

Abstract
Influenza A virus (IAV) is a major respiratory pathogen of both humans and animals. The lung is protected from pathogens by alveolar epithelial cells, tissue resident alveolar macrophages, dendritic cells, and mast cells. The role of alveolar epithelial cells, endothelial cells, and alveolar macrophages during IAV infection has been previously studied. Here we address the role of mast cells during IAV infection. Respiratory infection with A/WSN/33 causes significant disease and immunopathology in C57BL/6 mice, but not B6.Cg-KitW-sh mice that lack mast cells. During in vitro co-culture, A/WSN/33 caused mast cells to release histamine, secrete cytokines and chemokines, and produce leukotrienes. Moreover, when mast cells were infected with IAV, the virus did not replicate within mast cells. Importantly, human H1N1, H3N2, and influenza B virus isolates could also activate mast cells in vitro. Mast cell production of cytokines and chemokines occurs in a RIG-I/MAVS-dependent mechanism; in contrast, histamine production occurred through a RIG-I/MAVS-independent mechanism. Our data highlight that following IAV infection the response of mast cells is controlled by multiple receptors. In conclusion, we have identified a unique inflammatory cascade activated during IAV infection that could potentially be targeted to limit morbidity following IAV infection.
Introduction
Influenza A virus (IAV)2 is one of the most common respiratory infections in humans. IAV can cause a range of disease courses from asymptomatic or symptomatic seasonal outbreaks to severe forms of respiratory infection including acute respiratory distress and acute lung injury, which are observed during pandemic outbreaks. Typically seasonal IAV infections cause limited morbidity and mortality associated with specific patient populations. However, severe IAV infection such as with the avian H5N1 isolates and 1918 Spanish flu isolate causes pathological changes to the lung architecture (1). Recent evidence indicates that highly pathogenic strains of IAV lead to an uncontrolled inflammatory response characterized by excessive lung infiltration of macrophages and neutrophils and a dramatic ‘cytokine storm’, which participates in causing excessive lung damage (2–5). Dampening the ‘cytokine storm’ response can significantly enhance the survival of mice during IAV infection (6). Thus it is imperative that we understand the mechanisms of the early events triggered by IAV infection which result in inflammatory cell infiltration and initiation of the ‘cytokine storm’.
The initial lines of defense in the respiratory tract include alveolar epithelial cells, endothelial cells, tissue resident alveolar macrophages, dendritic cells, and mast cells. However, the role of mast cells during respiratory infections is an understudied area (7). Mast cells are tissue sentinel cells of hematopoietic origins found in most vasculature tissue, but are enriched tissues that are at environmental interfaces such as skin, gastrointestinal tract, urinary tract, and lungs (8). This localization is significant because mast cells are poised to play an important role in the early immunosurveillence for pathogens. Mast cells express numerous molecules which can recognize pathogens including CD48, FcR, complement receptors, TLR, Retinoic acid inducible gene I (RIG-I)-like receptors (RLR), Nod-like receptors (NLR), and C-type lectin receptors (CLR) (9). After activation the mast cell response can be separated into two distinct phases: 1) immediate degranulation and secretion of stored mediators and 2) delayed secretion of de novo synthesized mediators. The immediate response is characterized by release of histamine, serotonin, tryptases, chymases, and TNFα, while the delayed response includes secretion of leukotrienes, prostaglandins, cytokines, chemokines, and growth factors (10). Interestingly, mast cells do not respond uniformly to all stimuli. Stimulation of TLR4 by LPS causes mast cells to facilitate a strong inflammatory cytokine response, but not degranulation; in contrast, TLR2 activation results in both inflammatory cytokine release and degranulation by mast cells (11). Thus, the mast cell response is extremely adaptable, which enables them to have dramatic effects on the composition and regulation of subsequent inflammatory responses.
