

Abstract
Overexpression of the unfolded protein response (UPR) master regulator GRP78 is associated with poor prognosis and therapeutic resistance in numerous human cancers, yet its role in endometrial cancers (EC) is undefined. To better understand the contribution of GRP78 to EC, we examined its expression levels in EC patient samples and EC cell lines. We demonstrate that GRP78 overexpression occurs more frequently in EC tissues compared to that found in normal endometrium, and that GRP78 expression occurs in most EC cell lines examined. Functional analysis demonstrated that GRP78 is inducible by cisplatin in EC cells, and siRNA knockdown of GRP78 augments chemotherapy-mediated cell death. Examination of AKT and GRP78 expression demonstrated that inhibition of AKT activity by MK2206 blocks GRP78 expression in EC cells. SiRNA studies also revealed that knockdown of GRP78 reduces but does not abrogate AKT activity, demonstrating that GRP78 is required for optimal AKT activity. In the presence of MK2206, siRNA knockdown of GRP78 does not augment AKT mediated survival in response to cisplatin treatment, suggesting that GRP78’s anti-apoptosis functions are part of the AKT survival pathway. Targeted therapies that reduce GRP78 expression or activity in cancers may serve to increase the effectiveness of current therapies for EC patients.
INTRODUCTION
Endometrial cancer (EC) affects over 47,000 women annually making it the most common gynecologic cancer in the United States 1. While early stage EC is typically treated with surgery, advanced and/or recurrent EC typically requires systemic chemotherapy regimens and/or radiotherapy 2. Unfortunately most current chemotherapeutic regimens used in advanced EC patients have only modest activity 3. While new therapies are being tested, the development of additional treatments that can improve the efficacy of existing therapies may have the greatest immediate benefit to patients with advanced or recurrent EC. Elucidation of the mechanisms in the unfolded protein response (UPR) suggests that modulating the function of specific UPR components may augment the cytotoxic effects of current chemotherapeutic regimens 4.
The UPR primarily functions to correct misfolded proteins which accumulate in the endoplasmic reticulum (ER) during cellular stress. Regulation of the UPR, which serves to restore normal protein processing, is mediated by members of the ER chaperone family. While a number of the ER chaperones cooperate in the stress response, the glucose regulated protein 78 (GRP78) is the most abundant and is an essential UPR regulator 5. During ER stress GRP78 promotes PERK and IRE1 activation by dissociating from these ER signaling molecules and subsequently coupling with misfolded proteins to aid in trafficking 5. While the UPR/GRP78 can be induced during normal physiologic processes, such as adipogenesis, many pathological states are associated with GRP78 overexpression and UPR activation 5, 6. Evidence demonstrates that GRP78 overexpression occurs in numerous human cancers, including breast, prostate, lung, ovarian, and colorectal carcinoma 7–11. Furthermore, GRP78 overexpression in these cancers is strongly associated with increased malignancy, poor patient outcome, and chemoresistance 12–16.
How GRP78 expression contributes to resistance in response to cytotoxic chemotherapies is not fully understood, yet reducing GRP78 expression restores sensitivity in some models 4. Recent reports suggest that in addition to its ER protein trafficking role, GRP78 also promotes cell survival by interacting with and blocking the pro-apoptotic functions of BIK and caspase-7 15, 17. Other studies show that while the majority of GRP78 exist within the ER, some GRP78 resides on the cell membrane acting as a co- receptor that regulates the MAPK and PI3K/AKT survival/proliferation pathways 4, 18–20. The ability of GRP78 to regulate AKT would appear to be important in the development of chemoresistance; studies examining the effects of cisplatin on kinase signaling in cancer cells suggest that AKT activity is critical for attenuating chemotherapy-mediated cytotoxicity 21–24. Furthermore, data suggest that the acquisition of chemoresistance by cancers, such as lung, osteosarcomas, and ovarian, results from increases in activation of the PI3K/mTOR/AKT pathway 25–27. This may have particular importance in endometrial cancers where loss of PTEN activity with resultant constitutive activation of the PI3K/mTOR/AKT pathways, has been reported to occur in upwards of 60% of patient tumors 28–30.
To better understand the role of GRP78 in chemotherapeutic resistance in endometrial cancers, we analyzed its expression in patient tumor samples. Immunohistochemical analysis showed that GRP78 overexpression occurs more frequently in malignant tissues compared to that in normal endometrium. In vitro examination demonstrated that EC cell lines grown under normal conditions have differential expression of GRP78. Treatment of EC cell lines with cisplatin is capable of inducing GRP78 expression, and loss of GRP78 significantly augments cisplatin-mediated cytotoxicity by enhancing the cleavage of apoptotic markers, Poly (ADP-ribose) polymerase (PARP) and caspase-3. Examination of AKT and MAPK activity revealed that only AKT phosphorylation changed with cisplatin treatment, and preceded GRP78-induction by 24h. Use of the small molecule pan-AKT inhibitor MK2206, which reduced AKT activity in all lines tested, blocked constitutive GRP78 expression and cisplatin-mediated induction of GRP78. Further examination also demonstrated that GRP78 knockdown by siRNA reduced AKT activity. This suggests that in EC cells, GRP78 may be both an upstream- and downstream regulator of AKT, whereby GRP78 expression is dependent upon AKT activity, and that GRP78 expression in turn further enhance AKT activity. Studies with MK2206 showed that at concentrations where AKT activity was blocked, there was no additive cytotoxic effects in response to cisplatin with GRP78 knockdown compared to controls, suggesting that GRP78-mediated survival functions are part of the AKT cascade. Collectively our data highlight the importance of GRP78 in the response of EC cells to chemotherapeutic treatments. Whether directly targeting GRP78 itself, or blocking AKT activity and reduction of GRP78 expression proves to have the greatest impact potentiating current EC treatments merits further study.
