

Abstract
The purpose of this review is to provide those interested in glycosylation analysis with the most updated information on the availability of automated tools for MS characterization of N-linked and O-linked glycosylation types. Specifically, this review describes software tools that facilitate elucidation of glycosylation from MS data on the basis of mass alone, as well as software designed to speed the interpretation of glycan and glycopeptide fragmentation from MS/MS data. This review focuses equally on software designed to interpret the composition of released glycans and on tools to characterize N-linked and O-linked glycopeptides. Several websites have been compiled and described that will be helpful to the reader who is interested in further exploring the described tools.
INTRODUCTION
Overview of Glycosylation
The addition of monosaccharide residues onto a protein or lipid, known as glycosylation, serves an important function in many cellular signaling and communication events, including those involving host-pathogen interactions.1–4 It has long been understood that protein-carbohydrate interactions play a participatory role in many processes affecting disease progression.1, 4–7 Furthermore, experimental evidence demonstrates that the identity of the attached glycans change during these events.6, 8–10 For example, aberrant glycosylation is often present in individuals experiencing cancer, diabetes, and inflammation.4, 8–11 As such, accurate characterization of a glycoprotein’s glycan substituents has been shown to be crucial in the development of potential biomarkers, protein-based vaccine candidates, and pharmaceutical treatments.5,9,12,13
In studies involving protein glycosylation, mass spectrometry (MS) is currently considered the most utilized instrument for the characterization of glycan substituents, both attached or released.11 Accordingly, this review focuses on those automated analysis tools that can be applied to more efficiently analyze glycosylation profiles through interrogation by MS and MSn methods.
Glycosylation Heterogeneity
Unlike DNA replication and protein transcription, glycosylation is a “non-template”-driven process,5,14 where the sugar residues form a multitude of arrangements.5,12 The monosaccharides that comprise the glycan may be long or short, branched or linear, and linked in a variety of ways, creating a large degree of variability.3,5,12 This heterogeneity is described in two ways: Glycan differences at different sites of attachment (macroheterogeneity), or within the same site (microheterogeneity).12 The large amount of heterogeneity presents a challenging obstacle to researchers attempting to elucidate structural, as well as compositional, information on a protein’s glycan population, especially when samples are mixtures of proteins.
Types of Protein Glycosylation
Over half of all proteins expressed are predicted to be glycosylated.15 In addition to the established forms of protein glycosylation, including N-linked, O-linked, and C-linked forms,1, 16–18 rarer configurations such as S-linked glycosylation, are also being discovered.19 Although a variety of types exist, the two most common types of glycosylation are N-linked and O-linked.1,7,20 In N-linked glycosylation, the addition of a glycan may occur at the asparagine residue when the consensus sequence Asn-Xaa-Ser/Thr occurs, where Xaa is any amino acid except proline.12,18,21 The inclusion of this pattern is a fundamental requirement for N-linked glycosylation to occur, though it is not a guarantee that a glycosylation site will be occupied.21 With O-linked glycosylation, the glycan addition may occur at any Ser or Thr residue within the protein sequence,4,11 though a very low percentage of these sites are actually occupied.15 In contrast to N-linked glycans, O-linked glycans have less defined sequence patterns, 12,18,22 and may consist of several distinct core arrangements.11 For these reasons, both the prediction and determination of O-linked glycosylation characteristics has advanced slower than N-linked glycosylation analysis.11
Glycosylation Analysis
In the determination of glycan structural information, mass spectrometry (MS) has shown to be a powerful tool, as a large part of the successful interrogation of glycan composition and structures has been accomplished utilizing MS experiments.18,23,24 There are two main strategies for elucidating glycosylation information using MS techniques: 1) Characterization of a protein’s glycans after they are released from glycoproteins and 2) Characterization of glycopeptides after proteolytic digestion of a glycoprotein.11,18,23 The study of released glycans is particularly useful when rapid analysis of glycan composition is desired. Though N- and O-linked glycan populations can be studied independently through the use of different cleavage procedures,11,12 no information on where the individual glycans were attached along the protein is obtained when the glycans are cleaved a priori. In order to obtain glycosylation site-specific information for individual glycoforms, the second method, glycopeptide analysis, which requires digestion of the protein using a protease such as trypsin, is necessary.18,23,25 This method is generally advantageous because it provides information about both glycan composition and the site of the glycan’s attachment.23 Despite the challenges mentioned in this review, techniques that allow for complete profiling of a peptide’s glycan population have advanced greatly in the past decade, especially with respect to site-specific glycopeptide analysis.
In the study of protein glycosylation by either of these techniques, a number of resources, including databases providing information on known glycosylation structures or site occupancy, as well as collections of experimental data, are currently available.26–33 For instance, researchers needing to identify occupied N-linked glycosylation sites on a specific protein can access UniProtKB,26 while those wanting statistics specific to proteins modified by O-GlcNAc could visit dbOGAP.33 Although this repertoire of information is greater for proteins modified by N-and O-linked glycan types, databases that contain entries on C-glycosylated proteins, such as dbPTM,27 are available as well. A current list and description of these database resources are provided in Table 1.
Table 1.
Glycosylation Databases.
| Database | Link to Database | Type | Description |
|---|---|---|---|
| UniProtKB | http://www.uniprot.org/ |
N-glycos. O glycos. C-glycos. |
Contains annotation of N-, O-, and C-linked glycosylation, as well as glycation. Both mammalian and non-mammalian entries are provided. |
| dbPTM | http://dbptm.mbc.nctu.edu.tw/ |
N-glycos. O-glycos. C-glycos. |
Contains a combinational repertoire of protein PTMs from other databases, including experimentally obtained data on site of modification. |
| GlycomeDB | http://www.glycome-db.org/ |
N-glycos. O-glycos. C-glycos. |
Contains over 30,000 carbohydrate structures from all major taxonomies. |
| GlycoSuiteDB | http://glycosuitedb.expasy.org/glycosuite/glycodb |
N-glycos. O-glycos. |
Contains over 9400 entries of curated and annotated glycans from various organisms. |
| O-GlycBase | http://www.cbs.dtu.dk/databases/OGLYCBASE/ |
O-glycos. C-glycos. |
Contains over 2000 entries of protein glycosylation sites, the majority of which are O-linked. |
| UniPep | http://www.unipep.org/ | N-glycos. | Contains over 1500 entries of human protein N-linked glycosylation sites. |
| GlycoBase | http://glycobase.univ-lille1.fr/base/ | N-glycos. | Contains HPLC elution positions for 2-AB labeled N-linked glycans from exoglycosidase sequencing and LC-MS data. |
| dbOGAP | http://cbsb.lombardi.georgetown.edu/hulab/OGAP.html | O-glycos. | Contains over 1100 entries on O-GlcNAcylation sites. |
AUTOMATED ANALYSIS OF RELEASED GLYCANS
Characterization of High Resolution MS data for Oligosaccharides
Often, the easiest way to identify a protein’s glycan population is by enzymatically cleaving the glycan substituents and analyzing the monosaccharide residues directly.25 N- and O-linked glycans from the same protein can be independently characterized in this manner, as in the method described by Goetz et al. where β-elimination is used to release O-linked glycans, which are simultaneously permethylated.34 Once cleaved, automated analysis tools that determine glycan composition from MS data may be used.
