirajan et al. (2008), Schlisio et al. (2008) |
| MDM2 | Mdm2 p53-binding protein homolog | 12q14.3–q15 | Oncogene | Amplification | Corvi et al. (1995a) |
| ABCB1 | Multidrug resistance protein 1 (MRP1) | 7q21.12 | Multidrug resistance | High expression | Bourhis et al. (1989), Chan et al. (1991), Dhooge et al. (1997), Kutlik et al. (2002) |
| Mir34a | MicroRNA 34a | 1p36.22 | Micro RNA/tumor suppressor | Deletion/low expression | Welch et al. (2007), Chen and Stallings (2007) |
| Mir17–92 | MicroRNA cluster 17–92 (mir17, 18, 19, 20, 92) | 13q31.3 | Micro RNA/Oncogene | High expression | Schulte et al. (2008) |
| MMP2, MMP | Matrix metallopeptidase 2 | 16q13–q21 | Proteinase/metastasis | High expression | Sugiura et al. (1998), Ribatti et al. (2004) |
| MMP9 | Matrix metallopeptidase 9 | 20q11.2–q13.1 | Proteinase/metastasis | High expression | (Sugiura et al. (1998), Ribatti et al. (2004)) |
| ABCC1 | Multidrug resistance-associated protein 1 | 16p13.1 | Multidrug resistance | High expression | Norris et al. (1996, 2005), Haber et al. (2006), Goto et al. (2000), De Cremoux et al. (2007) |
| MYCL | Myc-related gene from lung cancer | 1p34.2 | Oncogene | Amplification | Jinbo et al. (1989) |
| MYCN | Neuroblastoma MYC oncogene | 2p24.1 | Oncogene | Amplification | Schwab et al. (1983, 1984), Kohl et al. (1983), Pelengaris et al. (2002), Reiter and Brodeur (1996, 1998) |
| NME1 | Metastasis inhibition factor NM23 | 17q22 | Nucleoside kinase/antimetastasis | Overexpression | Godfried et al. (2002), Almgren et al. (2004), Chang et al. (1996) |
| PHOX2B | Paired-like homeobox 2b | 4p13 | Neuron development | Mutation | Trochet et al. (2004), Mosse et al. (2004) |
| TERT | Telomerase reverse transcriptase | 5p15.33 | Telomere maintenance | High expression | Hiyama et al. (1997), Reynolds et al. (1997), Ohali et al. (2006) |
| NTRK1 | Neurotrophic tyrosine kinase receptor 1 (TrKA) | 1q21–q22 | Receptor tyrosine kinase | Inverse correlation with Mycn | Nakagawara et al. (1992), Nakagawara (1993), Tacconelli et al. (2004), Kim et al. (1999) |
| NTRK2 | Neurotrophic tyrosine kinase receptor 2 (TrKB) | 9q22.1 | Receptor tyrosine kinase | Strong correlation with Mycn | Nakagawara et al. (1994), Nakagawara (1994), Douma et al. (2004), Ho et al. (2002) |
| NTRK3 | Neurotrophic tyrosine kinase receptor 3 (TrKC) | 15q25 | Receptor tyrosine kinase | Coexpression with TrkA | Yamashiro et al. (1996), Svensson et al. (1997) |
| CADM1 | Tumor suppressor in lung cancer 1 (TSLC1) | 11q23.2 | Cell adhesion/Tumor suppressor | Deletion/low expression | Gomyo et al. (1999), Ando et al. (2008) |
| TWIST1 | Twist homolog 1 | 7p21.2 | Apoptosis/metastasis | High expression | Valsesia-Wittmann et al. (2004) |
Acknowledgments
We apologize to all of our colleagues whose work was either not cited or was cited in a review article. We thank the members of the Lahti lab, especially Judith Hyle, for their input. This work was funded by NIH grants R01 CA067938 to JML, Comprehensive Cancer Center Support Grant P01 CA021765 and the American Syrian Lebanese Associated Charities ALSAC.
References
- Abe M, Ohira M, Kaneda A, Yagi Y, Yamamoto S, Kitano Y, Takato T, Nakagawara A, Ushijima T. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Res. 2005;65:828–834. [PubMed] [Google Scholar]
- Abe M, Westermann F, Nakagawara A, Takato T, Schwab M, Ushijima T. Marked and independent prognostic significance of the CpG island methylator phenotype in neuroblastomas. Cancer Lett. 2007;247:253–258. doi: 10.1016/j.canlet.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Abel F, Sjoberg RM, Nilsson S, Kogner P, Martinsson T. Imbalance of the mitochondrial pro- and anti-apoptotic mediators in neuroblastoma tumours with unfavourable biology. Eur J Cancer. 2005;41:635–646. doi: 10.1016/j.ejca.2004.12.021. [DOI] [PubMed] [Google Scholar]
- Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- Alenina N, Bashammakh S, Bader M. Specification and differentiation of serotonergic neurons. Stem Cell Rev. 2006;2:5–10. doi: 10.1007/s12015-006-0002-2. [DOI] [PubMed] [Google Scholar]
- Almgren MA, Henriksson KC, Fujimoto J, Chang CL. Nucleoside diphosphate kinase A/nm23-H1 promotes metastasis of NB69-derived human neuroblastoma. Mol Cancer Res. 2004;2:387–394. [PubMed] [Google Scholar]
- Alvarado CS, London WB, Look AT, Brodeur GM, Altmiller DH, Thorner PS, Joshi VV, Rowe ST, Nash MB, Smith EI, Castleberry RP, Cohn SL. Natural history and biology of stage A neuroblastoma: A Pediatric Oncology Group Study. J Pediatr Hematol Oncol. 2000;22:197–205. doi: 10.1097/00043426-200005000-00003. [DOI] [PubMed] [Google Scholar]
- Anderson DJ. Molecular control of cell fate in the neural crest: The sympathoadrenal lineage. Annu Rev Neurosci. 1993;16:129–158. doi: 10.1146/annurev.ne.16.030193.001021. [DOI] [PubMed] [Google Scholar]
- Anderson DJ, Axel R. A bipotential neuroendocrine precursor whose choice of cell fate is determined by NGF and glucocorticoids. Cell. 1986;47:1079–1090. doi: 10.1016/0092-8674(86)90823-8. [DOI] [PubMed] [Google Scholar]
- Anderson DJ, Carnahan JF, Michelsohn A, Patterson PH. Antibody markers identify a common progenitor to sympathetic neurons and chromaffin cells in vivo and reveal the timing of commitment to neuronal differentiation in the sympathoadrenal lineage. J Neurosci. 1991;11:3507–3519. doi: 10.1523/JNEUROSCI.11-11-03507.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, Ohira M, Ozaki T, Nakagawa A, Akazawa K, Suenaga Y, Nakamura Y, Koda T, Kamijo T, Murakami Y, Nakagawara A. Expression of TSLC1, a candidate tumor suppressor gene mapped to chromosome 11q23, is downregulated in unfavorable neuroblastoma without promoter hypermethylation. Int J Cancer. 2008;123:2087–2094. doi: 10.1002/ijc.23776. [DOI] [PubMed] [Google Scholar]
- Astuti D, Agathanggelou A, Honorio S, Dallol A, Martinsson T, Kogner P, Cummins C, Neumann HP, Voutilainen R, Dahia P, Eng C, Maher ER, et al. RASSF1A promoter region CpG island hypermethylation in phaeochromocytomas and neuroblastoma tumours. Oncogene. 2001;20:7573–7577. doi: 10.1038/sj.onc.1204968. [DOI] [PubMed] [Google Scholar]
- Bader SA, Fasching C, Brodeur GM, Stanbridge EJ. Dissociation of suppression of tumorigenicity and differentiation in vitro effected by transfer of single human chromosomes into human neuroblastoma cells. Cell Growth Differ. 1991;2:245–255. [PubMed] [Google Scholar]
- Bagchi A, Papazoglu C, Wu Y, Capurso D, Brodt M, Francis D, Bredel M, Vogel H, Mills AA. CHD5 is a tumor suppressor at human 1p36. Cell. 2007;128:459–475. doi: 10.1016/j.cell.2006.11.052. [DOI] [PubMed] [Google Scholar]
- Banelli B, Casciano I, Croce M, di Vinci A, Gelvi I, Pagnan G, Brignole C, Allemanni G, Ferrini S, Ponzoni M, Romani M. Expression and methylation of CASP8 in neuroblastoma: Identification of a promoter region. Nat Med. 2002;8:1333–1335. doi: 10.1038/nm1202-1333. [DOI] [PubMed] [Google Scholar]
- Banerjee SA, Hoppe P, Brilliant M, Chikaraishi DM. 5′ flanking sequences of the rat tyrosine hydroxylase gene target accurate tissue-specific, developmental, and transsynaptic expression in transgenic mice. J Neurosci. 1992;12:4460–4467. doi: 10.1523/JNEUROSCI.12-11-04460.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Beckwith JB, Perrin EV. In situ neuroblastomas: A contribution to the natural history of neural crest tumors. Am J Pathol. 1963;43:1089–1104. [PMC free article] [PubMed] [Google Scholar]
- Beierle EA, Nagaram A, Dai W, Iyengar M, Chen MK. VEGF-mediated survivin expression in neuroblastoma cells. J Surg Res. 2005;127:21–28. doi: 10.1016/j.jss.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Benailly HK, Lapierre JM, Laudier B, Amiel J, Attie T, De Blois MC, Vekemans M, Romana SP. PMX2B, a new candidate gene for Hirsch-sprung’s disease. Clin Genet. 2003;64:204–209. doi: 10.1034/j.1399-0004.2003.00105.x. [DOI] [PubMed] [Google Scholar]
- Betters E, Liu Y, Kjaeldgaard A, Sundstrom E, Garcia-Castro M. Analysis of early human neural crest development. Dev Biol. 2010 doi: 10.1016/j.ydbio.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke AJ, Weiner JA, Chun J. Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J Comp Neurol. 1998;396:39–50. doi: 10.1002/(sici)1096-9861(19980622)396:1<39::aid-cne4>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15:725–731. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Bourdeaut F, Trochet D, Janoueix-Lerosey I, Ribeiro A, Deville A, Coz C, Michiels JF, Lyonnet S, Amiel J, Delattre O. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Cancer Lett. 2005;228:51–58. doi: 10.1016/j.canlet.2005.01.055. [DOI] [PubMed] [Google Scholar]