Materials and Methods
Clinical endometrial tissue specimens, histologic preparation, and examination
Formalin-fixed, paraffin-embedded endometrial tissue micro arrays (TMAs # EMC1501, #EMC1502) consisting of normal endometrium and endometrioid adenocarcinomas samples were purchased from Pantomics (Richmond, California, www.pantomics). TMAs were deparaffinized and rehydrated in declining grades of ethanol. Antigen retrieval was performed using the Retrievagen A kit (BD Pharmingen, Carpenteria, CA) in a pressure cooker. Endogenous peroxidases were blocked with 3% H2O2-PBS (Thermo Fisher Scientific, Pittsburg, PA) for 12 min at room temperature (RT). Non-specific epitopes were blocked with 5% normal horse serum for 20 min at RT. Sections were incubated with primary antibodies (GRP78, 1:200, ab21685, Abcam, Cambridge, MA) at 4°C overnight with appropriate negative controls. Vectastain Elite avidin-biotin complex (ABC) kit (Vector Laboratories, Burlingame, CA) was used with the species-specific secondary antibodies (45 min) per manufacturer instructions followed by 3,3′-diaminobenzidine (DAB, Phoenix Biotechnologies, Huntsville, AL) and counterstaining with Gill’s No. 3 hematoxylin (Sigma-Aldrich, St. Louis, MO). GRP78 overexpression was evaluated in all samples by a board-certified gynecologic pathologist (PMF) and scored using a semi-quantitative scale 13.
Cell lines and culture conditions
All human endometrial cancer cell lines were a generous gift from Dr. Gottfried Konecny (UCLA). AN3CA, ECC-1, Ishikawa, and MFE-296, were maintained in Dulbecco essential media supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, penicillin, and streptomycin (Life Technologies, Grand Island, NY). ARK1, Hec-1A, Hec-1B, SMG-11, Spec-L cells were grown in RPMI supplemented with 10% FBS, 2 mM L-glutamine, penicillin, and streptomycin. All cell lines were authenticated by mitrochondial sequencing (7.1.11) and frozen stocks created for each verified cell lines. All cell lines discarded after an additional 20–25 passages and replenished with the initial verified frozen stock.
Transient and Lentiviral siRNA knockdowns
The siRNA sequences used for transient transfections were previously described 31: siGRP78 is 5′-ggagcgcauugauacuagatt-3′ and siCon is 5′-aaggagacguauagcaacggu-3′. Transfection was performed using the INTERFERin siRNA transfection reagent (Polyplus, New York, NY USA) according to the manufacturer’s protocol. Briefly, 1×106 cells were plated per well in a 6-well plate. The next day, 25 ng of siRNA oligo’s were mixed with 10uL of INTERFERin reagent in 200uL of serum free media, incubated for 15 minutes and added to cells. Media was replaced 24h later, and cells were treated with cisplatin and/or MK2206 (Sellick chemicals, Boston, MA) as detailed in the methods. For all EC50 analyses siRNA lentiviruses for GRP78 and control were utilized (Santa Cruz Biotechnology, siGRP78 lenti: sc-44261-V, control; siCon sc-108080) due to the length of experiments and the need for extended knock down. For lentiviral transfections, 0.5×105 cells were plated per well in a 12-well plate, and 24h later, cells were pretreated with 1 mL of media with Polybrene (5ug/mL). Following a 5-minute incubation, cells were infected with the lentiviral constructs (MOI=5). Fresh media was placed on each infected well 24h later, and after an additional 48h incubation, cells were trypsinized and utilized for calculation of EC50 studies.
Quantitative Western analysis
Cells were solubilized in 20 mM Tris-Cl (pH 8.0), 137 mM NaCl, 1% Triton X-100, 1 mM Na3VO4, and 2 mM EDTA with protease inhibitor (Roche Diagnostics). Cell lysates were separated by SDS/Polyacrylamide gel electrophoresis on 4–20% pre-cast NuPage Tris-Glycine gradient gels (Invitrogen) and then transferred to PVDF membranes. Membranes were blocked with Odyssey blocking buffer (Li-COR) and incubated with primary antibodies. All antibodies, unless otherwise noted were used at 1:1000 dilution. Membranes were probed with antibodies against GRP78, pAKT, AKT, poly (ADP-ribose) polymerase (PARP), pERK1/2, caspase-3, and caspase-9 (all from Cell Signaling Technology, Danvers, MA), and vinculin (1:10,000, Serotec, Raleigh, NC). Odyssey secondary antibodies Goat anti-rabbit IRDye 680 and Goat anti-mouse IRDye 800) were added at 1:2000 according to manufacturer’s instructions (Blots were imaged using an Odyssey Infrared Imaging System (Li-COR Biosciences). Scans were imaged at 169μm. Quantification was performed on single channels with the analysis software provided.
MTT analysis of cell proliferation and EC50 determination
In vitro proliferation was assessed with tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as described previously. Briefly, 1,000 cells of each cell type (transfected or parental) were plated per well onto 96-well microtiter plates in minimum essential medium (MEM) with 10% FBS. One platewas developed immediately after cells had adhered (at approximately 4 h), and other plates were developed every 24 hours for 5 days. For EC50 calculations, 1,000 cells were plated per well onto 96-well microtiter plates in MEM with 10% FBS. Cells were treated 24h later and allowed to grow for 5 additional days. Cisplatin treatment was performed with a 9-point 3-fold dilution (100μM – 0.046uM) dose, with untreated as a control. Assays were done by incubating each plate with 20 μL of MTT substrate for 2 h followed by removal of medium and addition of 200 μL of dimethylsulfoxide. Plates were read at a wavelength of 570 nm. EC50s were calculated using Graphpad Prism 5.0 non-linear regression analysis.
Statistical analyses
Statistical analyses were performed with Graphpad Prism 5.0 software (GraphPad Software, San Diego, CA), with Student’s t tests or Fisher’s exact tests as appropriate. Significance (P < 0.05) was determined with 95% confidence intervals.
