

Abstract
Platelets are most recognized as the cellular mediator of thrombosis, but they are increasingly appreciated for their immunomodulatory roles, including responses to Plasmodium infection. Platelet interactions with endothelial cells and leukocytes contribute significantly to the pathogenesis of experimental cerebral malaria (ECM). Recently it has been suggested that platelets not only have an adverse role in cerebral malaria, but platelets may also be protective in animal models of uncomplicated malaria. We now demonstrate that these diverse and seemingly contradictory roles for platelets extend to cerebral malaria models and are dependent on the timing of platelet activation during infection. Our data shows that platelets are activated very early in ECM and have a central role in initiation of the acute phase response to blood stage infection. Unlike platelet depletion or inhibition post infection, pre-infection platelet depletion or treatment with a platelet inhibitor is not protective. Additionally, we show that platelet driven acute phase responses have a major role in protecting mice from ECM by limiting parasite growth. Our data now suggests that platelets have a complex role in ECM pathogenesis: platelets help limit parasite growth early post infection, but with continued platelet activation as the disease progresses, platelets contribute to ECM associated inflammation.
Introduction
Malaria is a leading cause of child morbidity and mortality worldwide. In 2011 the World Health Organization (WHO) estimated there were 216 million cases of malaria with 655,000 deaths, 86% of which were children less than 5 years of age (1). Cerebral malaria (CM) is a severe complication of Plasmodium falciparum infection characterized by neurocognitive deficits, stroke-like cerebral vascular lesions and often death. CM has a predilection for children under 5 years of age and accounts for a significant percentage of malaria related mortality. CM is typified by sequestration of infected RBCs (iRBCs) in cerebral microvessels with post mortem brains showing petechial hemorrhage and vascular lesions with iRBCs, leukocytes and platelets (2). Despite its potentially devastating outcome, the pathogenesis of CM is still not well understood.
Platelets have important roles in many vascular inflammatory diseases, including CM (3). Platelets are best known for their role as the cellular mediator of hemostasis, but platelets also have important immunomodulatory effects (4). Platelets interact with an intact, inflamed endothelial cell layer, leading to platelet activation and further vascular inflammation. An activated endothelium expresses selectins and adhesion molecules (ICAM, VCAM) that mediate platelet tethering, rolling and eventually adhesion. Platelet activation leads to degranulation and the release of cytokines and chemokines, as well as the secretion of newly synthesized immune mediators, such as thromboxane and IL-1β, leading to more vascular inflammation and leukocyte recruitment. This contributes to the obstruction of vessels in CM by platelets and infiltrating leukocytes. Platelet derived immune mediators also exert effects distant from the site of vascular injury. Because platelets are so numerous and are packed with large numbers of immune mediators, platelet activation leads to high plasma concentrations of chemokines and cytokines such as PF4, RANTES, CXCL7, and IL-1β (5).
Vascular inflammation, immune stimulation and vascular obstruction that typify CM are driven in large part by platelets (3). We have previously described a mechanism in which the platelet derived chemokine PF4, contributes to CM pathogenesis by recruiting and activating monocytes and T cells at sites distant from cerebral vascular inflammation, helping to drive CM pathology (6). There are numerous reports demonstrating a mechanistic role for platelets in driving the pathology associated with CM (2, 3, 7, 8). There are recent reports indicating that platelets may also have a protective role in uncomplicated malaria by killing Plasmodium in iRBCs (9). This coupled with other conflicting reports on the benefits of antiplatelet therapy in humans (10, 11) prompted us to explore the complex role of platelets in CM development.
The acute phase response (APR) is the earliest stage of the innate immune responses to injury or infection and results in the production of acute phase proteins (APP) from the liver. Major APPs include C reactive protein (CRP), serum amyloids A and P (SAA and SAP) and complement proteins. Pro-inflammatory cytokines produced early in inflammation activate APP production from hepatocytes (12). Major stimulators of APP production include IL-6, IL-1β and TGF-β. Although IL-6 is not found in platelets, IL-1β and TGF-β are abundant platelet derived immune mediators. Activated platelets express adhesion molecules (eg. P-selectin) and hepatic sinusoids have lectin receptors that may interact with the activated platelets to induce the production of APPs, demonstrating the potential for platelets to have a major role in the induction of the APR.
Platelets are immune modulators in ECM, but the mechanisms and cell interactions have not been fully elucidated. Because the timing and phase of the response is key in immune pathogenesis, this immune stimulatory role for platelets may account for the seemingly conflicting reports on the roles of platelets in Plasmodium related disease pathogenesis. We now demonstrate that platelets are activated very early during blood stage infection with P berghei and platelets initiate the acute phase response. Furthermore, we demonstrate that adverse versus protective roles for platelets depend on the timing during infection and the activation of the acute phase response.
Materials & Methods
Reagents
Enzyme linked immune-absorbent assays (ELISAs) for mouse IL-1β, mouse PF4 and human CRP were purchased from R&D Systems. Mouse SAA and SAP ELISAs were purchased from Invitrogen and Immunology Consultants Lab Inc respectively. Platelet depleting as well as fluorescent platelet labeling antibodies were purchased from Emfret Analytics. SYBRGreen DNA labeling dye was purchased from Invitrogen. Clopidogrel was obtained from Bristol Myers Squib/Sanofi Partnership. Murine recombinant SAP (rSAP) and murine recombinant IL-1β (mr IL-1β) were purchased from R & D Systems.
