

Abstract
Endotoxin tolerance is a complex phenomenon characterized primarily by decreased production of proinflammatory cytokines, chemokines, and other inflammatory mediators, while other genes are induced or unchanged in expression. Endotoxin tolerance is induced by prior exposure of murine macrophages/human monocytes, experimental animals, or people to TLR ligands. Although recent studies have reported a possible relationship between endotoxin tolerance and differentiation of alternatively activated macrophages (AA-Mφ or M2), we show herein that LPS pretreatment of IL-4Rα−/− and STAT6−/− macrophages, that fail to develop into AA-Mφ, resulted in tolerance of proinflammatory cytokines as well as molecules and chemokines previously associated with AA-Mφ (e.g., arginase-1, mannose receptor, CCL2, CCL17, and CCL22). In contrast to LPS, wild-type (WT) macrophages pretreated with IL-4, the prototype inducer of AA-Mφ, did not induce endotoxin tolerance with respect to proinflammatory cytokines, AA-Mφ-associated chemokines, negative regulators, NF-κB binding and subunit composition, and MAPKs, and conversely, IL-13−/− macrophages were tolerized equivalently to WT macrophages by LPS pretreatment. Further, IL-4Rα-deficiency did not affect the reversal of endotoxin tolerance exerted by histone deacetylase inhibitor, TSA. Like WT mice, 100% of LPS-tolerized IL-4Rα-deficient mice survived LPS + D-galactosamine-induced lethal toxicity and exhibited decreased serum levels of proinflammatory cytokines and AA-Mφ-associated chemokines induced by LPS challenge when compared to non-tolerized mice. These data indicate that the signaling pathways leading to endotoxin tolerance and differentiation of AA-Mφ are dissociable.
Introduction
Pathogen-associated molecular patterns (PAMPs), including bacterial and viral components, are immunostimulatory and are recognized by Toll-like receptors (TLRs) (1, 2), a family of closely related pattern recognition receptors. Upon ligand engagement, TLR activation of intracellular signaling cascades results in robust production of proinflammatory mediators including cytokines and chemokines (3, 4). Lipopolysaccharide (LPS), an integral outer membrane component of Gram negative bacteria, is the prototype PAMP recognized by the TLR4/MD2 complex and has long been associated with the strong inflammatory response associated with endotoxin shock (3, 5). However, after an initial exposure of monocytes/macrophages, experimental animals, or people to LPS, a transient period of “endotoxin tolerance,” a state of LPS hyporesponsiveness, is observed (6–8). Endotoxin tolerance has long been associated with a down-regulation of proinflammatory cytokine production (e.g., TNF-α, IL-6, IL-12, and IFN-β) due to alterations in signaling cascades that affect NF-κB, mitogen-activated protein kinases (MAPKs), and interferon regulatory factors (IRFs) (9–11). In addition, endotoxin tolerance has also been associated with increased expression of variety of negative regulators of TLR signaling such as IRAK-M, ST2, SHIP-1, MyD88s, and A20 (12, 13). A similar phenomenon has been observed in septic patients where LPS hyporesponsiveness is observed after the initial cytokine storm. While protecting the host from the ill effects of inflammation, the diminished capacity of sepsis survivors to respond to LPS is thought to underlie increased susceptibility to secondary infection that is common is such patients (6, 14).
Macrophages are heterogeneous innate immune cells that exhibit great “plasticity” (15). Macrophages are involved in homeostasis and innate immunity, and through antigen uptake, processing, and presentation, serve to initiate the adaptive immune response (16). Based on their responses to environmental stimuli, macrophages possess the ability to differentiate into functionally distinct subsets that have been termed “classically activated” (CA-Mφ or M1) or “alternatively activated” (AA-Mφ or M2). CA-Mφ macrophage polarization is typically mediated by strong inflammatory stimuli such as IFN-γ and bacterial products, including LPS, and is characterized by increased production of proinflammatory cytokines, nitric oxide (NO), and reactive oxygen species (ROS) that mediate microbicidal activities and induce cellular immunity (16–19). AA-Mφ polarization is mediated by the Type I (IL-4Rα + γc) or the Type II (IL-4Rα + IL-13Rα1) receptor complexes by IL-4 or by IL-4 or IL-13 engagement, respectively (20). Upon ligand binding, both the Type I and Type II IL-4 receptors activate the STAT6 signaling pathway (17, 18, 21). The AA-Mφ macrophage phenotype has been characterized by expression of mannose receptor (MR) (22), intracellular expression of Arginase-1 (Arg-1), secretion of chitinases (e.g., Ym1) (23), and anti-inflammatory cytokines (21). AA-Mφ macrophages are strongly associated with helminthic infections and tissue repair (17).
Recent studies revealed a possible relationship between endotoxin tolerance and AA-Mφ polarization (24, 25). For example, Porta et al. reported that CCL2, CCL17, and CCL22, chemokines that attract Th2 cells and have been associated previously with the AA-Mφ phenotype (26), were upregulated in endotoxin-tolerized macrophages in an NF-κB p50-dependent manner (25). However, since both LPS and IL-4, prototype inducers of CA-Mφ and AA-Mφ phenotypes, respectively, induce expression of these chemokines (27), it is difficult to know whether they are truly AA-Mφ differentiation markers. Therefore, for the remainder of this report, we will refer to these chemokines as “AA-Mφ-associated.”
To determine the fate of these chemokines, as well as other well-known AA-Mφ markers, during endotoxin tolerance, we used IL-4Rα- and STAT6-deficient macrophages to determine if AA-Mφ differentiation plays a role in LPS-tolerized macrophages. We show that while both LPS and IL-4 induce expression of AA-Mφ-associated chemokine mRNA and protein, the IL-4Rα-STAT6 signaling pathway is not required for induction of endotoxin tolerance, but is necessary for induction of AA-Mφ by IL-4. Mechanistically, these two states of macrophage differentiation were distinguishable by NF-κB and MAPK activation, NF-κB subunit composition, modulation of negative inhibitors, and sensitivity to a histone deacetylase inhibitor. Our findings further indicate that LPS-tolerized, IL-4Rα-deficient mice, like WT mice, were comparably refractory in vivo to challenge with LPS/D-galactosamine and produced decreased cytokine/chemokine production upon LPS challenge in vivo. Taken collectively, these findings dissociate induction of endotoxin tolerance and the differentiation pathway that leads to AA-Mφ macrophages.
Materials and Methods
Reagents and mice
Protein-free Escherichia coli K235 LPS (<0.008% protein) was prepared as described previously (28). TLR grade LPS from Salmonella abortus equi S-form was purchased from Enzo Life Sciences, Inc. Murine recombinant IL-4 was purchased from R & D Systems. Antibodies directed against phospho-ERK1/2, ERK, phospho-JNK1/2, JNK, Rel B, STAT3, phospho-STAT3, STAT6, phospho-STAT6, IκBα, and β-actin were purchased from Cell Signaling Technology, Inc. and anti-phospho-p38 antibody from Promega Corporation. Trichostatin A (TSA) was obtained from Calbiochem EMD-Millipore. Wild-type (WT) C57BL/6J and BALB/cByJ mice were purchased from The Jackson Laboratory. C57BL/6 TLR4−/− and IL-13−/− mice and BALB/c IL-4Rα−/−, IL-4Rα−/−/Rag2−/−, and STAT6−/− mice were bred at the University of Maryland, School of Medicine, animal facility. All animal studies were carried out with institutional approval.
Cell culture
Peritoneal exudate cells were obtained by peritoneal lavage from 6 to 8 wk old mice 4 days after i.p. injection with sterile thioglycollate (Remel) as described previously (29). Macrophages were enriched by adherence and extensive washing and were cultured in RPMI 1640 supplemented with 2% FBS, 2 mM glutamine, 1% penicillin, and streptomycin as described previously (29).
The murine macrophage cell line, RAW 264.7 (ATCC), was maintained in DMEM supplemented with 10% FBS, 2 mM glutamine, 1% penicillin, and streptomycin as described previously (30).