Results
GRP78 is elevated in EC patient samples and is present in most EC cell lines
Immunohistochemical analysis of GRP78 overexpression in clinical samples revealed that GRP78 overexpression was detectable in a significantly greater number of malignant tissue samples compared to normal uninvolved endometrium tissue samples. In 30% (9 of 30) of normal endometrium GRP78 overexpression was detected compared to 73.8% (192 of 260) of the malignant samples examined (p= 0.0235). Analysis of basal protein levels in nine EC cell lines demonstrated GRP78 expression occurred at some level during non-stressed growth conditions. Cell lines with minimal GRP78 expression included ECC-1, HEC-1A, and Ishikawa. In comparison, the EC cell lines AN3CA, ARK1, and SMG-11 had elevated basal levels of GRP78 (Fig. 1B).
Figure 1. Assessment of GRP78 expression in human endometrial tissues and cell lines.

A, GRP78 patient tissue analysis. Graphical representation of normal endometrium and EC tissues samples strained for GRP78 expression. Samples where strong GRP78 detection occurred were considered positive. B, Western blot analysis of GRP78 and pTEN expression in nine endometrial cancer cell lines. Vinculin was used as an internal loading control. C, Western blot time course analysis of the ability of cisplatin (1 uM) to induce GRP78 expression in the EC cell lines Ishikawa, ECC-1, AN3PCA, and ARK1. Vinculin was used as an internal loading control. Thapsigargin, a known GRP78 inducer was used as a positive control (TG).
Examination of the cell lines for the presence of the PTEN protein demonstrated that GRP78 expression does not appear to correlate with PTEN expression in EC cells. High GRP78 expression occurred in the PTEN positive lines SMG-11 and MFE-296, as well as in the PTEN-deficient lines AN3CA and SPEC-1S (Fig. 1B).
Cisplatin induces GRP78 expression in EC cells
To determine if cisplatin treatment is capable of inducing GRP78, EC cells were treated with cisplatin (1 μM) for 24h, 48h, and 72h and analyzed by Western blot. As shown in Figure 1C, cells with minimal basal levels (Ishikawa, ECC-1) showed an increase in GRP78 protein levels after 48h of cisplatin treatment that was comparable to that seen after thapsigargin (Tg) treatment. In cell lines that expressed high basal levels of GRP78 (AP3CA and ARK1), treatment by cisplatin (or thapsigargin) only modestly induced GRP78 in the ARK1 cell line above levels observed prior to chemotherapy treatment (Fig. 1C). This data demonstrates that cisplatin is capable of inducing GRP78 in endometrial cancer cells.
GRP78 knockdown augments cisplatin mediated toxicity and apoptosis in endometrial cancer cells
While cisplatin can induce GRP78 expression in EC cells, we sought to determine if GRP78 expression enhances survival in response to cisplatin treatment. Using siRNA targeting, GRP78 levels were reduced by greater than 80% in Ishikawa (Fig. 2A, left panel) and AN3CA cells (Fig 2A, right panel) compared to parental or siRNA controls. Comparison of the effects of the knockdown on each cell line showed that loss of GRP78 augmented the cytotoxicity mediated by cisplatin chemotherapy (Fig. 2B, left panel). In Ishikawa cells, where GRP78 was induced by cisplatin treatment, knockdown of GRP78 decreased the EC50 of cisplatin by greater than 5-fold (1.56 μM to 0. 26μM, p=0.029) (Fig. 2B, left panel, Table 1). In AN3CA cells, which have high basal levels of GRP78, knockdown of GRP78 caused a similar reduction of the cisplatin EC50, from 1.05 μM to 0.20 μM (p=0.038) (Fig. 2B, right panel, Table 1).
Figure 2. Effect GRP78 knockdown on cisplatin mediated cytotoxicity and apoptosis in human endometrial cancer cells.

A, Western blot analysis of GRP78 levels in lentivirus infected siRNA controls (siCon) and siRNA-GRP78 knockdown cells (siGRP78) in Ishikawa (left) and AN3CA cells (right). Cells were grown for the same time length as those in EC50 studies to ensure extended knockdown. B, EC50 assay examining the effectiveness of cisplatin, cisplatin+SiCon and cisplatin + siGRP78 cytotoxicity in Ishikawa (left) and AN3CA cells (right) lentiviral infected cells. C, Western blot analysis of apoptotic regulatory proteins in Ishikawa (left) and AN3CA cells (right). Cells were transient transfected with control siRNA scrambled (siCon) or siRNA-GRP78 specific sequences (siGRP78) and levels of GRP78, PARP, cleaved PARP, Caspase-3, and cleaved caspase-3 determined after treating with cisplatin (1uM) for the times indicated. Vinculin was used as an internal loading control.
Table 1.
