

Abstract
Objectives
To explore the potential prognostic role of family history (FH) of prostate cancer and prostate cancer risk single nucleotide polymorphisms (SNPs) in patients undergoing active surveillance (AS) for prostate cancer.
This is the first study to date, which has investigated the potential prognostic role of SNP profiles in an AS cohort
Patients and methods
FH data were collected from patients in the Royal Marsden Hospital AS study.
In all, 39 prostate cancer-risk SNPs identified from published genome wide association studies (GWAS) were genotyped using the Sequenom Platform and TaqMan™ assays from available DNA.
The cumulative genetic-risk scores for each patient were then calculated using the weighted effect estimated from previous GWAS (log-additive model).
FH status and the genetic-risk scores were assessed against adverse outcomes in AS, time to treatment and adverse histology on repeat biopsy, using univariable and multivariable Cox regression models to address time to treatment; and binary logistic regression to address biopsy upgrade.
Results
Of 471 patients, 55 (13.6%) had adverse histology on repeat biopsies and 145 (30.8%) had deferred treatment.
On univariate analysis, there was no significant relationship between FH of prostate cancer in any degree of relation, and adverse histology or time to treatment.
For risk score analyses, 386 patients’ DNA was studied; and there was also no relationship found between the calculated genetic risk scores and adverse histology or time to treatment (P = 0.573 and P = 0.965, respectively).
The retrospective study design and the few events was the main limitation of the study.
Conclusions
There is currently insufficient data to support the use of FH status or prostate cancer SNP profile-risk scores as prognostic factors in AS and these should not be used to influence management decisions.
As more genetic variants are discovered this may change and should be reassessed in multicentre AS cohorts.
Keywords: active surveillance, family history, genetic risk profiles, prostate cancer
Introduction
Prostate cancer is one of the most heritable cancers and a positive family history (FH) of prostate cancer increases risk to first-degree relatives by over two-fold [1]. Mutations that have moderate to high penetrance for prostate cancer, including BRCA2 and HOXB13, have been implicated in some families, but these rare variants are unlikely to explain the inherited genetic predisposition to prostate cancer in the vast majority of the population [2, 3]. The search for further causative genetic variants has led to genome-wide association studies (GWAS), which have so far identified 47 susceptibility loci associated with prostate cancer that occur more commonly but are of a lower penetrance. Following on from the discovery of these variants, efforts are now underway to evaluate their potential clinical application.
Epidemiological studies have reported differences in outcomes amongst races in prostate cancer [4], which persist despite correcting for socio-economic factors, e.g. access to healthcare and screening. Retrospective cohort studies have also shown familial aggregation of fatal prostate cancer cases [5]. These results suggest a potential role of inherited factors as prognostic markers for worse disease. However, the GWAS susceptibility loci have so far not been conclusively linked to disease aggressiveness and survival [2]. Their clinical utility as prognostic markers is therefore still undefined. The ability to predict disease aggressiveness or response to treatment, using inherited variables including FH and genetic variants would be invaluable, allowing clinicians to tailor treatment according to risks. A cohort where this might prove useful is in active surveillance (AS) cohorts where early identification of disease progression is vital.
AS is an accepted management option for early prostate cancer. Several groups have reported results from AS programmes, which support its use [6]. For favourable-risk cancers, AS is seen as a potential option to avoid toxic therapies. The challenge is to identify disease progression to ensure prompt treatment before local advancement of the cancer or the development of distant metastases [6]. Men are monitored regularly with PSA testing and repeat biopsies to look for evidence of progression. However, there exists a certain subset of men, who have early disease progression and spread, despite their low-risk disease. Over the years, several advances have been reported that improve our ability to identify men early who are at risk of disease progression including, MRI scanning techniques and transperineal prostate biopsy mapping, which improves tumour sampling by reducing the random and systematic errors of TRUS-guided biopsy [7]. PSA kinetics as prognostic markers have also been investigated by different groups with conflicting outcomes [8, 9]. The Transatlantic Consensus Group’s recommendations, were published recently, which emphasised the need for further studies to define an optimal approach for AS with regard to determination of disease progression and trigger for active treatment [7]. The roles of genetic variants or FH as predictors for disease progression in this cohort have not been clearly defined. If patients with a positive FH or those with specific genetic profiles are at risk for worse outcomes, these men should potentially be discouraged from AS. Here, we report a study exploring the potential prognostic roles of FH or genetic-risk scores in patients managed by AS.