- Bourhis J, Benard J, Hartmann O, Boccon-Gibod L, Lemerle J, Riou G. Correlation of MDR1 gene expression with chemotherapy in neuroblastoma. J Natl Cancer Inst. 1989;81:1401–1405. doi: 10.1093/jnci/81.18.1401. [DOI] [PubMed] [Google Scholar]
- Bown N, Cotterill S, Lastowska M, O’Neill S, Pearson AD, Plantaz D, Meddeb M, Danglot G, Brinkschmidt C, Christiansen H, Laureys G, Speleman F, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med. 1999;340:1954–1961. doi: 10.1056/NEJM199906243402504. [DOI] [PubMed] [Google Scholar]
- Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–7854. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- Breit S, Schwab M. Suppression of MYC by high expression of NMYC in human neuroblastoma cells. J Neurosci Res. 1989;24:21–28. doi: 10.1002/jnr.490240105. [DOI] [PubMed] [Google Scholar]
- Brodeur GM. Neuroblastoma: Biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:203–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- Brodeur GM, Minturn JE, Ho R, Simpson AM, Iyer R, Varela CR, Light JE, Kolla V, Evans AE. Trk receptor expression and inhibition in neuroblastomas. Clin Cancer Res. 2009;15:3244–3250. doi: 10.1158/1078-0432.CCR-08-1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner-Fraser M. Neural crest cell migration in the developing embryo. Trends Cell Biol. 1993;3:392–397. doi: 10.1016/0962-8924(93)90089-j. [DOI] [PubMed] [Google Scholar]
- Bronner-Fraser M, Sieber-Blum M, Cohen AM. Clonal analysis of the avian neural crest: Migration and maturation of mixed neural crest clones injected into host chicken embryos. J Comp Neurol. 1980;193:423–434. doi: 10.1002/cne.901930209. [DOI] [PubMed] [Google Scholar]
- Caren H, Abel F, Kogner P, Martinsson T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem J. 2008;416:153–159. doi: 10.1042/bj20081834. [DOI] [PubMed] [Google Scholar]
- Caron H, van SP, Buschman R, Pereira do TR, Maes P, Beks L, de KJ, Voute PA, Vergnaud G, Westerveld A, Slater R, Versteeg R. Allelic loss of the short arm of chromosome 4 in neuroblastoma suggests a novel tumour suppressor gene locus. Hum Genet. 1996;97:834–837. doi: 10.1007/BF02346199. [DOI] [PubMed] [Google Scholar]
- Castle VP, Heidelberger KP, Bromberg J, Ou X, Dole M, Nunez G. Expression of the apoptosis-suppressing protein bcl-2, in neuroblastoma is associated with unfavorable histology and N-myc amplification. Am J Pathol. 1993;143:1543–1550. [PMC free article] [PubMed] [Google Scholar]
- Castleberry RP, Pritchard J, Ambros P, Berthold F, Brodeur GM, Castel V, Cohn SL, De BB, Cks-Mireaux C, Frappaz D, Haase GM, Haber M, et al. The International Neuroblastoma Risk Groups (INRG): A preliminary repor. Eur J Cancer. 1997;33:2113–2116. doi: 10.1016/s0959-8049(97)00202-5. [DOI] [PubMed] [Google Scholar]
- Chambery D, Mohseni-Zadeh S, de GB, Babajko S. N-myc regulation of type I insulin-like growth factor receptor in a human neuroblastoma cell line. Cancer Res. 1999;59:2898–2902. [PubMed] [Google Scholar]
- Chan HS, Haddad G, Thorner PS, DeBoer G, Lin YP, Ondrusek N, Yeger H, Ling V. P-glycoprotein expression as a predictor of the outcome of therapy for neuroblastoma. N Engl J Med. 1991;325:1608–1614. doi: 10.1056/NEJM199112053252304. [DOI] [PubMed] [Google Scholar]
- Chang CL, Strahler JR, Thoraval DH, Qian MG, Hinderer R, Hanash SM. A nucleoside diphosphate kinase A (nm23–H1) serine 120–> glycine substitution in advanced stage neuroblastoma affects enzyme stability and alters protein-protein interaction. Oncogene. 1996;12:659–667. [PubMed] [Google Scholar]
- Charron J, Malynn BA, Fisher P, Stewart V, Jeannotte L, Goff SP, Robertson EJ, Alt FW. Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev. 1992;6:2248–2257. doi: 10.1101/gad.6.12a.2248. [DOI] [PubMed] [Google Scholar]
- Chen Y, Stallings RL. Differential patterns of microRNA expression in neuroblastoma are correlated with prognosis, differentiation, and apoptosis. Cancer Res. 2007;67:976–983. doi: 10.1158/0008-5472.CAN-06-3667. [DOI] [PubMed] [Google Scholar]
- Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–974. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- Chen L, Iraci N, Gherardi S, Gamble LD, Wood KM, Perini G, Lunec J, Tweddle DA. p53 is a direct transcriptional target of MYCN in neuroblastoma. Cancer Res. 2010;70:1377–1388. doi: 10.1158/0008-5472.CAN-09-2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Ott GR. Anaplastic lymphoma kinase as a therapeutic target in anaplastic large cell lymphoma, non-small cell lung cancer and neuroblastoma. Anticancer Agents Med Chem. 2010;10:236–249. doi: 10.2174/1871520611009030236. [DOI] [PubMed] [Google Scholar]
- Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, Mosseri V, Simon T, et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J Clin Oncol. 2009;27:289–297. doi: 10.1200/JCO.2008.16.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combaret V, Gross N, Lasset C, Frappaz D, Peruisseau G, Philip T, Beck D, Favrot MC. Clinical relevance of CD44 cell-surface expression and N-myc gene amplification in a multicentric analysis of 121 pediatric neuroblastomas. J Clin Oncol. 1996;14:25–34. doi: 10.1200/JCO.1996.14.1.25. [DOI] [PubMed] [Google Scholar]
- Coppola D, Ouban A, Gilbert-Barness E. Expression of the insulin-like growth factor receptor 1 during human embryogenesis. Fetal Pediatr Pathol. 2009;28:47–54. doi: 10.1080/15513810802679498. [DOI] [PubMed] [Google Scholar]
- Corvi R, Savelyeva L, Amler L, Handgretinger R, Schwab M. Cytogenetic evolution of MYCN and MDM2 amplification in the neuroblastoma LS tumour and its cell line. Eur J Cancer. 1995a;31A:520–523. doi: 10.1016/0959-8049(95)00031-d. [DOI] [PubMed] [Google Scholar]
- Corvi R, Savelyeva L, Schwab M. Duplication of N-MYC at its resident site 2p24 may be a mechanism of activation alternative to amplification in human neuroblastoma cells. Cancer Res. 1995b;55:3471–3474. [PubMed] [Google Scholar]
- Coulter DW, Wilkie MB, Moats-Staats BM. Inhibition of IGF-I receptor signaling in combination with rapamycin or temsirolimus increases MYC-N phosphorylation. Anticancer Res. 2009;29:1943–1949. [PubMed] [Google Scholar]
- Cui H, Li T, Ding HF. Linking of N-Myc to death receptor machinery in neuroblastoma cells. J Biol Chem. 2005;280:9474–9481. doi: 10.1074/jbc.M410450200. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: Critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- De Zio D, Giunta L, Corvaro M, Ferraro E, Cecconi F. Expanding roles of programmed cell death in mammalian neurodevelopment. Semin Cell Dev Biol. 2005;16:281–294. doi: 10.1016/j.semcdb.2004.12.003. [DOI] [PubMed] [Google Scholar]
- De Cremoux P, Jourdan-Da-Silva N, Couturier J, Tran-Perennou C, Schleiermacher G, Fehlbaum P, Doz F, Mosseri V, Delattre O, Klijanienko J, Vielh P, Michon J. Role of chemotherapy resistance genes in outcome of neuroblastoma. Pediatr Blood Cancer. 2007;48:311–317. doi: 10.1002/pbc.20853. [DOI] [PubMed] [Google Scholar]
- Degoutin J, Brunet-de CN, Cifuentes-Diaz C, Vigny M. ALK (Anaplastic Lymphoma Kinase) expression in DRG neurons and its involvement in neuron-Schwann cells interaction. Eur J Neurosci. 2009;29:275–286. doi: 10.1111/j.1460-9568.2008.06593.x. [DOI] [PubMed] [Google Scholar]
- D’Ercole AJ, Ye P, Calikoglu AS, Gutierrez-Ospina G. The role of the insulin-like growth factors in the central nervous system. Mol Neurobiol. 1996;13:227–255. doi: 10.1007/BF02740625. [DOI] [PubMed] [Google Scholar]
- Dhooge CR, De Moerloose BM, Benoit YC, Van RN, Philippe, Laureys GG. Expression of the MDR1 gene product P-glycoprotein in childhood neuroblastoma. Cancer. 1997;80:1250–1257. doi: 10.1002/(sici)1097-0142(19971001)80:7<1250::aid-cncr8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Dole M, Nunez G, Merchant AK, Maybaum J, Rode CK, Bloch CA, Castle VP. Bcl-2 inhibits chemotherapy-induced apoptosis in neuroblastoma. Cancer Res. 1994;54:3253–3259. [PubMed] [Google Scholar]
- Dole MG, Jasty R, Cooper MJ, Thompson CB, Nunez G, Castle VP. Bcl-xL is expressed in neuroblastoma cells and modulates chemotherapy-induced apoptosis. Cancer Res. 1995;55:2576–2582. [PubMed] [Google Scholar]