| Condition | Ishikawa EC50 | AN3CA EC50 |
|---|---|---|
| Cis (=Cisplatin) | 1.56 +/−0.30 uM | 1.05 +/−0.29 uM |
| Cis+siCon | 1.62 +/−0.32 uM | 1.15 +/−0.29 uM |
| Cis+siGRP78 | 0.26 +/−0.20 uM | 0.20 +/−0.15 uM |
| Cis+SiCon+MK2206 (5 nM) | 1.59 +/−0.40 uM | 0.92 +/−0.22 uM |
| Cis+SiGRP78+MK2206 (5 nM) | 0.25 +/−0.13 uM | 0.25 +/−0.05 uM |
| Cis+SiCon+MK2206 (50 nM) | 0.07 +/−.01 uM | 0.05 +/−0.01 uM |
| Cis+GRP78+MK2206 (50 nM) | 0.05 +/−0.02 uM | 0.05 +/−0.02 uM |
To ensure that the shifts in EC50’s observed with cisplatin treatment and GRP78 knockdown were a result of enhanced cell survival as opposed to changes in cell proliferation, cell doubling times were calculated in the absence of cisplatin. Reduced GRP78 levels in Ishikawa cells resulted in no significant change in proliferation times (32.5+/− 1.5h for controls vs. 33.5 +/− 2.0h for GRP78 knockdowns, p>0.05, data not shown). While a slight reduction in doubling time was observed in AN3CA cells that had reduced GRP78 protein levels, the change in proliferation rates between cells with GRP78 knockdown and controls was not significant (23.0 +/− 3.1 h vs. 26 +/− 1.8 h, p>0.05, data not shown). While these data suggest that GRP78 regulates cell survival during cisplatin exposure, it is uncertain if GRP78 regulates growth or survival mechanisms during chemotherapy treatment. To better understand the role of GRP78 in response to cisplatin, Ishikawa and AN3CA cell lines with GRP78 knockdown were treated with cisplatin and examined by western analysis. In Ishikawa cells, reduction of GRP78 levels by siRNA targeting itself did not result in detectable changes in levels of the apoptotic markers PARP and caspase-3 prior to cisplatin treatment, demonstrating that under normal growth conditions, a reduction of GRP78 levels had little effect upon apoptotic pathway activation (Fig. 2C). In response to cisplatin treatment, loss of GRP78 expression in Ishikawa cells led to a more rapid onset and increased levels of cleaved PARP. Quantitative analysis revealed that the loss of GRP78 expression results in a 80% increase in cleaved PARP by 48h after cisplatin treatment, compared to no detected cleaved PARP levels at a similar time point in cells with GRP78 (Fig. 2C, right panel). At 72h after cisplatin treatment PARP cleavage becomes detectable in control cells. However, cells with GRP78 siRNA knockdown continued to have greater levels of cleaved PARP (60% vs. 85% percent, respectively). Examination of cleaved-caspase 3 in Ishikawa cells showed a similar trend to that of PARP cleavage; loss of GRP78, when accompanied with cisplatin treatment, was associated with earlier and greater levels of cleaved caspase-3 compared cells with intact GRP78 protein (Fig 2C). Similar to our results in Ishikawa cells, knockdown of GRP78 in AN3CA cells resulted in changes in both PARP and caspase-3 levels in response to cisplatin (data not shown). Combined, these data demonstrate that GRP78 expression promotes cell survival in EC cells in response to chemotherapeutic agents.
GRP78 protein levels are regulated by AKT activity in endometrial cancer cells
Next, we sought to determine if AKT phosphorylation was induced by cisplatin in EC cells, as reported in other cancers, and if so, whether or not this activity was required for the changes observed in GRP78 expression 21. As shown in Fig. 3, Ishikawa and AN3CA cells were subjected to cisplatin treatment and changes in AKT activity were determined by western blot. In Ishikawa cells. AKT activity increased at 24h and 48h post-treatment following an initial decrease in AKT activity at 8h (Fig. 3A). The observed increase in AKT activity preceded the induction of GRP78 expression by 24h (Fig. 3A). To determine if the increase in AKT activity in Ishikawa cells is a prerequisite for increased GRP78 expression, we utilized the AKT-specific inhibitor, MK2206, to block AKT activation. As expected, Ishikawa cells pretreated with MK2206 (50nM) demonstrated a loss of basal phospho-AKT levels compared to control cells prior to cisplatin treatment (Fig 3B, lanes 1 and 2). In addition to the loss of phospho-AKT, however, basal levels of GRP78 were reduced greater than 85% compared to untreated cells (Fig. 3B). Cisplatin treatment, which increased GRP78 levels over 2-fold in control Ishikawa cells was unable to induce GRP78 in the setting of AKT inhibition (Fig. 3B). Similar results were observed in the ECC-1 cell line with MK2206 treatment; AKT inhibition again blocked cisplatin-mediated GRP78 induction (data not shown). In AN3CA cells, which have high basal levels of GRP78 expression cisplatin treatment only modestly increased phospho-AKT above untreated levels (1.0 vs 1.2), and appeared to cause minimal changes in GRP78 expression levels above that in untreated control cells (Fig. 3C). Treatment of AN3CA cells with MK2206 reduced both basal AKT activity and constitutive GRP78 expression prior to and with the addition of cisplatin (Fig. 3D). Similar results with MK2206-induced GRP78 reduction where observed in the ARK1 cell line, which similarly to AN3CA also expresses high basal levels of GRP78 (data not shown). To ensure that these results were not specific to MK2206, we also treated Ishikawa and AN3CA cells with the pan-AKT inhibitor GSK690693. Similar to that seen with MK2206, pharmacologic inhibition of AKT with GSK690693 abrogated basal and cisplatin-induced GRP78 expression by 78% and 98%, respectively (data not shown). Our data here demonstrate that AKT activity is required for both maintaining basal levels and for inducing GRP78 expression in response to cisplatin.
Figure 3. Requirement for AKT activity in GRP78 expression in endometrial cancer cells.
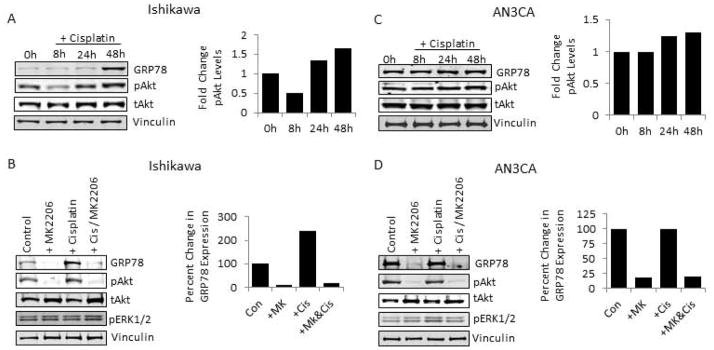
A, Western blot analysis of cisplatin (1uM) treated Ishikawa cells (left) and AN3CA cells (right) for levels of GRP78, phospho-AKT, and total AKT, and with graphical representation of image analysis of changes in pAKT activity. Vinculin was used as an internal loading control. B, Analysis of Ishikawa cells (left) and AN3CA cells (right) pretreated with or without the AKT inhibitor MK2206 (50nM) followed by cisplatin treatment (1uM). Levels of GRP78, phospho-AKT, total AKT, and phospho-ERK 1/2 were determined by western analysis. Changes in GRP78 levels accompanying AKT inhibition represented by graphical representation of image analysis for GRP78 are displayed. Vinculin was used as an internal loading control.