Animals
Wild type mice on a C57BL6/J background between 6–8 weeks of age were purchased from Jackson Labs. Mice transgenic for rabbit CRP on a B6 background greater than 10 generations are described elsewhere(13) and were a kind gift from Philip Shaul, UT Southwestern. All experimental procedures using mice were approved by the University of Rochester School of Medicine Institutional Animal Care and Use Committee.
Platelet depletion
Mice were platelet depleted by an intraperitoneal (i.p) injection with 4 μg/g of platelet depleting antibody (directed against GPIbα; CD42b) or control IgG (isotype control).
Murine Experimental Cerebral Malaria Model
Mice were injected i.p with 0.5–0.7 × 106 P. berghei ANKA iRBCs (clone 2.34 provided by Dr. Fidel Zavala, The Johns Hopkins University School of Public Health). Survival was determined within 10 days post P. berghei ANKA inoculation and mice were monitored for experimental cerebral malaria (ECM) symptoms: cachexia, deviation of the head, prostanation and coma. Mice exhibiting 2 or more of these symptoms were deemed ECM+ and sacrificed if unable to be aroused from a prostate position. Parasitemia was determined by SYBRGreen labeling of iRBCs (14) and confirmed by Diff Quik staining of thin blood smears.
Cytokine and Chemokine Analyses
Mouse blood was collected by retro-orbital sinus into EDTA coated tubes and plasma obtained by subsequent centrifugation at 3200 rpm for 10 minutes. The plasma was then used in ELISAs per manufacturer’s instructions.
Platelet Isolation
Murine blood was obtained through the retro-orbital plexus into heparinized tyrodes buffer (134 mM NaCl; 0.34 mM Na2HPO4; 2.9 mM KCl; 12 mM NaHCO3; 20 mM Hepes; pH 7.0; 5 mM glucose; 0.35% (w/v) bovine serum albumin) in eppendorf tubes. The blood was then centrifuged at 1000 rpm for 5 minutes at room temperature to obtain platelet rich plasma (PRP) which was then transferred into new tubes containing tyrodes buffer (with 1% PGE2 to prevent platelet activation). Platelets were pelleted by a subsequent 2600 rpm centrifugation for 5 minutes at room temperature and resuspended in tyrodes buffer and used for further experiments within 2 hours.
Platelet trafficking by fluorescent microscopy
Mice were infected and 24 hours post infection, mice were injected i.v with fluorescently labeled anti-platelet antibody (X488, Emfret). Mice were then euthanized, livers thinly sliced (<1mm sections) and imaged with a Nikon Ti microscope and still images captured using Elements software. Platelet foci from 10 fields were analyzed and quantified using ImageJ software.
Transmission Electron Microscopy
Mice were infected with P. berghei, euthanized 24 hours later and livers excised into PBS:BSA buffer without perfusion. The excised livers were then thinly sectioned and immersion fixed in a combination fixative containing 4.0% paraformaldehyde/2.5% glutaraldehyde in 0.1M sodium cacodylate buffer (pH 7.4). After 24 hours of primary fixation the tissue was rinsed in the same buffer and post-fixed for one hour in buffered 1.0% osmium tetraoxide, dehydrated in a graded series of ethanol to 100%, transitioned into propylene oxide, infiltrated with EPON/Araldite resin overnight, embedded into molds and polymerized for 48 hours at 60°C. Using a glass knife, epoxy embedded liver blocks were sectioned at one micron and stained with Toluidine Blue to determine areas for thin sectioning. A diamond knife inserted into the ultramicrotome was used to cut 70nm thin sections which were placed onto carbon coated nickel grids. The grids were stained with aqueous uranyl acetate and lead citrate and examined using a Hitachi 7650 TEM with an attached Gatan 11 megapixel Erlangshen digital camera and Digitalmicrograph software.
SAP reconstitution in vivo
Mice were platelet depleted or treated with isotype control as above. Platelet depleted mice were given murine recombinant SAP (1mg/kg; ip) from day 1–3 post P berghei infection and survival monitored. Data Analysis. Data is expressed as mean ± standard deviations. Statistical significance of values was determined by using unpaired Students’ two-tailed t-tests; P-values of 0.05 or less were deemed significant. Survival data were analyzed by Log-Rank tests using Prism Graphpad software. P values of 0.05 or less were deemed significant.
Results
The pathogenic effect of platelets in ECM is time dependent
Several reports have shown that platelets have an adverse role in ECM pathogenesis (7, 8) and that platelet depletion post-infection is protective (6, 15). However, there is also a report of increased parasitemia in platelet deficient mice and platelet mediated killing of intraerythrocytic Plasmodium parasites in a mouse model of uncomplicated malaria (9). We therefore sought to determine whether the timing of platelet activation influences the platelet dependent outcome in ECM. C57BL6/J (B6) mice were infected with 0.5×106 Plasmodium berghei (ANKA strain, PbA) iRBCs. As a marker of platelet activation, plasma concentrations of the platelet derived chemokine PF4 were measured daily. Platelets were activated within 48 hours of infection (Figure 1A) and platelet activation increased over time independent of blood collection (Supplementary Figure S1). Platelet activation correlated with parasitemia within the first three days post infection before mice presented with symptoms of ECM (Figure 1A). These data indicate that platelet activation may be a direct consequence of parasite burden.
Figure 1. The Role of Platelets in ECM is Time Dependent.