One such tool developed to analyze MS data of glycans is Cartoonist, as described by Goldberg et al.35 Cartoonist works to increase the speed of compositional determination in permethylated N-linked glycans from matrix laser desorption/ionization-mass spectrometry (MALDI-MS) data through identification and annotation of MALDI-TOF spectra.35 The most likely glycan compositions are selected using precursor mass data.35 The program automatically labels MALDI peaks with cartoons of the most probable oligosaccharide structure, as determined by the program’s algorithm, from a library of 300 generated mammalian N-linked glycans.35 Recently, Goldberg et al. extended this concept by developing an automated tool for the analysis of O-linked glycans from MS/MS data,36 as described below in MS/MS approaches for glycan analysis section of this review. To date, neither program is publicly available.
Another software solution useful for the identification of glycans from MS data is SysBioWare, described by Vakhrushev et al.37 SysBioWare takes raw MS1 data that a user uploads and performs baseline adjustment and denoising, wavelet analysis, and peak detection before grouping isotopes of detected peaks.37 The isotopic grouping is also performed automatically, which enables the program to deduce monoisotopic m/z values and precursor charge states without the need of manual input by the user.37 The software then determines monosaccharide compositions on the basis of mass.37 Currently, the SysBioWare program is being updated to include analysis of MS/MS data for glycans as well.38 SysBioWare is not freely available to the public at this time.
Similar to SysBioWare is GlycoWorkbench. GlycoWorkbench evaluates glycan compositions (which are proposed by the user) by searching the spectral peak list of user-input MS data for matches between calculated theoretical glycan masses and corresponding m/z values.39 The GlycanBuilder tool, designed to interface with GlycoWorkbench, enables the drawing of glycan structure representations, with all stereochemical information on the monosaccharides depicted as specified by the user.40 Both analysis tools, GlycanBuilder and GlycoWorkbench, are available online free of charge, as described in Table 2.
Table 2.
Tools available online to facilitate glycan characterization from MS and MS/MS data.
| MS Analysis Tool | Link to Tool | Concept and Data Type |
|---|---|---|
| GlycoWorkbench | http://download.glycoworkbench.org/ | Identifies and annotates MS and MS/MS data with appropriate glycan compositions or fragments. |
| GlycanBuilder | http://live.glycanbuilder.org/ | Drawing tool interfacing with GlycoWorkbench that displays different stereochemical representations of glycans. |
| GlycoSpectrumScan | http://www.glycospectrumscan.org. | Quantitatively identifies N- and O-linked glycoforms within a protein using LC-MS Data. |
| GlycoFragment | http://www.glycosciences.de/tools/GlycoFragments/fragment.php4 | Identifies and displays the main product ions expected for oligosaccharide MS/MS data. |
| GlycoSearchMS | http://www.glycosciences.de/database/start.php?action=form_ms_search | Compares experimental MS/MS data to product ions calculated from an extensive library of N-and O-linked glycans. |
| SimGlycan | http://www.premierbiosoft.com/glycan/index.html | Predicts the structure of glycans from MS/MS data by matching spectra to a built-in database. |
GlycoSpectrumScan, another freely available program, was developed by Deshpande et al. and works to identify N-and O-linked glycoforms using MS1 data.41 This software is capable of analyzing both singly or multiply charged ions directly from raw data, and accepts the input of both ESI and MALDI spectra.41 GlycoSpectrumScan also determines the relative abundance of N-and O-linked glycoforms that are identified for each glycosylation site.41 However, the user must enter the N- and/or O-linked glycan compositions potentially present in the sample, as well as the in silico peptide masses of the digested glycoprotein.41 GlycoSpectrumScan is available online (see Table 2).
MS/MS Approaches for Glycan Characterization
Until recently, when automated software tools and scoring algorithms became available, the identification of accurate glycan or glycopeptide assignments from MS/MS data was a key bottle-neck, due to the need for extensive manual data analysis. STAT, designed by Gaucher et al., is one of the first automated tools for the determination of glycan composition using tandem MS.42 STAT is designed for glycans of up to ten monosaccharide residues, and has the ability to quickly analyze relevant N-glycan compositions.42 STAT also lists the most likely structures in order of probability to provide a ranking system when more than one candidate glycan matches the fragmentation profile of the data being analyzed.42 Unfortunately, this program is no longer publicly accessible.
An early analysis tool capable of evaluating O-linked glycan fragmentation is the OSCAR algorithm.43 OSCAR, as developed by Ashline et al., is specifically designed for the annotation of permethylated O-linked oligosaccharides from MSn data.43 OSCAR is part of a collection of software tools termed Glyspy, which is not currently accessible to the public.43 Although innovative, the use of OSCAR is limited to direct infusion experiments, as the software cannot effectively process data from LC-MS methods.43
A program contemporary to OSCAR and also developed to handle glycan MS/MS data is StrOligo.44 This instrument-specific program was designed by Ethier et al. for the determination of N-linked glycan structures from MALDI MS/MS data.44 In published research, StrOligo successfully assigned the correct glycan structure in 24 out of 28 cases.44 Although the results of these two programs are promising, neither program is freely accessible online.
Several alternative glycan analysis tools are freely available online. One of the earliest of these was reported by Lohmann et al. in 2004.45 The authors describe the web tools GlycoFragment and GlycoSearchMS, which were developed for glycan structural determination.45 The theoretical fragmentation patterns of carbohydrate structures are calculated using GlycoFragment, which displays theoretical b- and y-fragments as well as c-, z-, a- and x-ions.45 GlycoSearchMS works to analyzes experimental glycan data by comparing it against a library of theoretical spectra from N-linked and O-linked glycan fragmentation entries extracted from SweetDB.45 The GlycoFragment program has been validated on both N-linked and O-linked glycan classes, and, used in conjunction with GlycoSearchMS, enables researchers to determine the most probable glycan composition according to the information from the combined algorithms.45,46 Both GlycoFragment and GlycoSearchMS are freely available. See Table 2 for more information.
Another heavily used, free, online tool for glycoform analysis is GlycoWorkbench, which has shown to be a resourceful tool not only for analysis of MS1 data, as mentioned previously, but in the identification of glycans from MS/MS data as well.39 To utilize the glycan fragmentation analysis feature, a user must first input/define the possible glycan compositions and spectral peak list.39 The software then calculates expected glycan fragmentation and relative m/z values, and annotates peaks of the uploaded data with the most probable identity (shown in red to distinguish it), of all compositions tested.39 As previously stated, GlycoWorkbench is available for free online.
In addition to the freely available tools mentioned above, several other MS/MS analysis tools for glycans are available for researchers, either for purchase or by special request to the tools’ developers. Two of these are GlyCH and Glyquest.47,48 GlyCH was developed by Tang et al. to perform automated interpretation of oligosaccharide tandem mass spectra.47 The algorithm has a scoring function built in to allow researchers to compare compositions when more than one is determined to be possible.47 The GlyCH algorithm, which has so far been tested on released N-glycans, is also capable of de novo analysis, providing no more than 10 monosaccharide residues comprise the glycan chain.47 Although not freely accessible online; the program is available upon request from the authors.47 More recently, Gao et al. developed Glyquest, an automated analysis program that takes a different approach to determine compositions of intact N-linked glycans.48 This software utilizes a database in conjunction with an integrated search engine to determine the composition of peptide-attached N-glycans from CID MS/MS data.48 After the program algorithmically determines the molecular weight of the protonated peptide within a given spectrum; candidate N-glycan compositions are selected and fragmented in silico to generate a theoretical spectrum that is then compared to the experimental spectrum.48 The glycan compositions with fragmentation profiles that are most similar to the experimental fragmentation are determined to be the most probable candidates.48 Glyquest is not freely available to the public.