GRP78 knockdown reduces AKT activity in EC cells
While we demonstrated that AKT activity regulates GRP78 expression in endometrial cancer cells, other studies suggest that GRP78 regulates AKT and/or MEK/ERK 1/2 19, 31–33. To discern if GRP78 also regulates AKT expression in endometrial cancer cells, GRP78 knockdown was performed by siRNA and changes in AKT and ERK 1/2 activity were determined. In Ishikawa cells, which have low basal levels of GRP78 expression, GRP78 knockdown resulted in a modest decrease in constitutive AKT activity under normal growing conditions (Fig. 4A, lanes 1 and 5). With cisplatin treatment, control cells showed increased AKT activity by 24h, followed by increased GRP78 levels at 48h after treatment (Fig. 4A lanes 1–4). Contrary to this, Ishikawa cells with GRP78 knockdown had no increase in AKT activity at any time point during cisplatin treatment (Fig. 4A, lanes 5–8). ERK 1/2 activity remained unchanged in Ishikawa cells with GRP78 knockdown compared to controls cells prior to and during cisplatin treatment. These data suggest that GRP78 contributes to increased AKT activity in response to cisplatin, and the loss of GRP78 has no discernible effect on MEK/ERK activity in Ishikawa cells. In AN3CA cells, which have high basal levels of GRP78 expression, knockdown of GRP78 expression reduced constitutive AKT activity by 50% compared to controls with endogenous GRP78 (Fig. 4B, lanes 1 and 5). While treatment of controls cells with cisplatin increased AKT activity slightly, in knockdown cells AKT phosphorylation remained reduced compared to cell with GRP78 (Fig. 4B). ERK activity, similar to that observed in Ishikawa cells, remained unchanged with GRP78 knockdown. These data demonstrate that GRP78 contributes to, but is not a requirement, for AKT phosphorylation in EC cells.
Figure 4. Effect of GRP78 knockdown on AKT and ERK 1/2 signaling in endometrial cancer cells.

A, Western analysis of siRNA controls (siCon) and GRP78 knockdowns (siGRP78) transiently transfected as described and treated with and without cisplatin for 24, 48 and 72 hours. Ishikawa cells (upper) and AN3CA cells (lower) were probed for levels of GRP78, phospho-AKT, phospho-ERK, and total AKT. Vinculin was used as an internal loading control.
Small molecule inhibition of AKT blocks the contribution of GRP78 to cisplatin resistance
To better understand the relationship of GRP78 expression and AKT activity in mediating survival in EC cells, we treated GRP78 siRNA control or GRP78 knockdown cells with the AKT inhibitor MK2206 at different concentrations and subjected them to cisplatin treatment. As expected, at concentrations where MK2206 (5nM) did not inhibit AKT or GRP78 expression there was no difference between similar groups in the response to cisplatin with or without MK2206. All cells with intact GRP78 expression had comparable EC50s (Parental and siRNA control), whereas cells with GRP78 knockdown were more sensitive to cisplatin than cells with GRP78 as previously observed (Fig. 5B, Table 1). At concentrations where MK2206 (50nM) reduced AKT activity and the substituent loss of GRP78 was observed, both siRNA control cells and siRNA GRP78 targeted cells had comparable EC50s in response to cisplatin, suggesting that MK2206 effectively blocked GRP78 expression and its contribution to cisplatin resistance (cisplatin+siCon+MK2206 (50nM); EC50=0.07 uM+/− 0.01uM, cisplatin+siGRP78+MK2206 (50nM); 0.05uM +/− 0.02 uM, (Fig. 5B, Table 1). In AN3CA cells we found similar results, at concentrations of MK2206 (5nM) where no loss of AKT activity and GRP78 expression occurred siRNA to GRP78 effectively sensitized cells to cisplatin. However at levels where MK2206 (50nM) blocked AKT activity and GRP78 expression was reduced, both control cells and GRP78 knockdown cells had similar IC50s. Again suggesting that for in vitro models AKT inhibition can effectively block GRP78s contribution to survival in response to cisplatin in EC cell by negating its expression.
Figure 5. Effect of AKT inhibition and GRP78 knockdown on cisplatin mediated cytotoxicity.

A, Western analysis examining the dose response MK2206 on AKT and GRP78 in Ishikawa cells. B EC50 assays examining the cytotoxic effects of cisplatin on Ishikawa as described in materials and methods. Treatment groups were parental control (Cisplatin), or cisplatin with siRNA control (Cisplatin+siCon), or cisplatin+GRP78 knockdown (Cisplatin +siGRP78) with or without combination with MK2206 (5nM, 50nM). Graph is representative of one of five independent experiments. C, Western analysis examining the dose response MK2206 on AKT and GRP78 in AN3CA cells. B EC50 assays examining the cytotoxic effects of cisplatin on AN3CA as described in materials and methods. Treatment groups were parental control (Cisplatin), or cisplatin with siRNA control (Cisplatin+siCon), or cisplatin+GRP78 knockdown (Cisplatin +siGRP78) with or without combination with MK2206 (5nM, 50nM). Graph is representative of one of five independent experiments.
Discussion
Cisplatin comprises the backbone for most chemotherapy regimens for EC even though they typically only show modest anti-tumor activity in advanced stage disease 34. The development of additional targeted combination therapies that can improve the efficacy of traditional platinum-based chemotherapeutics, such as cisplatin, may prove to have the greatest immediate benefit for EC patients. While the list of potential target candidates for these therapies is expanding, members of the unfolded protein response, including GRP78, are being actively explored 5. Data supporting potential GRP78 targeting include cancers, such as melanoma, breast, prostate, and glioma, where its expression is associated with poor prognosis and disappointing responses to existing therapies, 10, 13, 35, 36. However, the extent of GRP78 expression and its contribution to disease progression and chemoresistance in EC remains unclear.