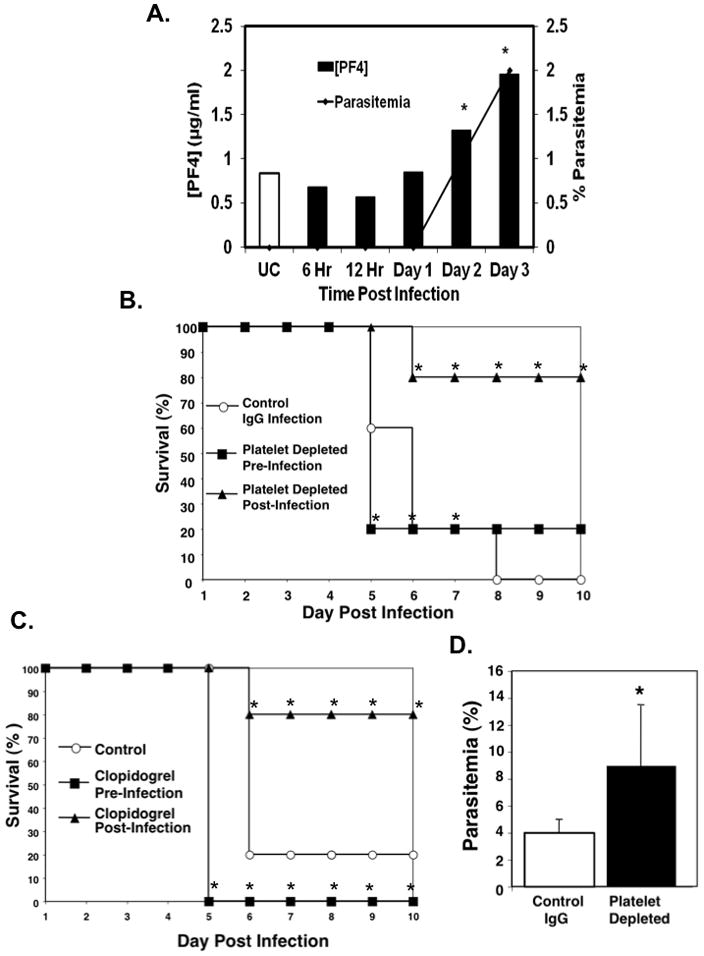
A. Platelets are activated early in ECM. Mice were infected with P. berghei and plasma PF4 concentration measured by ELISA (bars). The corresponding parasitemia (line) was also determined at the same time points. (N=5 per timepoint, mean ± SD,*P<0.05).
B. Platelet depletion pre-infection is not ECM protective. Mice were platelet depleted 24 hrs before or after P. berghei ANKA infection and survival monitored Mice depleted of platelets post infection, but not pre-infection had improved survival. (N=5,*P<0.05, log-rank test).
C. Platelet inhibition pre-infection is also not protective. Mice were treated with clopidogrel beginning 24 hours pre- or post P. berghei infection and survival monitored. (N=10,* P<0.05 log-rank test).
D. Mice platelet depleted pre-infection have in increased parasitemia. Mice were platelet depleted 24 hours prior to P. bergehi infection and the number of infected RBCs compared with non platelet-depleted controls was determined at day 5 (N=5, mean ± S.D, *P≤0.05).
To determine whether platelet mediated ECM outcome is dependent on the timing of blood stage infection, we platelet depleted mice using an anti-GPIb antibody either 24 hrs before or 24 hrs after PbA infection and survival was monitored. Platelet depletion pre-infection did not protect mice from ECM (Figure 1B). However as we previously reported (6), mice platelet depleted post infection had increased survival (Fig. 1B). Platelet depleted mice also had higher parasitemia compared to isotype control IgG treated mice on day 5 post infection (Figure 1D). Similarly, pre-treatment of P berghei infected mice with the platelet inhibitor clopidogrel beginning 24 hours before infection failed to protect mice from ECM (Figure 1C), but mice treated with clopidogrel beginning 24 hours after infection had increased survival compared to controls (Figure 1C). These data demonstrate that platelet activation begins early post P berghei infection, and this early platelet activation may be protective, in part, by reducing parasite burden. However, platelet activation beyond this very early protective phase may be deleterious.
Platelets drive the acute phase response to P berghei infection
The acute phase response (APR) is the rapidly induced first stage of response to infection or tissue injury. In contrast to mice platelet depleted post-infection, mice platelet depleted pre-infection had a loss of protection from ECM. To determine whether platelet dependent induction of the APR may have a protective role, and account in part for the loss of protection from ECM when mice are platelet depleted pre-infection, we first measured the plasma concentrations of serum amyloids A (SAA) and P (SAP) as markers of the murine APR. SAP and SAA were significantly decreased in mice platelet depleted pre-infection compared to IgG control mice (Figure 2A and B). Plasma concentrations of SAP and SAA in post-infection platelet depleted mice were not significantly different from IgG control infected mice, indicating that by 24 hrs post-infection platelets have contributed greatly to induction of the acute phase response to blood stage P berghei infection (Figure 2C and D respectively).
Figure 2. Platelets drive the acute phase response to P. berghei infection.