SimGlycan is another tool that can be used to increase throughput of glycan analysis.49,50 More information is available online (http://www.premierbiosoft.com/). This for purchase tool is useful for determining glycan structures from MS/MS data obtained from many different mass spectrometers, once an acquisition file is converted into mzXML format.50 A user uploads an MS/MS data file, and the software utilizes a built-in database with theoretical fragmentation profiles of nearly 10,000 glycan structures to provide the most likely structural candidates.49 One unique feature of SimGlycan is that no filtering of biologically relevant structures is provided, which can be advantageous for identifying novel glycan structures, but disadvantageous in that it returns a user many structures which are not relevant.49 However, commercially available software such as SimGlycan are expensive, which potentially limit their use.
A more recent program developed specifically for the compositional interpretation of O-linked glycan fragmentation is CartoonistTwo, as described by Goldberg et al.36 CartoonistTwo was designed using CID data acquired on an FTICR-MS, and validated using data from a test set of 34 spectra acquired from Xenopus egg jelly.36 Unfortunately, the program is not freely accessible to the public.
AUTOMATED ANALYSIS OF GLYCOPEPTIDES
For researchers performing site-specific glycosylation analysis, the initial step toward accomplishing the characterization of attached glycoforms at unique sites within a digested protein is to identify potential glycosylation sites within that protein; the tools to facilitate this step are described in Table 1 of this review. In addition to these, programs that utilize algorithms to predict the likelihood of site-occupancy by examination of the amino acid residues surrounding the potential glycosylation site have also been developed.51–59 These prediction tools, along with a description and link to each tool, are provided in Table 3.
Table 3.
Glycosylation Site Prediction Tools.
| Database | Link to Prediction Tool | Type | Overview |
|---|---|---|---|
| EnsembleGly | http://turing.cs.iastate.edu/EnsembleGly/ |
N-glycos. O-glycos. C-glycos. |
Uses ensemble learning to predict N-, O-, and C-linked sites, as well as O-linked glycan types. |
| GlySeq | http://www.glycosciences.de/tools/glyseq/ |
N-glycos. O-glycos. |
Uses the PDB and SwissProt to perform statistical analysis of glycosylation sites. |
| GPP | http://comp.chem.nottingham.ac.uk/glyco/ |
N-glycos. O-glycos. |
Algorithmically predicts N-linked and O-linked glycosylation. |
| NetNGlyc | http://www.cbs.dtu.dk/services/NetNGlyc/ | N-glycos. | Uses consensus sequence to predict N-glycosylation in human proteins. |
| NetCGlyc | http://www.cbs.dtu.dk/services/NetCGlyc/ | C-glycos. | Predicts sites in mammalian proteins for C-mannosylation attachment. |
| NetOGlyc | http://www.cbs.dtu.dk/services/NetOGlyc/ | O-glycos. | Predicts mucin-type GalNAc O-glycosylation in mammalian proteins. |
| CKSAAP_OGlysite | http://bioinformatics.cau.edu.cn/zzd_lab/CKSAAP_OGlysite | O-glycos. | Predicts mucin-type O-glycosylation sites in mammalian proteins. |
| OGPET | http://ogpet.utep.edu/ | O-glycos. | Predicts occurrence of mucin-type O-glycosylation in eukaryotic proteins. |
| YinOYang | http://www.cbs.dtu.dk/services/YinOYang/ | O-glycos. | Predicts O-β-GlcNAc attachment sites in eukaryotic proteins. |
| DictyOGlyc | http://www.cbs.dtu.dk/services/DictyOGlyc/ | O-glycos. | Predicts sites for O-GlcNAc attachment in Dictyostelium discoideum proteins. |
Experimental Data Requirements
After the resultant glycopeptides are obtained from the proteolytic digest, two types of data are generally used to accurately characterize the identity of a glycopeptide. First, high resolution MS data of the glycopeptide is used to infer possible glycopeptide compositions; second, tandem MS data is acquired to distinguish between isomers and isobars.60 Figure 1 provides a schematic of this analysis process.
Figure 1.
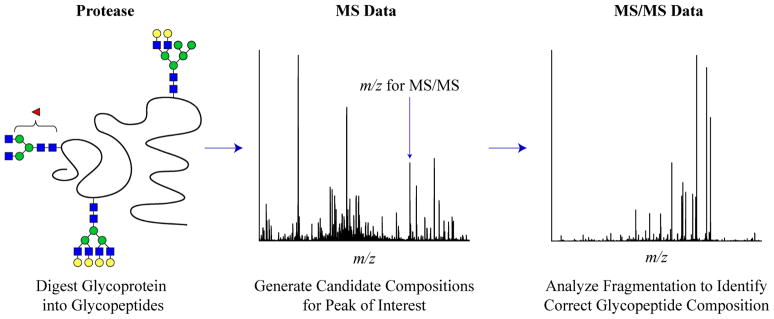
Flow chart outlining the use of MS and MS/MS data for glycopeptide identification.
N-LINKED GLYCOPEPTIDES
Although N-linked glycoforms share a common core structure, the rest of the glycan follows one of three distinct arrangements. Based on the arrangement pattern, N-linked glycans compositions are classified into three main types, those with: 1) High mannose type glycans 2) Complex type glycans and 3) Hybrid type glycans.12,18 This information is useful when deciphering glycopeptide compositions from MS experiments, specifically from CID MS/MS data.61
N-linked Glycopeptide MS Data
A variety of automated and semi-automated analysis tools have been created to aid in the interpretation of N-linked glycopeptide MS data. The key objective of these tools is to provide glycopeptide compositions that are consistent with the high resolution MS data. Researchers then typically use MS/MS analysis, described later in this review, to determine which of the compositions is correct for each given ion. Three of these tools are accessible to the public: GlycoMod (http://web.expasy.org/glycomod/), GlycoPep DB (http://hexose.chem.ku.edu/glycop.htm), and the previously mentioned GlycoSpectrumScan.41,62,63 GlycoMod, the earliest and most heavily used tool, accepts a protein sequence, possible monosaccharide building blocks, and experimental mass data as inputs, and it calculates all possible glycopeptide compositions that fall within the mass tolerance.62 One restriction in the capacity of GlycoMod to analyze glycopeptide data is the inability to handle multiply-charged precursors.62
Programs such as GlycoPep DB and GlycoSpectrumScan were designed to overcome some of the limitations in GlycoMod. GlycoPep DB, developed by Go et al. limits its output by restricting the potential glycans in the glycopeptide to a database of biologically relevant glycoforms that have been previously identified in MS data.63 It also accepts precursor ions in multiple charge states.63 The disadvantage of using this approach, however, is that if the glycan in the spectrum is not in the GlycoPep DB database, then the software will not be effective at providing the correct assignment for the peak.63 GlycoSpectrumScan is a more recent program, developed by Deshpande et al. that also interprets MS data on both N- and O-linked glycopeptides.41 Like GlycoPep DB, this program has the ability to handle input for both singly-and multiply-charged data.41 GlycoSpectrumScan is described in detail below for O-linked linked MS data analysis. Regardless of which tool is used for assigning the high resolution data, these assignments must be supported by MS/MS data, to provide high confidence assignments.60
N-linked Glycopeptide MS/MS Data
Each common N-linked glycan type (complex, hybrid, or high mannose) has a signature fragmentation profile that is present when a glycopeptide is subjected to MS/MS experiments.25,61 These characteristic fragmentation profiles are useful for determining the correct composition of an N-linked glycopeptide when isobaric candidate compositions are possible.61 However, as manual interpretation of these data are challenging, software is required to speed analysis time.