In vitro endotoxin tolerance
Primary macrophages and RAW 264.7 cells were cultured at 1–1.5 × 106 cells/well in 12-well plates for gene expression and ELISA experiments. For signaling experiments, 3.0 × 106 cells/well in 6-well plates were used. Cells were initially stimulated with medium only or with LPS (100 ng/ml) for 20–24 h, washed, and then “challenged” by treating the macrophages with medium only or with LPS (100 ng/ml) for the times indicated in the figures and legends. Macrophages that were treated first with medium then challenged with medium are designated as M/M (medium/medium), cells stimulated with one dose of LPS after medium pretreatment are designated as M/L (medium/LPS), cells stimulated with LPS for 24 h then with medium as L/M (LPS/medium), and cells stimulated with LPS for 24 h and challenged with LPS are designated as L/L (LPS/LPS). Similarly, in some studies, cells were pretreated for 24 h with IL-4 (40 ng/ml) and challenged with medium or LPS and are indicated as IL-4/M and IL-4/L, respectively. For cytokine ELISAs, culture supernatants were collected after pretreating the cells with medium or LPS for 24 h and challenged with medium or LPS for an additional 16–18 h. For TSA experiments, macrophage cultures were pretreated with medium or LPS in the absence or presence of TSA (50 nM) for 16 h then challenged with medium or LPS in the absence or presence of TSA for 3 h. For detection of activated MAPKs and other signaling proteins by Western analysis (below), cell lysates were prepared from macrophages stimulated with medium, LPS, or IL-4 for 24 h and challenged with medium or LPS for 30 min using cell lysis buffer (Cell Signaling Technology, Inc.).
RNA and cDNA
Total RNA was isolated using High Pure RNA Isolation Kit from Roche (Indianapolis, IN) according to manufacturer’s instructions. The RNA concentration was determined using a NanoDrop spectrophotometer. One μg total RNA was reverse transcribed using iScript cDNA synthesis kit according to manufacturer’s instructions (Bio-Rad).
Quantitative real-time PCR (qRT-PCR)
Differential gene expression of cytokines and chemokines was analyzed by quantitative real-time PCR (qRT-PCR) using SyBR green per the manufacturer’s guidelines in the 7000HT Fast Real-time PCR system (Applied Biosystems). Briefly, PCR was conducted in a 25 μl reaction volume containing 20 ng cDNA template and 3 μM murine genes specific primer mix. Primers for detection of IL-1β, TNF-α, IL-6, IL-12 p40, IL-4, IL-13, IFN-β, Arginase 1, MR, FIZZ 1, YM1 and HPRT were designed using the Primer Express 2.0 program (Applied Biosystems) and have been published elsewhere (21). Primer sequences for CCL2, CCL17, and CCL22 were obtained from Porta et al. (25). Relative gene expression was calculated by normalizing to HPRT as a housekeeping gene.
Western analysis
Protein estimation from cell lysates was carried out using BCA Protein assay reagents (Thermo Scientific/Pierce). The proteins (25 – 40 μg) in the lysate were boiled in Laemmli buffer for 5 min, resolved by 10% SDS-PAGE in Tris/glycine/SDS buffer (Bio-Rad) and transferred onto a PVDF membrane at constant voltage (100 volts) for 2 h in the cold room. After blocking for 2 h in TBST (20 mM Tris-HCl, 150 mM NaCl, 0.1% Tween 20) containing 5% fat-free milk and probed overnight at 4°C with the respective Abs according to the manufacturer’s instructions. Following extensive washing (4–5 times) in TBST, membranes were incubated with secondary HRP-conjugated anti-rabbit IgG from Jackson ImmunoResearch (1:10,000) for 1 h at room temperature. Membranes were washed 4–5 times in TBST and bands were detected using ECL plus reagents (Amersham Pharmacia Biotech).
Preparation of nuclear extracts and EMSA
Nuclear extracts were prepared using a nuclear extraction kit (Active Motif) according to the manufacturer’s instructions. NF-κB consensus sequence was 32P-end-labeled with T4 polynucleotide kinase (Invitrogen Life Technologies), as recommended by the manufacturer and EMSA was carried out as described previously (31). Supershift assays were performed using anti-p65 and anti-p50 antibodies as described previously (31). The polyacrylamide gels were dried at 80°C for 2 h and exposed to a phosphor screen overnight and the images were visualized using a Storm 680 scanner (Molecular Dynamics).
ELISAs
Cytokine/chemokine levels in the control and tolerized culture supernatants were analyzed by ELISA in the Cytokine Core Facility (University of Maryland, School of Medicine).
In vivo endotoxin tolerance
For mouse survival studies, groups (5–6 mice/group) of WT or IL-4Rα−/− mice received PBS or LPS (25 μg)/mouse i.p. After 24–30 h mice were challenged i.p. with LPS (1 μg) plus D-galactosamine (16 mg)/mouse. Mouse survival was monitored every 6 h for 3–4 days.
For analysis of serum cytokines and chemokines, WT and IL-4Rα−/− mice (5–6/group) were treated with PBS or LPS (25 μg) i.p. and then challenged 3 days later with LPS (25 μg) i.p. Two hours after challenge, mice were bled and the sera prepared. Cytokine and chemokine levels in sera were determined, using Multiplex beads at the Cytokine Core Lab (University of Maryland, Baltimore).
Statistical analysis
One-way analysis of variance (ANOVA) with Student Newman-Keuls (SNK) post hoc test was performed to assess statistical significance (p values < 0.05) using GraphPad PRISM 4.0 (GraphPad Software).
Results
Effect of endotoxin tolerance on classically activated (M1) and alternatively activated (M2) macrophage-specific gene expression profiles
Previous studies reported that murine and human AA-Mφ-specific genes were upregulated during endotoxin tolerance, and therefore, it was concluded that endotoxin tolerance and alternative activation were, in fact, related states of macrophage differentiation (24, 25). To confirm and extend these findings, we initially sought to validate the expression of CA-Mφ- and AA-Mφ-specific genes induced during endotoxin tolerance. To accomplish this, C57BL/6J primary peritoneal macrophage cultures were stimulated for 24 h with medium (M), as a control, or protein-free E. coli K235 LPS (L) to induce a state of “endotoxin tolerance,” and then the cells were washed and challenged with M or L for 3, 6, or 24 h. Figure 1A shows that CA-Mφ proinflammatory cytokine gene mRNA (e.g., TNF-α, IL-1β, IL-6, IL-12 p40, and IFN-β) was induced by LPS stimulation of medium-pretreated macrophages, but was poorly induced in LPS-pretreated (i.e., tolerized) cells. For each of these genes, maximal induction occurred at the 3 and/or 6 h time points. Next, we examined the induction of mRNA for two well-characterized AA-Mφ markers, Arg-1 and MR, as well as three chemokine genes, i.e., CCL2, CCL17, and CCL22, that have more recently been AA-Mφ–associated (25). Figure 1B shows that, in contrast to LPS-inducible CA-Mφ-associated genes, the protype AA-Mφ genes, Arg-1 and MR, were poorly induced (<10-fold) by LPS and only after 24 h stimulation (M/L); nonetheless, expression of both genes was down-regulated upon subsequent LPS stimulation (i.e., these genes are also “tolerizable”). Finally, like the proinflammatory CA-Mφ genes, the AA-Mφ associated genes encoding the chemokines CCL2, CCL17, and CCL22 were also detectable early (at 3 and 6 h after LPS stimulation (M/L)), and CCL2 expression remained elevated as late as 24 h after LPS stimulation (Figure 1C). The genes that encode these chemokines were also tolerizable, as evidenced by the failure of LPS restimulation (L/L) to increase their expression much above baseline (M/M). Thus, two well characterized AA-Mφ markers (16), as well as chemokine genes previously associated with AA-Mφ differentiation (25), are down-regulated by LPS pretreatment that results in tolerance of CA-Mφ genes. Similar experiments using S. abortus equi S-form LPS, as utilized by Porta et al. (25), were also performed and the results mirrored those obtained using E. coli LPS as the stimulant (Supplementary Figure 1A). Both LPS preparations were highly purified as evidenced by their failure to induce gene expression in TLR4−/− macrophages (Supplementary Figure 1A).
Figure 1. CA-Mφ and AA-Mφ marker profiles in control and endotoxin-tolerized murine macrophages.