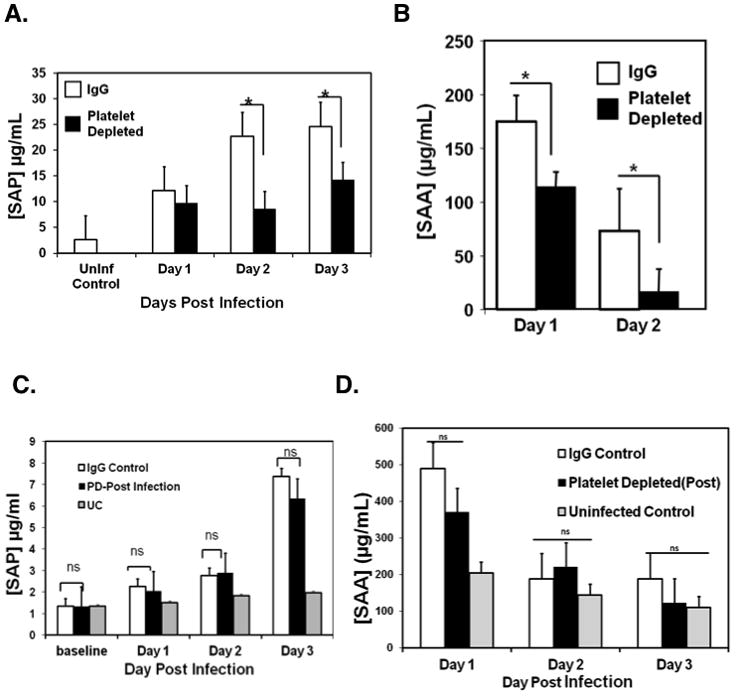
A. Mice were platelet-depleted or treated with control IgG 24 hrs pre-infection and plasma SAP measured by ELISA (N=5, mean ± S.D.*P<0.05).
B. Mice were platelet-depleted or treated with IgG 24 hrs pre-infection and plasma serum amyloid A (SAA) measured by ELISA (n=5, mean ± S.D.*P<0.03).
C. Mice were platelet-depleted or treated with IgG 24 hours post-infection and plasma serum amyloid P (SAP) measured by ELISA (n=5, mean ± S.D).
D. Mice were platelet-depleted or treated with IgG 24 hours post-infection and plasma serum amyloid A (SAA) measured by ELISA (n=5, mean ± S.D.*P<0.03).
Acute phase proteins (APP) are primarily liver derived and induced by cytokines such as IL-6, IL-1β and TGF-β. Platelets synthesize and secrete IL-1β upon activation (16). To determine whether the platelet dependent APR to infection involves IL-1β, we measured IL-1β in the plasma of mice platelet depleted before and after infection. Mice platelet depleted pre-infection had decreased plasma IL-1β compared to non-depleted controls (Figure 3A). In contrast mice platelet depleted post-infection had similar levels of circulating plasma IL-1β as non-platelet depleted IgG controls (Figure 3B). As noted in Figure 2, plasma SAP was also decreased by pre-infection depletion, but plasma SAP levels were restored with recombinant murine IL-1β (mrIL-1β) treatment within the first 72 hours of infection (Figure 3C). These data indicate that platelets are central to IL-1β response to infection. To demonstrate that platelets are a direct source of IL-1β, washed murine platelets were permeabilized and intracellular IL-1β measured by flow cytometry. Intracellular IL-1β was significantly increased in infected mice 24 hours post infection compared to uninfected controls (Figure 3D).
Figure 3. Platelets are a major source of IL-1β.
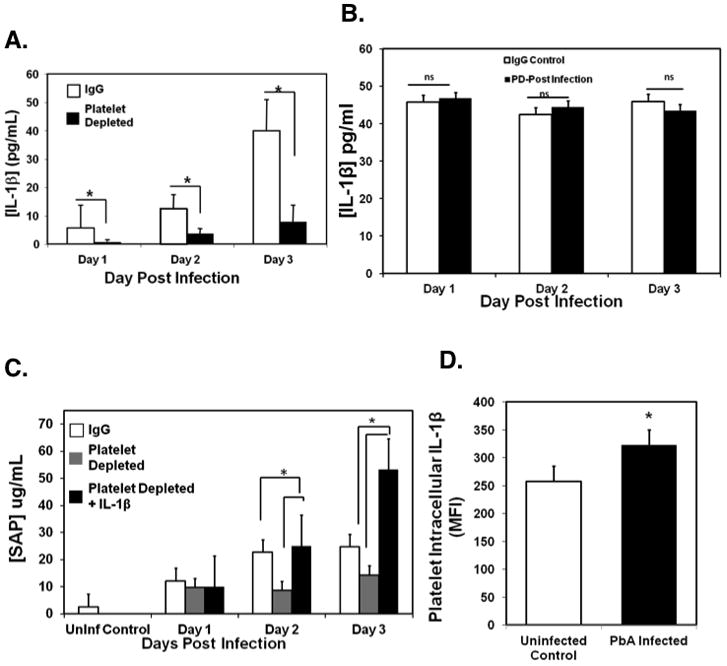
A. Platelets are a primary mediator of IL-1β during the acute phase response to ECM. Mice were platelet depleted or IgG treated before infection and plasma IL1-β measured by ELISA (N=5, mean ± SD,*P≤ 0.01).
B. Plasma IL-1β in post-infection platelet depleted mice. Mice were platelet depleted or control IgG treated 24 hours post-infection and plasma IL1-β measured by ELISA (N=5, mean ± SD).
C. IL-1β reconstitution restores APP production in pre-infection platelet depleted mice. Mice were platelet depleted or treated with control IgG 24 hrs pre-infection; platelet depleted mice were treated with control buffer (PBS) or mrIL-1β at the time of infection, 24 and 48 hrs post infection. Plasma SAP levels were measured by ELISA (N=5, mean ± SD, *P<0.05).