Our analysis of normal endometrium and endometrioid adenocarcinomas patient samples demonstrates that GRP78 overexpression occurs more frequently in malignant tissue than in normal endometrium (p=0.0265). Examination of GRP78 levels also demonstrates that the ER chaperone is detectable in most EC cell lines, and its expression does not appear to correlate with PTEN status. Because of the association of GRP78 with chemoresistance in other cancers we initially investigated the effects of cisplatin on EC cells. Similar to results in observed in other cancers, cisplatin treatment increases AKT activity in EC cells 37. We also show cisplatin is capable of inducing GRP78 in EC cells, suggesting that GRP78 may act in EC to counter chemotherapeutic mediated cytotoxicity through enhancing AKT activity or other mechanisms yet undefined. Indeed knockdown of GRP78 by siRNA targeting in EC cells increased cisplatin mediated apoptosis. This suggests that therapeutic blockade of GRP78 function may have a clinical impact for the treatment of EC 38. This has been further postulated following the identification of cell membrane-associated GRP78, which reportedly acts as a multifunctional co-receptor that promotes PI3K/AKT activity in some cancer cells 19, 39, 40. Treatment of prostate cancer cells with anti-GRP78 antibodies, which targets the fraction of membrane-associated GRP78, has inhibitory effects on cell survival 19, 41, 42. While these results are encouraging, auto-GRP78 antibodies from prostate cancer patients block cancer cell apoptosis, suggesting that distinct domains on membrane-associated GRP78 elicit dramatically different responses 43. Since the majority of GRP78 exists in the ER, therapies that readily cross the plasma membrane and block GRP78’s ER-functions directly, or regulatory pathways that control global GRP78 expression, may represent more potent therapeutic strategies 44.
As we have shown, AKT is critical for inducing and maintaining GRP78 expression in EC cells. Blocking AKT activity with a small molecule pan-AKT inhibitor (MK2206) prevented cisplatin-mediated GRP78 induction and reduced constitutive expression of the ER chaperone. Furthermore, treatment of EC cells with MK2206 at concentrations where AKT activity was inhibited, established that GRP78 contribution to cisplatin mediated resistance could effectively be negated. Work in other models also suggest a dependence upon AKT activity for GRP78 regulation; in HEK293 cells PI3K/AKT activity is necessary for GRP78 induction in response thapsigargin, and in murine embryonic fibroblast IGF mediated induction of GRP78 also requires PI3K/AKT activity 45, 46. This suggests that GRP78 is a downstream target/regulator of the AKT signaling network, and AKT may be one of primary regulators of GRP78 expression. If true, the association of GRP78 with other pathological conditions may suggest that interrupting the PI3K/AKT signaling axis may extend beyond cancer specific treatments.
In addition we also demonstrate not only is GRP78 regulated by AKT, but we also show that GRP78 expression promotes AKT activity and is required to enhance AKT activity in response to cisplatin. Other studies examining the relationship of GRP78 and AKT activation have suggested that AKT activity is mediated in large by GRP78 31–33. We show, however, that while GRP78 expression contributes to AKT activity, loss of GRP78 expression by siRNA targeting does not completely abrogate AKT activity. Therefore, we propose that in addition to being a downstream target of AKT, GRP78 expression contributes to and enhances AKT activity in EC cells. Since AKT is a central survival regulator, the ability of GRP78 to maintain AKT activity and promote survival during times of stress, such as those encountered during chemotherapy treatment, serves to enhance drug resistance. It is generally accepted that AKT activity contributes to drug resistance 47, 48. Given the fact that AKT activity also controls GRP78 levels, this regulatory feedback loop would logically in turn serve to maintain GRP78 expression, increase AKT function, and sustain GRP78-mediated survival functions, possibly by blocking the pro-apoptotic functions of BIK and caspase-7 15, 17.
In summary, our study here demonstrates that GRP78 is an important mediator in EC cell survival and may represent a promising therapeutic target in tumors where its expression occurs. We show for the first time that GRP78 expression is associated with malignant disease in EC patients. Furthermore our results demonstrate that GRP78 contributes to cisplatin resistance, a critical aspect of EC progression. The development of therapies to block GRP78 function(s), either directly or indirectly, offers the potential to substantially improve existing treatments and provide a unique opportunity to circumvent the ability of GRP78-expressing tumors to overcome current therapies.
Significance and novelty.
We show for the first time that GRP78 expression is associated with malignant disease and chemoresistance in endometrial cancers (EC). Our data demonstrate that cisplatin induces AKT activity and subsequent GRP78 expression. Inhibition of AKT activity blocks GRP78 expression, showing that GRP78 expression is AKT-dependent. Furthermore reduction of GRP78 levels by siRNA-targeting augments cisplatin-mediated cytotoxicity. Targeted therapies that reduce GRP78 expression or activity may increase the effectiveness of therapies in EC.
Acknowledgments
The project described was supported in part by award number P30CA014089 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. YGL supported in part by the Stop Cancer Research Career Development Award. ASL supported by National Cancer Institute grant CA027607.