Gene expression profiles for (A) CA-Mφ macrophage cytokines, (B) AA-Mφ macrophage markers, (C) AA-Mφ-associated chemokines, and (D) IL-4 and IL-13 were analyzed in C57BL/6J thioglycollate-elicted macrophage cultures pretreated for 24 h with medium (M) or LPS (L; 100 ng/ml), washed, and then challenged with medium (M) or LPS (L; 100 ng/ml) for 3, 6, or 24 h. The X-axis indicates the primary/challenge treatments. Results represent the mean ± SEM from 3 independent experiments. (*p < 0.05 between non-tolerized (M/L) and tolerized (L/L) groups; #p < 0.05 between non tolerized (M/L) and tolerized (L/M) groups).
Since IL-4 and IL-13 play a key role in AA-Mφ polarization, we also examined the expression of these genes during endotoxin tolerance. Interestingly, LPS failed to modulate expression of IL-4 mRNA (Figure 1D). However, IL-13 mRNA expression was strongly upregulated in medium-pretreated macrophages by primary LPS stimulation (M/L) at 3 and 6 h, but was inhibited in LPS-pretreated cells (Figure 1D), indicating that it too is a “tolerizable” gene. However, in supernatants derived from macrophage cultures following a prolonged (24 h) post-treatment period, neither IL-4 nor IL-13 protein levels were detectably modulated above levels seen in supernatants from medium-treated macrophages (data not shown).
Endotoxin-tolerant and AA-Mφ exhibit marked differences in CA-Mφ and AA-Mφ gene expression
Although it has been previously reported the expression of AA-Mφ-associated chemokines were not “tolerizable” by LPS (24, 25), these studies did not directly compare endotoxin-tolerant and alternatively activated macrophage populations for gene expression. IL-4 or IL-13 is necessary and sufficient for macrophage polarization towards an AA-Mφ phenotype (17, 32, 33). To compare directly the responses of cells rendered endotoxin-tolerant by LPS-pretreatment vs. alternatively activated by IL-4 pretreatment, peritoneal exudate macrophage cultures were initially treated with medium, LPS, or IL-4 for 24 h, then washed and restimulated cells with medium, LPS, or IL-4 for 3 h. Consistent with Figure 1, all CA-Mφ cytokine genes (Figure 2A) and AA-Mφ-associated chemokine genes (Figure 2B) were upregulated in medium-pretreated macrophages 3 h after LPS stimulation (M/L) and were tolerized by 24 h LPS-pretreatment, without or with LPS challenge (L/M or L/L). In contrast, IL-4 failed to induce any of the CA-Mφ genes and IL-4 pretreatment of macrophages did not tolerize against LPS challenge for expression of these genes (Figure 2A). IL-4 treatment for 3 h or 24 h failed to induce CCL2. However, IL-4 only induced CCL17 (Figure 2B) and CCL22 (data not shown) when Mφ were treated for 24 h. Similar to CA-Mφ genes, IL-4 failed to induce tolerance to LPS for each of these AA-Mφ-associated chemokine genes (Figure 2B). On the other hand, Arg1 and MR (data not shown) mRNA were poorly induced by LPS treatment for 3 h or 24 h, while IL-4 pretreatment for 24 h resulted in strong Arg1 and MR expression that was refractory to LPS stimulation (IL-4/L vs. IL-4/L) (Figure 2B).
Figure 2. IL-4 pretreatment of macrophages failed to induce LPS tolerance, modulate NF-κB and MAPK activation, or induce negative regulators associated with tolerance.

Gene expression profiles of (A) CA-Mφ proinflammatory cytokines, (B) AA-Mφ-associated chemokines and AA-Mφ macrophage marker, Arg-1 were analyzed in C57BL/6J thioglycollate-elicited macrophage cultures pretreated for 24 h with medium (M), LPS (L; 100 ng/ml) or IL-4 (40 ng/ml), washed, and then challenged with medium (M), LPS (100 ng/ml) or IL-4 (40 ng/ml) for 3 h. (C, D, F) Whole cell lysates and nuclear extracts were prepared from PEC pretreated for 24 h with medium-, LPS-, or IL-4, then challenged with medium, LPS, or IL-4 for 30 min and 1 h, respectively. (C) Nuclear extracts were pre-incubated with p65 and p50 antisera and analyzed for supershift assay by EMSA using an NF-κB consensus sequence and (D) whole cell lysates were subjected to Western analysis for MAPK activity. (E) Gene expression profile of negative regulators of LPS signaling, IRAK-M and, SHIP1 was analyzed as described for A and B. (F) I-κBα, RelB, p-STAT3, STAT3 and p-STAT6 protein expression was analyzed in whole cell lysates as described for D. Results represent the mean ± SEM from 2–3 independent experiments. (*p < 0.05 between non-tolerized (M/L) and tolerized (L/L) groups; #p < 0.05 between L/L and IL-4/L groups; ‡p < 0.05 between M/M and IL-4/M groups).
Endotoxin tolerant and AA-Mφ exhibit marked differences in NF-κB binding and MAPK activation
NF-κB is the key transcription factor involved in the induction of inflammatory mediator genes expression during LPS signaling. A switch in the preponderance of p65/p50 heterodimers to p50/p50 homodimers in endotoxin-tolerized murine and human cells has been well established (34). Accordingly, we next confirmed by EMSA with supershifts that formation of p50/p50 homodimers was increased in endotoxin-tolerized macrophages (Figure 2C). In contrast, IL-4-pretreated (IL-4/L and IL-4/M) cells failed to upregulate expression of p50/p50 homodimers and failed to alter the pattern of NF-κB p65 or p50 binding induced by LPS (compare M/L to IL-4/L) (Figure 2C). This indicates that IL-4 does not induce a state of LPS tolerance with respect to altered expression of the relative composition of p65 and p50 proteins. Hence, induction of AA-Mφ differentiation by IL-4 does not render macrophages endotoxin-tolerant by increasing expression of negative regulators, including p50/p50 homodimers.
The data provided in Figure 2D further support the conclusion that induction of LPS-tolerance does not affect IL-4 signaling and that differentiation of AA-Mφ with IL-4 does not interfere with LPS signaling. Specifically, medium-pretreated cells show robust phosphorylation of MAPKs in response to a 30 min exposure to LPS (M/L), whereas inhibition of MAPK activation was observed in macrophages pretreated with LPS, as evidenced by decreased phosphorylation of MAPKs in response to a 30 min LPS stimulation (L/L; Figure 2D). LPS-induced MAPK activity was not affected in IL-4-pretreated AA-Mφ macrophages (IL-4/L). IL-4 (M/IL-4) did not induce MAPK activity at 30 min and overnight exposure of macrophages to IL-4 failed to tolerize against LPS-induced MAPK (IL-4/L).
Endotoxin tolerant and AA-Mφ exhibit marked differences in expression of negative regulators of LPS signaling
Endotoxin tolerance has been associated with the production of many negative regulators (12). For example, IRAK-M was previously implicated as a mediator of tolerance as evidenced by the resistance of IRAK-M−/− mice to LPS-induced tolerance (35). Endotoxin-tolerized, LPS-restimulated macrophages (L/L) exhibited a significant increase in IRAK-M gene expression when compared to medium-pretreated, LPS-stimulated cells (Figure 2E). Boone et al. (36) have shown that A20 is rapidly induced by LPS stimulation and negatively regulates LPS signaling. However, this same group found that knockdown of A20 in mouse macrophages failed to affect tolerance induced by LPS (36). Similarly, we found that A20 gene expression was induced by LPS challenge (M/L) and was down-regulated in endotoxin-tolerized macrophages (L/L) (Figure 2E). The observed gene expression of A20 also correlated with protein expression (data not shown). In the case of both IRAK-M and A20, IL-4 stimulation of macrophages for either 3 (M/IL-4) or 24 (IL-4/M) h failed to elicit any of the changes induced by LPS pretreatment and failed to alter the response to LPS stimulation (IL-4/L) (Figure 2E)
IκBα, the cytosolic inhibitor of NF-κB, is degraded upon LPS stimulation, thereby allowing for NF-κB p65/p50 to translocate to the nucleus where it can induce many proinflammatory genes (37). As expected, IκBα was degraded in response to LPS in medium-pretreated cells (Figure 2F), and was not degraded in LPS-tolerized cells, as previously reported (38). RelB protein, an NF-κB family member previously associated with tolerance (39), was upregulated in LPS-tolerized cells (Figure 2F). IL-4 pretreatment of macrophages also failed to modulate expression of RelB.