Many of the available tools to analyze glycopeptides are software expansions of tools that have been developed previously to analyze released glycans. One disadvantage of expanding glycan analysis tools to glycopeptides is that these tools generally lack capabilities for analyzing and scoring the peptide component of the glycopeptides. SimGlycan is one such example. Available for purchase, SimGlycan has been updated to perform fragmentation analysis for glycopeptides, in addition to glycans.49,50 As stated previously, SimGlycan uses a database of over 9,000 glycan structures that could be consistent with the MS/MS data to identify the most appropriate composition for the acquired spectrum.50 SimGlycan may be purchased online (http://www.premierbiosoft.com/).
Many other publicly available tools to elucidate glycosylation profiles of glycopeptides have also emerged out of glycan analysis software. GlycoWorkbench and Glyco-Peakfinder both can annotate glycan fragmentation in glycopeptide data, although the peptide portion of the glycopeptide must be determined by some means other than the use of these tools.39,64 On the positive side, Glyco-Peakfinder is useful for de novo calculation and annotation of glycan fragment ions within tandem mass spectra.64 Users may allow constraints on the oligosaccharide such as size and attachment of other substituents (such as acetate, phosphate, and sulfate), and the program is capable of annotating multiply-charged ions (− 4 to + 4).64 Additionally, glycan fragmentation is analyzed across multiple charge states, and across multiple charge carriers (cationic carriers), within the same spectrum.64
A completely different approach is used in GlycoPep ID.65 GlycoPep ID is a web-based tool developed by Go et al. to interpret MS/MS data of glycopeptides and to identify the peptide component of glycopeptides through analysis of expected product ions.65 The URL to access this program is listed in Table 4. Although this program is useful for identification of the peptide portion of the glycopeptide in complex LC-MS samples, it does not contain a scoring algorithm to identify the most probable glycopeptide match.65
Table 4.
Freely Available N-linked Glycopeptide Analysis Tools.
| Analysis Tool | Link to Automated Program | Overview |
|---|---|---|
| GlycoMod | http://web.expasy.org/glycomod/ | GlycoMod determines potential glycopeptide compositions, on the basis of mass, from MS data. |
| GlycoPep DB | http://hexose.chem.ku.edu/glycop.htm | GlycoPep DB deduces possible biologically relevant glycan compositions from glycopeptide MS data using a “smart search”. |
| GlycoSpectrumScan | http://www.glycospectrumscan.org. | GlycoSpectrumScan searches LC-MS data to identify glycopeptides and determine glycoform location. |
| GlycoWorkbench | http://download.glycoworkbench.org/ | GlycoWorkbench was designed for the annotation of glycopeptide MS/MS data through fragmentation analysis and scoring of only the glycan portion. |
| Glyco-Peakfinder | http://glyco-peakfinder.org/ | Glyco-Peakfinder is capable of de novo glycopeptide analysis through glycan fragmentation profiling for spectra after the peptide sequence is input by the user. |
| GlycoPep ID | http://hexose.chem.ku.edu/predictiontable2.php | GlycoPep ID works to analyze glycopeptides from MS/MS spectra of complex mixtures by identifying the peptide portion based on expected product ions. |
| GlycoMiner | http://www.chemres.hu/ms/glycominer/tutorial.html | GlycoMiner is designed to identify glycopeptides from qTOF MS/MS data, and assigns composition for spectra of quality containing specific marker ions. |
| GPS | http://edwardslab.bmcb.georgetown.edu/software/GlycoPeptideSearch.html | GPS utilizes a glycan database to generate glycopeptide compositions after searching LC-MS/MS data of purified proteins and matching fragmentation patterns. |
| GlypID | http://www.cbs.dtu.dk/services/DictyOGlyc/ | GlypID is designed to identify glycopeptides from LC-MS/MS experiments using a combination of MS1 and MS2 data. |
| GlypID 2.0 | http://mendel.informatics.indiana.edu/~chuyu/glypID/software.html | GlypID 2.0 uses CID and HCD information to identify glycan type, monosaccharide composition, and attachment site for N-linked glycopeptide MS/MS data. |
| GPG | http://glycopro.chem.ku.edu/GPGHome.php | GPG scores glycopeptide candidates after searching MS/MS data for the predicted product ions for each composition tested. |
Software with the ability to score potential compositions is especially useful to researchers. Often, more than one glycan or glycopeptide composition could correspond to a given spectrum within the accepted range of mass tolerance. Therefore, programs that have a scoring function to evaluate each of those possible matches, and return which of them is the most likely structure, greatly improves the efficiency of the analysis. For programs that lack this feature, a user must spend time manually determining which of the mathematically possible predictions is the best match for the data
Some alternative, unique strategies have been developed with the goal of scoring MS/MS data against potential glycopeptide compositions, such as those described using Peptoonist, Medicel Integrator, the Branch-and-Bound algorithm, GlycoMaster, Sweet Substitute, and GlyDB.66,67,68,69,70,71 Unfortunately, none of these programs are currently publicly available.
To address the need for publicly accessible tools specifically designed to interpret and score fragmentation of glycopeptides, GlycoMiner was developed by Ozohanics et al.72 In the analysis of 3132 spectra, the program was reported to have found 338 that corresponded to MS/MS data of glycopeptides (versus peptides).72 Designed using qTOF data, the software is capable of assigning glycopeptide compositions when both the peptide and glycan components portions are unknown.72 However, the program is only capable of performing compositional analysis when the spectra are of good quality.72 The program fails when spectral quality is low, as evidenced by the program’s identification of glycan composition in only 196/338 glycopeptide spectra.72 Although this tool is a great advancement towards automated interpretation of glycopeptide MS/MS data, GlycoMiner often generates multiple plausible compositions and fails to rank the correct glycopeptide as the top candidate.72 In addition, the program requires spectra containing a low S/N ratio as well as the presence of low-mass marker ions, which are not typically present in data collected on ion trap instruments.72 Available online, GlycoMiner is free to download and use; see Table 4.
Similar to GlycoMiner, GlycoPeptide Search (GPS) is a recently developed program by Chandler et al. for the determination of glycopeptide composition from CID data.73 Designed for purified glycoprotein samples analyzed by LC-MS/MS, GPS utilizes GlycomeDB, a glycan database in conjunction with the peptide file, which is supplied by the user, to generate an Excel file of glycopeptide matches based on fragmentation evidence.73 To generate the peptide-glycan pairs, GPS must find both low mass oxonium, and N-glycan core-containing, product ions.73 GPS is freely available online as well.73 For further information, see Table 4.