D. Platelets from P. berghei infected mice have increased IL-1β. Mice were infected with P. berghei, platelets isolated and intracellular IL-1β analyzed by flow cytometry (N=5, mean ± SD *P≤0.05).
Because the liver is the major site of APP production we determined whether platelets are recruited to the liver early in P. berghei blood stage infection. 24 hrs post infection mice were injected with a fluorescent platelet labeling antibody, livers were removed, cut into thin sections and imaged on a fluorescent microscope (Figure 4A). Mice infected with P berghei had approximately 3 fold more platelet foci in liver sections compared to uninfected controls, indicating an increase in platelet trafficking to the liver during APR induction (Figure 4B). We further examined platelet localization to the liver using electron microscopy. Transmission electron micrographs (TEMs) of murine livers 24 hours post infection also showed increased platelet-rich foci within the liver microvasculature of infected mice compared to uninfected controls (Figure 4C top). In addition, there were multiple platelets with extended lamellipodia and platelet interactions with liver sinusoidal endothelial cells in infected mice that were not seen in control mice (LSECs; Figure 4C bottom right). Platelets present in the lumen of hepatic microvessels of uninfected control mice appeared quiescent and were not interacting with the sinusoid endothelium (Figure 4C bottom left).
Figure 4. Platelets are recruited to the liver early post P berghei infection.
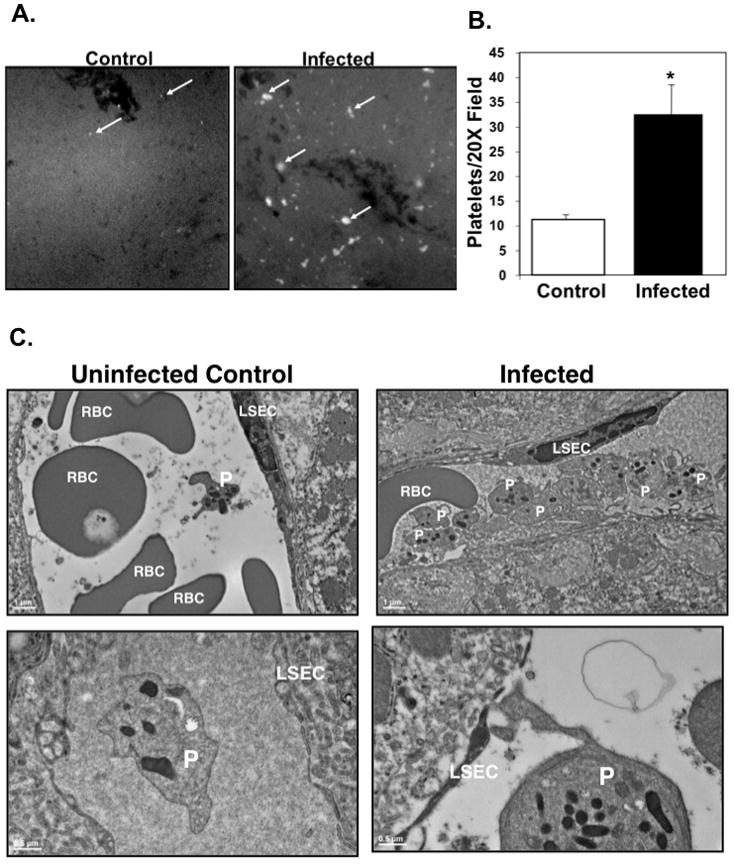
A. Platelets are recruited to the liver post-infection. 24 hrs after infection mice were injected with anti-platelet fluorescent antibody and sacrificed. Livers were imaged with a fluorescent microscope (Representative Images).
B. Platelet foci were quantified from 10 different fields. (N=5, mean ± S.D., *P≤0.01, unpaired Student’s t-test).
C. Platelets (P) interact with liver sinusoidal endothelial cells (LSEC) during infection (representative TEM images). Ultrathin liver sections from uninfected controls (left) and infected mice (right) 24 hrs post P berghei infection.
Taken together, these results indicate that platelets induce the acute phase response and that platelet contact dependent mechanisms as well as platelet derived or induced IL-1β have central roles in eliciting the APR.
The acute phase response is protective in ECM
Based on our findings that platelets initiate the APR and production of acute phase proteins, we determined whether the main mouse and human APPs, SAP and CRP respectively, are protective in the ECM model. Mice were platelet depleted pre-infection and given exogenous recombinant murine SAP (rSAP) or control buffer (PBS) on days 1–3 post infection. Platelet depleted mice treated with rSAP had a delay in death compared to platelet depleted mice (Figure 5A). The survival of the rSAP treated mice was similar to control mice indicating that loss in platelet induction of SAP may in part account for the pre-infection platelet depletion phenotype (Figure 5A).
Figure 5. The Acute Phase Response is Protective in ECM.