STAT3 has been shown to regulate LPS-induced SOCS3 via IL-10 production and increased LPS-induced cytokine secretion was observed in STAT3-deficient macrophages (40, 41). While phospho-STAT3 was induced by 30 min LPS treatment, it was not significantly modulated by LPS-pretreatment. IL-4 pre- or post-treatment failed to phosphorylate STAT3 and had no effect on STAT3 phosphorylation induced by LPS (Figure 2F).
IL-4 receptor engagement is known to induce STAT6 (42) and, indeed, IL-4 strongly induced phospho-STAT6; however, phospho-STAT6 was not activated by LPS challenge in either control (M/L) or LPS-tolerized (L/L) macrophages, nor did LPS stimulation alter the response induced by IL-4 (Figure 2F).
A summary of the gene expression profile of cytokines and chemokines, M2 markers, and negative regulators induced by LPS in naïve or endotoxin-tolerized macrophages, in the absence or presence of IL-4, is presented in Table I.
Table I.
Induction of cytokines, chemokines, M2 markers, and negative regulators by LPS in naïve vs. tolerized macrophages and the effect of IL-4 pre-treatment on LPS signaling (Pretreatment = 24 h/Challenge = 3 h)
| M/M | M/L | L/M | L/L | IL-4/L | L/IL-4 | M/IL-4 | IL-4/M | ||
|---|---|---|---|---|---|---|---|---|---|
| Cytokines | TNF-α | − | +++ | +/− | + | +++ | − | − | − |
| IL-1β | − | +++ | +/− | + | +++ | − | − | − | |
| IL-6 | − | +++ | +/− | + | +++ | − | − | − | |
| Chemokines | CCL2 | − | +++ | +/− | + | +++ | + | − | − |
| CCL17 | − | +++ | +/− | +/− | ++++ | + | − | ++ | |
| CCL22 | − | +++ | +/− | +/− | ++++ | + | − | ++ | |
| M2 markers | Arg1 | − | − | − | − | +++ | + | + | +++ |
| MR | − | − | − | − | +++ | + | + | +++ | |
| Negative regulators | IRAK-M | − | ++ | ++ | +++ | ++ | ++ | − | − |
| A20 | − | +++ | +/− | + | +++ | − | − | − | |
| I-κBα* | +++ | − | +++ | +++ | − | +++ | +++ | +++ | |
| RelB* | − | − | +++ | +++ | − | +++ | − | − | |
−, no induction; +/−, mild/no induction; +, mild induction; ++, moderate induction; +++, high induction; ++++, very high induction; *, protein expression.
IL-4Rα, STAT6, and IL-13 are not required to establish endotoxin tolerance in macrophages
The IL-4Rα-mediated STAT6 signaling pathway plays an essential role in AA-Mφ polarization in vitro and in vivo (16, 17, 42). Since 24 h pretreatment of macrophages with IL-4 failed to induce endotoxin tolerance (Figure 2), we next used IL-4Rα−/−, STAT6−/− and IL-13−/− macrophages to confirm whether this pathway plays a role in LPS-induced tolerance. IL-4Rα is necessary for signaling by both IL-4 and IL-13 through the shared type II IL-4R (42) and STAT6 is activated downstream of IL-4/IL-13-mediated signaling through this receptor complex (20, 43). Arg-1 (Figure 3A) and MR (data not shown) mRNA levels were measured as positive controls for the AA-Mφ phenotype and were strongly induced by IL-4 in WT BALB/cByJ macrophages, but not in IL-4Rα−/− macrophages (Figure 3A). Consistent with the findings in C57BL/6 macrophages, both Arg1 and MR gene expression were increased only ~5-fold in endotoxin-tolerized WT BALB/cByJ macrophages (Figure 3B; L/M and L/L), whereas and LPS-induced Arg-1 and MR mRNA expression were no greater than cells treated with medium in IL-4Rα−/− macrophages (Figure 3B). This indicates that the weak induction of Arg-1 and MR mRNA detected in endotoxin-tolerized macrophages requires IL-4Rα signaling. However, we did not observe any difference in AA-Mφ-associated chemokine mRNA (Figure 3C) or protein (Figure 3D) levels during endotoxin tolerance. Like WT macrophages, IL-4Rα−/− macrophages become tolerant to LPS following 24 h exposure to endotoxin (L/M and L/L). As an additional positive control, LPS-induced tolerance was shown to be induced both at the level of mRNA (Figure 3E) and protein (Figure 3F) for the proinflammatory CA-Mφ markers analyzed. A nearly identical pattern of endotoxin tolerance was observed in LPS-tolerized STAT6−/− and IL-13−/− macrophages (Supplementary Figure 2). Collectively, these data indicate that AA-Mφ polarization and endotoxin tolerance are dissociable pathways and that the LPS-induced AA-Mφ-associated chemokines are not affected by the lack of IL-4Rα mediated signaling or by the lack of IL-13.
Figure 3. IL-4Rα signaling is not required for endotoxin tolerance.

(A) Thioglycollate-elicted macrophage cultures from WT BALB/cByJ and IL-4Rα−/− mice were pretreated with medium (Med) or IL-4 (40 ng/ml) for 24 h and mRNA was analyzed for Arg-1 mRNA. (B–F) Macrophages from WT BALB/c ByJ and IL-4Rα−/− mice were pretreated with medium (M) or LPS (L) for 24 h, washed, and then challenged with medium (M) or LPS (L) for 3 h. (B, C, E) Analysis of gene expression for (B) AA-Mφ markers; (C) AA-Mφ-associated chemokines; and (E) CA-Mφ cytokines, measured by qRT-PCR. (D, F) Analysis of AA-Mφ-associated chemokine protein and CA-Mφ cytokines in supernatants derived from cultures in (C) and (E), respectively, by ELISA. Results represent the mean ± SEM from 2 independent experiments. (*p < 0.05 between non-tolerized (M/L) and tolerized (L/L) groups; #p < 0.05 between non-tolerized (M/L) and tolerized (L/M) groups).
Since attenuation of TLR4-dependent signaling is a hallmark of endotoxin tolerance, we sought to determine whether such loss of TLR4 signaling was observed in macrophages that cannot be alternatively activated due to a lack of IL-4Rα or STAT6. Lack of IL-4Rα (Figure 4A) or STAT6 (Figure 4B) did not alter the ability of LPS to activate MAPKs, nor did it restore MAPK signaling in endotoxin-tolerized macrophages. These findings correlate well with gene expression and protein levels of proinflammatory cytokines during tolerance observed both in IL-4Rα−/− and STAT6−/− macrophages (Figures 3 and Supplementary Figure 2).
Figure 4. IL-4Rα or STAT6 deficiency does not affect the inhibition of LPS-driven MAPK activity seen in endotoxin-tolerized cells.
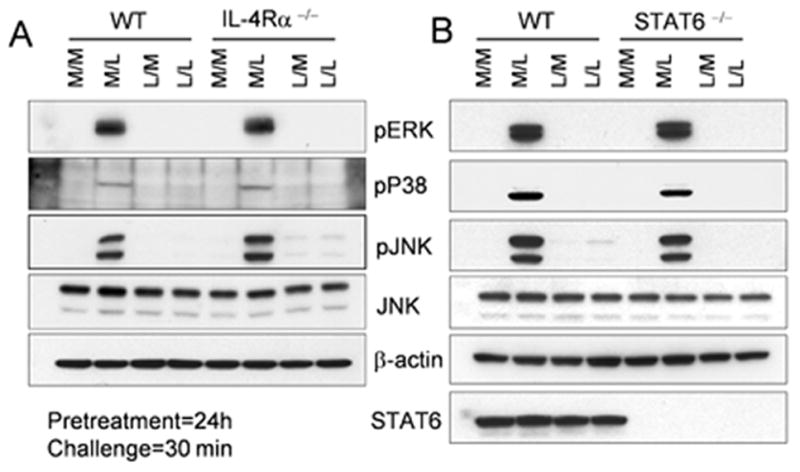
Thioglycollate-elicited macrophage cultures from (A) WT BALB/cByJ and IL-4Rα−/− or (B) WT BALB/cByJ and STAT6−/− mice were pretreated with medium (M) or LPS (L) for 24 h, washed, and challenged with medium (M) or LPS (L) for 30 min. Whole cell lysates from these cells were analyzed for MAPKs by Western blotting. Results represent the results of a single representative experiment of 2 independent experiments.