It is well documented that mast cells play a crucial role in immunity against certain parasitic and bacterial infections (reviewed in (8, 9, 12)). More recently, the role of mast cells during viral infections has been explored. In vitro, mast cells have been shown to be capable of responding to vesicular stomatitis virus, Sendai virus, Hantavirus, dengue virus, and reovirus (13–17). However, there is a limited understanding about the in vivo relevance of mast cells during viral infections. In a peritonitis model of Newcastle disease virus infection, mast cells were shown to be important in inflammatory cell infiltration in a TLR3-dependent manner (18). During cutaneous dengue virus infection mast cells have been shown to play an important role in immunosurveillance through RIG-I and Mda5-dependent recognition of the virus (19, 20). In humans, dengue shock syndrome has recently been associated with elevated serum levels of mast cell-derived VEGF and proteases (21). Additionally, mast cells have been shown to play a protective role during skin vaccinia virus infection (22). However, the in vivo relevance of mast cells during respiratory virus infections remains understudied. IAV has been shown to enhance IgE-mediated histamine release from basophilic leukocytes, but IAV alone caused minimal histamine release (23). Moreover, IAV infections can sensitize mice leading to flu-specific cutaneous anaphylaxis (24). Together these data demonstrate that IAV infection can have effects on mast cells, but whether mast cells are important in the inflammatory response to respiratory IAV infection remains unresolved.
Here we specifically demonstrate that mast cells play an important role in the pathological response during A/WSN/33 infection of mice. Importantly, mast cell activation was also observed with human influenza virus isolates from the H1N1 IAV, H3N2 IAV, and influenza B virus families. The ability of IAV isolates to activate mast cells correlated with their ability to infect those cells in vitro. Interestingly, upon infection cytokine and chemokine production by mast cells were entirely dependent on RIG-I signaling, while mast cell degranulation occurred even in the absence of RIG-I signaling. Thus, we demonstrate that mast cells can play a central role in the inflammatory pathology induced by IAV infection.
Materials and Methods
Viral strains
Allantoic fluid containing the A/Puerto Rico/8/34 virus was purchased from Charles River. Allantoic fluid containing the A/WSN/33 virus was originally obtained from Dr. David Topham (University of Rochester) and was subsequently grown in embryonated chicken eggs. Human influenza virus isolates used in Figure 5 were obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH.
Figure 5. Human influenza virus isolates can activate mast cells.

BMCMC were derived by culturing total bone marrow with IL-3 for 5 weeks, supplementing with stem cell factor for the last 2 weeks. 2.5×105 FcRε1+ and CD117+ mast cells were treated with media or 50μL of each virus for 6 hours at which time supernatants were collected and analyzed for cytokine and chemokine analysis with Milliplex™ multiplex analysis (A–D). Expression of IL-6 is shown for each virus: Mouse adapted strains of IAV H1N1: A/WSN/33 and A/PR/8/34 (EID50/ml log10=8.8); Human derived isolates of IAV H1N1 which are either seasonal or pandemic viruses: A/Solomon Island/03/2006 (EID50/ml log10=7.2), A/FtMonmouth/1/47 (EID50/ml log10=7.4), A/CA/04/2009 (EID50/ml log10=6.9), A/CA/07/2009 (EID50/ml log10=8.4), A/Netherlands/2290/2009 (EID50/ml log10=7.4), and A/WI/629-D02452/2009 (EID50/ml log10=7.2); Human derived isolates of IAV H3N2: A/Brisbane/10/2007 (EID50/ml log10=8.4), A/Sydney/5/97 (EID50/ml log10=7.9), and A/Hong Kong/8/68 (EID50/ml log10=8.4); and Human derived isolates of influenza B virus B/FL/4/2006 (EID50/ml log10=8.4). Similar results were observed with CCL2 and CCL4 (data not shown). Data are representative of two independent experiments. All IAV isolates, except A/PR/8/34, induced an IL-6 response which was statistically significantly different than the medium control, as determined by a one-way ANOVA (p<0.05).
Mouse strains and infectious protocol
B6.Cg-kitW-sh mice were original purchased from Jackson Laboratories and subsequently bred in house. C57BL/6J mice were bred in house. Specific knock-out bone marrow was kindly provided by multiple investigators: RIG-I by Dr. Michael Gale (University of Washington), MAVS by Dr. Mathias Schnell (Thomas Jefferson University), MYPS/STING by Dr. John Cambier (National Jewish Health), CARD9 by Dr. Tobias Hohl (Fred Hutchinson Cancer Center), and STAT6 by Dr. Daniel Campbell (Benoyra Institute).