- Douma S, Van LT, Zevenhoven J, Meuwissen R, Van GE, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430:1034–1039. doi: 10.1038/nature02765. [DOI] [PubMed] [Google Scholar]
- Dupin E, Calloni G, Real C, Goncalves-Trentin A, Le Douarin NM. Neural crest progenitors and stem cells. C R Biol. 2007;330:521–529. doi: 10.1016/j.crvi.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Ejeskar K, Aburatani H, Abrahamsson J, Kogner P, Martinsson T. Loss of heterozygosity of 3p markers in neuroblastoma tumours implicate a tumour-suppressor locus distal to the FHIT gene. Br J Cancer. 1998;77:1787–1791. doi: 10.1038/bjc.1998.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuel BS, Balaban G, Boyd JP, Grossman A, Negishi M, Parmiter A, Glick MC. N-myc amplification in multiple homogeneously staining regions in two human neuroblastomas. Proc Natl Acad Sci USA. 1985;82:3736–3740. doi: 10.1073/pnas.82.11.3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson CA, Turley EA. The effects of epidermal growth factor on neural crest cells in tissue culture. Exp Cell Res. 1987;169:267–279. doi: 10.1016/0014-4827(87)90189-3. [DOI] [PubMed] [Google Scholar]
- Ernsberger U, Esposito L, Partimo S, Huber K, Franke A, Bixby JL, Kalcheim C, Unsicker K. Expression of neuronal markers suggests heterogeneity of chick sympathoadrenal cells prior to invasion of the adrenal anlagen. Cell Tissue Res. 2005;319:1–13. doi: 10.1007/s00441-004-0996-1. [DOI] [PubMed] [Google Scholar]
- Evans AE, D’Angio GJ, Randolph J. A proposed staging for children with neuroblastoma. Children’s cancer study group A. Cancer. 1971;27:374–378. doi: 10.1002/1097-0142(197102)27:2<374::aid-cncr2820270221>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–1129. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5:876–885. doi: 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
- Fontaine-Perus JC, Chanconie M, Le Douarin NM. Differentiation of peptidergic neurones in quail-chick chimaeric embryos. Cell Differ. 1982;11:183–193. doi: 10.1016/0045-6039(82)90065-3. [DOI] [PubMed] [Google Scholar]
- Fulda S, Debatin KM. 5-Aza-2′-deoxycytidine and IFN-gamma cooperate to sensitize for TRAIL-induced apoptosis by upregulating caspase-8. Oncogene. 2006;25:5125–5133. doi: 10.1038/sj.onc.1209518. [DOI] [PubMed] [Google Scholar]
- Fulda S, Lutz W, Schwab M, Debatin KM. MycN sensitizes neuroblas-toma cells for drug-induced apoptosis. Oncogene. 1999;18:1479–1486. doi: 10.1038/sj.onc.1202435. [DOI] [PubMed] [Google Scholar]
- Fulda S, Kufer MU, Meyer E, van Valen F, Dockhorn-Dworniczak B, Debatin KM. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene. 2001;20:5865–5877. doi: 10.1038/sj.onc.1204750. [DOI] [PubMed] [Google Scholar]
- Gebeshuber CA, Zatloukal K, Martinez J. miR-29a suppresses tristetraprolin, which is a regulator of epithelial polarity and metastasis. EMBO Rep. 2009;10:400–405. doi: 10.1038/embor.2009.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George RE, Kenyon RM, McGuckin AG, Malcolm AJ, Pearson AD, Lunec J. Investigation of co-amplification of the candidate genes ornithine decarboxylase, ribonucleotide reductase, syndecan-1 and a DEAD box gene, DDX1, with N-myc in neuroblastoma. United Kingdom Children’s Cancer Study Group. Oncogene. 1996;12:1583–1587. [PubMed] [Google Scholar]
- George RE, Sanda T, Hanna M, Frohling S, Luther W, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–978. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert F, Feder M, Balaban G, Brangman D, Lurie DK, Podolsky R, Rinaldt V, Vinikoor N, Weisband J. Human neuroblastomas and abnormalities of chromosomes 1 and 17. Cancer Res. 1984;44:5444–5449. [PubMed] [Google Scholar]
- Godfried MB, Veenstra M, Sluis P, Boon K, Asperen R, Hermus MC, Schaik BD, Voute TP, Schwab M, Versteeg R, Caron HN. The N-myc and c-myc downstream pathways include the chromosome 17q genes nm23-H1 and nm23-H2. Oncogene. 2002;21:2097–2101. doi: 10.1038/sj.onc.1205259. [DOI] [PubMed] [Google Scholar]
- Gomyo H, Arai Y, Tanigami A, Murakami Y, Hattori M, Hosoda F, Arai K, Aikawa Y, Tsuda H, Hirohashi S, Asakawa S, Shimizu N, et al. A 2-Mb sequence-ready contig map and a novel immunoglobulin superfamily gene IGSF4 in the LOH region of chromosome 11q23.2. Genomics. 1999;62:139–146. doi: 10.1006/geno.1999.6001. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gomez P, Bello MJ, Inda MM, Alonso ME, Arjona D, Aminoso C, Lopez-Marin I, de Campos JM, Sarasa JL, Castresana JS, Rey JA. Deletion and aberrant CpG island methylation of Caspase 8 gene in medulloblastoma. Oncol Rep. 2004;12:663–666. [PubMed] [Google Scholar]
- Goto H, Keshelava N, Matthay KK, Lukens JN, Gerbing RB, Stram DO, Seeger RC, Reynolds CP. Multidrug resistance-associated protein 1 (MRP1) expression in neuroblastoma cell lines and primary tumors. Med Pediatr Oncol. 2000;35:619–622. doi: 10.1002/1096-911x(20001201)35:6<619::aid-mpo28>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Grenet J, Teitz T, Wei T, Valentine V, Kidd VJ. Structure and chromosome localization of the human CASP8 gene. Gene. 1999;226:225–232. doi: 10.1016/s0378-1119(98)00565-4. [DOI] [PubMed] [Google Scholar]
- Grimmer MR, Weiss WA. Childhood tumors of the nervous system as disorders of normal development. Curr Opin Pediatr. 2006;18:634–638. doi: 10.1097/MOP.0b013e32801080fe. [DOI] [PubMed] [Google Scholar]
- Guo C, White PS, Weiss MJ, Hogarty MD, Thompson PM, Stram DO, Gerbing R, Matthay KK, Seeger RC, Brodeur GM, Maris JM. Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene. 1999;18:4948–4957. doi: 10.1038/sj.onc.1202887. [DOI] [PubMed] [Google Scholar]
- Guo J, Dong Q, Fang Z, Chen X, Lu H, Wang K, Yin Y, Cai X, Zhao N, Chen J, Zen K, Zhang J, et al. Identification of miRNAs that are associated with tumor metastasis in neuroblastoma. Cancer Biol Ther. 2010;9:446–452. doi: 10.4161/cbt.9.6.10894. [DOI] [PubMed] [Google Scholar]
- Haase GM, Perez C, Atkinson JB. Current aspects of biology, risk assessment, and treatment of neuroblastoma. Semin Surg Oncol. 1999;16:91–104. doi: 10.1002/(sici)1098-2388(199903)16:2<91::aid-ssu3>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Haber M, Bordow SB, Gilbert J, Madafiglio J, Kavallaris M, Marshall GM, Mechetner EB, Fruehauf JP, Tee L, Cohn SL, Salwen H, Schmidt ML, et al. Altered expression of the MYCN oncogene modulates MRP gene expression and response to cytotoxic drugs in neuroblastoma cells. Oncogene. 1999;18:2777–2782. doi: 10.1038/sj.onc.1202859. [DOI] [PubMed] [Google Scholar]
- Haber M, Smith J, Bordow SB, Flemming C, Cohn SL, London WB, Marshall GM, Norris MD. Association of high-level MRP1 expression with poor clinical outcome in a large prospective study of primary neuroblastoma. J Clin Oncol. 2006;24:1546–1553. doi: 10.1200/JCO.2005.01.6196. [DOI] [PubMed] [Google Scholar]
- Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH, Meyerson M, Weinberg RA. Inhibition of telomerase limits the growth of human cancer cells. Nat Med. 1999;5:1164–1170. doi: 10.1038/13495. [DOI] [PubMed] [Google Scholar]
- Harada K, Toyooka S, Shivapurkar N, Maitra A, Reddy JL, Matta H, Miyajima K, Timmons CF, Tomlinson GE, Mastrangelo D, Hay RJ, Chaudhary PM, et al. Deregulation of caspase 8 and 10 expression in pediatric tumors and cell lines. Cancer Res. 2002;62:5897–5901. [PubMed] [Google Scholar]
- Harris RG, White E, Phillips ES, Lillycrop KA. The expression of the developmentally regulated proto-oncogene Pax-3 is modulated by N-Myc. J Biol Chem. 2002;277:34815–34825. doi: 10.1074/jbc.M109609200. [DOI] [PubMed] [Google Scholar]
- Hatzi E, Murphy C, Zoephel A, Ahorn H, Tontsch U, Bamberger AM, Yamauchi-Takihara K, Schweigerer L, Fotsis T. N-myc oncogene overexpression down-regulates leukemia inhibitory factor in neuroblastoma. Eur J Biochem. 2002;269:3732–3741. doi: 10.1046/j.1432-1033.2002.03066.x. [DOI] [PubMed] [Google Scholar]
- Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y, Takahashi T. A polycistronic micro-RNA cluster, miR-17–92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005;65:9628–9632. doi: 10.1158/0008-5472.CAN-05-2352. [DOI] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, He X, Lim LP, de SE, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Henson ES, Gibson SB. Surviving cell death through epidermal growth factor (EGF) signal transduction pathways: Implications for cancer therapy. Cell Signal. 2006;18:2089–2097. doi: 10.1016/j.cellsig.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Hirai M, Yoshida S, Kashiwagi H, Kawamura T, Ishikawa T, Kaneko M, Ohkawa H, Nakagawara A, Miwa M, Uchida K. 1q23 gain is associated with progressive neuroblastoma resistant to aggressive treatment. Genes Chromosom Cancer. 1999;25:261–269. doi: 10.1002/(sici)1098-2264(199907)25:3<261::aid-gcc8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Hiyama E, Hiyama K, Ohtsu K, Yamaoka H, Ichikawa T, Shay JW, Yokoyama T. Telomerase activity in neuroblastoma: Is it a prognostic indicator of clinical behaviour? Eur J Cancer. 1997;33:1932–1936. doi: 10.1016/s0959-8049(97)00226-8. [DOI] [PubMed] [Google Scholar]
- Ho R, Eggert A, Hishiki T, Minturn JE, Ikegaki N, Foster P, Camoratto AM, Evans AE, Brodeur GM. Resistance to chemotherapy mediated by TrkB in neuroblastomas. Cancer Res. 2002;62:6462–6466. [PubMed] [Google Scholar]
- Ho R, Minturn JE, Hishiki T, Zhao H, Wang Q, Cnaan A, Maris J, Evans AE, Brodeur GM. Proliferation of human neuroblastomas mediated by the epidermal growth factor receptor. Cancer Res. 2005;65:9868–9875. doi: 10.1158/0008-5472.CAN-04-2426. [DOI] [PubMed] [Google Scholar]
- Hopkins-Donaldson S, Bodmer JL, Bourloud KB, Brognara CB, Tschopp J, Gross N. Loss of caspase-8 expression in neuroblastoma is related to malignancy and resistance to TRAIL-induced apoptosis. Med Pediatr Oncol. 2000;35:608–611. doi: 10.1002/1096-911x(20001201)35:6<608::aid-mpo25>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Hopkins-Donaldson S, Ziegler A, Kurtz S, Bigosch C, Kandioler D, Ludwig C, Zangemeister-Wittke U, Stahel R. Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation. Cell Death Differ. 2003;10:356–364. doi: 10.1038/sj.cdd.4401157. [DOI] [PubMed] [Google Scholar]
- Howard MJ, Stanke M, Schneider C, Wu X, Rohrer H. The transcription factor dHAND is a downstream effector of BMPs in sympathetic neuron specification. Development. 2000;127:4073–4081. doi: 10.1242/dev.127.18.4073. [DOI] [PubMed] [Google Scholar]
- Huber K. The sympathoadrenal cell lineage: Specification, diversification, and new perspectives. Dev Biol. 2006;298:335–343. doi: 10.1016/j.ydbio.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Huber K, Franke A, Bruhl B, Krispin S, Ernsberger U, Schober A, von Bohlen und HO, Rohrer H, Kalcheim C, Unsicker K. Persistent expression of BMP-4 in embryonic chick adrenal cortical cells and its role in chromaffin cell development. Neural Dev. 2008;3:28. doi: 10.1186/1749-8104-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley SP, Clary DO, Copie V, Lefcort F. Anaplastic lymphoma kinase is dynamically expressed on subsets of motor neurons and in the peripheral nervous system. J Comp Neurol. 2006;495:202–212. doi: 10.1002/cne.20887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlin PJ. N-Myc functions in transcription and development. Birth Defects Res C Embryo Today. 2005;75:340–352. doi: 10.1002/bdrc.20059. [DOI] [PubMed] [Google Scholar]
- Ichimiya S, Nimura Y, Kageyama H, Takada N, Sunahara M, Shishikura T, Nakamura Y, Sakiyama S, Seki N, Ohira M, Kaneko Y, McKeon F, et al. p73 at chromosome 1p36.3 is lost in advanced stage neuroblastoma but its mutation is infrequent. Oncogene. 1999;18:1061–1066. doi: 10.1038/sj.onc.1202390. [DOI] [PubMed] [Google Scholar]
- Ikegaki N, Katsumata M, Tsujimoto Y, Nakagawara A, Brodeur GM. Relationship between bcl-2 and myc gene expression in human neuroblastoma. Cancer Lett. 1995;91:161–168. doi: 10.1016/0304-3835(95)03746-j. [DOI] [PubMed] [Google Scholar]
- Islam A, Kageyama H, Takada N, Kawamoto T, Takayasu H, Isogai E, Ohira M, Hashizume K, Kobayashi H, Kaneko Y, Nakagawara A. High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene. 2000;19:617–623. doi: 10.1038/sj.onc.1203358. [DOI] [PubMed] [Google Scholar]
- Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14:439–449. doi: 10.1038/sj.onc.1200849. [DOI] [PubMed] [Google Scholar]
- Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, De PL, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, Valteau-Couanet D, Frebourg T, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- Jinbo T, Iwamura Y, Kaneko M, Sawaguchi S. Coamplification of the L-myc and N-myc oncogenes in a neuroblastoma cell line. Jpn J Cancer Res. 1989;80:299–301. doi: 10.1111/j.1349-7006.1989.tb02309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen JI, Pettersen I, Ponthan F, Sveinbjornsson B, Flaegstad T, Kogner P. Synergistic induction of apoptosis in neuroblastoma cells using a combination of cytostatic drugs with interferon-gamma and TRAIL. Int J Oncol. 2004;25:1849–1857. [PubMed] [Google Scholar]
- Johnsen JI, Kogner P, Albihn A, Henriksson MA. Embryonal neural tumours and cell death. Apoptosis. 2009;14:424–438. doi: 10.1007/s10495-009-0325-y. [DOI] [PubMed] [Google Scholar]
- Kang Y, Massague J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell. 2004;118:277–279. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Vaux DL. Alterations in the apoptotic machinery and their potential role in anticancer drug resistance. Oncogene. 2003;22:7414–7430. doi: 10.1038/sj.onc.1206945. [DOI] [PubMed] [Google Scholar]
- Kelleher FC, McDermott R. The emerging pathogenic and therapeutic importance of the anaplastic lymphoma kinase gene. Eur J Cancer. 2010;46:2357–2368. doi: 10.1016/j.ejca.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Keshelava N, Zuo JJ, Chen P, Waidyaratne SN, Luna MC, Gomer CJ, Triche TJ, Reynolds CP. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001;61:6185–6193. [PubMed] [Google Scholar]
- Kim CJ, Matsuo T, Lee KH, Thiele CJ. Up-regulation of insulin-like growth factor-II expression is a feature of TrkA but not TrkB activation in SH-SY5Y neuroblastoma cells. Am J Pathol. 1999;155:1661–1670. doi: 10.1016/S0002-9440(10)65481-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Lee JW, Soung YH, Park WS, Kim SY, Lee JH, Park JY, Cho YG, Kim CJ, Jeong SW, Nam SW, Kim SH, et al. Inactivating mutations of caspase-8 gene in colorectal carcinomas. Gastroenterology. 2003;125:708–715. doi: 10.1016/s0016-5085(03)01059-x. [DOI] [PubMed] [Google Scholar]
- Klefstrom J, Verschuren EW, Evan G. c-Myc augments the apoptotic activity of cytosolic death receptor signaling proteins by engaging the mitochondrial apoptotic pathway. J Biol Chem. 2002;277:43224–43232. doi: 10.1074/jbc.M206967200. [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699–2712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl NE, Kanda N, Schreck RR, Bruns G, Latt SA, Gilbert F, Alt FW. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell. 1983;35:359–367. doi: 10.1016/0092-8674(83)90169-1. [DOI] [PubMed] [Google Scholar]
- Krelin Y, Zhang L, Kang TB, Appel E, Kovalenko A, Wallach D. Caspase-8 deficiency facilitates cellular transformation in vitro. Cell Death Differ. 2008;15:1350–1355. doi: 10.1038/cdd.2008.88. [DOI] [PubMed] [Google Scholar]
- Kulesa P, Ellies DL, Trainor PA. Comparative analysis of neural crest cell death, migration, and function during vertebrate embryogenesis. Dev Dyn. 2004;229:14–29. doi: 10.1002/dvdy.10485. [DOI] [PubMed] [Google Scholar]
- Kutlik MT, Ayhan A, Gogus S, Yalcin B, Caglar M, Buyukpamukcu M. Glutathione S-transferase and P-glycoprotein expressions in neuroblastoma. Pediatr Hematol Oncol. 2002;19:337–345. doi: 10.1080/08880010290057354. [DOI] [PubMed] [Google Scholar]
- LaBrosse EH, Comoy E, Bohuon C, Zucker JM, Schweisguth O. Catecholamine metabolism in neuroblastoma. J Natl Cancer Inst. 1976;57:633–638. doi: 10.1093/jnci/57.3.633. [DOI] [PubMed] [Google Scholar]
- Lahti JM, Teitz T, Stupack DG. Does integrin-mediated cell death confer tissue tropism in metastasis? Cancer Res. 2006;66:5981–5984. doi: 10.1158/0008-5472.CAN-06-0131. [DOI] [PubMed] [Google Scholar]
- Lallier TE, Bronner-Fraser M. A spatial and temporal analysis of dorsal root and sympathetic ganglion formation in the avian embryo. Dev Biol. 1988;127:99–112. doi: 10.1016/0012-1606(88)90192-3. [DOI] [PubMed] [Google Scholar]
- Lasorella A, Boldrini R, Dominici C, Donfrancesco A, Yokota Y, Inserra A, Iavarone A. Id2 is critical for cellular proliferation and is the oncogenic effector of N-myc in human neuroblastoma. Cancer Res. 2002;62:301–306. [PubMed] [Google Scholar]