Patients and methods
Patients were selected from a prospective clinical cohort study of AS (National Institute for Health Research [NIHR] 1592; http://prostate.icr.ac.uk/ActiveSurveillance.htm) that was initiated at the Royal Marsden Hospital, and were enrolled from April 2002 to May 2011. The outcomes of patients in this AS cohort have been reported by Selvadurai et al. (paper in preparation). All patients gave informed consent and had histologically confirmed prostate cancer, stage T1/2a, N0, M0, Gleason score 3+3, PSA level of < 15 ng/mL with cancer present in <50% of the total number of biopsy cores. Gleason 3+4 was only allowed if patients were aged > 65 years. All biopsies were reviewed centrally. Patients were aged between 50 and 80 years and fit for radical treatment but chose AS as their initial management.
All patients underwent an AS programme consisting of 3-monthly serum PSA level testing in the first year, 4-monthly in the second year, and 6-monthly thereafter. TRUS-guided octant prostate biopsies were taken after 18–24 months on AS, and every 2 years thereafter. Patients who have adverse features on the AS protocol, i.e. PSA velocity of > 1 ng/mL/year, based on a minimum of four values taken over 6 months, or adverse histology were recommended for radical treatment. Adverse histology was defined as >50% of cores involved, or any increase in primary Gleason score, or an increase in composite Gleason score of ≥8, on any repeat biopsy after consent. An exception is made for baseline Gleason scores of <3+3, which are considered as equivalent to a baseline score of 3+3 (Selvadurai et al., paper in preparation).
FH data acquisition
For the FH analyses, all those enrolled in the Royal Marsden Hospital AS study were eligible. FH was obtained using detailed questionnaires. For those with missing or incomplete data, hospital records were used. Patients were then classified into three prognostic groups to further stratify their inherited genetic risk:
One or more first-degree relatives with prostate cancer (with or without any number of second-degree relatives with prostate cancer);
One or more second-degree relative(s) but no first-degree relatives with prostate cancer;
No first- or second-degree relative with prostate cancer;
Genotyping methods
For the DNA analyses, in addition to the criteria described above, patients also had to be enrolled in the United Kingdom Genetic Prostate Cancer Study (UKGPCS; NIHR869; www.icr.ac.uk/ukgpcs), which is an on-going prospective cohort study investigating genes predisposing to prostate cancer. This ensures the identification of only patients for whom consent has been given for DNA analyses.
Single nucleotide polymorphism (SNP) genotyping was done using the Sequenom MassARRAY iPLEX Platform, which is a high throughput genotyping technique using a standard widely-published protocol [10]. At the time of genotyping (May 2011), 39 SNPs were identified from GWAS publications (Table 1) [14, 23–34], which were fitted into two plexes for analyses. A single SNP (rs5945619) could not fit into either plex and this was genotyped using the TaqMan™ assay technique [11]. Standard quality control was employed using internal negative controls, which were 2% per 384-well plate, and 2% duplicates were genotyped as external controls with a requirement for at least a 98% concordance of called genotypes. The cumulative risk scores for each patient were then calculated by summing risk alleles for each loci using the weighted effect, as estimated in the previous published GWAS studies. This is the log-additive model, which has been previously reported and used [12]. The risk scores were analysed as a continuous variable. In addition the risk scores were used to generate a risk distribution curve and the outcomes of the highest and lowest extremes of the curve compared to evaluate for worse outcomes.
Table 1.