Mice were intranasally infected with 1500 plaque forming units (PFU) of A/PR/8/34 or A/WSN/33 under 2,2,2-tribromoethanol (Avertin) anesthesia. At the indicated times after IAV infection, mice were given a lethal overdose of pentobarbital. Broncheoalveolar lavage fluid (BALF) was collected by washing the lungs with 2 ml of PBS containing 50 mM EDTA. Lungs were saved for viral titers and stored at −80°C. BALF was spun down, cells were analyzed by cytospins, and BALF supernatant was analyzed using a lactate dehydrogenase kit (Promega) and BCA assay (Thermo Scientific). For weight loss studies, mice were infected as previously stated and weighed daily. All animal protocols were approved by the Montana State University Institutional Animal Care and Use Committee.
IAV plaque assay
IAV viral titers in the lungs were quantified using a standard plaque assay. Briefly, lungs were homogenized in 2 ml of medium using a dounce homogenizer. Next, 10-fold serial dilutions of the lung homogenates were plated in duplicate on MDCK cells (ATCC) in a 6-well plate (dilutions 10−1–10−6). Virus was left to adhere to the cells for 1 h at 35°C, tipping every 10 minutes. Cells were overlayed with 2 ml of 0.8% SeaKem® agarose (Lonza) in DMEM containing 0.5 μg TPCK trypsin (Worthington) and 0.2% bovine serum albumin (Millipore). Plates were incubated at 35°C for 3 days afterwhich plates were fixed overnight by adding 3 ml of 1:1 methanol:acetone. After fixation, monolayers were stained with 0.1% crystal violet.
Histological analysis of lungs
Lungs were inflated with 10% buffered formalin phosphate, placed in the same solution for at least 24 hours, then paraffin embedded. 5μm sections were then stained with hematoxylin and eosin (H&E) to assess lung inflammatory cell infiltration. H&E stained lungs were observed on an upright 80i eclipse microscope (Nikon) and images were captured with a DS Ri1 color camera (Nikon).
Growth of bone marrow cultured mast cells (BMCMC) and generation of mast cell knock-in mice
BMCMC were grown as previously described (25). Briefly, femurs from 4–8 week old mice were removed and bone marrow cells were collected by centrifugation for 30 seconds at 5000 rpm. Cells were resuspended in RPMI supplemented with 1% non-essential amino acids, 1mM sodium pyruvate, 1% HGPG, 10 mM HEPES, 10% fetal bovine serum, and 0.1% 2-mercaptoethanol. For the first three weeks, the cells were incubated with 10 ng/mL recombinant murine IL-3 (Peprotech). In subsequent weeks, 25 ng/mL recombinant murine stem cell factor (Peprotech) was added along with IL-3. After five weeks, the purity of the population was >90% mast cells as determined by flow cytometry analysis using anti-FcεR1α (BioLegend) and anti-CD117 (BioLegend). To generate BMCMC ‘knock-in’ mice, we followed the methods previously used by others (26–28). Briefly, BMCMC were generated as described above. Mice 3–4 weeks of age were reconstituted with 5×106 BMCMC via intravenous injection and rested for ~8–10 weeks before use.
In vitro mast cell activation assay
For activation assays, 2.5×105 BMCMC were plated per well in a 96 well U-bottom plate. Virus was added at an MOI of ~1 and the final volume was brought to 100 μl and incubated for 4–6 hours. As positive controls BMCMC were stimulated with LPS (5 μg/ml; List Biologicals Laboratory) for cytokine/chemokine release or the calcium ionophore A23187 (40 nM; Fisher Scientific) for leukotriene synthesis and mast cells degranulation. Additionally, naive allantoic fluid could not activate the BMCMC similar to medium alone (Supplemental Figure 1).
Luminex assay for cytokine and chemokine secretion
In vitro BMCMC activation assay supernatants from 4–6 h post-treatment and BALF from the indicated time points were analyzed for cytokine and chemokines according to the manufacturer’s instructions. Custom Milliplex® plates were used for cytokine and chemokine analyses (Millipore), while murine IFNα and IFNβ were measured using Procarta® 2-plex assay (Affymetrix). Plates were read using a BioPlex® 200 (Bio-Rad).
Histamine and leukotriene B4 enzyme immunoassays (EIA)
Histamine and leukotriene B4 EIA assays were conducted following the manufacturer’s instructions (Cayman Chemical). Briefly, the samples were incubated with an acetylcholinesterase linked histamine or leukotriene B4 tracer for 24 hours or overnight, respectively. Plates were washed and Ellman’s Reagent was added to detect the tracer labeled histamine or leukotriene B4. Obtained results are inversely proportional to free histamine or leukotriene B4 present in the well.