The targeted MS/MS approach utilizing the computational tool GlypID recently described by Wu et al. aims to characterize N-linked glycopeptides through the combined use of MS1 and MS2 information extracted from LC-MS/MS experiments.74 One of the benefits to the method is that no prior knowledge of the potential glycosylation or identity of the glycopeptide is necessary.74 Instead, GlypID assigns a cluster of glycopeptides in the “same family” (microheterogeneities) based on observed mass.74 In addition, the approach utilizes an isotope deconvolution algorithm to assign ion charges along with monoisotopic ions.74 This information is then added to the inclusion list of “prioritized precursor ions” for the MS/MS analysis that follows.74 Next, the resultant CID data is searched for the longest series of glycosidic bond cleavage series.74 These product ions are used to determine the oligosaccharide sequence tag, which is used to verify whether or not the spectrum is from a glycopeptide.74 A score is assigned to the CID spectrum based on this sequence tag.74 MS data is used to evaluate and score the relative probability of a glycopeptide by examining the clusters of peptide glycoforms, or those glycopeptides with the same peptide backbone that co-elute within a specific time range.74 The glycoform is then identified using the mass of the attached N-linked glycan, though the most current version of GlypID allows the entry of user-defined glycan compositions as well.74 A limitation to the program is that when low resolution data is used, there is a significant increase in the number of false-positive identifications of glycopeptide microheterogeneities within a cluster. Although the new targeted MS/MS approach has been optimized for FT-MS instrumentation and data, the original GlypID algorithm was designed using LC-MS ion trap data.75 A publicly accessible version of the computational tool is currently available online, free of charge to users (see Table 4).
Mayampurath et al. have now modified the GlypID algorithm with a scoring function that works to determine glycopeptide composition from high-energy C-trap dissociation (HCD) MS/MS data.76 The new software tool, GlypID 2.0, uses high resolution MS1 data along with CID and HCD scan information to improve the accuracy of N-linked glycopeptide identification.76 Like the original GlypID, GlypID 2.0 also works to score CID spectra independently on MS systems that do not contain the HCD instrument option.76 GlypID 2.0 is freely available to download, as listed in Table 4.
Woodin et al., have also developed a freely accessible web-based tool, GlycoPep Grader (GPG), to assign a glycopeptide composition to MS/MS data in an automated fashion.61 This tool is specifically designed for data collected in an ion trap mass spectrometer, and it features a novel algorithm that enables users to identify the correct glycopeptide composition from a pool of candidate compositions of the same nominal mass.61
GPG utilizes the MS/MS data by calculating, scoring, and searching for the expected product ions of potential glycopeptide candidate compositions.61 The algorithm scores the glycopeptide candidate composition through detection of two types of product ions: 1) Ions that contain the peptide portion and some portion of the pentasaccharide core, or [peptide + core component] ions, and 2) Ions formed via neutral loss of monosaccharide residues from the precursor ion, or [precursor − monosaccharide] ions.61 The algorithm that powers GPG has been shown to assign the correct glycopeptide candidate after performing the MS/MS peak list search with a very high degree of accuracy.61
One advantage to the algorithm behind GPG is that the precursor ion’s charge state is included in the input data, so all product ions can be searched for in a charge state specific manner.61 Secondly, no spectral transformation (to singly charged ions) needs to be performed prior to using the program as GPG automatically searches for product ions in a charge-specific fashion, bypassing the need for additional processing software.61 A disadvantage of the program is that the user must utilize a separate program, such as GlycoMod, to obtain potential matches for the high resolution MS data, prior to assigning the MS2 data with GPG.61 GPG can be found online, and is free to use.
O-LINKED GLYCOPEPTIDES
The analysis of O-linked glycoforms is particularly challenging, as no single consensus sequence exists to predict the site of glycan attachment.7,77 Further adding to the difficulty of analysis, factors that affect the efficiency of glycosylation at N-linked sites are different than those affecting O-glycosylation efficiency. For example, the presence of aromatic residues near an O-linked site inhibits glycosylation; whereas the presence of an aromatic residue near an N-linked site increases the likelihood of site-occupancy.22
Mucin-type O-linked Glycosylation
The most prevalent form of O-linked glycosylation to occur in eukaryotic organisms is mucin-type O-glycosylation, which occurs where glycans are attached to a protein by the addition of α-N-acetylgalactosamine (GalNAc) residues to the hydroxyl group of Ser/Thr side chains (commonly referred to as the Tn antigen).2,7 Though still in the infancy stage, analysis tools have recently been created to assist researchers in the determination of O-linked glycoforms, many of which are mucin in type, from MS data.
O-linked Glycopeptide Characterization from MS Data
Recently, Deshpande et al. advanced the MS data analysis of N- and O-linked glycopeptides with the advent of the GlycoSpectrumScan program.41 GlycoSpectrumScan is designed to analyze LC-MS data of intact glycopeptides from proteolytic digests.41 The program utilizes MS1 data to determine glycopeptide composition, along with the relative distribution of glycoforms at each of the sites.41 In addition, the algorithm behind the program offers a few distinct advantages in that it handles multiply charged ions, making it amenable to both MALDI and ESI data, and is currently freely available online (www.glycospectrumscan.org.).41
GlycoX and GlycoMod, described earlier in the analysis of N-linked glycopeptides, are capable of O-linked glycopeptide data interpretation as well.62,78 Unlike GlycoMod (http://web.expasy.org/glycomod/), GlycoX is not publically-available, though it is available upon request from the authors.78 GlycoWorkbench, also described previously, performs automation of O-linked glycopeptide MS1 data to elucidate the most likely composition from an experimental peak list in the same manner for N-linked glycopeptide spectra.39 GlycoWorkbench is freely available online (http://download.glycoworkbench.org/).39
O-linked Glycopeptide Characterization from MS/MS Data
Currently, there are no freely available stand-alone programs designed to automate the analysis of O-linked glycopeptide CID MS/MS data through evaluation of both unknown portions of a glycopeptide, the peptide and glycan. The GlycoWorkbench program is capable of annotating glycans in CID fragmentation data of glycopeptides.39 However, as described for the MS/MS characterization of N-linked glycopeptides, the identity of the peptide portion must already be known, as GlycoWorkbench solely evaluates the fragmentation of the glycan-containing portion of a glycopeptide.39
There are promising advances being made in the compositional determination of glycopeptides using electron transfer dissociation (ETD) fragmentation techniques,11,24 or a combination of CID and ETD, particularly in the study of O-linked species.79 A recent method described by Daracula et al. in which MS1, CID, and ETD data are used in conjunction with Protein Prospector v5.3 for the identification of SA1-10GalGalNAc-containing O-linked glycopeptides enriched from bovine serum, demonstrates the potential for automated analysis through a combination of these techniques and database searches.79 However, this process is only semi-automated and works only in the case of simple carbohydrate structures are present in the sample.79 Hopefully, the compositional information gained between the two complementary fragmentation methods of CID and ETD will enable researchers to develop algorithms and gain insight into creating automated programs to speed the analysis of O-linked MS/MS glycopeptide data as well.