Common susceptibility loci for prostate cancer identified through GWAS that was genotyped
| Locus | SNP | RAF* | Per allele OR (95% CI)* | Nearby genes | Reference |
|---|---|---|---|---|---|
| 2p11 | rs10187424 | 0.41 | 0.92 (0.89–0.94) | GGCX/VAMP8 | [14] |
| 2p15 | rs721048 | 0.19 | 1.15 (1.10–1.21) | Intronic in EHBP1 | [23] |
| 2p21 | rs1465618 | 0.23 | 1.08 (1.03–1.12) | Intronic in THADA | [24] |
| 2q31 | rs12621278 | 0.06 | 0.75 (0.70–0.80) | Intronic in ITGA6 | [24] |
| 2q37 | rs2292884 | 0.25 | 1.14 (1.09–1.19) | MLPH | [14] |
| 3p12 | rs2660753 | 0.11 | 1.18 (1.06–1.31) | [25] | |
| 3q21 | rs10934853 | 0.28 | 1.12 (1.08–1.16) | Intronic in EEFSEC | [26] |
| 3q23 | rs6763931 | 0.45 | 1.04 (1.01–1.07) | Intronic in ZBTB38 | [14] |
| 3q26 | rs10936632 | 0.48 | 0.90 (0.88–0.93) | CLDN11/SKIL | [14] |
| 4q22 | rs17021918 | 0.34 | 0.90 (0.87–0.93) | Intronic in PDLIM5 | [24] |
| 4q22 | rs12500426 | 0.46 | 1.08 (1.05–1.12) | Intronic in PDLIM5 | [24] |
| 4q24 | rs7679673 | 0.45 | 0.91 (0.88–0.94) | TET2 | [24] |
| 5p12 | rs2121875 | 0.34 | 1.05 (1.02–1.08) | Intronic in FGF10 | [14] |
| 5p15 | rs2242652 | 0.19 | 0.87 (0.84–0.90) | Intronic in TERT | [14] |
| 6p21 | rs130067 | 0.21 | 1.05 (1.02–1.09) | Missense coding in CCHCR1 | [14] |
| 6q25 | rs9364554 | 0.29 | 1.17 (1.08–1.26) | Intronic in SLC22A3 | [25] |
| 7p15 | rs10486567 | 0.77 | 0.74 (0.66–0.83) | Intronic in JAZF1 | [27] |
| 7q21 | rs6465657 | 0.46 | 1.12 (1.05–1.20) | Intronic in LMTK2 | [25] |
| 8p21 | rs2928679 | 0.42 | 1.05 (1.01–1.09) | SLC25A37 | [24] |
| 8p21 | rs1512268 | 0.45 | 1.18 (1.14–1.22) | NKX3.1 | [24] |
| 8q24 | rs1447295 | 0.11 | 1.62 | [28] | |
| 8q24 | rs6983267 | 0.50 | 1.26 (1.13–1.41) | [29] | |
| 8q24 | rs16901979 | 0.09 | 1.79 (1.36–2.34) | [30] | |
| 8q24 | rs10086908 | 0.30 | 0.87 (0.81–0.94) | [31] | |
| 8q24 | rs12543663 | 0.31 | 1.08 (1.00–1.16) | [31] | |
| 8q24 | rs620861 | 0.37 | 0.90 (0.84–0.96) | [31] | |
| 9q33 | rs1571801 | 0.25 | 1.27 (1.10–1.48) | Intronic in DAB21P | [32] |
| 10q11 | rs10993994 | 0.40 | 1.25 (1.17–1.34) | Promoter of MSMB | [25,27] |
| 10q26 | rs4962416 | 0.27 | 1.20 (1.07–1.34) | Intronic in CTBP2 | [27] |
| 11p15 | rs7127900 | 0.20 | 1.22 (1.17–1.27) | [24] | |
| 11q13 | rs7931342 | 0.49 | 0.84 (0.79–0.90) | [25, 27] | |
| 12q13 | rs10875943 | 0.31 | 1.07 (1.04–1.10) | TUBA1C/PRPH | [14] |
| 17q12 | rs4430796 | 0.49 | 1.22 (1.15–1.30) | Intronic in HNF1B | [33] |
| 17q12 | rs11649743 | 0.80 | 1.28 (1.07–1.52) | Intronic in HNF1B | [34] |
| 17q24 | rs1859962 | 0.46 | 1.20 (1.14–1.27) | [33] | |
| 19q13 | rs2735839 | 0.15 | 0.83 (0.75–0.91) | KLK2/KLK3 | [25] |
| 22q13 | rs5759167 | 0.47 | 0.86 (0.83–0.88) | BIL/TTLL1 | [24] |
| Xp11 | rs5945619 | 0.36 | 1.19 (1.07–1.31) | NUDT11 | [23, 25] |
| Xq12 | rs5919432 | 0.19 | 0.94 (0.89–0.98) | AR | [14] |
Data for RAF (risk allele frequency) and per allele OR (odds ratio) are taken from the original publications. 95% CIs are given in brackets where available.