Intracellular staining for viral proteins
To measure virus infectivity of mast cells, we stained BMCMC looking for intracellular expression of the NS-1 protein. Briefly, 2.5×105 BMCMC were plated per well in a 96 well U-bottom plate. Virus was added at MOI ~1 and the final volume was brought to 100 μl and incubated for 4–6 hours. The cells were then fixed with 100 μl BD Cytofix/Cytoperm (BD Biosciences) for 30 minutes. Cells were then stained with a mouse monoclonal antibody against the IAV NS1 protein (NS1-1A7; BEI Resources) for 30 minutes in BD Perm-wash buffer. Subsequently, the cells were washed with BD Perm-wash buffer and then stained with PE-labeled goat anti-mouse IgG F(ab′) fragments (Jackson ImmunoResearch Laboratories). Cell were then washed with BD Perm-wash buffer and resuspended in FACS Buffer. Samples were collected using a FACS Calibur and analyzed via FlowJo.
Statistical analysis
Statistical significance was determined by either a Mann-Whitney U-test or one-way ANOVA using Prism 5 (Graphpad Software). Significance was set as p < 0.05.
Results
Mast cells are critical for inducing the pulmonary and systemic inflammatory disease induced by A/WSN/33 infection of mice
Mast cells have been found to be crucial participants in the immune responses to parasitic and bacterial infections (8, 12). Recently, the role of mast cells during respiratory infection with bacterial pathogens has begun to be explored (29–31), but the role of mast cells during respiratory infection with viral pathogens remains understudied. IAV has been reported to enhance IgE-mediated histamine secretion from basophilic leukocytes, but those studies failed to observe any direct activation of mast cells by IAV alone (23). Furthermore, genetic analysis of lungs from mice infected with a model strain of the 2009 IAV pandemic virus demonstrated an enrichment of genes associated with mast cells (32). Thus, we looked to address the role mast cells might play during IAV infections. To assess the role of mast cells during respiratory infection with IAV, 10–12 week old B6.Cg-KitW-sh mice, which are known to have a mast cell deficiency (26), and C57BL/6 mice were infected with 1500 PFU of A/WSN/33. Body weight of individual animals was longitudinally monitored for 2 weeks. When infected with A/WSN/33, B6.Cg-KitW-sh mice did not lose substantial amounts of their initial body weight, while C57BL/6 mice displayed significant weight loss (Figure 1a). Weight loss during A/WSN/33 infection correlated with the amount of vascular leakage and tissue damage observed in the lungs of A/WSN/33 infected mice as measured by total protein (Figure 1b) or lactate dehydrogenase (data not shown) levels present in the BALF. Moreover, B6.Cg-KitW-sh mice infected with A/WSN/33 had reduced numbers of inflammatory cells infiltrating into the BALF 7 days after infection (Figure 1c). The reduced BALF cell numbers in the B6.Cg-KitW-sh mice were observed in all cell populations (macrophages, neutrophils, and lymphocytes). Reduced inflammatory cells infiltrating the BALF was not due to significantly decreased cell numbers found in the BALF of naïve B6.Cg-KitW-sh mice (C57BL/6 = 1.00×105 ± 0.48×105 versus B6.Cg-KitW-sh = 0.77×105 ± 0.54×105; p=0.29) or BALF composition which was >90% alveolar macrophages in both naïve C57BL/6 and B6.Cg-KitW-sh mice (data not shown). Moreover, leukocyte numbers are largely normal in B6.Cg-KitW-sh mice, if anything those mice display neutrophilia (33). Histologically analysis of lungs from A/WSN/33 infected C57BL/6 mice display robust interstitial inflammatory infiltrates on both day 7 and 10 post-infection; additionally, by day 10 airway infiltration was seen by histology in the infected C57BL/6 mice (Figure 1e). In contrast, B6.Cg-KitW-sh mice displayed minimal interstitial inflammatory infiltrates at either time-point. These data track with the amount of IAV-induced immunopathology observed as measured by protein in the BALF (Figure 1b). The reduction in IAV-induced immunopathology in B6.Cg-KitW-sh mice was not the result of decreased viral growth in the respiratory tract because viral titers were similar after A/WSN/33 infection in C57BL/6 and B6.Cg-KitW-sh mice and both mouse strains cleared the IAV infection by 10 dpi (Figure 1d).