- Lastowska M, Nacheva E, McGuckin A, Curtis A, Grace C, Pearson A, Bown N. Comparative genomic hybridization study of primary neuroblastoma tumors. United Kingdom Children’s Cancer Study Group, Genes Chromosomes. Cancer. 1997;18:162–169. [PubMed] [Google Scholar]
- Le Dourin NM, Kalcheim G. The Neural Crest. 2. Cambridge University Press; 1999. [Google Scholar]
- Lee WH, Murphree AL, Benedict WF. Expression and amplification of the N-myc gene in primary retinoblastoma. Nature. 1984;309:458–460. doi: 10.1038/309458a0. [DOI] [PubMed] [Google Scholar]
- Lutz W, Stohr M, Schurmann J, Wenzel A, Lohr A, Schwab M. Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene. 1996;13:803–812. [PubMed] [Google Scholar]
- Lutz W, Fulda S, Jeremias I, Debatin KM, Schwab M. MycN and IFNgamma cooperate in apoptosis of human neuroblastoma cells. Oncogene. 1998;17:339–346. doi: 10.1038/sj.onc.1200201. [DOI] [PubMed] [Google Scholar]
- Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- Mac SM, D’Cunha CA, Farnham PJ. Direct recruitment of N-myc to target gene promoters. Mol Carcinog. 2000;29:76–86. [PubMed] [Google Scholar]
- Maris JM. The biologic basis for neuroblastoma heterogeneity and risk stratification. Curr Opin Pediatr. 2005;17:7–13. doi: 10.1097/01.mop.0000150631.60571.89. [DOI] [PubMed] [Google Scholar]
- Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17:2264–2279. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- Maris JM, Weiss MJ, Mosse Y, Hii G, Guo C, White PS, Hogarty MD, Mirensky T, Brodeur GM, Rebbeck TR, Urbanek M, Shusterman S. Evidence for a hereditary neuroblastoma predisposition locus at chromosome 16p12–13. Cancer Res. 2002;62:6651–6658. [PubMed] [Google Scholar]
- Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- Marshall B, Isidro G, Martins AG, Boavida MG. Loss of heterozygosity at chromosome 9p21 in primary neuroblastomas: Evidence for two deleted regions. Cancer Genet Cytogenet. 1997;96:134–139. doi: 10.1016/s0165-4608(96)00300-7. [DOI] [PubMed] [Google Scholar]
- Martin DM, Yee D, Carlson RO, Feldman EL. Gene expression of the insulin-like growth factors and their receptors in human neuroblastoma cell lines. Brain Res Mol Brain Res. 1992;15:241–246. doi: 10.1016/0169-328x(92)90114-q. [DOI] [PubMed] [Google Scholar]
- Martinez R, Setien F, Voelter C, Casado S, Quesada MP, Schackert G, Esteller M. CpG island promoter hypermethylation of the pro-apoptotic gene caspase-8 is a common hallmark of relapsed glioblastoma multiforme. Carcinogenesis. 2007;28:1264–1268. doi: 10.1093/carcin/bgm014. [DOI] [PubMed] [Google Scholar]
- Matsuo N, Shiraha H, Fujikawa T, Takaoka N, Ueda N, Tanaka S, Nishina S, Nakanishi Y, Uemura M, Takaki A, Nakamura S, Kobayashi Y, et al. Twist expression promotes migration and invasion in hepatocellular carcinoma. BMC Cancer. 2009;9:240. doi: 10.1186/1471-2407-9-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay KK. Neuroblastoma: A clinical challenge and biologic puzzle. CA Cancer J Clin. 1995;45:179–192. doi: 10.3322/canjclin.45.3.179. [DOI] [PubMed] [Google Scholar]
- Matthay KK, Perez C, Seeger RC, Brodeur GM, Shimada H, Atkinson JB, Black CT, Gerbing R, Haase GM, Stram DO, Swift P, Lukens JN. Successful treatment of stage III neuroblastoma based on prospective biologic staging: A Children’s Cancer Group study. J Clin Oncol. 1998;16:1256–1264. doi: 10.1200/JCO.1998.16.4.1256. [DOI] [PubMed] [Google Scholar]
- Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, Swift P, Shimada H, Black CT, Brodeur GM, Gerbing RB, Reynolds CP. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N Engl J Med. 1999;341:1165–1173. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- McConkey DJ, Choi W, Marquis L, Martin F, Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A, Siefker-Radtke A, Dinney C. Role of epithelial-to-mesenchymal transition (EMT) in drug sensitivity and metastasis in bladder cancer. Cancer Metastasis Rev. 2009;28:335–344. doi: 10.1007/s10555-009-9194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan R, Teddy JM, Kasemeier-Kulesa JC, Romine MH, Kulesa PM. Vascular endothelial growth factor (VEGF) regulates cranial neural crest migration in vivo. Dev Biol. 2010;339:114–125. doi: 10.1016/j.ydbio.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meddeb M, Danglot G, Chudoba I, Venuat AM, Benard J, vet-Loiseau H, Vasseur B, Le PD, Terrier-Lacombe MJ, Hartmann O, Bernheim A. Additional copies of a 25 Mb chromosomal region originating from 17q23.1–17qter are present in 90% of high-grade neuroblastomas, Genes Chromosomes. Cancer. 1996;17:156–165. doi: 10.1002/(SICI)1098-2264(199611)17:3<156::AID-GCC3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Mejia MC, Navarro S, Pellin A, Castel V, Llombart-Bosch A. Study of bcl-2 protein expression and the apoptosis phenomenon in neuroblastoma. Anticancer Res. 1998;18:801–806. [PubMed] [Google Scholar]
- Meltzer SJ, O’Doherty SP, Frantz CN, Smolinski K, Yin J, Cantor AB, Liu J, Valentine M, Brodeur GM, Berg PE. Allelic imbalance on chromosome 5q predicts long-term survival in neuroblastoma. Br J Cancer. 1996;74:1855–1861. doi: 10.1038/bjc.1996.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, Kaneko M, London WB, Matthay KK, Nuchtern JG, von SD, Simon T, et al. The International Neuroblastoma Risk Group (INRG) staging system: An INRG Task Force report. J Clin Oncol. 2009;27:298–303. doi: 10.1200/JCO.2008.16.6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora J, Cheung NK, Chen L, Qin J, Gerald W. Loss of heterozygosity at 19q13.3 is associated with locally aggressive neuroblastoma. Clin Cancer Res. 2001;7:1358–1361. [PubMed] [Google Scholar]
- Morales AV, Barbas JA, Nieto MA. How to become neural crest: From segregation to delamination. Semin Cell Dev Biol. 2005;16:655–662. doi: 10.1016/j.semcdb.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, Witte DP. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK) Oncogene. 1997;14:2175–2188. doi: 10.1038/sj.onc.1201062. [DOI] [PubMed] [Google Scholar]
- Mosse YP, Laudenslager M, Khazi D, Carlisle AJ, Winter CL, Rappaport E, Maris JM. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet. 2004;75:727–730. doi: 10.1086/424530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosse YP, Wood A, Maris JM. Inhibition of ALK signaling for cancer therapy. Clin Cancer Res. 2009;15:5609–5614. doi: 10.1158/1078-0432.CCR-08-2762. [DOI] [PubMed] [Google Scholar]
- Motegi A, Fujimoto J, Kotani M, Sakuraba H, Yamamoto T. ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. J Cell Sci. 2004;117:3319–3329. doi: 10.1242/jcs.01183. [DOI] [PubMed] [Google Scholar]
- Muhlethaler-Mottet A, Balmas K, Auderset K, Joseph JM, Gross N. Restoration of TRAIL-induced apoptosis in a caspase-8-deficient neuroblastoma cell line by stable re-expression of caspase-8. Ann NY Acad Sci. 2003;1010:195–199. doi: 10.1196/annals.1299.033. [DOI] [PubMed] [Google Scholar]
- Munchar MJ, Sharifah NA, Jamal R, Looi LM. CD44s expression correlated with the International Neuroblastoma Pathology Classification (Shimada system) for neuroblastic tumours. Pathology. 2003;35:125–129. [PubMed] [Google Scholar]
- Munirajan AK, Ando K, Mukai A, Takahashi M, Suenaga Y, Ohira M, Koda T, Hirota T, Ozaki T, Nakagawara A. KIF1Bbeta functions as a haploinsufficient tumor suppressor gene mapped to chromosome 1p36.2 by inducing apoptotic cell death. J Biol Chem. 2008;283:24426–24434. doi: 10.1074/jbc.M802316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawara A. Neural crest development and neuroblastoma: The genetic and biological link. Prog Brain Res. 2004;146:233–242. doi: 10.1016/s0079-6123(03)46015-9. [DOI] [PubMed] [Google Scholar]
- Nakagawara A, Ohira M. Comprehensive genomics linking between neural development and cancer: Neuroblastoma as a model. Cancer Lett. 2004;204:213–224. doi: 10.1016/S0304-3835(03)00457-9. [DOI] [PubMed] [Google Scholar]
- Nakagawara A, Arima M, Azar CG, Scavarda NJ, Brodeur GM. Inverse relationship between trk expression and N-myc amplification in human neuroblastomas. Cancer Res. 1992;52:1364–1368. [PubMed] [Google Scholar]
- Nakagawara A, rima-Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, Brodeur GM. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med. 1993;328:847–854. doi: 10.1056/NEJM199303253281205. [DOI] [PubMed] [Google Scholar]
- Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994a;14:759–767. doi: 10.1128/mcb.14.1.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawara A, rima-Nakagawara M, Azar CG, Scavarda NJ, Brodeur GM. Clinical significance of expression of neurotrophic factors and their receptors in neuroblastoma. Prog Clin Biol Res. 1994b;385:155–161. [PubMed] [Google Scholar]
- Nieminen AI, Partanen JI, Klefstrom J. c-Myc blazing a trail of death: Coupling of the mitochondrial and death receptor apoptosis pathways by c-Myc. Cell Cycle. 2007;6:2464–2472. doi: 10.4161/cc.6.20.4917. [DOI] [PubMed] [Google Scholar]
- Norris MD, Bordow SB, Marshall GM, Haber PS, Cohn SL, Haber M. Expression of the gene for multidrug-resistance-associated protein and outcome in patients with neuroblastoma. N Engl J Med. 1996;334:231–238. doi: 10.1056/NEJM199601253340405. [DOI] [PubMed] [Google Scholar]
- Norris MD, Smith J, Tanabe K, Tobin P, Flemming C, Scheffer GL, Wielinga P, Cohn SL, London WB, Marshall GM, Allen JD, Haber M. Expression of multidrug transporter MRP4/ABCC4 is a marker of poor prognosis in neuroblastoma and confers resistance to irinotecan in vitro. Mol Cancer Ther. 2005;4:547–553. doi: 10.1158/1535-7163.MCT-04-0161. [DOI] [PubMed] [Google Scholar]
- Nowak K, Kerl K, Fehr D, Kramps C, Gessner C, Killmer K, Samans B, Berwanger B, Christiansen H, Lutz W. BMI1 is a target gene of E2F-1 and is strongly expressed in primary neuroblastomas. Nucleic Acids Res. 2006;34:1745–1754. doi: 10.1093/nar/gkl119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai H, Takenobu H, Nakagawa A, Yamaguchi Y, Kimura M, Ohira M, Okimoto Y, Fujimura Y, Koseki H, Kohno Y, Nakagawara A, Kamijo T. Bmi1 is a MYCN target gene that regulates tumorigenesis through repression of KIF1Bbeta and TSLC1 in neuroblastoma. Oncogene. 2010;29:2681–2690. doi: 10.1038/onc.2010.22. [DOI] [PubMed] [Google Scholar]
- Ohali A, Avigad S, Ash S, Goshen Y, Luria D, Feinmesser M, Zaizov R, Yaniv I. Telomere length is a prognostic factor in neuroblastoma. Cancer. 2006;107:1391–1399. doi: 10.1002/cncr.22132. [DOI] [PubMed] [Google Scholar]
- Okabe-Kado J, Kasukabe T, Honma Y, Hanada R, Nakagawara A, Kaneko Y. Clinical significance of serum NM23-H1 protein in neuroblastoma. Cancer Sci. 2005;96:653–660. doi: 10.1111/j.1349-7006.2005.00091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osajima-Hakomori Y, Miyake I, Ohira M, Nakagawara A, Nakagawa A, Sakai R. Biological role of anaplastic lymphoma kinase in neuroblastoma. Am J Pathol. 2005;167:213–222. doi: 10.1016/S0002-9440(10)62966-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RH, Vernersson E, Grabbe C, Hallberg B. Anaplastic lymphoma kinase: Signalling in development and disease. Biochem J. 2009;420:345–361. doi: 10.1042/BJ20090387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JR, Eggert A, Caron H. Neuroblastoma: Biology, prognosis, and treatment. Pediatr Clin North Am. 2008;55:97–120. doi: 10.1016/j.pcl.2007.10.014. (x) [DOI] [PubMed] [Google Scholar]
- Passoni L, Longo L, Collini P, Coluccia AM, Bozzi F, Podda M, Gregorio A, Gambini C, Garaventa A, Pistoia V, Del GF, Tonini GP, et al. Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Cancer Res. 2009;69:7338–7346. doi: 10.1158/0008-5472.CAN-08-4419. [DOI] [PubMed] [Google Scholar]
- Pearson AD, Pinkerton CR, Lewis IJ, Imeson J, Ellershaw C, Machin D. High-dose rapid and standard induction chemotherapy for patients aged over 1 year with stage 4 neuroblastoma: A randomised trial. Lancet Oncol. 2008;9:247–256. doi: 10.1016/S1470-2045(08)70069-X. [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Khan M, Evan G. c-MYC: More than just a matter of life and death. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- Perini G, Diolaiti D, Porro A, Della VG. In vivo transcriptional regulation of N-Myc target genes is controlled by E-box methylation. Proc Natl Acad Sci USA. 2005;102:12117–12122. doi: 10.1073/pnas.0409097102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perri P, Longo L, Cusano R, McConville CM, Rees SA, Devoto M, Conte M, Ferrara GB, Seri M, Romeo G, Tonini GP. Weak linkage at 4p16 to predisposition for human neuroblastoma. Oncogene. 2002;21:8356–8360. doi: 10.1038/sj.onc.1206009. [DOI] [PubMed] [Google Scholar]
- Pingoud-Meier C, Lang D, Janss AJ, Rorke LB, Phillips PC, Shalaby T, Grotzer MA. Loss of caspase-8 protein expression correlates with unfavorable survival outcome in childhood medulloblastoma. Clin Cancer Res. 2003;9:6401–6409. [PubMed] [Google Scholar]
- Raabe EH, Laudenslager M, Winter C, Wasserman N, Cole K, LaQuaglia M, Maris DJ, Mosse YP, Maris JM. Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene. 2008;27:469–476. doi: 10.1038/sj.onc.1210659. [DOI] [PubMed] [Google Scholar]
- Ramani P, Lu QL. Expression of bcl-2 gene product in neuroblastoma. J Pathol. 1994;172:273–278. doi: 10.1002/path.1711720308. [DOI] [PubMed] [Google Scholar]
- Reed JC. Proapoptotic multidomain Bcl-2/Bax-family proteins: Mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 2006;13:1378–1386. doi: 10.1038/sj.cdd.4401975. [DOI] [PubMed] [Google Scholar]
- Reed JC, Tomaselli KJ. Drug discovery opportunities from apoptosis research. Curr Opin Biotechnol. 2000;11:586–592. doi: 10.1016/s0958-1669(00)00148-8. [DOI] [PubMed] [Google Scholar]
- Reissmann E, Ernsberger U, Francis-West PH, Rueger D, Brickell PM, Rohrer H. Involvement of bone morphogenetic protein-4 and bone morpho-genetic protein-7 in the differentiation of the adrenergic phenotype in developing sympathetic neurons. Development. 1996;122:2079–2088. doi: 10.1242/dev.122.7.2079. [DOI] [PubMed] [Google Scholar]
- Reiter JL, Brodeur GM. High-resolution mapping of a 130-kb core region of the MYCN amplicon in neuroblastomas. Genomics. 1996;32:97–103. doi: 10.1006/geno.1996.0081. [DOI] [PubMed] [Google Scholar]
- Reiter JL, Brodeur GM. MYCN is the only highly expressed gene from the core amplified domain in human neuroblastomas. Genes Chromosom Cancer. 1998;23:134–140. doi: 10.1002/(sici)1098-2264(199810)23:2<134::aid-gcc6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Reynolds CP, Zuo JJ, Kim NW, Wang H, Lukens JN, Matthay KK, Seeger RC. Telomerase expression in primary neuroblastomas. Eur J Cancer. 1997;33:1929–1931. doi: 10.1016/s0959-8049(97)00287-6. [DOI] [PubMed] [Google Scholar]
- Ribatti D, Marimpietri D, Pastorino F, Brignole C, Nico B, Vacca A, Ponzoni M. Angiogenesis in neuroblastoma. Ann NY Acad Sci. 2004;1028:133–142. doi: 10.1196/annals.1322.014. [DOI] [PubMed] [Google Scholar]
- Ruffins S, Artinger KB, Bronner-Fraser M. Early migrating neural crest cells can form ventral neural tube derivatives when challenged by transplantation. Dev Biol. 1998;203:295–304. doi: 10.1006/dbio.1998.8973. [DOI] [PubMed] [Google Scholar]
- Saito-Ohara F, Imoto I, Inoue J, Hosoi H, Nakagawara A, Sugimoto T, Inazawa J. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res. 2003;63:1876–1883. [PubMed] [Google Scholar]
- Salvesen GS, Abrams JM. Caspase activation—Stepping on the gas or releasing the brakes?. Lessons from humans and flies. Oncogene. 2004;23:2774–2784. doi: 10.1038/sj.onc.1207522. [DOI] [PubMed] [Google Scholar]
- Sawada T. Past and future of neuroblastoma screening in Japan. Am J Pediatr Hematol Oncol. 1992;14:320–326. [PubMed] [Google Scholar]
- Sawai S, Shimono A, Hanaoka K, Kondoh H. Embryonic lethality resulting from disruption of both N-myc alleles in mouse zygotes. New Biol. 1991;3:861–869. [PubMed] [Google Scholar]
- Sawai S, Shimono A, Wakamatsu Y, Palmes C, Hanaoka K, Kondoh H. Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development. 1993;117:1445–1455. doi: 10.1242/dev.117.4.1445. [DOI] [PubMed] [Google Scholar]
- Scaruffi P, Valent A, Schramm A, Astrahantseff K, Eggert A, Tonini GP. Application of microarray-based technology to neuroblastoma. Cancer Lett. 2005;228:13–20. doi: 10.1016/j.canlet.2005.01.052. [DOI] [PubMed] [Google Scholar]
- Schlisio S, Kenchappa RS, Vredeveld LC, George RE, Stewart R, Greulich H, Shahriari K, Nguyen NV, Pigny P, Dahia PL, Pomeroy SL, Maris JM, et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008;22:884–893. doi: 10.1101/gad.1648608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ML, Lukens JN, Seeger RC, Brodeur GM, Shimada H, Gerbing RB, Stram DO, Perez C, Haase GM, Matthay KK. Biologic factors determine prognosis in infants with stage IV neuroblastoma: A prospective Children’s Cancer Group study. J Clin Oncol. 2000;18:1260–1268. doi: 10.1200/JCO.2000.18.6.1260. [DOI] [PubMed] [Google Scholar]