Statistical methods
FH status and the SNP-risk scores were analysed against defined adverse outcomes in AS, including adverse histology on repeat biopsy and time to treatment, to determine their prognostic value.
For adverse histology, the analyses were performed using adverse histology as a binary outcome, i.e. yes/no, and also as a continuous variable, using the time to adverse histology. When predicting for adverse histology on repeat biopsy, only patients with at least one repeat biopsy were included.
Time to treatment was described in the population using the Kaplan–Meier method. Time was measured from the date of consent into the AS study, to the first treatment date. Possible modes of treatment included: external beam radiotherapy, brachytherapy, prostatectomy, and androgen-deprivation therapy. Patients were censored at the date of last follow-up/death. Patients who died without treatment were censored at date of death. Patients who withdrew from the trial before treatment (e.g. in favour of watchful waiting) were censored at date of withdrawal.
FH status and the SNP-risk scores were analysed separately (univariate analysis) using either Cox regression (time to adverse histology, time to treatment), or binary logistic regression (adverse histology). If either variable was significant in univariate analysis, further evaluation in a multivariate model using stepwise model selection techniques for potential confounding factors was used.
Results
In all, 471 men were enrolled in the AS trial and were eligible for analyses. The baseline characteristics are listed in Table 2. In all, 416 were of Caucasian ethnicity (88.3%), 24 Black-Caribbean (5.1%), 10 Asian Indian (2.1%), eight Black-African (1.7%), eight Asian Other (1.7%) and five Unknown (1.1%). The median (range) follow-up was 5.6 (0.3–9.1) years. In all, 412 men underwent repeat biopsy, of whom 56 (13.6%) had histological upgrade on repeat biopsies. Overall, 145 (30.8%) men from the total cohort have undergone deferred treatment.
Table 2.
Baseline characteristics at diagnosis
| Variable | Value |
|---|---|
| Median (range): | |
| Age, years | 66 (51–79) |
| PSA level, ng/mL | 6.0 (0.2–36) |
| Max % cancer in any biopsy core | 10 (1–95) |
| N (%): | |
| Gleason score: | |
| ≤3+3 | 439 (93.2) |
| 3+4 | 32 (6.8) |
| T stage: | |
| T1 | 400 (84.9) |
| T2 | 71 (15.1) |
| FH: | |
| 0 first/second-degree relative | 350 (82.7) |
| ≥1 first-degree relative | 54 (12.8) |
| ≥1 second-degree relative | 19 (4.5) |
In the FH analyses, 423 patients had FH recorded. When analysing FH against adverse histology, the results were not significant (P = 0.154; Table 3). For time to adverse histology, the analysis also did not show any significant association with a first-degree relative with prostate cancer (hazard ratio [HR] 0.3, 95% CI 0.1–1.4) or with a second-degree relative with prostate cancer (HR 1.3, 95% CI0.4–4.2). When analysed against time to treatment, there was again no significant association with a FH of a first-degree relative with prostate cancer (HR 1.1, 95% CI 0.7–1.9) or a second-degree relative (HR 0.8, 95% CI 0.3–2.1; Table 4.)
Table 3.
Impact of FH on biopsy upgrade analysed as a binary outcome.
| Relatives with prostate cancer | Number with biopsy upgrade (%) | Number with no biopsy upgrade (%) | Total number |
|---|---|---|---|
| 0 first/second-degree relative | 45 (14.2) | 271 (85.8) | 316 |
| ≥1 first-degree relative | 2 (4.3) | 44 (95.7) | 46 |
| ≥1 second-degree relative (no first degree) | 3(17.6) | 14 (82.4) | 17 |
P = 0.154 (chi-squared test)
Table 4.