Footnotes
Conflict of Interest Disclosure: The authors listed above have no commercial or financial relationships relevant to the subject of this manuscript
References
- 1.Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, Lin C, Leach C, et al. Cancer treatment and survivorship statistics. CA: A Cancer Journal for Clinicians. 2012 doi: 10.3322/caac.21149. n/a-n/a. [DOI] [PubMed] [Google Scholar]
- 2.Wright JD, Barrena Medel NI, Sehouli J, Fujiwara K, Herzog TJ. Contemporary management of endometrial cancer. Lancet. 2012;379:1352–60. doi: 10.1016/S0140-6736(12)60442-5. [DOI] [PubMed] [Google Scholar]
- 3.Dedes KJ, Wetterskog D, Ashworth A, Kaye SB, Reis-Filho JS. Emerging therapeutic targets in endometrial cancer. Nat Rev Clin Oncol. 2011;8:261–71. doi: 10.1038/nrclinonc.2010.216. [DOI] [PubMed] [Google Scholar]
- 4.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2012 doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfaffenbach KT, Lee AS. The critical role of GRP78 in physiologic and pathologic stress. Curr Opin Cell Biol. 2011;23:150–6. doi: 10.1016/j.ceb.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sha H, He Y, Chen H, Wang C, Zenno A, Shi H, Yang X, Zhang X, Qi L. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 2009;9:556–64. doi: 10.1016/j.cmet.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xing X, Li Y, Liu H, Wang L, Sun L. Glucose regulated protein 78 (GRP78) is overexpressed in colorectal carcinoma and regulates colorectal carcinoma cell growth and apoptosis. Acta Histochem. 2011;113:777–82. doi: 10.1016/j.acthis.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 8.Uramoto H, Sugio K, Oyama T, Nakata S, Ono K, Yoshimastu T, Morita M, Yasumoto K. Expression of endoplasmic reticulum molecular chaperone Grp78 in human lung cancer and its clinical significance. Lung Cancer. 2005;49:55–62. doi: 10.1016/j.lungcan.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez PM, Tabbara SO, Jacobs LK, Manning FC, Tsangaris TN, Schwartz AM, Kennedy KA, Patierno SR. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res Treat. 2000;59:15–26. doi: 10.1023/a:1006332011207. [DOI] [PubMed] [Google Scholar]
- 10.Pootrakul L, Datar RH, Shi SR, Cai J, Hawes D, Groshen SG, Lee AS, Cote RJ. Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin Cancer Res. 2006;12:5987–93. doi: 10.1158/1078-0432.CCR-06-0133. [DOI] [PubMed] [Google Scholar]
- 11.Delie F, Petignat P, Cohen M. GRP78 Protein Expression in Ovarian Cancer Patients and Perspectives for a Drug-Targeting Approach. J Oncol. 2012;2012:468615. doi: 10.1155/2012/468615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang LW, Lin CY, Lee CC, Liu TZ, Jeng CJ. Overexpression of GRP78 Is Associated with Malignant Transformation in Epithelial Ovarian Tumors. Appl Immunohistochem Mol Morphol. 2012 doi: 10.1097/PAI.0b013e3182434113. [DOI] [PubMed] [Google Scholar]
- 13.Lee E, Nichols P, Spicer D, Groshen S, Yu MC, Lee AS. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006;66:7849–53. doi: 10.1158/0008-5472.CAN-06-1660. [DOI] [PubMed] [Google Scholar]
- 14.Chen TC. GRP78/BiP modulation of GRP78/BiP in altering sensitivity to chemotherapy. Methods Enzymol. 2011;491:25–36. doi: 10.1016/B978-0-12-385928-0.00002-X. [DOI] [PubMed] [Google Scholar]
- 15.Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007;67:3734–40. doi: 10.1158/0008-5472.CAN-06-4594. [DOI] [PubMed] [Google Scholar]
- 16.Mann MJ, Hendershot LM. UPR activation alters chemosensitivity of tumor cells. Cancer Biol Ther. 2006;5:736–40. doi: 10.4161/cbt.5.7.2969. [DOI] [PubMed] [Google Scholar]
- 17.Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–24. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 18.Lin Y, Wang Z, Liu L, Chen L. Akt is the downstream target of GRP78 in mediating cisplatin resistance in ER stress-tolerant human lung cancer cells. Lung Cancer. 2011;71:291–7. doi: 10.1016/j.lungcan.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Kelber JA, Panopoulos AD, Shani G, Booker EC, Belmonte JC, Vale WW, Gray PC. Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways. Oncogene. 2009;28:2324–36. doi: 10.1038/onc.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434:181–8. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belyanskaya LL, Hopkins-Donaldson S, Kurtz S, Simoes-Wust AP, Yousefi S, Simon HU, Stahel R, Zangemeister-Wittke U. Cisplatin activates Akt in small cell lung cancer cells and attenuates apoptosis by survivin upregulation. Int J Cancer. 2005;117:755–63. doi: 10.1002/ijc.21242. [DOI] [PubMed] [Google Scholar]
- 22.Peng DJ, Wang J, Zhou JY, Wu GS. Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells. Biochem Biophys Res Commun. 2010;394:600–5. doi: 10.1016/j.bbrc.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayakawa J, Ohmichi M, Kurachi H, Kanda Y, Hisamoto K, Nishio Y, Adachi K, Tasaka K, Kanzaki T, Murata Y. Inhibition of BAD phosphorylation either at serine 112 via extracellular signal-regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res. 2000;60:5988–94. [PubMed] [Google Scholar]
- 24.Hoekstra AV, Ward EC, Hardt JL, Lurain JR, Singh DK, Buttin BM, Schink JC, Kim JJ. Chemosensitization of endometrial cancer cells through AKT inhibition involves FOXO1. Gynecol Oncol. 2008;108:609–18. doi: 10.1016/j.ygyno.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 25.Fraser M, Bai T, Tsang BK. Akt promotes cisplatin resistance in human ovarian cancer cells through inhibition of p53 phosphorylation and nuclear function. Int J Cancer. 2008;122:534–46. doi: 10.1002/ijc.23086. [DOI] [PubMed] [Google Scholar]