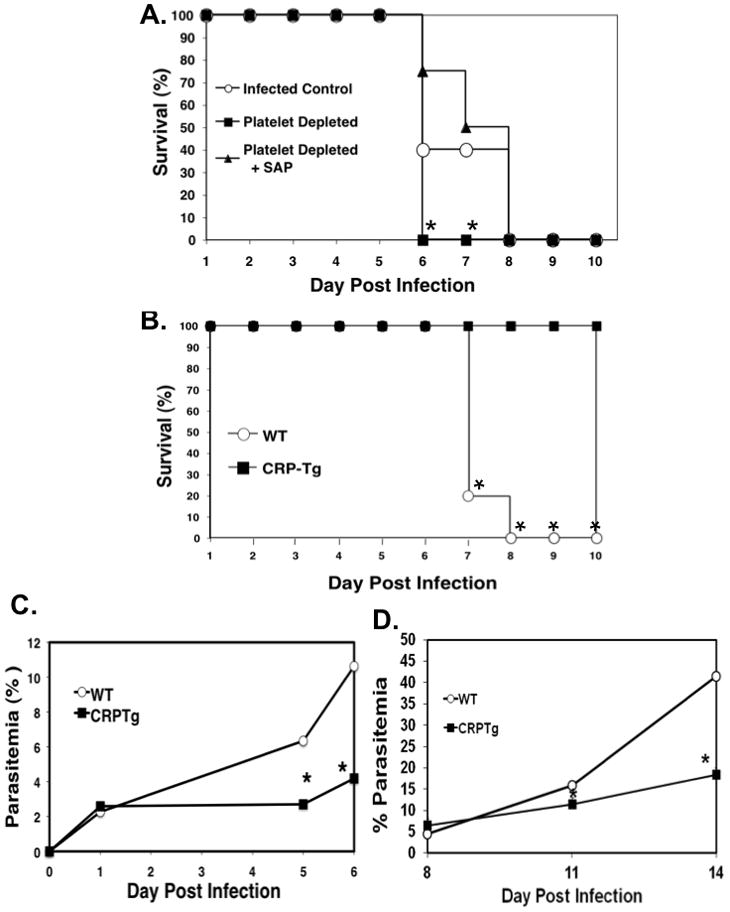
A. Exogenous SAP administration of platelet depleted P. berghei infected mice returns them to control survival levels. Mice were platelet depleted 24 hours pre-infection or platelet depleted and treated with rSAP for the first 3 days post infection. Survival (N=5, *P<0.05 log-rank test vs platelet depleted+SAP).
B. CRP-Tg mice are protected from ECM. CRP-Tg or WT mice were infected with P. berghei and survival monitored (N=5,*P<0.05 log-rank test).
C. CRP reduces the parasite burden in ECM. Mice were infected with P. berghei and parasitemia determined by flow cytometry analysis of SybrGreen labeled iRBCs (N=5, *P<0.05 unpaired Students t-test).
D. CRP reduces the parasite burden in P yoelii infections. Mice were infected with P yoelii and parasitemia determined by flow cytometry analysis of SyBrGreen stained iRBCs (N=8, mean ± SD, *P<0.05, unpaired Students t-test).
Mice do not utilize CRP as an acute phase protein. We therefore used CRP transgenic mice (CRP-Tg) (13) in the ECM model to determine the effects of the dominant human acute phase protein in ECM pathogenesis. CRP-Tg mice were infected and found to be completely protected from ECM death (Figure 5B), and had significantly reduced parasitemia compared to WT control mice (Figure 5C). CRP-Tg mice also had a significantly reduced parasite burden in mice infected with a non-lethal P yoelii strain (Figure 5D). These data indicate a protective role for acute phase proteins in ECM, in part by reducing parasitemia.
Discussion
Our data demonstrate a novel time dependent role for platelets in the pathogenesis of ECM. Our prior studies (6, 17), and those of many other investigators (2, 3, 7, 8), have demonstrated that platelets have a central role in cerebral vascular injury leading to ECM. When platelets were depleted, or mice treated with platelet inhibitors post infection, ECM survival was greatly improved (6, 15, 18). In this study we show that platelets have a more complex role in ECM than previously thought. Very early in infection platelets are activated and initiate the acute phase response that is protective by limiting the development of and death due to ECM. Depletion of platelets or the use of platelet inhibitors pre-infection was not survival protective and increased parasitemia. The acute phase response is initiated in the liver, and we demonstrated that platelets are recruited to the liver upon P berghei infection and that platelets are needed to increase the plasma concentration of a major acute phase initiator IL-1β. This may in part be a direct platelet effect, as we noted increased intracellular platelet IL-1β in P berghei infected mice; or the effect of platelet interaction with other IL-1β producing cells such as monocytes. The acute phase response is protective as CRP transgenic mice and mice treated with exogenous SAP had greatly improved survival and delayed onset of death respectively. Platelets are thus a critical mediator of protection in ECM through initiation of the acute phase response.
The adverse role for platelets in ECM is largely due to platelet inflammatory functions, the capacity for platelets to mediate iRBC sequestration, as well as platelet secretion of chemokines that lead to leukocyte recruitment. As infection progresses platelets therefore establish an inflammatory environment that drives and exacerbates severe disease. But evidence of a more complex role for platelets in malaria, particularly in uncomplicated malaria, came with the discovery that platelets may bind to and kill intra-erythrocytic parasites through yet to be understood mechanisms. Our new findings for a time dependent protective role for platelets indicate that this paradigm may extend to severe malaria in the very early stages of infection. The acute phase response, an ancient defense mechanism, is the earliest phase of the innate immune response to infection and trauma and occurs within 24–72 hours after stimulus. There is a robust acute phase response in human malaria infection both to the liver (19) and erythrocytic stages (20). We have found an early platelet activation response to infection and a decrease in acute phase proteins when platelets are depleted or inhibited, indicating an important role for platelets in activating the acute phase response. Together this work and our past studies establish a new important paradigm: platelets are activated by Plasmodium infection leading to a protective acute phase response. However, with continued platelet activation there is an adverse innate and acquired immune response as well as thrombosis(21) that accelerates ECM.