CONCLUSION
For the study of protein glycosylation, there are two main approaches used by researchers: Glycan analysis and glycopeptide analysis. The less challenging mode of analysis is to release the glycans from a glycoprotein and analyze them independently. However, the most informative approach is to utilize a protease and cleave the glycoprotein into glycopeptides, retaining information on where each glycan is attached within the protein sequence. The generation of automated MS and MS/MS analysis tools to assist in the characterization of both glycans and glycopeptides is emerging as an effort to facilitate more rapid characterization of glycosylation by either method. Without these analysis tools, data interpretation is a difficult and tedious task. Currently available and recently developed programs for various glycan types, along with their associated advantages and disadvantages, are discussed within this review. With access to the automated tools described here, research in glycomics and glycoproteomics can be greatly facilitated.
Acknowledgments
The authors acknowledge financial support from the National Institutes of Health (RO1RR026061) and an NSF Career award (0645120) to HD, an NSF Fellowship (DGE-0742523) and Pfizer Award to CW, and a Seo Scholarship to MM.
References
- 1.Spiro RG. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology. 2002;12:43R–56R. doi: 10.1093/glycob/12.4.43r. [DOI] [PubMed] [Google Scholar]
- 2.Hang H, Bertozzi CR. The chemistry and biology of mucin-type O-linked glycosylation. Bioorg Med Chem. 2005;13:5021–5034. doi: 10.1016/j.bmc.2005.04.085. [DOI] [PubMed] [Google Scholar]
- 3.Murrell MP, Yarema KJ, Levchenko A. The systems biology of glycosylation. Chem Biochem. 2004;5:1334–1347. doi: 10.1002/cbic.200400143. [DOI] [PubMed] [Google Scholar]
- 4.Van den Steen P, Rudd PM, Dwek RA, Opdenakker G. Concepts and principles of O-linked glycosylation. Crit Rev Biochem Mol Biol. 1998;33:151–208. doi: 10.1080/10409239891204198. [DOI] [PubMed] [Google Scholar]
- 5.Bertozzi CR, Kiessling LL. Chemical glycobiology. Science. 2001;291:2357–2364. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- 6.Dennis JW, Granovsky M, Warren C. Protein glycosylation in development and disease. BioEssays. 1999;21:412–421. doi: 10.1002/(SICI)1521-1878(199905)21:5<412::AID-BIES8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 7.Tian E, Ten Hagen KG. Recent insights into the biological roles of mucin-type O-glycosylation. Glycoconjugate J. 2009;26:325–334. doi: 10.1007/s10719-008-9162-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wada Y, Tajiri M, Ohshima S. Quantitation of saccharide compositions of O-glycans by mass spectrometry of glycopeptides and its application to rheumatoid arthritis. J Proteome Res. 2010;9:1367–1373. doi: 10.1021/pr900913k. [DOI] [PubMed] [Google Scholar]
- 9.Dube DH, Bertozzi CR. Glycans in cancer and inflammation Potential for therapeutics and diagnostics. Nat Rev Drug Discovery. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 10.Lefebvre T, Dehennault V, Guinez C, Olivier S, Drougart L, Mir AM, Mortuaire M, Vercoutter-Edouart AS, Michalski JC. Dysregulation of the nutrient/stress sensor O-GlcNAcylation is involved in the etiology of cardiovascular disorders, type-2 diabetes and Alzheimer’s disease. J Biochim Biophys Acta. 2010;1800:67–79. doi: 10.1016/j.bbagen.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 11.Jensen PH, Kolarich D, Packer NH. Mucin-type O-glycosylation Putting the pieces together. FEBS J. 2010;277:81–94. doi: 10.1111/j.1742-4658.2009.07429.x. [DOI] [PubMed] [Google Scholar]
- 12.Marino K, Bones J, Kattla JJ, Rudd PM. A systematic approach to protein glycosylation analysis: A path through the maze. Nat Chem Biol. 2010;6:713–723. doi: 10.1038/nchembio.437. [DOI] [PubMed] [Google Scholar]
- 13.Budnik BA, Lee RS, Steen JAJ. Global methods for protein glycosylation analysis by mass spectrometry. J Biochim Biophys Acta. 2006;1764:1870–1880. doi: 10.1016/j.bbapap.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Raman R, Raguram S, Venkataraman G, Paulson JC, Sasiekharan R. Glycomics: An integrated systems approach to structure-function relationships of glycans. Nat Methods. 2005;2:817–824. doi: 10.1038/nmeth807. [DOI] [PubMed] [Google Scholar]
- 15.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. J Biochim Biophys Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 16.Brazier-Hicks M, Evans KM, Gershater MC, Puschmann H, Steel PG, Edwards R. The C-glycosylation of flavonoids in cereals. J Biol Chem. 2009;284:17926–17934. doi: 10.1074/jbc.M109.009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hofsteenge J, Muller DR, de Beer T, Loffler A, Richter WJ, Vliegenthart JFG. New type of linkage between a carbohydrate and a protein: C-glycosylation of a specific tryptophan residue in human RNase Us. Biochemistry. 1994;33:13524–13530. doi: 10.1021/bi00250a003. [DOI] [PubMed] [Google Scholar]
- 18.Morelle W, Canis K, Chirat F, Faid V, Michalski JC. The use of mass spectrometry for the proteomic analysis of glycosylation. Proteomics. 2006;6:3993–4015. doi: 10.1002/pmic.200600129. [DOI] [PubMed] [Google Scholar]
- 19.Stepper J, Shasti S, Loo TS, Preston JC, Novak P, Man P, Moore CH, Havli ek V, Patchett ML, Norris GE. Cysteine S-glycosylation, a new post-translational modification found in glycopeptide bacteriocins. FEBS Lett. 2011;585:645–650. doi: 10.1016/j.febslet.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 20.Mazola Y, Chinea G, Mussacchio A. Integrating bioinformatics tools to handle glycosylation. PLOS Comput Biol. 2011;7 doi: 10.1371/journal.pcbi.1002285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones J, Krag SS, Betenbaugh MJ. Controlling N-glycan site occupancy. Biochim Biophys Acta. 2005;1726:121–137. doi: 10.1016/j.bbagen.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Christlet THT, Veluraja K. Database analysis of O-glycosylation sites in proteins. Biophys J. 2001;80:952–960. doi: 10.1016/s0006-3495(01)76074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dalpathado DS, Desaire H. Glycopeptide analysis by mass spectrometry. Analyst. 2008;133:731–738. doi: 10.1039/b713816d. [DOI] [PubMed] [Google Scholar]