Inhibition of histone deacetylation induced by endotoxin tolerance in macrophages is not affected by IL-4Rα deficiency
Histone modification by acetylation and deacetylation of chromatin are important modifications that regulate transcription (44). In general, histone acetylation is a positive regulator, while deacetylation is a negative regulator of transcription (44). It has been reported previously that the down-regulated expression of certain “tolerizable” genes can be reversed and their expression up-regulated in the presence of Trichostatin A (TSA), an inhibitor of histone deacetylase (45, 46). Therefore, macrophages were pretreated with medium or LPS (as described in Figure 1) in the absence or presence of TSA. Proinflammatory (Figure 5A) and M2-associated chemokine (Figure 5B) gene expression was measured. TSA profoundly inhibited IL-6 mRNA expression in M/L-treated macrophages, and to a lesser extent, IL-1β and TNF-α mRNA expression. In LPS-tolerized (L/M or L/L) macrophages, TSA led to a reversal of the tolerance phenotype only with respect to IL-1β gene expression. Importantly, IL-4Rα deficiency had no effect on this pattern of gene expression.
Figure 5. IL-4Rα deficiency does not affect the endotoxin driven histone modification in endotoxin tolerized macrophages.

Thioglycollate-elicited macrophage cultures from WT BALB/cByJ and IL-4Rα−/−/Rag2−/− mice were pretreated with medium (M) or LPS (L) in presence or absence of TSA for 16–18 h, washed, and challenged with medium (M) or LPS (L) in the presence of absence of TSA for 3 h. (A, B) Analysis of gene expression for (A) AA-Mφ-associated chemokines and (B) CA-Mφ cytokines, measured by qRT-PCR. (‡p < 0.05 between non tolerized groups (M/L) in the absence or presence of TSA; *p < 0.05 between non-tolerized (M/L) and tolerized (L/L) groups; #p < 0.05 between tolerized (L/L) groups in the absence or presence of TSA).
TSA also suppressed expression of M2-associated chemokines (CCL2, CCL17 and CCL22) (Figure 5B) induced by LPS in non-tolerized macrophages (M/L). In LPS-tolerized macrophages, TSA reversed the suppression of all three M2-associated chemokines (L/L; Figure 5B), but failed to increase expression of CCL2 mRNA unless the cells were re-stimulated with LPS. Again, IL-4Rα deficiency did not alter the effect of TSA on endotoxin tolerance, consistent with the data observed in Figure 3.
LPS-pretreated WT and IL-4Rα-deficient animals are comparably tolerant to LPS + D-galactosamine-induced lethal shock and exhibit decreased LPS-induced cytokine levels in vivo
To extend the in vitro results we observed in WT and IL-4Rα-deficient macrophages, WT and knockout mice were pretreated with PBS or LPS, 24–30 h prior to challenge with LPS + D-galactosamine. All of the tolerized WT and IL-4Rα−/− mice survived an otherwise lethal challenge (Figure 6A). Non-tolerized (PBS-pretreated) control animals died within 24–30 h of LPS + D-galactosamine administration (Figure 6A). The level of serum cytokines/chemokines in LPS-tolerized (L/L) and non-tolerized (P/L) mice was also determined. IL-4Rα−/− mice were comparably LPS-responsive to WT mice. Also, the levels of TNF-α, IL-6 (Figure 6B), CCL2, and CCL22 (Figure 6C) were equivalently down-regulated both in WT and IL-4Rα−/− mice that were pretreated with LPS, consistent with the survival data (Figure 6A) as well as in vitro cytokines and chemokines we observed throughout the study. While CCL17 levels were slightly lower in sera of LPS-tolerized mice challenged with LPS, these did not achieve statistical significance when compared to PBS-pretreated, LPS-challenged values.
Figure 6. WT and IL-4Rα−/− mice are rendered comparably LPS-tolerant to LPS + D-galactosamine-induced lethal shock and LPS-induced cytokine production.
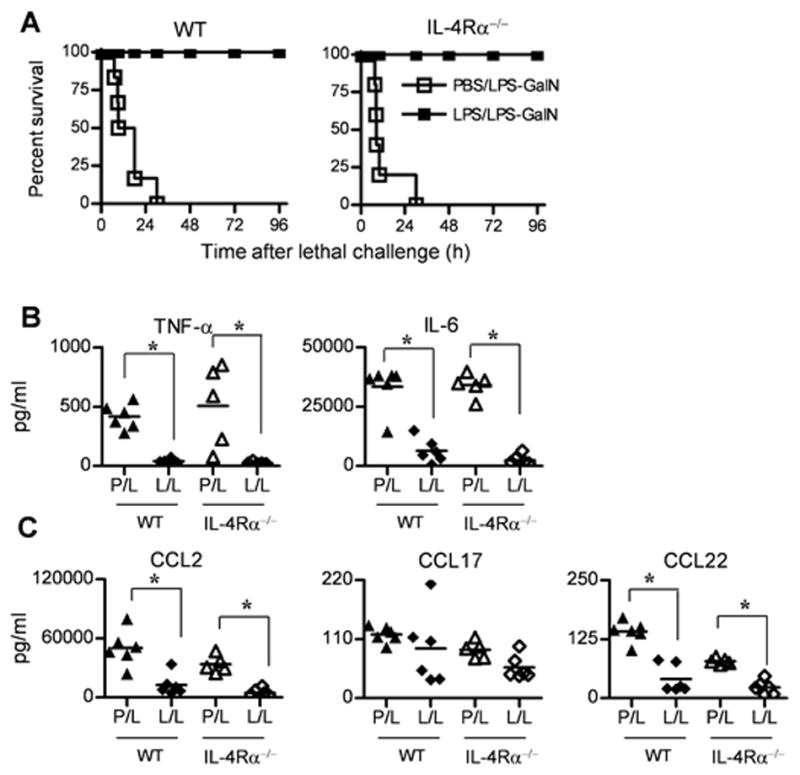
(A) Survival of WT BALB/cByJ and IL-4Rα−/− mice pretreated with PBS or LPS (25 μg/mouse) and then challenged 1 day later with LPS-D-GalN. Serum (B) cytokine and (C) chemokine levels were measured by ELISA in non-tolerized (PBS-pretreated) and tolerized (LPS-pretreated; 25 μg/mouse) WT and IL-4Rα−/− mice challenged with 25 μg LPS for 2 h. Each point represents an individual mouse.
Discussion
Endotoxin tolerance has been recognized for more than 70 years, but remains an enigma mechanistically. Because endotoxin tolerance is not a global shutdown of gene expression, i.e., some genes are repressed, while others are either up-regulated or unchanged (47), the use of the word “reprogramming” was coined by Morrison and coworkers to describe the mixed hyporesponsive/responsive transcriptional phenotype (48, 49). The mechanisms implicated in the induction of endotoxin tolerance are also complex: failure to recruit TLRs to rafts (50), failure to assemble the TLR signaling complex or recruit downstream adapter molecules (10, 51, 52), diminished NF-κB, AP-1, MAPK, and other signaling pathways (10, 53), an increase in p50/p50 homodimers (54) and RelB (39), and chromatin remodeling (45, 55) have all been implicated in the induction of the tolerant state. During sepsis, acute and prolonged stimulation of macrophages and monocytes by LPS or other TLR ligands may lead to excessive and pathological inflammation. It has been suggested that the hyporesponsiveness develops in septic patients following the “cytokine storm” is an adaptive mechanism to protect the host against inflammatory damage; however, this TLR hyporesponsiveness may also render the host more susceptible to secondary bacterial infection (56).