Upon activation platelets undergo numerous changes including the release of inflammatory molecules that have effector functions both locally and at sites distant from their site of release. IL-1β is a major platelet derived cytokine(16, 22) with important roles in fever and acute phase responses to infection(23). IL-1β knockout mice have a severely attenuated acute phase response(24) and low doses of exogenous IL-1β have been shown to protect mice against lethal cerebral malaria (25). Human studies have demonstrated that low levels of IL-1β are associated with increased severe malaria anemia (26). Our studies indicate a potentially important role for platelet IL-1β in protective immune responses, however as platelet depletion did not completely abrogate plasma IL-1β in infected mice, other cells likely also contribute IL-1β or are stimulated by activated platelets to contribute to the total circulating IL-1β.
Acute phase proteins are commonly used as markers of infection, injury or trauma, but also have immune effector functions. For example, during S. pneumoniae infection, SAP has been found to be protective by aiding complement activation(27). In patients presenting with P falciparum infection in Papua New Guinea, acute uncomplicated malaria infection was positively correlative to high serum CRP levels, whereas patients with severe or fatal disease had low levels of CRP(28). CRP was also increased in experimental human malaria infections (20). In another study, the absence of SAP in mice resulted in increased parasitemia in P. chabaudi infected mice and the addition of SAP to P falciparum cultures inhibited in vitro growth(29). Our work and these prior studies indicate a protective function of platelet induced APPs. Further studies are needed to determine how APPs mediate reduced parasite burden and ECM protection.
Platelets are becoming more appreciated for their non-hemostatic roles, particularly their immune functions. Our studies describe an early protective immune function for platelets in ECM, but with continued platelet activation platelets promote cerebral vascular lesions. There is a need for more study of the time dependent role of platelets in CM, as well as many other inflammatory diseases with platelet involvement. The judicious use of platelet inhibitors may be best approached with a better understanding of platelets in all phases of immune responses.
Supplementary Material
Figure 6. Platelet Mediated Acute Phase Response in ECM.
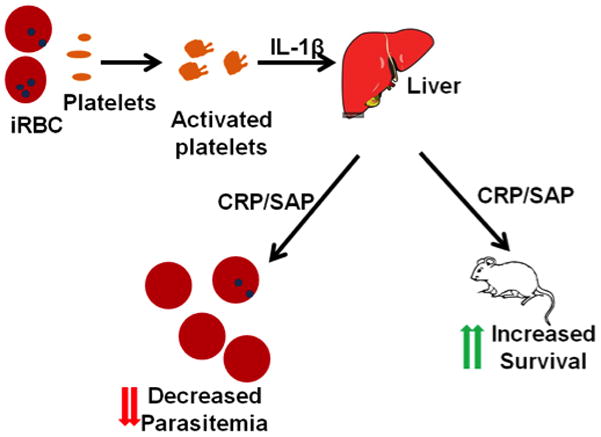
Platelets are activated by infected RBCs and in an IL-1β dependent manner induce the production of acute phase proteins such as CRP and SAP. Acute phase proteins decrease parasitemia and mediate survival in ECM.
Acknowledgments
Special thanks to Karen Bentley and Gayle Schneider of the URMC Electron Microscopy Core for assistance with the TEMs, Philip Shaul of UT Southwestern for the CRP transgenic mice and Munekazu Yamakuchi of the Aab CVRI for helpful discussions.
AAA is supported by F31AI094955 from NIAID. CNM is supported by R01HL093179, R01HL093179-02S109, and R01HL094547 from the NIH.
Footnotes
Author Contributions:
AAA: designed research, performed research, analyzed data, wrote the paper
DJF: performed research
KS: performed research
CNM: designed research, performed research, analyzed data, and wrote the paper
Disclosures: The authors have nothing to disclose.
References
- 1.WHO. World Malaria Report: 2010. World Health Organization; Geneva, Switzerland: 2011. [Google Scholar]
- 2.Grau GE, Mackenzie CD, Carr RA, Redard M, Pizzolato G, Allasia C, Cataldo C, Taylor TE, Molyneux ME. Platelet accumulation in brain microvessels in fatal pediatric cerebral malaria. J Infect Dis. 2003;187:461–466. doi: 10.1086/367960. [DOI] [PubMed] [Google Scholar]
- 3.van der Hyde HC. A unified hypothesis for the genesis of cerebral malaria. Trends In Parasitology. 2006;22:503–508. doi: 10.1016/j.pt.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Wagner D. Platelets in Inflammation and Thrombosis. Arterioscler Thromb Vasc Biol. 2003;23:2131–2137. doi: 10.1161/01.ATV.0000095974.95122.EC. [DOI] [PubMed] [Google Scholar]
- 5.Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115:3378–3384. doi: 10.1172/JCI27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srivastava K, I, Cockburn A, Swaim A, Thompson LE, Tripathi A, Fletcher CA, Shirk EM, Sun H, Kowalska MA, Fox-Talbot K, Sullivan D, Zavala F, Morrell CN. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell Host Microbe. 2008;4:179–187. doi: 10.1016/j.chom.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pain A, et al. Platelet Mediated Clumping of Plasmodium falciparum-Infected Erythrocytes is a Common Adhesive Phenotype and is accociated with severe malaria. PNAS. 2001;98:1805–1810. doi: 10.1073/pnas.98.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wassmer C, et al. Platelets Potentiate Brain Endothelial Alterations Induced by Plasmosium Falciparum. Infect Immun. 2006;74:645–653. doi: 10.1128/IAI.74.1.645-653.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McMorran BJ, V, Marshall M, de Graaf C, Drysdale KE, Shabbar M, Smyth GK, Corbin JE, Alexander WS, Foote SJ. Platelets kill intraerythrocytic malarial parasites and mediate survival to infection. Science. 2009;323:797–800. doi: 10.1126/science.1166296. [DOI] [PubMed] [Google Scholar]