Figure 1. B6.Cg-KitW-sh is less sensitive to A/WSN/33.

Age-matched C57BL/6 (B6) or B6.Cg-KitW-sh (Wsh) mice were infected nasally with 1500 PFU of A/WSN/33 (WSN). (A) Body weights were measured daily. Each graph is normalized to each mouse’s starting body weight. (B) Damage to the lung was assessed by measuring the total protein levels in the BALF at the indicated time points. (C) Total leukocyte recruitment to the BALF was measured day 7 post infection. (D) Lung viral titers were determined at indicated time points post-infection by plaque assay on MDCK cells. Dotted line represents the limit of detection of the plaque assay (LOD = 2 PFU/mL). N.D. = none detected. (E) Formalin fixed lungs were paraffin embedded, then sectioned and stained with H&E for analysis by microscopy. Pictures of representative lung sections were taken using 10x magnification objectives from naïve and A/WSN/33 infected (7d or 10d) C57BL/6 and Wsh mice. Data are representative of 2–4 independent experiments consisting of 4–8 mice per group. Statistically significant differences were determined using a Mann-Whitney U-test (*p<0.05).
It appears that mice devoid of mast cells are highly resistant to IAV-induced inflammatory disease (Figure 1). However, B6.Cg-KitW-sh mice have other abnormalities in addition to the mast cell deficiency (26, 33). To ensure the defect in the B6.Cg-KitW-sh mice was within the mast cell compartment we made mast cell knock-in mice as previously described (26–28). Selective reconstitution of the mast cell compartment of B6.Cg-KitW-sh mice with 5×106 C57BL/6-derived bone marrow cultured mast cells (BMCMC) 8–10 weeks prior to IAV infection was able to complement the reduced weight loss and inflammatory cell recruitment observed in B6.Cg-KitW-sh mice (Figure 2 a & b). Thus, our data demonstrate that during respiratory infection with IAV, mast cells are crucial participants in the pathological innate immune response.
Figure 2. B6.Cg-KitW-sh mice reconstituted with BMCMC were susceptible to WSN.

(A) Age-matched C57BL/6 (B6), B6.Cg-KitW-sh (Wsh), or B6.Cg-KitW-sh mice reconstituted with 3×106 BMCMC 10 weeks prior were infected nasally with 1500 PFU of A/WSN/33 (WSN). Body weights were measured daily. († = mouse had to be euthanized). The graph is normalized to each mouse’s starting body weight. (B) Age-matched C57BL/6 mice and B6.Cg-KitW-sh mice, reconstituted with either 1×106 or 5×106 BMCMC, were infected nasally 10 weeks later with 1500 PFU of A/WSN/33 (WSN) and total lymphocyte, neutrophil, and macrophage counts in the BALF was measured 7 days after infection. Data are representative of two independent experiments consisting of 4–8 mice per group. Statistically significant differences were determined using a Mann-Whitney U-test (A) or one-way ANOVA (B) (*p<0.05).
Altered inflammatory milieu in B6.Cg-KitW-sh mice during A/WSN/33 infection
Because our data demonstrated a dramatic reduction in the number of inflammatory cells infiltrating into the BALF of B6.Cg-KitW-sh mice (Figures 1 & 2), we next wanted to assess the inflammatory cytokine and chemokine production pattern in C57BL/6 and B6.Cg-KitW-sh mice after A/WSN/33 infection. To do this, C57BL/6 and B6.Cg-KitW-sh mice were infected with 1500 PFU of A/WSN/33. BALF was collected from individual mice 7 days after A/WSN/33 infection and analyzed by multiplex bead-based assays. We found that there was a marked alteration of the inflammatory milieu found in the BALF of B6.Cg-KitW-sh mice after A/WSN/33 infection as compared with C57BL/6 mice. Specifically, there was a reduction in TNFα, CCL2, CCL3, CCL4, CXCL2, and CXCL10 at this time point in the B6.Cg-KitW-sh mice (Figure 3 and data not shown). Moreover, there was a trend toward reduced levels of IL-6 and IFNγ (Figure 3 and data not shown). However, not all cytokine expression was lost in B6.Cg-KitW-sh mice because there was equivalent expression of CCL5, CCL12, IL-5, IL-16, LIF, G-CSF, and CX3CL1 (Figure 3 and data not shown). All cytokine and chemokines were below the limit of detection in naïve BALF from C57BL/6 mice, except IL-16 (36.9 ± 5.9 pg/ml) and CX3CL1 (140.6 ± 79.3 pg/ml). Thus, our data suggest that mast cells can be important for initiating the inflammatory milieu found during IAV infection.