Impact of FH on time to treatment.
| Relatives with prostate cancer | Total number of patients | Number of time to treatment events | HR (95% CI) |
|---|---|---|---|
| 0 first/second-degree relative | 350 | 106 | 1 |
| ≥1 first-degree relative | 54 | 16 | 1.1 (0.6–1.9) |
| ≥1 second-degree relative (no first degree) | 19 | 4 | 0.8 (0.3–2.1) |
For the genetic prostate cancer-risk score analyses, 386 patients were eligible. The mean (range) calculated risk score was 1.400 (0.192–5.393). The risk score distribution is shown in Figure 1. On univariate analysis, there was no significant association between the calculated genetic-risk scores and adverse histology (P = 0.573), time to adverse histology (HR 0.921, 95% CI 0.748–1.134) or time to treatment (HR 0.965, 95% CI 0.798–1.168). When analysing differences between the higher and lower genetic-risk groups (defined as the top 25% and lowest 25% of the genetic risk distribution), there was again no relationship between time to adverse histology and time to treatment using chi-squared testing (P = 0.767 and P = 0.481, respectively). Figure 2 shows the overlapping risk scores’ distributions between those who did or did not have adverse histology (2a), and those who did or did not receive treatment (2b), highlighting the statistically non-significant findings, as no separation of the curves was seen. We were not able to evaluate the risk distribution in smaller percentiles due to the low numbers encountered. Multivariate analyses were not carried out in view of the non-significant findings.
Figure 1.

Risk score distribution generated from SNP genotyping of the AS cohort.
Figure 2.
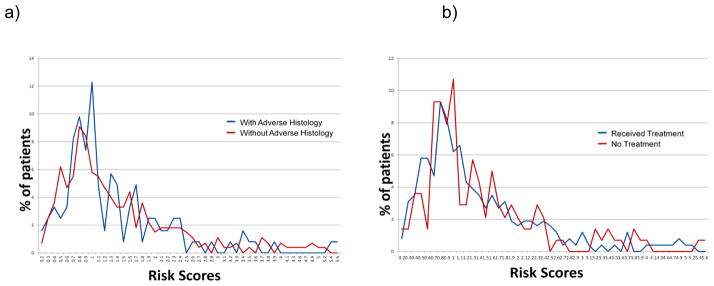
Risk score distributions for patients with or without adverse histology (a) and for patients who did or did not receive active treatment (b).
Subgroup analyses were also conducted using risk scores generated from loci thought to be associated with aggressiveness, which were SNPs in 8q24 region [13] and the androgen receptor SNP (X-chromosome SNP rs5919432) [14]. These also showed no significant correlation for all the adverse outcome endpoints.
As GWAS risk SNPs have been shown to be associated with PSA level [15], further analyses on the effect of SNP-risk scores on PSA kinetics were also performed. Both PSA velocity and PSA-doubling time at 2 years were analysed for this cohort, but again, there were no significant correlations found with the risk scores.
Discussion
To our knowledge, this is the first study to date, which has investigated the potential prognostic role of SNP profiles in an AS cohort. It is the larger of just two known studies that have reported on the impact of FH as an independent prognosticator in AS. The present results were consistent with the study reported by San Francisco et al. [16]. Despite having more patients, we also found no significant association between FH and adverse outcomes in AS. We also further stratified our cohort according to the degree of relation, to reflect the associated higher genetic risks encountered with closer relatives [1]. However, this did not alter the non-significant association observed.
For the risk SNP analyses, there was no significant association with outcome. McGuire et al. [17] recently reported that some of the risk SNPs were more significantly prevalent in patients with unfavourable post-prostatectomy pathology compared with the baseline biopsy, and could potentially be used to screen AS patients to identify and exclude these men from this management strategy. This finding was not replicated in the present study. The disparity of the results could be due to the differences in the cohorts used. Their study was a retrospective evaluation of patients with prostate cancer in a prostatectomy cohort, who, based on their baseline pathology, was suitable for AS. The present study used a prospective cohort actually undergoing AS and only 44 of our cohort had radical surgery. Adverse histology was assessed from biopsies and is subject to potential sampling error, which might reduce our ability to detect a true association. To avoid this potential bias, analyses could be done by restricting the analyses of adverse histology events to only patients who have undergone radical prostatectomy. This method would allow a thorough assessment of their pathology. The few patients in the present cohort did not allow for meaningful analyses using surgical pathology outcomes. However, the present study concurs with previous reports of a lack of predictive use of the GWAS risk SNPs [2]. There is emerging evidence that some rarer genetic variants are associated with more aggressive disease outcome [18]. As further genetic variants are found that are associated with risk this may alter. Further validation is therefore still needed.