Recently, several studies have reported that the AA-Mφ differentiation state resembles endotoxin tolerized macrophages phenotypically in both human monocytes and murine thioglycollate-elicited or bone marrow-derived macrophages, suggesting a common induction pathway (19). Specifically, Porta et al. (25) reported that induction of tolerance by exposure of murine and human macrophages to LPS-induced gene expression profiles that were consistent with AA-Mφ polarization (as evidenced by expression of CCL2, CCL17 and CCL22), and that the expression of these markers is associated with increased NF-κB p50, consistent with the findings of Ziegler-Heitbrock et al. that p50/p50 homodimers predominate in endotoxin-tolerized macrophages (54). However, Porta et al. also reported that LPS restimulation of LPS-pretreated macrophages resulted in sustained expression, rather than limited expression, of these same chemokines. In an effort to extend their findings, we first sought to replicate their protocol exactly: thioglycollate-elicited C57BL/6 macrophages were tolerized by overnight treatment in vitro with our protein-free E. coli K235 LPS. Tolerance induction was confirmed by the finding that expression of proinflammatory genes (e.g., TNF-α, IL-β, IL-6, IL-12 p40 and IFN-β) were, indeed, strongly induced in response to primary exposure to LPS, but clearly repressed upon secondary exposure to LPS (Figure 1). Two markers frequently associated with the AA-Mφ phenotype, Arg-1 and MR, were also poorly induced by LPS pre- or post-exposure (<10-fold; Figure 1B); however, both were down-regulated upon LPS restimulation in C57BL/6 macrophages, indicating that they, too, were subject to the effects of LPS-induced tolerance (Figure 1). While our data completely mirrored that of Porta et al. in that the AA-Mφ-associated chemokine genes were strongly upregulated by LPS, we observed that expression of these chemokine genes was strongly inhibited in endotoxin-tolerized cells upon LPS restimulation. At this time, we cannot account for these discrepancies; however, we observed similar patterns of responsiveness in thioglycollate-elicited BALB/c macrophages, bone marrow-derived C57BL/6J macrophages (data not shown), and in the RAW 264.7 cell line, which was derived from BALB/c mice (Supplementary Figure 1B). Thus, it is unlikely that the differences observed are due to strain differences or the source of macrophages.
Importantly, pretreatment of macrophages with IL-4 to induce an AA-Mφ state of macrophage differentiation (as evidenced by robust induction of Arg-1 mRNA; Figure 2B and MR mRNA; data not shown) failed to diminish LPS-induced expression of proinflammatory genes (e.g., TNF-α, IL-1β, and IL-6) (Figure 2A), the AA-Mφ-associated chemokines, CCL2, CCL17, and CCL22 (Figure 2B), NF-κB binding or relative expression of p65 and p50 as observed in EMSA and supershift assays (Figure 2C) or MAPK activation (Figure 2D) and negative regulators of LPS signaling (Figures 2E and F), in response to LPS challenge, in contrast to macrophages pretreated with LPS. Collectively, these data suggest that IL-4 stimulation of macrophages engagement does not induce the well-characterized signaling changes previously associated with LPS-induced tolerance. Since IL-4/IL-13-induced STAT6 activation is critical for the establishment of AA-Mφ (23), we reasoned that examining the requirement for IL-4Rα (which is utilized by both IL-4 and IL-13 in the type II IL-4R) and/or STAT6 (which is downstream of both IL-4/IL-13 signaling) might shed light on a possible relationship between these the AA-Mφ and tolerant states. We confirmed that induction of both Arg-1 and MR mRNA by LPS was entirely dependent on both the IL-4Rα chain and STAT6 signaling (Figures 3B and Supplementary Figure 2B). Although LPS induced IL-13, but not IL-4, mRNA, we were unable to detect a measurable increase in IL-13 protein in supernatants from LPS-stimulated macrophages (data not shown). However, the finding that IL-13−/− macrophages are fully tolerizable by LPS pre-treatment (Supplemental Figure 2G), but not by IL-4 pretreatment, further supports the hypothesis that neither LPS-induced IL-4 nor IL-13 mediate tolerance. Interestingly, the lack of IL-13 does not preclude expression of CCL17 mRNA induced by LPS, suggesting that an alternate pathway for induction of AA-Mφ associated markers exists. The low level of Arg1 mRNA induced by LPS was IL-4Rα- and STAT6-dependent. Qualls et al. (57) showed that in M. tuberculosis-infected macrophages, Arg1 was induced in the absence of STAT6 by autocrine stimulation by cytokines (IL-6, IL-10 and G-CSF) in a manner that was STAT3- and partially C/EBPβ dependent. We observed a low level of phospho-STAT3 in response to LPS, although this did not differ in WT and IL-4Rα−/− macrophages (data not shown). Moreover, STAT3 activation was slightly increased in response to LPS, but not IL-4 (Figure 2F), in WT macrophages. Thus, it seems unlikely that the low level of Arg1 mRNA detected in LPS-stimulated macrophages is due to the STAT3-dependent mechanism described by Qualls et al. However, lack of IL-4R, STAT6, or IL-13 failed to affect the response to LPS for the AA-Mφ-associated chemokine or traditional proinflammatory gene and protein expression (Figures 3C-F and Supplementary Figures 2C-G). This was also the case for MAPK activation (Figure 4). These data further support our central conclusion that the ability of macrophages to differentiate into AA-Mφ is not a prerequisite for endotoxin tolerance. Moreover, this same pattern of responsiveness to LPS in naïve vs. tolerized macrophages was observed in both C57BL/6 and BALB/c backgrounds for proinflammatory and chemokine genes.
A significant issue highlighted in our work concerns the relative plasticity and responsiveness of the AA-Mφ and endotoxin tolerant phenotypes. AA-Mφ induced by IL-4 remained competent to respond to TLR ligands, and can robustly induce the full array of classical pro-inflammatory cytokines. As such, this AA-Mφ phenotype can be readily reversed by TLR4 signaling. The signaling and epigenetic changes conferred in endotoxin tolerance appear to be far more durable, as exposure to subsequent TLR stimulation or to IL-4 does not restore either the M1 or M2 polarization, respectively. It is also important to note that the molecular differences we describe in the regulation of TLR4 signaling between AA-Mφ and endotoxin tolerant macrophages may reflect the different biological contexts in which these populations accumulate in vivo. As has been elegantly described in a recent publication by Jenkins et al. (58), classically activated, and subsequently endotoxin tolerant, macrophages rapidly accumulate at sites of microbial infection through the induced recruitment of Ly6Chi “inflammatory” monocytes from the blood. In contrast, alternatively activated macrophages appear to be generated in situ via IL-4/13-dependent cell division in response to chronic “sterile” inflammation such as generated during nematode infection or in cases of wound repair. Additionally, our data indicates that these AA-Mφs retain the ability to respond to microbial PAMPs via TLR signaling.
At the core of the present study and those of others is the molecular definition of the AA-Mφ phenotype. There are relatively few studies that address why there are so few AA-Mφ markers in common between murine vs. human cells. Perhaps, this is attributable to differences in the way the macrophages are prepared (unelicited, elicited, or bone marrow-derived in the case of mouse vs. differentiated overnight or longer, treated with growth promoting agents or not, in the case of human monocytes/macrophages) prior to treatment with AA-Mφ-inducing agents. There is a clear need for systematic examination of different cell types for their responsiveness to IL-4 or IL-13 over time to determine whether or not there is a truly common set of “markers” that defines the AA-Mφ phenotype in both species. For example, there does not appear to be consensus in the literature about the suitability of CCL chemokines as macrophage alternative activation markers. We observed that these previously AA-Mφ-associated CCL chemokines were also strongly induced by LPS with kinetics that strongly resemble proinflammatory, CA-Mφ genes, and that the LPS-dependent induction of the CCL chemokines was blunted in LPS-tolerized macrophages. This same observation was reported by Foster et al. who used microarray analysis to show that CCL22 was categorized as an endotoxin-tolerizable gene (45) and Carmody et al. who showed that CCL2 gene expression was also repressed during tolerance (59). However, the fate of CCL17 gene expression during endotoxin tolerance has not been reported previously, although the induction of its gene expression by LPS has been previously reported (27, 60).