- 10.Greenbaum DC, FitzGerald GA. Platelets, pyrexia, and plasmodia. N Engl J Med. 2009;361:526–528. doi: 10.1056/NEJMcibr0903050. [DOI] [PubMed] [Google Scholar]
- 11.English M, Marsh V, Amukoye E, Lowe B, Murphy S, Marsh K. Chronic salicylate poisoning and severe malaria. Lancet. 1996;347:1736–1737. doi: 10.1016/s0140-6736(96)90809-0. [DOI] [PubMed] [Google Scholar]
- 12.Gabay C, Kushner I. Acute phase response and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 13.Lin CS, Xia D, Yun JS, Wagner T, Magnuson T, Mold C, Samols D. Expression of rabbit C-reactive protein in transgenic mice. Immunol Cell Biol. 1995;73:521–531. doi: 10.1038/icb.1995.82. [DOI] [PubMed] [Google Scholar]
- 14.Izumiyama S, Omura M, Takasaki T, Ohmae H, Asahi H. Plasmodium falciparum: Development and validation of a measure of intraerythrocytic growth using SYBR Green I in a flow cytometer. Experimental Parasitology. 2009;121:144–150. doi: 10.1016/j.exppara.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 15.van der Hyde HC, et al. Platelet depletion by anti-CD41 mAb injection early but not late in the course of disease protects against Plasmodium berghei pathogenesis by altering the levels of pathogenic cytokines. Blood. 2005;105:1956–1963. doi: 10.1182/blood-2004-06-2206. [DOI] [PubMed] [Google Scholar]
- 16.Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, Weyrich AS. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. 2001;154:485–490. doi: 10.1083/jcb.200105058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srivastava K, Field DJ, Aggrey A, Yamakuchi M, Morrell CN. Platelet factor 4 regulation of monocyte KLF4 in experimental cerebral malaria. PloS one. 2010;5:e10413. doi: 10.1371/journal.pone.0010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun G, et al. Inhibition of platelet adherence to brain microvasculature protects against severe Plasmodium berghei malaria. Infect Immun. 2003;71:6553–6561. doi: 10.1128/IAI.71.11.6553-6561.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pied S, Nussler A, Pontent M, Miltgen F, Matile H, Lambert PH, Mazier D. C-reactive protein protects against preerythrocytic stages of malaria. Infect Immun. 1989;57:278–282. doi: 10.1128/iai.57.1.278-282.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harpaz R, Edelman R, Wasserman SS, Levine MM, Davis JR, Sztein MB. Serum cytokine profiles in experimental human malaria. Relationship to protection and disease course after challenge. J Clin Invest. 1992;90:515–523. doi: 10.1172/JCI115889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dorovini-Zis K, Schmidt K, Huynh H, Fu W, Whitten RO, Milner D, Kamiza S, Molyneux M, Taylor TE. The neuropathology of fatal cerebral malaria in malawian children. Am J Pathol. 2011;178:2146–2158. doi: 10.1016/j.ajpath.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, Yost CC, Rubner FJ, Albertine KH, Swoboda KJ, Fratto CM, Tolley E, Kraiss LW, McIntyre TM, Zimmerman GA, Weyrich AS. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005;122:379–391. doi: 10.1016/j.cell.2005.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dinarello C. Biologic basis for interleuken-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 24.Zheng H, Fletcher D, Kozak W, Jiang M, Hofmann KJ, Conn CA, Soszynski D, Grabiec C, Trumbauer ME, Shaw A, et al. Resistance to fever induction and impaired acute-phase response in interleukin-1 beta-deficient mice. Immunity. 1995;3:9–19. doi: 10.1016/1074-7613(95)90154-x. [DOI] [PubMed] [Google Scholar]
- 25.Curfs JHvdM, Sauerwein JW, Eling RW, WM Low dosages of interleukin1 protect mice against lethal cerebral malaria. J Exp Med. 1990;172:1287–1291. doi: 10.1084/jem.172.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ouma C, Davenport GC, Awandare GA, Keller CC, Were T, Otieno MF, Vulule JM, Martinson J, Ong’echa JM, Ferrell RE, Perkins DJ. Polymorphic variability in the interleukin (IL)-1beta promoter conditions susceptibility to severe malarial anemia and functional changes in IL-1beta production. J Infect Dis. 2008;198:1219–1226. doi: 10.1086/592055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuste J, et al. Serum amyloid P aids complement-mediated immunity to Streptococcus pneumoniae. PlosPathogens. 2007;3:e120. doi: 10.1371/journal.ppat.0030120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Donnell A, Fowkes FJ, Allen SJ, Imrie H, Alpers MP, Weatherall DJ, Day KP. The acute phase response in children with mild and severe malaria in Papua New Guinea. Trans R Soc Trop Med Hyg. 2009;103:679–686. doi: 10.1016/j.trstmh.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 29.Balmer P, McMonagle F, Alexander J, Stephen Phillips R. Experimental erythrocytic malaria infection induces elevated serum amyloid P production in mice. Immunol Lett. 2000;72:147–152. doi: 10.1016/s0165-2478(00)00180-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.