- 26.Gagnon V, Mathieu I, Sexton E, Leblanc K, Asselin E. AKT involvement in cisplatin chemoresistance of human uterine cancer cells. Gynecol Oncol. 2004;94:785–95. doi: 10.1016/j.ygyno.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 27.Abedini MR, Muller EJ, Bergeron R, Gray DA, Tsang BK. Akt promotes chemoresistance in human ovarian cancer cells by modulating cisplatin-induced, p53-dependent ubiquitination of FLICE-like inhibitory protein. Oncogene. 2010;29:11–25. doi: 10.1038/onc.2009.300. [DOI] [PubMed] [Google Scholar]
- 28.Kanamori Y, Kigawa J, Itamochi H, Shimada M, Takahashi M, Kamazawa S, Sato S, Akeshima R, Terakawa N. Correlation between loss of PTEN expression and Akt phosphorylation in endometrial carcinoma. Clin Cancer Res. 2001;7:892–5. [PubMed] [Google Scholar]
- 29.St-Germain ME, Gagnon V, Mathieu I, Parent S, Asselin E. Akt regulates COX-2 mRNA and protein expression in mutated-PTEN human endometrial cancer cells. Int J Oncol. 2004;24:1311–24. [PubMed] [Google Scholar]
- 30.Gagnon V, St-Germain ME, Parent S, Asselin E. Akt activity in endometrial cancer cells: regulation of cell survival through cIAP-1. Int J Oncol. 2003;23:803–10. [PubMed] [Google Scholar]
- 31.Wey S, Luo B, Tseng CC, Ni M, Zhou H, Fu Y, Bhojwani D, Carroll WL, Lee AS. Inducible knockout of GRP78/BiP in the hematopoietic system suppresses Pten-null leukemogenesis and AKT oncogenic signaling. Blood. 2012;119:817–25. doi: 10.1182/blood-2011-06-357384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P, Lee AS. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci U S A. 2008;105:19444–9. doi: 10.1073/pnas.0807691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Misra UK, Pizzo SV. Ligation of cell surface GRP78 with antibody directed against the COOH-terminal domain of GRP78 suppresses Ras/MAPK and PI 3-kinase/AKT signaling while promoting caspase activation in human prostate cancer cells. Cancer Biol Ther. 2010;9:142–52. doi: 10.4161/cbt.9.2.10422. [DOI] [PubMed] [Google Scholar]
- 34.Moxley KM, McMeekin DS. Endometrial carcinoma: a review of chemotherapy, drug resistance, and the search for new agents. Oncologist. 2010;15:1026–33. doi: 10.1634/theoncologist.2010-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pyrko P, Schonthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67:9809–16. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- 36.Martin S, Hill DS, Paton JC, Paton AW, Birch-Machin MA, Lovat PE, Redfern CP. Targeting GRP78 to enhance melanoma cell death. Pigment Cell Melanoma Res. 2010;23:675–82. doi: 10.1111/j.1755-148X.2010.00731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem. 2003;278:9100–6. doi: 10.1074/jbc.M210284200. [DOI] [PubMed] [Google Scholar]
- 38.Jiang L, Zhang L, Wang Q, Wang S. Down-regulation of GRP78 Enhances Chemotherapy Sensitivity to VP-16 in Lung Adenocarcinoma. Zhongguo Fei Ai Za Zhi. 2009;12:1159–63. doi: 10.3779/j.issn.1009-3419.2009.11.06. [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez-Gronow M, Selim MA, Papalas J, Pizzo SV. GRP78: a multifunctional receptor on the cell surface. Antioxid Redox Signal. 2009;11:2299–306. doi: 10.1089/ARS.2009.2568. [DOI] [PubMed] [Google Scholar]
- 40.Sato M, Yao VJ, Arap W, Pasqualini R. GRP78 signaling hub a receptor for targeted tumor therapy. Adv Genet. 2010;69:97–114. doi: 10.1016/S0065-2660(10)69006-2. [DOI] [PubMed] [Google Scholar]
- 41.Misra UK, Payne S, Pizzo SV. Ligation of prostate cancer cell surface GRP78 activates a proproliferative and antiapoptotic feedback loop: a role for secreted prostate-specific antigen. J Biol Chem. 2011;286:1248–59. doi: 10.1074/jbc.M110.129767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Misra UK, Mowery Y, Kaczowka S, Pizzo SV. Ligation of cancer cell surface GRP78 with antibodies directed against its COOH-terminal domain up-regulates p53 activity and promotes apoptosis. Mol Cancer Ther. 2009;8:1350–62. doi: 10.1158/1535-7163.MCT-08-0990. [DOI] [PubMed] [Google Scholar]
- 43.Gonzalez-Gronow M, Cuchacovich M, Llanos C, Urzua C, Gawdi G, Pizzo SV. Prostate cancer cell proliferation in vitro is modulated by antibodies against glucose-regulated protein 78 isolated from patient serum. Cancer Res. 2006;66:11424–31. doi: 10.1158/0008-5472.CAN-06-1721. [DOI] [PubMed] [Google Scholar]
- 44.Dong D, Stapleton C, Luo B, Xiong S, Ye W, Zhang Y, Jhaveri N, Zhu G, Ye R, Liu Z, Bruhn KW, Craft N, et al. A critical role for GRP78/BiP in the tumor microenvironment for neovascularization during tumor growth and metastasis. Cancer Res. 2011;71:2848–57. doi: 10.1158/0008-5472.CAN-10-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai RY, Chen SK, Yan DM, Chen R, Lui YP, Duan CY, Li J, He T, Li H. PI3K/Akt promotes GRP78 accumulation and inhibits endoplasmic reticulum stress-induced apoptosis in HEK293 cells. Folia Biol (Praha) 2010;56:37–46. doi: 10.14712/fb2010056020037. [DOI] [PubMed] [Google Scholar]
- 46.Pfaffenbach KT, Pong M, Morgan TE, Wang H, Ott K, Zhou B, Longo VD, Lee AS. GRP78/BiP is a novel downstream target of IGF-1 receptor mediated signaling. J Cell Physiol. 2012 doi: 10.1002/jcp.24090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, Lowe SW. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–7. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 48.Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, Basecke J, Stivala F, Donia M, Fagone P, Malaponte G, Mazzarino MC, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011;2:135–64. doi: 10.18632/oncotarget.240. [DOI] [PMC free article] [PubMed] [Google Scholar]