There could be several reasons for the lack of associations found. Firstly, we have a small sample size with a relatively short follow-up for a low-risk prostate cancer cohort. As such, the number of events was low decreasing the power of the present study. Potential solutions include re-analysis after a longer follow-up period to increase the number of events. However, it is likely that combined analyses with other research groups with similar AS cohorts will be needed to increase patient numbers and hence increase the power. An example of the power that can be achieved with collaborations is that, assuming that a genetic risk profile of SNPs is associated with a similar risk of adverse outcomes to that for prostate cancer risk, ≈1000 patients will provide >90% power to detect a difference in risk scores between cases who do or do not experience adverse outcomes in AS at P < 0.01. It is clear from published AS reports that single centres are unlikely to have sufficient numbers with enough follow-up data available to perform these analyses [6]. Collaborations are therefore needed and these could be done within the Clinical ELLIPSE (Elucidating Loci Involved In Prostate Cancer Susceptibility) Consortium, which is an National Institute of Health (NIH) funded post-GWAS initiative investigating the clinical application of the prostate cancer risk SNPs [19].
Secondly, the impact of environmental or lifestyle factors could play an important role. Cancer survivors have been shown to seek better diet, medication and lifestyle changes (e.g. physical activity, and statins intake) after a cancer diagnosis [20]. Their protective effects have been described [2]. The present analyses did not consider these factors, which could potentially be significant in an AS cohort, where they live with the impact of a cancer diagnosis for many years. These variables are not routinely collected, but it should be done to evaluate their impact in this setting.
Thirdly, we have still yet to discover the full extent of the genetic inheritability of prostate cancer, which has implications on the calculation of the risk scores. We included 39 SNPs that were known at the time of genotyping. GWAS continue to report novel SNPs with new studies due to be published, e.g. the Collaborative Oncological Gene-Environment Study (COGS) analysis [21]. These undiscovered genes might yet reveal potential links. Re-calculating the risk scores using more up to date SNP panels associated with prostate cancer risk might improve their predictive ability. The risk score calculation in the present study also resulted in a positively skewed distribution with a mean score close to one, i.e. close to the population average. The reason for this is unclear and could be a chance finding due to the small numbers. Alternatively, this could indicate that the risk scores are less predictive for the present low-risk prostate cancer cohort. A comparison of profiles between this cohort and a non-prostate cancer (control) cohort should be done to evaluate this further.
Finally, the present risk score analyses did include 11.7% of patients of non-Caucasian ethnicity. This could potentially reduce the power to detect any differences, as the GWAS risk SNPs were originally identified in Caucasian populations. However, recent analyses in the Multi-Ethnic Cohort study have suggested that a large fraction of the prostate cancer variants identified in populations of European ancestry are global markers of risk [22]. This would therefore suggest potential application across different populations. Confirmation in these cohorts is needed, but our low study numbers did not allow for further meaningful sub-analyses according to other ethnic groups.
In conclusion, although the present study has not shown FH status or prostate cancer risk SNP profiles to be prognostic factors, we were limited by the few events. Collaborative efforts in the form of combined analyses from multiple AS studies should be considered for future research. Current guidelines exist for clinicians and patients summarising evidence relating to the best outcomes of different prostate cancer treatment methods. FH and risk SNPs could potentially add to this stratification. The attraction of selecting the best treatment method based on FH status or SNP profiles is clear and studies like these are important to investigate this link further. Meta-analyses will be needed to define if this is appropriate in the clinical care pathway.
What’s known on the subject? and What does the study add?
Family history (FH) is a major risk factor for the development of prostate cancer. The search for genetic variants has led to genome-wide association studies (GWAS), which have so far reported 47 susceptibility loci that predispose men to prostate cancer. However, the use of genetics or FH status in predicting clinical outcomes after prostate cancer diagnosis remains uncertain. Guidelines currently exist for clinicians and patients summarising evidence relating to the best outcomes of different prostate cancer treatment methods. Genetics and FH could potentially add to this stratification.