IL-4 and IL-13 are considered to be anti-inflammatory cytokines that down-modulate expression of proinflammatory cytokines including TNF-α, IL-6, IL-8, and other inflammatory mediators such as iNOS and COX2 using IL-4Rα as a common receptor chain (61, 62). Of note, we observed slightly increased mRNA and protein levels of proinflammatory cytokines upon LPS stimulation in IL-4Rα−/− macrophages, indicating that the IL-4 signaling pathway plays an important role in keeping the inflammatory response induced by LPS in check. Interestingly, we did not observe increased proinflammatory cytokine gene or protein expression in STAT6−/− macrophages, consistent with published reports showing that suppression of proinflammatory cytokines mediated by IL-4 by both STAT6-dependent and STAT6-independent mechanisms (63). Other signaling pathways downstream of the IL-4 receptor complex, such as IRS2 and Shc, may suppress proinflammatory gene expression (64).
Additionally, another potential mediator of tolerance that we did not examine in the context of IL-4Rα−/− is IL-10. Previous studies have shown that IL-10 suppressed the proinflammatory response induced by LPS (65, 66). However, Berg et al. showed that IL-10 deficiency did not alter the endotoxin tolerance and that tolerized IL-10−/− mice nonetheless survived LPS-induced lethal shock (67). Finally, our molecular observations demonstrating a dissociation between endotoxin tolerance and alternative activation are directly supported by the in vivo LPS-induced lethality experiment in which both WT and IL-4Rα−/− mice survived a lethal exposure to LPS following the establishment of endotoxin tolerance. Based on the observed results reported herein, we believe that CCL2, CCL17 and CCL22 are not pure AA-Mφ markers (25), but rather, are inducible by both LPS and IL-4 and, perhaps, help to attract Th2 cells in vivo (26). Overall, our data indicate that endotoxin tolerance and AA-Mφ polarization are distinct pathways that are differentially regulated.
Supplementary Material
Acknowledgments
The authors thank Drs. Kari Ann Shirey, Meghan Pennini, Daniel Prantner, Mr. Pragnesh Mistry and Ms. Trystan Dyson for their help and advice throughout the study.
Abbreviations
- COX-2
Cyclooxygenase 2
- FIZZ1
Found in inflammatory zone 1
- HPRT
Hypoxanthine-guanine phosphoribosyltransferase
- iNOS
inducible NO synthase
- IRS
Insulin responsive substrate
- IRF
Interferon regulatory factor
- IRAK
Interleukin-1 (IL-1) receptor-associated kinase
- KO
Knockout
- MR
Mannose receptor
- PAMP
pathogen-associated molecular pattern
- qRT-PCR
quantitative real time-PCR
- ROS
reactive oxygen species
- TSA
tricostatin A
- WT
wild type
Footnotes
This work was supported in part by NIH grant #AI18797 (SNV) and AI038985 (ADK).
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 3.Daubeuf B, Mathison J, Spiller S, Hugues S, Herren S, Ferlin W, Kosco-Vilbois M, Wagner H, Kirschning CJ, Ulevitch R, Elson G. TLR4/MD-2 monoclonal antibody therapy affords protection in experimental models of septic shock. J Immunol. 2007;179:6107–6114. doi: 10.4049/jimmunol.179.9.6107. [DOI] [PubMed] [Google Scholar]
- 4.O’Neill LA. How Toll-like receptors signal: what we know and what we don’t know. Curr Opin Immunol. 2006;18:3–9. doi: 10.1016/j.coi.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 5.Beutler B, Poltorak A. Sepsis and evolution of the innate immune response. Crit Care Med. 2001;29:S2–6. doi: 10.1097/00003246-200107001-00002. discussion S6–7. [DOI] [PubMed] [Google Scholar]
- 6.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Dobrovolskaia MA, Vogel SN. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002;4:903–914. doi: 10.1016/s1286-4579(02)01613-1. [DOI] [PubMed] [Google Scholar]
- 8.West MA, Heagy W. Endotoxin tolerance: A review. Crit Care Med. 2002;30:S64–S73. [PubMed] [Google Scholar]
- 9.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 10.Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and toll-like receptor 2 and 4 gene expression. J Immunol. 2000;164:5564–5574. doi: 10.4049/jimmunol.164.11.5564. [DOI] [PubMed] [Google Scholar]
- 11.Perkins DJ, Qureshi N, Vogel SN. A Toll-like receptor-responsive kinase, protein kinase R, is inactivated in endotoxin tolerance through differential K63/K48 ubiquitination. MBio. 2010;1:e00239–10. doi: 10.1128/mBio.00239-10. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van ‘t Veer CPS, van den Pangaart MA, van Zoelen M, de Kruif RS, Birjmohun ES, Stroes AF, de Vos, van der Poll T. Induction of IRAK-M is associated with lipopolysaccharide tolerance in a human endotoxemia model. J Immunol. 2007;179:7110–7120. doi: 10.4049/jimmunol.179.10.7110. [DOI] [PubMed] [Google Scholar]
- 13.Xiong Y, Medvedev AE. Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J Leukoc Biol. 2011;90:1141–1148. doi: 10.1189/jlb.0611273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: tilting toward immunosuppression. Nat Med. 2009;15:496–497. doi: 10.1038/nm0509-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 17.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 18.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 19.Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 20.Heller NM, Qi X, Junttila IS, Shirey KA, Vogel SN, Paul WE, Keegan AD. Type I IL-4Rs selectively activate IRS-2 to induce target gene expression in macrophages. Sci Signal. 2008;1:ra17. doi: 10.1126/scisignal.1164795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shirey KA, Pletneva LM, Puche AC, Keegan AD, Prince GA, Blanco JC, Vogel SN. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunol. 2010;3:291–300. doi: 10.1038/mi.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–292. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raes G, De Baetselier P, Noel W, Beschin A, Brombacher F, Hassanzadeh GhG. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J Leukoc Biol. 2002;71:597–602. [PubMed] [Google Scholar]
- 24.Pena OM, Pistolic J, Raj D, Fjell CD, Hancock RE. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J Immunol. 2011;186:7243–7254. doi: 10.4049/jimmunol.1001952. [DOI] [PubMed] [Google Scholar]
- 25.Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, Dieli F, Ghisletti S, Natoli G, De Baetselier P, Mantovani A, Sica A. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A. 2009;106:14978–14983. doi: 10.1073/pnas.0809784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sekiya T, Miyamasu M, Imanishi M, Yamada H, Nakajima T, Yamaguchi M, Fujisawa T, Pawankar R, Sano Y, Ohta K, Ishii A, Morita Y, Yamamoto K, Matsushima K, Yoshie O, Hirai K. Inducible expression of a Th2-type CC chemokine thymus- and activation-regulated chemokine by human bronchial epithelial cells. J Immunol. 2000;165:2205–2213. doi: 10.4049/jimmunol.165.4.2205. [DOI] [PubMed] [Google Scholar]
- 27.El Chartouni C, Rehli M. Comprehensive analysis of TLR4-induced transcriptional responses in interleukin 4-primed mouse macrophages. Immunobiology. 2010;215:780–787. doi: 10.1016/j.imbio.2010.05.032. [DOI] [PubMed] [Google Scholar]
- 28.McIntire FC, Sievert HW, Barlow GH, Finley RA, Lee AY. Chemical, physical, biological properties of a lipopolysaccharide from Escherichia coli K-235. Biochemistry. 1967;6:2363–2372. doi: 10.1021/bi00860a011. [DOI] [PubMed] [Google Scholar]
- 29.Salkowski CA, Kopydlowski K, Blanco J, Cody MJ, McNally R, Vogel SN. IL-12 is dysregulated in macrophages from IRF-1 and IRF-2 knockout mice. J Immunol. 1999;163:1529–1536. [PubMed] [Google Scholar]
- 30.Medvedev AE, Blanco JC, Qureshi N, Vogel SN. Limited role of ceramide in lipopolysaccharide-mediated mitogen-activated protein kinase activation, transcription factor induction, and cytokine release. J Biol Chem. 1999;274:9342–9350. doi: 10.1074/jbc.274.14.9342. [DOI] [PubMed] [Google Scholar]