Figure 3. B6.Cg-KitW-sh mice had less detection of cytokines in the lung during A/WSN/33 infection.

Age-matchedC57BL/6 (B6) or B6.Cg-KitW-sh (Wsh) mice were infected nasally with 1500 PFU of A/WSN/33 (WSN). Cytokine and chemokine levels in the BALF were assessed 7 days after infection using Milliplex™ multiplex assays. Statistically significant differences were determined using a Mann-Whitney U-test (*p<0.05). Data are representative of two independent experiments consisting of 4–6 mice per group.
In vitro activation of mast cells in response A/WSN/33 treatment
Next we wanted to assess whether IAV had the potential to directly activate mast cells in vitro. To do this, 2.5×105 BMCMC were treated with medium or A/WSN/33 for 6 hours or at the indicated times. BMCMC supernants were then analyzed for mast cell mediators, which included histamine, leukotriene B4 (LTB4), and cytokines. Medium treated BMCMC showed minimal expression of all three mediators (Figure 4a–c). Additionally, uninfected allantoic fluid did not cause mast cell mediator release greater than medium alone (Supplemental Figure 1). A/WSN/33 treatment of BMCMC resulted in histamine release, CCL2 secretion, and LTB4 production (Figure 4a–c). Specifically, we found that mast cells released histamine in two waves: the first peaking ~30 minutes after A/WSN/33 treatment and the second beginning ~4 hours after stimulation (Figure 4a). Additionally, 6 hours after A/WSN/33 inoculation mast cells produced substantial quantities of CCL2 (Figure 4b) and LTB4 (Figure 4c). In a broader analysis of the cytokines and chemokines secreted by BMCMCs after A/WSN/33 activation, we found that mast cells produced CCL2, CCL3, CCL4, CCL5, CXCL2, CXCL9, CXCL10, IL-6, and TNFα, but not CXCL1, IFNα, IFNβ, IFNγ, IL-1β, IL-5, IL-9, IL-15, and VEGF in response to A/WSN/33 (Supplemental Table 1). Furthermore, cytokine and chemokine expression began ~2 hours after IAV treatment (data not shown). Thus, at least in vitro, IAV-induced mast cell activation appears to occur through direct recognition of the virus.
Figure 4. In vitro BMCMC activation with A/WSN/33 results in mast cell activation.

BMCMC were derived by culturing total bone marrow with IL-3 for 5 weeks, supplementing with stem cell factor for the last 2 weeks. 2.5×105 FcRε1+ and CD117+ mast cells were treated with media, A/WSN/33 (WSN), or a positive control of calcium ionophore (Ca2+) at 40nM or LPS at 5μg/mL. (A) Histamine levels were measured by an EIA at the indicate time points. As a control histamine levels from unstimulated BMCMC were assayed after 6 hours of culture. Cytokine/chemokine (B) and LTB4 (C) secretion were measured 6 hours after stimulation by either Milliplex™ multiplex analysis or an EIA, respectively. Each virus was added at an MOI of 1. Data are representative of 2–4 independent experiments. N.D. = none detected.
Mast cells can be activated by human influenza virus isolates
Since A/WSN/33 is a highly mouse-adapted strain of IAV, we next wanted to assess whether human-derived isolates which had not been passed through mice could activate mast cells in vitro. To do this, 2.5×105 BMCMC were treated with 50 μl of each influenza virus isolate for 6 hours. BMCMC supernants were then analyzed for the cytokine and chemokine levels. In agreement with our earlier findings, A/WSN/33 treatment of BMCMC resulted in secretion of IL-6, CCL2, and TNFα (Figure 5, black bar and data not shown). Interestingly, another mouse adapted IAV strain, A/Puerto Rico/8/34 resulted in minimal activation of mast cells (Figure 5), which corresponds with our in vivo observation that B6.Cg-KitW-sh mice are as susceptible as C57BL/6 to A/PR/8/34 infection (data not shown). Human-derived IAV isolations of both H1N1 (Figure 5, light gray bars) and H3N2 (Figure 5, dark gray bars) families were both able to activate murine BMCMC to produce cytokines and chemokines to varying levels. Furthermore, the influenza B virus isolate B/Florida/4/2006 could also activate murine BMCMC in vitro to produce these cytokines and chemokines (Figure 5, white bar). Our data support the notion that mast cell could play an important role in the inflammatory response to a range of influenza virus isolates.