Our study aimed to ascertain the potential prognostic roles of FH or genetic risk scores in patients managed by active surveillance.
Acknowledgments
The scientific development and funding of this project were in part supported by the Genetic Associations and Mechanisms in Oncology (GAME-ON) Initiative (NIH ELLIPSE grant: U19CA148537) and Cancer Research UK (Grant: C5047/A8385). We are grateful for the support from The Institute of Cancer Research Everyman Campaign. We acknowledge support from the NIHR to the Biomedical Research Centre at The Institute of Cancer Research and Royal Marsden Foundation NHS Trust.
Abbreviations
- AS
active surveillance
- FH
family history
- GWAS
genome-wide association studies
- HR
hazard ratio
- SNP
single nucleotide polymorphism
- NIHR
National Institute for Health Research
References
- 1.Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 2.Goh CL, Schumacher FR, Easton D, et al. Genetic Variants Associated With Predisposition to Prostate Cancer and Potential Clinical Implications. J Intern Med. 2012;271:353–65. doi: 10.1111/j.1365-2796.2012.02511.x. [DOI] [PubMed] [Google Scholar]
- 3.Karlsson R, Aly M, Clements M, et al. A Population-based Assessment of Germline HOXB13 G84E Mutation and Prostate Cancer Risk. Eur Urol. 2012 doi: 10.1016/j.eururo.2012.07.027. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 4.Evans S, Metcalfe C, Ibrahim F, Persad R, Ben-Shlomo Y. Investigating Black-White differences in prostate cancer prognosis: A systematic review and meta-analysis. Int J Cancer. 2008;123:430–5. doi: 10.1002/ijc.23500. [DOI] [PubMed] [Google Scholar]
- 5.Brandt A, Sundquist J, Hemminki K. Risk for incident and fatal prostate cancer in men with a family history of any incident and fatal cancer. Ann Oncol. 2012;23:251–6. doi: 10.1093/annonc/mdr056. [DOI] [PubMed] [Google Scholar]
- 6.Lawrentschuk N, Klotz L. Active surveillance for low-risk prostate cancer: an update. Nat Rev Urol. 2011;8:312–20. doi: 10.1038/nrurol.2011.50. [DOI] [PubMed] [Google Scholar]
- 7.Ahmed HU, Akin O, Coleman JA, et al. Transatlantic Consensus Group on active surveillance and focal therapy for prostate cancer. BJU Int. 2012;109:1636–47. doi: 10.1111/j.1464-410X.2011.10633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng MK, Van As N, Thomas K, et al. Prostate-specific antigen (PSA) kinetics in untreated, localized prostate cancer: PSA velocity vs PSA doubling time. BJU Int. 2009;103:872–6. doi: 10.1111/j.1464-410X.2008.08116.x. [DOI] [PubMed] [Google Scholar]
- 9.Ross AE, Loeb S, Landis P, et al. Prostate-specific antigen kinetics during follow-up are an unreliable trigger for intervention in a prostate cancer surveillance program. J Clin Oncol. 2010;(28):2810–6. doi: 10.1200/JCO.2009.25.7311. [DOI] [PubMed] [Google Scholar]
- 10.Gabriel S, Ziaugra L, Tabbaa D. SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr Protoc Hum Genet. 2009;Chapter 2(Unit 2):12. doi: 10.1002/0471142905.hg0212s60. [DOI] [PubMed] [Google Scholar]
- 11.Schleinitz D, Distefano JK, Kovacs P. Targeted SNP genotyping using the TaqMan(R) assay. Methods Mol Biol. 2011;700:77–87. doi: 10.1007/978-1-61737-954-3_6. [DOI] [PubMed] [Google Scholar]
- 12.Pereira TV, Patsopoulos NA, Salanti G, Ioannidis JP. Discovery properties of genome-wide association signals from cumulatively combined data sets. Am J Epidemiol. 2009;170:1197–206. doi: 10.1093/aje/kwp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cussenot O, Azzouzi AR, Bantsimba-Malanda G, et al. Effect of genetic variability within 8q24 on aggressiveness patterns at diagnosis and familial status of prostate cancer. Clin Cancer Res. 2008;14:5635–9. doi: 10.1158/1078-0432.CCR-07-4999. [DOI] [PubMed] [Google Scholar]