- 31.Polumuri SK, V, Toshchakov Y, Vogel SN. Role of phosphatidylinositol-3 kinase in transcriptional regulation of TLR-induced IL-12 and IL-10 by Fc gamma receptor ligation in murine macrophages. J Immunol. 2007;179:236–246. doi: 10.4049/jimmunol.179.1.236. [DOI] [PubMed] [Google Scholar]
- 32.Goerdt S, Politz O, Schledzewski K, Birk R, Gratchev A, Guillot P, Hakiy N, Klemke CD, Dippel E, Kodelja V, Orfanos CE. Alternative versus classical activation of macrophages. Pathobiology. 1999;67:222–226. doi: 10.1159/000028096. [DOI] [PubMed] [Google Scholar]
- 33.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 34.Ziegler-Heitbrock L. The p50-homodimer mechanism in tolerance to LPS. J Endotoxin Res. 2001;7:219–222. [PubMed] [Google Scholar]
- 35.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 36.Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, McNally E, Pickart C, Ma A. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 37.Carmody RJ, Chen YH. Nuclear factor-kappaB: activation and regulation during toll-like receptor signaling. Cell Mol Immunol. 2007;4:31–41. [PubMed] [Google Scholar]
- 38.Xiong Y, Qiu F, Piao W, Song C, Wahl LM, Medvedev AE. Endotoxin tolerance impairs IL-1 receptor-associated kinase (IRAK) 4 and TGF-beta-activated kinase 1 activation, K63-linked polyubiquitination and assembly of IRAK1, TNF receptor-associated factor 6, and IkappaB kinase gamma and increases A20 expression. J Biol Chem. 2011;286:7905–7916. doi: 10.1074/jbc.M110.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X, El Gazzar M, Yoza BK, McCall CE. The NF-kappaB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J Biol Chem. 2009;284:27857–27865. doi: 10.1074/jbc.M109.000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qin H, Roberts KL, Niyongere SA, Cong Y, Elson CO, Benveniste EN. Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 2007;179:5966–5976. doi: 10.4049/jimmunol.179.9.5966. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi M, Kweon MN, Kuwata H, Schreiber RD, Kiyono H, Takeda K, Akira S. Toll-like receptor-dependent production of IL-12p40 causes chronic enterocolitis in myeloid cell-specific Stat3-deficient mice. J Clin Invest. 2003;111:1297–1308. doi: 10.1172/JCI17085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dasgupta P, Chapoval SP, Smith EP, Keegan AD. Transfer of in vivo primed transgenic T cells supports allergic lung inflammation and FIZZ1 and Ym1 production in an IL-4Ralpha and STAT6 dependent manner. BMC Immunol. 2011;12:60. doi: 10.1186/1471-2172-12-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ford AQ, Heller NM, Stephenson L, Boothby MR, Keegan AD. An atopy-associated polymorphism in the ectodomain of the IL-4R(alpha) chain (V50) regulates the persistence of STAT6 phosphorylation. J Immunol. 2009;183:1607–1616. doi: 10.4049/jimmunol.0803266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 45.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 46.Nusinzon I, Horvath CM. Unexpected roles for deacetylation in interferon- and cytokine-induced transcription. J Interferon Cytokine Res. 2005;25:745–748. doi: 10.1089/jir.2005.25.745. [DOI] [PubMed] [Google Scholar]
- 47.Henricson BE, Benjamin WR, Vogel SN. Differential cytokine induction by doses of lipopolysaccharide and monophosphoryl lipid A that result in equivalent early endotoxin tolerance. Infect Immun. 1990;58:2429–2437. doi: 10.1128/iai.58.8.2429-2437.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shnyra A, Brewington R, Alipio A, Amura C, Morrison DC. Reprogramming of lipopolysaccharide-primed macrophages is controlled by a counterbalanced production of IL-10 and IL-12. J Immunol. 1998;160:3729–3736. [PubMed] [Google Scholar]
- 49.Zhang X, Morrison DC. Lipopolysaccharide structure-function relationship in activation versus reprogramming of mouse peritoneal macrophages. J Leukoc Biol. 1993;54:444–450. doi: 10.1002/jlb.54.5.444. [DOI] [PubMed] [Google Scholar]
- 50.Cuschieri J, Billigren J, Maier RV. Endotoxin tolerance attenuates LPS-induced TLR4 mobilization to lipid rafts: a condition reversed by PKC activation. J Leukoc Biol. 2006;80:1289–1297. doi: 10.1189/jlb.0106053. [DOI] [PubMed] [Google Scholar]
- 51.Nomura F, Akashi S, Sakao Y, Sato S, Kawai T, Matsumoto M, Nakanishi K, Kimoto M, Miyake K, Takeda K, Akira S. Cutting edge: endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J Immunol. 2000;164:3476–3479. doi: 10.4049/jimmunol.164.7.3476. [DOI] [PubMed] [Google Scholar]
- 52.Piao W, Song C, Chen H, Diaz MA, Wahl LM, Fitzgerald KA, Li L, Medvedev AE. Endotoxin tolerance dysregulates MyD88- and Toll/IL-1R domain-containing adapter inducing IFN-beta-dependent pathways and increases expression of negative regulators of TLR signaling. J Leukoc Biol. 2009;86:863–875. doi: 10.1189/jlb.0309189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobrovolskaia MA, Medvedev AE, Thomas KE, Cuesta N, Toshchakov V, Ren T, Cody MJ, Michalek SM, Rice NR, Vogel SN. Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR “homotolerance” versus “heterotolerance” on NF-kappa B signaling pathway components. J Immunol. 2003;170:508–519. doi: 10.4049/jimmunol.170.1.508. [DOI] [PubMed] [Google Scholar]
- 54.Ziegler-Heitbrock HW, Wedel A, Schraut W, Strobel M, Wendelgass P, Sternsdorf T, Bauerle PA, Haas JG, Riethmuller G. Tolerance to lipopolysaccharide involves mobilization of nuclear factor kappa B with predominance of p50 homodimers. J Biol Chem. 1994;269:17001–17004. [PubMed] [Google Scholar]
- 55.Chen J, Ivashkiv LB. IFN-γ abrogates endotoxin tolerance by facilitating Toll-like receptor-induced chromatin remodeling. Proc Natl Acad Sci U S A. 2010;107:19438–19443. doi: 10.1073/pnas.1007816107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 57.Qualls JE, Neale G, Smith AM, Koo MS, DeFreitas AA, Zhang H, Kaplan G, Watowich SS, Murray PJ. Arginine usage in mycobacteria-infected macrophages depends on autocrine-paracrine cytokine signaling. Sci Signal. 2010;3:ra62. doi: 10.1126/scisignal.2000955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS, Allen JE. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–1288. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. Negative regulation of toll-like receptor signaling by NF-kappaB p50 ubiquitination blockade. Science. 2007;317:675–678. doi: 10.1126/science.1142953. [DOI] [PubMed] [Google Scholar]
- 60.Liddiard K, Welch JS, Lozach J, Heinz S, Glass CK, Greaves DR. Interleukin-4 induction of the CC chemokine TARC (CCL17) in murine macrophages is mediated by multiple STAT6 sites in the TARC gene promoter. BMC Mol Biol. 2006;7:45. doi: 10.1186/1471-2199-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katz Y, Strunk RC. Enhanced synthesis of factor B induced by tumor necrosis factor in human skin fibroblasts is decreased by IL-4. J Immunol. 1990;144:4675–4680. [PubMed] [Google Scholar]
- 62.Cox GW, Chattopadhyay U, Oppenheim JJ, Varesio L. IL-4 inhibits the costimulatory activity of IL-2 or picolinic acid but not of lipopolysaccharide on IFN-gamma-treated macrophages. J Immunol. 1991;147:3809–3814. [PubMed] [Google Scholar]
- 63.Levings MK, Schrader JW. IL-4 inhibits the production of TNF-alpha and IL-12 by STAT6-dependent and -independent mechanisms. J Immunol. 1999;162:5224–5229. [PubMed] [Google Scholar]
- 64.Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD. Interleukin-4 and interleukin-13 signaling connections maps. Science. 2003;300:1527–1528. doi: 10.1126/science.1085458. [DOI] [PubMed] [Google Scholar]
- 65.de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O’Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–3822. [PubMed] [Google Scholar]
- 67.Berg DJ, Kuhn R, Rajewsky K, Muller W, Menon S, Davidson N, Grunig G, Rennick D. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J Clin Invest. 1995;96:2339–2347. doi: 10.1172/JCI118290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.