Mast cells are infected by A/WSN/33
We next wanted to ask whether mast cells were capable of being infected with IAV. To do this, 2.5×105 BMCMC were treated with medium, A/PR/8/34, or A/WSN/33 for 6 hours. Cells were then fixed, permeabilized, and stained for intracellular NS1 of IAV. NS1 is a non-structural protein which is not found in the IAV virion and therefore require de novo synthesis of viral proteins to detect protein. Uninfected BMCMC displayed minimal anti-NS1 staining over our secondary antibody alone (Figure 6, black histogram). Interestingly, A/WSN/33 treated BMCMC expressed intracellular NS1 (Figure 6, red histogram); while A/PR/8/34 treated BMCMC did not (Figure 6, blue histogram). However, we could not detect de novo production of infectious IAV virions over a 72 hour time period by BMCMC (data not shown). Thus, the A/WSN/33 strain of IAV is able to directly infect mast cells, which correlates with the virus’ ability to activate mast cells, but could not propagate IAV.
Figure 6. Mast cells are infected by A/WSN/33.
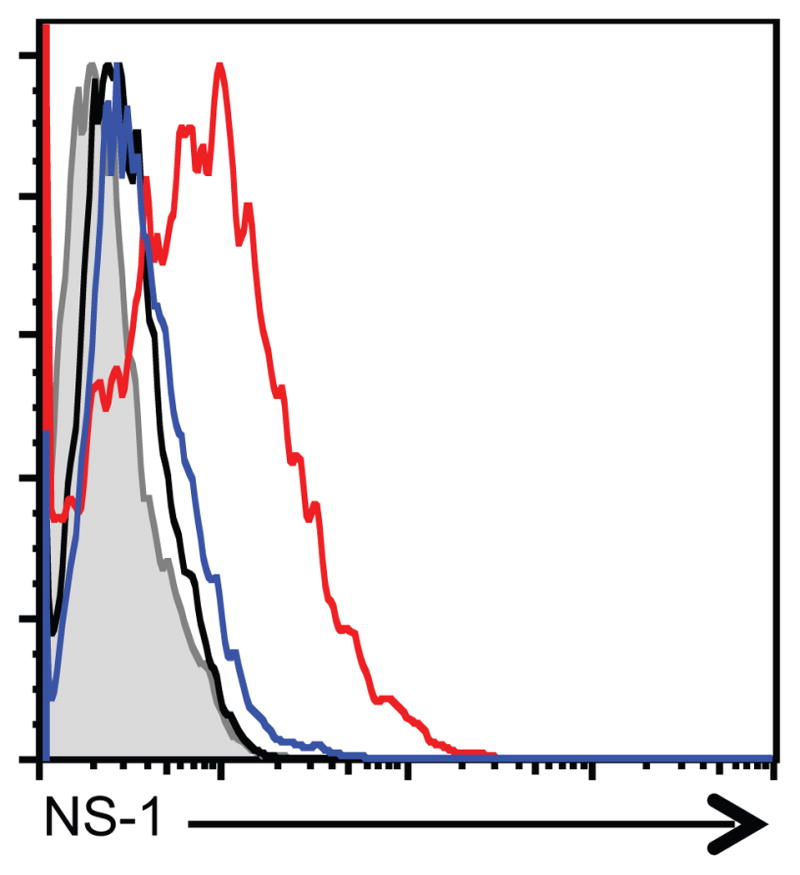
BMCMC were derived by culturing total bone marrow with IL-3 for 5 weeks, supplementing with stem cell factor for the last 2 weeks. 2.5×105 FcRε1+ and CD117+ mast cells were treated with medium, A/WSN/33, or A/PR/8/34. Each virus was added at an MOI of 1. BMCMC were harvested after 5 hours. BMCMC were fixed, permeabilized, and stained for intracellular NS-1 using mAb NS1-1A7 for 30 minutes. BMCMC were then stained with PE-labeled anti-mouse IgG F(ab′) fragments. Representative histograms are shown for 2 independent experiments. Shaded histogram = PE-labeled anti-mouse IgG F(ab′) fragments alone; Black histogram = media treated BMCMC; Blue histogram = A/PR/8/34 infected BMCMC; Red histogram = A/WSN/33 infected BMCMC.