- 24.North SJ, Hitchen PG, Haslam SM, Dell A. Mass spectrometry in the analysis of N-linked and O-linked glycans. Curr Opin Struct Biol. 2009;19:498–506. doi: 10.1016/j.sbi.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nwosu CC, Seipert RR, Strum JS, Hua SS, An HJ, Zivkovic AM, German BJ, Lebrilla CB. Simultaneous and extensive site-specific N- and O-glycosylation analysis in protein mixtures. J Proteome Res. 2011;10:2612–2624. doi: 10.1021/pr2001429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckermann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Mazumder R, O’Donovan C, Redaschi N, Suzek B. The Universal Protein Resource (UniProt): An expanding universe of protein information. Nucleic Acids Res. 2006;34:D187–D191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee TY, Huang HD, Hung JH, Huang HY, Yang YS, Wang TH. dbPTM: An informal repository of protein post-translational modification. Nucleic Acids Res. 2006;34:D622–D627. doi: 10.1093/nar/gkj083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ranzinger R, Herget S, Wetter T, von der Lieth CW. Glycome DB Integration of open-access carbohydrate structure search databases. BMC Bioinformatics. 2008;9 doi: 10.1186/1471-2105-9-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooper CA, Harrison MJ, Wilkins MR, Packer NH. GlycoSuiteDB: A new curated relational database of glycoprotein glycan structures and their biological sources. Nucleic Acids Res. 2001;29:332–335. doi: 10.1093/nar/29.1.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta R, Birch H, Rapacki K, Brunak S, Hansen JE. O-GLYCBASE version 4.0: A revised database of O-glycosylated proteins. Nucleic Acids Res. 1999;27:370–372. doi: 10.1093/nar/27.1.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H, Loriaux P, Eng J, Campbell D, Keller A, Moss P, Bonneau R, Zhang N, Zhou Y, Wollscheid B, Cooke K, Yi EC, Lee H, Peskind ER, Zhang J, Smith RD, Aebersold R. UniPep A database for human N-linked glycosites: a resource for biomarker discovery. Genome Biol. 2006;7 doi: 10.1186/gb-2006-7-8-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campbell MP, Roylez L, Radcliffe CM, Dwek RA, Rudd PM. GlycoBase and autoGU: Tools for HPLC-based glycan analysis. Bioinformatics. 2008;24:1214–1216. doi: 10.1093/bioinformatics/btn090. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Torii M, Liu H, Hart GW, Hu ZZ. dbOGAP An integrated bioinformatics resource for protein O-GlcNAcylation. BMC Bioinformatics. 2011;12 doi: 10.1186/1471-2105-12-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goetz JA, Novotny MV, Mechref Y. Enzymatic/chemical release of O-glycans allowing MS analysis at high sensitivity. Anal Chem. 2009;81:9546–9552. doi: 10.1021/ac901363h. [DOI] [PubMed] [Google Scholar]
- 35.Goldberg D, Sutton-Smith M, Paulson J, Dell A. Automatic annotation of matrix-assisted laser desorption/ionization N-glycan spectra. Proteomics. 2005;5:865–876. doi: 10.1002/pmic.200401071. [DOI] [PubMed] [Google Scholar]
- 36.Goldberg D, Bern M, Li B, Lebrilla CB. Automatic determination of O-glycan structure from fragmentation spectra. J Proteome Res. 2006;5:1429–1434. doi: 10.1021/pr060035j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vakhrushev SY, Dadimov D, Peter-Katalini J. Software platform for high-throughput glycomics. Anal Chem. 2009;81:3252–3260. doi: 10.1021/ac802408f. [DOI] [PubMed] [Google Scholar]
- 38.Vakhrushev SY, Dadimov D, Peter-Katalini J. SysBioWare: Structure assignment tool for automated glycomics. Glyco-Bioinformatics; Postdam, Germany: 2009. pp. 141–161. [Google Scholar]
- 39.Ceroni A, Maass K, Geyer H, Geyer R, Dell A, Haslam SM. GlycoWorkBench: A tool for the computer-assisted annotation of mass spectra of glycans. J Proteome Res. 2008;7:1650–1659. doi: 10.1021/pr7008252. [DOI] [PubMed] [Google Scholar]
- 40.Campbell MP, Royle L, Radcliffe CM, Dwek RA, Rudd PM. GlycoBase and autoGU: Tools for HPLC-based glycan analysis. Bioinformatics. 2008;24:1214–1216. doi: 10.1093/bioinformatics/btn090. [DOI] [PubMed] [Google Scholar]
- 41.Deshpande N, Jensen PH, Packer NH, Kolarich D. GlycoSpectrumScan: Fishing glycopeptides from MS spectra of protease digests of human colostrum sIgA. J Proteome Res. 2010;9:1063–1075. doi: 10.1021/pr900956x. [DOI] [PubMed] [Google Scholar]
- 42.Gaucher SP, Morrow J, Leary J. STAT: A saccharide topology analysis tool used in combination with tandem mass spectrometry. Anal Chem. 2000;72:2331–2336. doi: 10.1021/ac000096f. [DOI] [PubMed] [Google Scholar]
- 43.Ashline DJ, Lapadula AJ, Liu Y-H, Lin M, Grace M, Pramanik B, Reinhold VN. Carbohydrate structural isomers analyzed by sequential mass spectrometry. Anal Chem. 2007;79:3830–3842. doi: 10.1021/ac062383a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ethier M, Saba JA, Ens W, Standing KG, Perreault H. Automated structural assignment of derivatized complex N-linked oligosaccharides from tandem mass spectra. Rapid Commun Mass Spectrom. 2002;16:1743–1754. doi: 10.1002/rcm.779. [DOI] [PubMed] [Google Scholar]
- 45.Lohmann KK, von der Lieth CW. GlycoFragment and GlycoSearchMS: Web tools to support the interpretation of mass spectra of complex carbohydrates. Nucleic Acids Res. 2004;32:W261–W266. doi: 10.1093/nar/gkh392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lohmann KK, von der Lieth CW. GLYCO-FRAGMENT: A web tool to support the interpretation of mass spectra of complex carbohydrates. Proteomics. 2003;3:2028–2035. doi: 10.1002/pmic.200300505. [DOI] [PubMed] [Google Scholar]
- 47.Tang H, Mechref Y, Novotny M. Automated interpretation of MS/MS spectra of oligosaccharides. Bioinformatics. 2005;21:I431–I439. doi: 10.1093/bioinformatics/bti1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao HY. Generation of asparagine-linked glycan structure databases. J Am Soc Mass Spectrom. 2009;20:1739–1742. doi: 10.1016/j.jasms.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 49.Blow N. A spoonful of sugar. Nature. 2009;457:617–620. doi: 10.1038/457617a. [DOI] [PubMed] [Google Scholar]
- 50.Apte A, Meitei NS. Bioinformatics in glycomics: Glycan characterization with mass spectrometric data using SimGlycan. In: Li J, editor. Methods in Molecular Biology. Vol. 600. Humana Press; 2009. pp. 269–281. [DOI] [PubMed] [Google Scholar]
- 51.Caragea C, Sinapov J, Silvescu A, Dobbs D, Honavar V. Glycosylation site prediction using ensembles of support vector machine classifiers. BMC Bioinformatics. 2007:8. doi: 10.1186/1471-2105-8-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carbohydrate Structure Suite (CSS): analysis of carbohydrate 3D structures derived from the PDB. Nucleic Acids Res. 2005;33:D242–D246. doi: 10.1093/nar/gki013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hamby SE, Hirst JD. Prediction of glycosylation sites using random forests. BMC Bioinformatics. 2008;9 doi: 10.1186/1471-2105-9-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gupta R, Brunak S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput; Lyngby, Denmark. 2002. pp. 310–322. [PubMed] [Google Scholar]