- Schneider C, Wicht H, Enderich J, Wegner M, Rohrer H. Bone morphogenetic proteins are required in vivo for the generation of sympathetic neurons. Neuron. 1999;24:861–870. doi: 10.1016/s0896-6273(00)81033-8. [DOI] [PubMed] [Google Scholar]
- Schonherr C, Yang HL, Vigny M, Palmer RH, Hallberg B. Anaplastic lymphoma kinase activates the small GTPase Rap1 via the Rap1-specific GEF C3G in both neuroblastoma and PC12 cells. Oncogene. 2010;29:2817–2830. doi: 10.1038/onc.2010.27. [DOI] [PubMed] [Google Scholar]
- Schor NF. Neuroblastoma as a neurobiological disease. J Neurooncol. 1999;41:159–166. doi: 10.1023/a:1006171406740. [DOI] [PubMed] [Google Scholar]
- Schor NF. New approaches to pharmacotherapy of tumors of the nervous system during childhood and adolescence. Pharmacol Ther. 2009;122:44–55. doi: 10.1016/j.pharmthera.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte JH, Horn S, Otto T, Samans B, Heukamp LC, Eilers UC, Krause M, Astrahantseff K, Klein-Hitpass L, Buettner R, Schramm A, Christiansen H, et al. MYCN regulates oncogenic MicroRNAs in neuroblastoma. Int J Cancer. 2008;122:699–704. doi: 10.1002/ijc.23153. [DOI] [PubMed] [Google Scholar]
- Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, Brodeur G, Goldstein M, Trent J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. 1983;305:245–248. doi: 10.1038/305245a0. [DOI] [PubMed] [Google Scholar]
- Schwab M, Varmus HE, Bishop JM, Grzeschik KH, Naylor SL, Sakaguchi AY, Brodeur G, Trent J. Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature. 1984;308:288–291. doi: 10.1038/308288a0. [DOI] [PubMed] [Google Scholar]
- Seoane J, Le HV, Massague J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419:729–734. doi: 10.1038/nature01119. [DOI] [PubMed] [Google Scholar]
- Shah NM, Groves AK, Anderson DJ. Alternative neural crest cell fates are instructively promoted by TGFbeta superfamily members. Cell. 1996;85:331–343. doi: 10.1016/s0092-8674(00)81112-5. [DOI] [PubMed] [Google Scholar]
- Shi Y. Caspase activation: Revisiting the induced proximity model. Cell. 2004;117:855–858. doi: 10.1016/j.cell.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, Stram DO, Gerbing RB, Lukens JN, Matthay KK, Castleberry RP. The International Neuroblastoma Pathology Classification (the Shimada system) Cancer. 1999;86:364–372. [PubMed] [Google Scholar]
- Shimono R, Matsubara S, Takamatsu H, Fukushige T, Ozawa M. The expression of cadherins in human neuroblastoma cell lines and clinical tumors. Anticancer Res. 2000;20:917–923. [PubMed] [Google Scholar]
- Shohet JM, Hicks MJ, Plon SE, Burlingame SM, Stuart S, Chen SY, Brenner MK, Nuchtern JG. Minichromosome maintenance protein MCM7 is a direct target of the MYCN transcription factor in neuroblastoma. Cancer Res. 2002;62:1123–1128. [PubMed] [Google Scholar]
- Shtivelman E, Bishop JM. Expression of CD44 is repressed in neuroblastoma cells. Mol Cell Biol. 1991;11:5446–5453. doi: 10.1128/mcb.11.11.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon T, Spitz R, Faldum A, Hero B, Berthold F. New definition of low-risk neuroblastoma using stage, age, and 1p and MYCN status. J Pediatr Hematol Oncol. 2004;26:791–796. [PubMed] [Google Scholar]
- Slack A, Chen Z, Tonelli R, Pule M, Hunt L, Pession A, Shohet JM. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc Natl Acad Sci USA. 2005;102:731–736. doi: 10.1073/pnas.0405495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosic D, Olson EN. A new twist on twist–modulation of the NF-kappa B pathway. Cell Cycle. 2003;2:76–78. [PubMed] [Google Scholar]
- Soung YH, Lee JW, Kim SY, Jang J, Park YG, Park WS, Nam SW, Lee JY, Yoo NJ, Lee SH. CASPASE-8 gene is inactivated by somatic mutations in gastric carcinomas. Cancer Res. 2005;65:815–821. [PubMed] [Google Scholar]
- Souttou B, Carvalho NB, Raulais D, Vigny M. Activation of anaplastic lymphoma kinase receptor tyrosine kinase induces neuronal differentiation through the mitogen-activated protein kinase pathway. J Biol Chem. 2001;276:9526–9531. doi: 10.1074/jbc.M007333200. [DOI] [PubMed] [Google Scholar]
- Stanton BR, Perkins AS, Tessarollo L, Sassoon DA, Parada LF. Loss of N-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev. 1992;6:2235–2247. doi: 10.1101/gad.6.12a.2235. [DOI] [PubMed] [Google Scholar]
- Straub JA, Sholler GL, Nishi R. Embryonic sympathoblasts transiently express TrkB in vivo and proliferate in response to brain-derived neurotrophic factor in vitro. BMC Dev Biol. 2007;7:10. doi: 10.1186/1471-213X-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strieder V, Lutz W. E2F proteins regulate MYCN expression in neuroblastomas. J Biol Chem. 2003;278:2983–2989. doi: 10.1074/jbc.M207596200. [DOI] [PubMed] [Google Scholar]
- Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, Lahti JM, Cheresh DA. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. 2006;439:95–99. doi: 10.1038/nature04323. [DOI] [PubMed] [Google Scholar]
- Sugiura Y, Shimada H, Seeger RC, Laug WE, DeClerck YA. Matrix metalloproteinases-2 and -9 are expressed in human neuroblastoma: Contribution of stromal cells to their production and correlation with metastasis. Cancer Res. 1998;58:2209–2216. [PubMed] [Google Scholar]
- Suzuki T, Yokota J, Mugishima H, Okabe I, Ookuni M, Sugimura T, Terada M. Frequent loss of heterozygosity on chromosome 14q in neuroblastoma. Cancer Res. 1989;49:1095–1098. [PubMed] [Google Scholar]
- Svensson T, Ryden M, Schilling FH, Dominici C, Sehgal R, Ibanez CF, Kogner P. Coexpression of mRNA for the full-length neurotrophin receptor trk-C and trk-A in favourable neuroblastoma. Eur J Cancer. 1997;33:2058–2063. doi: 10.1016/s0959-8049(97)00290-6. [DOI] [PubMed] [Google Scholar]
- Tacconelli A, Farina AR, Cappabianca L, Desantis G, Tessitore A, Vetuschi A, Sferra R, Rucci N, Argenti B, Screpanti I, Gulino A, Mackay AR. TrkA alternative splicing: A regulated tumor-promoting switch in human neuroblastoma. Cancer Cell. 2004;6:347–360. doi: 10.1016/j.ccr.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Takita J, Hayashi Y, Takei K, Yamaguchi N, Hanada R, Yamamoto K, Yokota J. Allelic imbalance on chromosome 18 in neuroblastoma. Eur J Cancer. 2000;36:508–513. doi: 10.1016/s0959-8049(99)00342-1. [DOI] [PubMed] [Google Scholar]
- Takita J, Yang HW, Chen YY, Hanada R, Yamamoto K, Teitz T, Kidd V, Hayashi Y. Allelic imbalance on chromosome 2q and alterations of the caspase 8 gene in neuroblastoma. Oncogene. 2001;20:4424–4432. doi: 10.1038/sj.onc.1204521. [DOI] [PubMed] [Google Scholar]
- Tanabe KK, Ellis LM, Saya H. Expression of CD44R1 adhesion molecule in colon carcinomas and metastases. Lancet. 1993;341:725–726. doi: 10.1016/0140-6736(93)90490-8. [DOI] [PubMed] [Google Scholar]
- Teillet MA, Kalcheim C, Le Douarin NM. Formation of the dorsal root ganglia in the avian embryo: Segmental origin and migratory behavior of neural crest progenitor cells. Dev Biol. 1987;120:329–347. doi: 10.1016/0012-1606(87)90236-3. [DOI] [PubMed] [Google Scholar]
- Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med. 2000;6:529–535. doi: 10.1038/75007. [DOI] [PubMed] [Google Scholar]
- Teitz T, Lahti JM, Kidd VJ. Aggressive childhood neuroblastomas do not express caspase-8: An important component of programmed cell death. J Mol Med. 2001;79:428–436. doi: 10.1007/s001090100233. [DOI] [PubMed] [Google Scholar]
- Teitz T, Stupack DG, Lahti JM. Halting neuroblastoma metastasis by controlling integrin-mediated death. Cell Cycle. 2006;5:681–685. doi: 10.4161/cc.5.7.2615. [DOI] [PubMed] [Google Scholar]
- Tekautz TM, Zhu K, Grenet J, Kaushal D, Kidd VJ, Lahti JM. Evaluation of IFN-gamma effects on apoptosis and gene expression in neuroblastoma–preclinical studies. Biochim Biophys Acta. 2006;1763:1000–1010. doi: 10.1016/j.bbamcr.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Thompson EB. The many roles of c-Myc in apoptosis. Annu Rev Physiol. 1998;60:575–600. doi: 10.1146/annurev.physiol.60.1.575. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Seifried BA, Kyemba SK, Jensen SJ, Guo C, Maris JM, Brodeur GM, Stram DO, Seeger RC, Gerbing R, Matthay KK, Matise TC, et al. Loss of heterozygosity for chromosome 14q in neuroblastoma. Med Pediatr Oncol. 2001;36:28–31. doi: 10.1002/1096-911X(20010101)36:1<28::AID-MPO1008>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Treins C, Giorgetti-Peraldi S, Murdaca J, Monthouel-Kartmann MN, Van OE. Regulation of hypoxia-inducible factor (HIF)-1 activity and expression of HIF hydroxylases in response to insulin-like growth factor I. Mol Endocrinol. 2005;19:1304–1317. doi: 10.1210/me.2004-0239. [DOI] [PubMed] [Google Scholar]