- 14.Kote-Jarai Z, Olama AA, Giles GG, et al. Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nat Genet. 2011;43:785–91. doi: 10.1038/ng.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gudmundsson J, Besenbacher S, Sulem P, et al. Genetic correction of PSA values using sequence variants associated with PSA levels. Sci Transl Med. 2010;2:62ra92. doi: 10.1126/scitranslmed.3001513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.San Francisco IF, Werner L, Regan MM, Garnick MB, Bubley G, DeWolf WC. Risk stratification and validation of prostate specific antigen density as independent predictor of progression in men with low risk prostate cancer during active surveillance. J Urol. 2011;185:471–6. doi: 10.1016/j.juro.2010.09.115. [DOI] [PubMed] [Google Scholar]
- 17.McGuire BB, Helfand BT, Kundu S, et al. Association of prostate cancer risk alleles with unfavourable pathological characteristics in potential candidates for active surveillance. BJU Int. 2012;110:338–43. doi: 10.1111/j.1464-410X.2011.10750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castro E, Goh CL, Olmos D, Leongamornlert D, Saunders E, Tymrakiewicz M, Mahmud N, et al. Correlation of germline BRCA2 mutations with aggressive prostate cancer and outcome. J Clin Oncol. 2011;29(Suppl):abstract 1517. [Google Scholar]
- 19.National Institute of Health. [Accessed September 2012];ELLIPSE (Elucidating Loci Involved in Prostate Cancer Susceptibility) 2010 [updated December 2, 2010; cited 2011 July 1]; Available from: http://epi.grants.cancer.gov/pgwas/personnel.html#ellipse.
- 20.Davies NJ, Batehup L, Thomas R. The role of diet and physical activity in breast, colorectal, and prostate cancer survivorship: a review of the literature. Br J Cancer. 2011;105 (Suppl 1):S52–73. doi: 10.1038/bjc.2011.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.COGS. [Accessed September 2012];Collaborative Oncological Gene-environment Study. 2009 [cited 2011 July 1]; Available from: http://www.cogseu.org/
- 22.Waters KM, Le Marchand L, Kolonel LN, et al. Generalizability of associations from prostate cancer genome-wide association studies in multiple populations. Cancer Epidemiol Biomarkers Prev. 2009;18:1285–9. doi: 10.1158/1055-9965.EPI-08-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gudmundsson J, Sulem P, Rafnar T, et al. Common sequence variants on 2p15 and Xp11. 22 confer susceptibility to prostate cancer. Nat Genet. 2008;40:281–3. doi: 10.1038/ng.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eeles RA, Kote-Jarai Z, Al Olama AA, et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet. 2009;41:1116–21. doi: 10.1038/ng.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eeles RA, Kote-Jarai Z, Giles GG, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316–21. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 26.Gudmundsson J, Sulem P, Gudbjartsson DF, et al. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet. 2009;41:1122–6. doi: 10.1038/ng.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas G, Jacobs KB, Yeager M, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–5. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 28.Amundadottir LT, Sulem P, Gudmundsson J, et al. A common variant associated with prostate cancer in European and African populations. Nat Genet. 2006;38:652–8. doi: 10.1038/ng1808. [DOI] [PubMed] [Google Scholar]
- 29.Yeager M, Orr N, Hayes RB, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–9. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 30.Gudmundsson J, Sulem P, Manolescu A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–7. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 31.Al Olama AA, Kote-Jarai Z, Giles GG, et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat Genet. 2009;41:1058–60. doi: 10.1038/ng.452. [DOI] [PubMed] [Google Scholar]
- 32.Duggan D, Zheng SL, Knowlton M, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J Natl Cancer Inst. 2007;99:1836–44. doi: 10.1093/jnci/djm250. [DOI] [PubMed] [Google Scholar]
- 33.Gudmundsson J, Sulem P, Steinthorsdottir V, et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet. 2007;39:977–83. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 34.Sun J, Zheng SL, Wiklund F, et al. Evidence for two independent prostate cancer risk-associated loci in the HNF1B gene at 17q12. Nat Genet. 2008;40:1153–5. doi: 10.1038/ng.214. [DOI] [PMC free article] [PubMed] [Google Scholar]