- 55.Julenius K. NetCGlyc 1.0: Prediction of mammalian C-mannosylation sites. Glycobiology. 2007;17:868–876. doi: 10.1093/glycob/cwm050. [DOI] [PubMed] [Google Scholar]
- 56.Hansen JE, Lund O, Tolstrup N, Gooley AA, Williams KL, Brunak S. NetOglyc: Prediction of mucin type O-glycosylation sites based on sequence context and surface accessibility. Glycoconjugate J. 1998;15:115–130. doi: 10.1023/a:1006960004440. [DOI] [PubMed] [Google Scholar]
- 57.Chen YZ, Tang YR, Sheng ZY, Zhang Z. Prediction of mucin-type O-glycosylation sites in mammalian proteins using the composition of k-spaced amino acid pairs. BMC Bioinformatics. 2008;9 doi: 10.1186/1471-2105-9-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gerken TA, Jamison O, Perrine CL, Collette JC, Moinova H, Ravi L, Markowitz SD, Shen W, Patel H, Tabak LA. Emerging paradigms for the initiation of mucin-type protein O-glycosylation by the polypeptide GalNAc transferase family of glycosyltransferases. J Biol Chem. 2011;286:14493–14507. doi: 10.1074/jbc.M111.218701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gupta R, Jung E, Gooley AA, Williams KL, Brunak S, Hansen J. Scanning the available Dictyostelium discoideum proteome for O-linked GlcNAc glycosylation sites using neural networks. Glycobiology. 1999;9:1009–1022. doi: 10.1093/glycob/9.10.1009. [DOI] [PubMed] [Google Scholar]
- 60.Desaire H, Hua D. When can glycopeptides be assigned based solely on high-resolution mass spectrometry data? Int J Mass Spectrom. 2009;287:21–26. [Google Scholar]
- 61.Woodin CL, Hua D, Maxon M, Rebecchi KR, Go EP, Desaire H. GlycoPep Grader: A web-based utility for assigning the composition of N-linked glycopeptides. Anal Chem. 2012;84:4821–4829. doi: 10.1021/ac300393t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cooper CA, Gasteiger E, Packer NH. GlycoMod A software tool for determining glycosylation compositions from mass spectrometric data. Proteomics. 2001;1:340–349. doi: 10.1002/1615-9861(200102)1:2<340::AID-PROT340>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 63.Go EP, Rebecchi KR, Dalpathado DS, Bandu ML, Zhang Y, Desaire H. GlycoPep DB: A tool for glycopeptide analysis using a “smart search”. Anal Chem. 2007;79:1708–1713. doi: 10.1021/ac061548c. [DOI] [PubMed] [Google Scholar]
- 64.Maass K, Ranzinger R, Geyer H, von der Lieth CW, Geyer R. “Glyco-peakfinder” De novo composition analysis of glycoconjugates. Proteomics. 2007;7:4435–4444. doi: 10.1002/pmic.200700253. [DOI] [PubMed] [Google Scholar]
- 65.Irungu J, Go EP, Dalpathado DS, Desaire H. Simplification of mass spectral analysis of acidic glycopeptides using GlycoPep ID. Anal Chem. 2007;79:3065–3074. doi: 10.1021/ac062100e. [DOI] [PubMed] [Google Scholar]
- 66.Goldberg D, Bern M, Parry S, Sutton-Smith M, Panico M, Morris HR, Dell A. Automated N-glycopeptide identification using a combination of single- and tandem-MS. J Proteome Res. 2007;6:3995–4005. doi: 10.1021/pr070239f. [DOI] [PubMed] [Google Scholar]
- 67.Joenvaara S, Ritamo I, Peltoniemi H, Renkonen R. N-glycoproteomics An automated workflow approach. Glycobiology. 2008;18:339–349. doi: 10.1093/glycob/cwn013. [DOI] [PubMed] [Google Scholar]
- 68.Peltoniemi H, Joenvaara S, Renkonen R. De novo glycan structure search with the CID MS/MS spectra of native N-glycopeptides. Glycobiology. 2009;19:707–714. doi: 10.1093/glycob/cwp034. [DOI] [PubMed] [Google Scholar]
- 69.Shan B, Ma B, Zhang K. Complexities and algorithms for glycan sequencing using tandem mass spectrometry. J Bioinform Comput Biol. 2008;6:77–91. doi: 10.1142/s0219720008003291. [DOI] [PubMed] [Google Scholar]
- 70.Clerens S, Van den Ende W, Verhaet P, Geenen L, Archens L. Sweet Substitute: A software tool for in silico fragmentation of peptide-linked N-glycans. Proteomics. 2004;4:629–632. doi: 10.1002/pmic.200300572. [DOI] [PubMed] [Google Scholar]
- 71.Ren JM, Rejtar T, Li L, Karger BL. N-glycan structure annotation of glycopeptides using a linearized glycan structure database (GlyDB) J Proteome Res. 2007;6:3162–3173. doi: 10.1021/pr070111y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ozohanics O, Krenyacz J, Ludanyi K, Pollreisz F, Vekey K, Drahos L. GlycoMiner: A new software tool to elucidate glycopeptide composition. Rapid Commun Mass Spectrom. 2008;22:3245–3254. doi: 10.1002/rcm.3731. [DOI] [PubMed] [Google Scholar]
- 73.Pompach P, Chandler K, Lan R, Edwards N, Goldman R. Semi-automated identification of N-glycopeptides by hydrophilic interaction chromatography, nano-reverse-phase LC-MS/MS, and glycan database search. J Proteome Res. 2012;11:1728–1740. doi: 10.1021/pr201183w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu Y, Mechref Y, Klouckova I, Mayampurath AM, Novotny MV, Tang H. Mapping site-specific protein N-glycosylations through liquid chromatography/mass spectrometry and targeted tandem mass spectrometry. Rapid Commun Mass Spectrom. 2010;24:965–972. doi: 10.1002/rcm.4474. [DOI] [PubMed] [Google Scholar]
- 75.Wu Y, Mechref Y, Klouckova I, Novotny MV, Tang H. A computational approach for the identification of site-specific protein glycosylations through ion-trap mass spectrometry. In: Ideker T, Bafna V, editors. Systems Biology and Computational Proteomics. Vol. 4532. Springer Berlin/Heidelberg; 2007. pp. 96–107. [Google Scholar]
- 76.Mayampurath AM, Wu Y, Segu ZM, Mechref Y, Tang H. Improving confidence in detection and characterization of protein N-glycosylation sites and microheterogeneity. Rapid Commun Mass Spectrom. 2011;25:2007–2019. doi: 10.1002/rcm.5059. [DOI] [PubMed] [Google Scholar]
- 77.Rakus JF, Mahal LK. New technologies for glycomic analysis: Toward a systematic understanding of the glycome. In: Cooks RG, Yeung ES, editors. Annual Reviews of Analytical Chemistry. Vol. 4. Annual Reviews; Palo Alto: 2011. pp. 367–392. [DOI] [PubMed] [Google Scholar]
- 78.An HJ, Tillinghast JS, Woodruff DL, Rocke DM, Lebrilla CB. A new computer program (GlycoX) to determine simultaneously the glycosylation sites and oligosaccharide heterogeneity of glycoproteins. J Proteome Res. 2006;5:2800–2808. doi: 10.1021/pr0602949. [DOI] [PubMed] [Google Scholar]
- 79.Darula Z, Chalkley RJ, Baker P, Burlingame AL, Medzihradszky KF. Mass spectrometric analysis, automated identification and complete annotation of O-linked glycopeptides. Eur J Mass Spectrom. 2010;16:421–428. doi: 10.1255/ejms.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]