- Trochet D, Bourdeaut F, Janoueix-Lerosey I, Deville A, de PL, Schleiermacher G, Coze C, Philip N, Frebourg T, Munnich A, Lyonnet S, Delattre O, et al. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet. 2004;74:761–764. doi: 10.1086/383253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trochet D, Hong SJ, Lim JK, Brunet JF, Munnich A, Kim KS, Lyonnet S, Goridis C, Amiel J. Molecular consequences of PHOX2B missense, frameshift and alanine expansion mutations leading to autonomic dysfunction. Hum Mol Genet. 2005;14:3697–3708. doi: 10.1093/hmg/ddi401. [DOI] [PubMed] [Google Scholar]
- Trochet D, Mathieu Y, Pontual L, Savarirayan R, Munnich A, Brunet JF, Lyonnet S, Goridis C, Amiel J. In vitro studies of non poly alanine PHOX2B mutations argue against a loss-of-function mechanism for congenital central hypoventilation. Hum Mutat. 2009;30:E421–E431. doi: 10.1002/humu.20923. [DOI] [PubMed] [Google Scholar]
- Tweddle DA, Pearson AD, Haber M, Norris MD, Xue C, Flemming C, Lunec J. The p53 pathway and its inactivation in neuroblastoma. Cancer Lett. 2003;197:93–98. doi: 10.1016/s0304-3835(03)00088-0. [DOI] [PubMed] [Google Scholar]
- Valdes-Mora F, Gomez Del PT, Bandres E, Cejas P, Ramirez De MA, Perez-Palacios R, Gallego-Ortega D, Garcia-Cabezas MA, Casado E, Larrauri J, Nistal M, Gonzalez-Baron M, et al. TWIST1 overexpression is associated with nodal invasion and male sex in primary colorectal cancer. Ann Surg Oncol. 2009;16:78–87. doi: 10.1245/s10434-008-0166-x. [DOI] [PubMed] [Google Scholar]
- Valentinis B, Baserga R. IGF-I receptor signalling in transformation and differentiation. Mol Pathol. 2001;54:133–137. doi: 10.1136/mp.54.3.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valsesia-Wittmann S, Magdeleine M, Dupasquier S, Garin E, Jallas AC, Combaret V, Krause A, Leissner P, Puisieux A. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell. 2004;6:625–630. doi: 10.1016/j.ccr.2004.09.033. [DOI] [PubMed] [Google Scholar]
- van Golen CM, Schwab TS, Kim B, Soules ME, Su OS, Fung K, van Golen KL, Feldman EL. Insulin-like growth factor-I receptor expression regulates neuroblastoma metastasis to bone. Cancer Res. 2006;66:6570–6578. doi: 10.1158/0008-5472.CAN-05-1448. [DOI] [PubMed] [Google Scholar]
- van Noesel MM, Versteeg R. Pediatric neuroblastomas: Genetic and epigenetic ‘danse macabre’. Gene. 2004;325:1–15. doi: 10.1016/j.gene.2003.09.042. [DOI] [PubMed] [Google Scholar]
- van Noesel MM, van Bezouw S, Salomons GS, Voute PA, Pieters R, Baylin SB, Herman JG, Versteeg R. Tumor-specific down-regulation of the tumor necrosis factor-related apoptosis-inducing ligand decoy receptors DcR1 and DcR2 is associated with dense promoter hypermethylation. Cancer Res. 2002;62:2157–2161. [PubMed] [Google Scholar]
- Van RN, Forus A, Myklebost O, Cheng NC, Versteeg R, Speleman F. Identification of two distinct chromosome 12-derived amplification units in neuroblastoma cell line NGP. Cancer Genet Cytogenet. 1995;82:151–154. doi: 10.1016/0165-4608(95)00034-m. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, Van RN, Van GM, Laureys G, Ambros P, Heimann P, Devalck C, Schuuring E, Brock P, Otten J, Gyselinck J, De PA, et al. Genetic heterogeneity of neuroblastoma studied by comparative genomic hybridization. Genes Chromosom Cancer. 1998;23:141–152. doi: 10.1002/(sici)1098-2264(199810)23:2<141::aid-gcc7>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernersson E, Khoo NK, Henriksson ML, Roos G, Palmer RH, Hallberg B. Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene Expr Patterns. 2006;6:448–461. doi: 10.1016/j.modgep.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Vitali R, Mancini C, Cesi V, Tanno B, Mancuso M, Bossi G, Zhang Y, Martinez RV, Calabretta B, Dominici C, Raschella G. Slug (SNAI2) down-regulation by RNA interference facilitates apoptosis and inhibits invasive growth in neuroblastoma preclinical models. Clin Cancer Res. 2008;14:4622–4630. doi: 10.1158/1078-0432.CCR-07-5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada RK, Seeger RC, Reynolds CP, Alloggiamento T, Yamashiro JM, Ruland C, Black AC, Rosenblatt JD. Cell type-specific expression and negative regulation by retinoic acid of the human N-myc promoter in neuroblastoma cells. Oncogene. 1992;7:711–717. [PubMed] [Google Scholar]
- Wagner LM, Danks MK. New therapeutic targets for the treatment of high-risk neuroblastoma. J Cell Biochem. 2009;107:46–57. doi: 10.1002/jcb.22094. [DOI] [PubMed] [Google Scholar]
- Wakamatsu Y, Watanabe Y, Nakamura H, Kondoh H. Regulation of the neural crest cell fate by N-myc: Promotion of ventral migration and neuronal differentiation. Development. 1997;124:1953–1962. doi: 10.1242/dev.124.10.1953. [DOI] [PubMed] [Google Scholar]
- Wartiovaara K, Barnabe-Heider F, Miller FD, Kaplan DR. N-myc promotes survival and induces S-phase entry of postmitotic sympathetic neurons. J Neurosci. 2002;22:815–824. doi: 10.1523/JNEUROSCI.22-03-00815.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT, Tsang P, Zhang Q, Thiele CJ, Slack A, Shohet J, Khan J. The MYCN oncogene is a direct target of miR-34a. Oncogene. 2008;27:5204–5213. doi: 10.1038/onc.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16:2985–2995. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007;26:5017–5022. doi: 10.1038/sj.onc.1210293. [DOI] [PubMed] [Google Scholar]
- WESTON JA. A radioautographic analysis of the migration and localization of trunk neural crest cells in the chick. Dev Biol. 1963;6:279–310. doi: 10.1016/0012-1606(63)90016-2. [DOI] [PubMed] [Google Scholar]
- Witt O, Deubzer HE, Lodrini M, Milde T, Oehme I. Targeting histone deacetylases in neuroblastoma. Curr Pharm Des. 2009;15:436–447. doi: 10.2174/138161209787315774. [DOI] [PubMed] [Google Scholar]
- Xue C, Haber M, Flemming C, Marshall GM, Lock RB, MacKenzie KL, Gurova KV, Norris MD, Gudkov AV. p53 determines multidrug sensitivity of childhood neuroblastoma. Cancer Res. 2007;67:10351–10360. doi: 10.1158/0008-5472.CAN-06-4345. [DOI] [PubMed] [Google Scholar]
- Yamashiro DJ, Nakagawara A, Ikegaki N, Liu XG, Brodeur GM. Expression of TrkC in favorable human neuroblastomas. Oncogene. 1996;12:37–41. [PubMed] [Google Scholar]
- Yang QW, Liu S, Tian Y, Salwen HR, Chlenski A, Weinstein J, Cohn SL. Methylation-associated silencing of the thrombospondin-1 gene in human neuroblastoma. Cancer Res. 2003;63:6299–6310. [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Yang Q, Kiernan CM, Tian Y, Salwen HR, Chlenski A, Brumback BA, London WB, Cohn SL. Methylation of CASP8, DCR2, and HIN-1 in neuroblastoma is associated with poor outcome. Clin Cancer Res. 2007;13:3191–3197. doi: 10.1158/1078-0432.CCR-06-2846. [DOI] [PubMed] [Google Scholar]
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman S, Chen HX, Matthay KK, Cohn SL, Maris JM, Sondel P. A phase III randomized trial of the chimeric anti-GD2 antibody ch14.18 with GM-CSF and IL2 as immunotherapy following dose intensive chemotherapy for high-risk neuroblastoma: Children’s Oncology Group (COG) study ANBL0032. J Clin Oncol. 2009;27:15s. (suppl; abstr 10067z) [Google Scholar]
- Zage PE, Kletzel M, Murray K, Marcus R, Castleberry R, Zhang Y, London WB, Kretschmar C. Outcomes of the POG 9340/9341/9342 trials for children with high-risk neuroblastoma: A report from the Children’s Oncology Group. Pediatr Blood Cancer. 2008;51:747–753. doi: 10.1002/pbc.21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li Y, Lai M. The microRNA network and tumor metastasis. Oncogene. 2010;29:937–948. doi: 10.1038/onc.2009.406. [DOI] [PubMed] [Google Scholar]
- Zimmerman KA, Yancopoulos GD, Collum RG, Smith RK, Kohl NE, Denis KA, Nau MM, Witte ON, Toran-Allerand D, Gee CE, et al. Differential expression of myc family genes during murine development. Nature. 1986;319:780–783. doi: 10.1038/319780a0. [DOI] [PubMed] [Google Scholar]