RIG-I detection of IAV is involved in mast cell cytokine, chemokine, and leukotriene production, but not degranulation
Numerous pattern recognition receptors have been shown to play a role in sensing IAV infection. These include the TLR3, TLR7, NLRP3, and RIG-I receptors (34–43). Because BMCMC were productively infected with A/WSN/33 we hypothesized that detection of cytosolic genomic RNA might be crucial for mast cell activation. The major cytosolic receptor for IAV RNA is RIG-I, which subsequently docks on its adaptor MAVS (also known as IPS-1, VISA, and Cardif) to initiate IRF3- and NFκB-dependent signaling (44–47). We first decided to examine the role of RIG-I in the activation of mast cells by A/WSN/33. To do this 2.5×105 BMCMC from either C57BL/6 or RIG-I−/− mice were treated with medium, A/WSN/33, or LPS for 6 hours. In agreement with our previous findings, A/WSN/33 treatment of C57BL/6-derived BMCMCs resulted in significant production of histamine, IL-6, and LTB4 (Figure 7a–c). In contrast, RIG-I−/−-derived BMCMCs had significantly blunted IL-6 and LTB4 responses after A/WSN/33 treatment (Figure 7a & b). However, RIG-I−/−-derived BMCMCs produced equivalent levels of histamine when compared with A/WSN/33 treated C57BL/6-derived BMCMCs (Figure 7c). Similarly, when BMCMCs from MAVS−/− mice were used cytokine and chemokine production was significantly reduced, while histamine release was normal (data not shown). Importantly, RIG-I−/− BMCMC responded similarly to C57BL/6 BMCMC to an irrelevant stimulus, such as LPS or A23187 (data not shown). Our data demonstrate that RIG-I/MAVS signaling is crucial for de novo generated mediators, but not the immediate degranulation response of mast cells during IAV treatment.
Figure 7. Mast cell activation by A/WSN/33 is dependent on RIG-I.
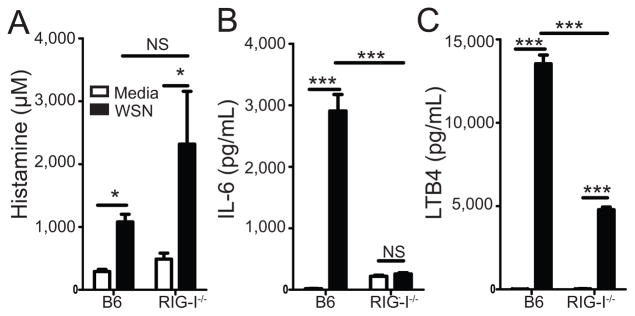
BMCMC were generated by culturing total bone marrow from either C57BL/6 or RIG-I−/− mice with IL-3 for 5 weeks, supplementing with stem cell factor for the last 2 weeks. 2.5×105 FcRε1+ and CD117+ mast cells were treated with media or A/WSN/33 (WSN). Virus was added at an MOI of 1. Six hours after inoculation IL-6 (A), LTB4 (B), and histamine (C) levels were measured by Milliplex™ multiplex analysis or EIA assays. Similar data was observed for CCL2 and CCL4 expression (data not shown). Statistically significant differences were determined using a one-way ANOVA (***p<0.001; *p<0.05; NS=not significant). Data are representative of 2–4 independent experiments. White bars = Media treated; Black bars = A/WSN/33 treated.
Cytokine and chemokine expression by mast cells in response to A/WSN/33 treatment is partially STING and STAT6 dependent
Our knowledge about the signaling networks downstream of RIG-I recognition of ssRNA has expanded to include STING, CARD9, and STAT6. RIG-I/MAVS interactions result in the activation of IRF3 and NF-κB (44–47). Recently, CARD9 and MALT10 were shown to be necessary for activation of NF-κB, but not IRF3 in response to synthetic RIG-I ligands or VSV infection (48). STING is an ER resident protein that has been shown to be important in the RIG-I dependent anti-viral response to VSV (49). Additionally, others have found that STAT6 is recruited to the STING-MAVS-RIGI complex during Sendai virus or VSV infection to mediate CCL2 and CCL20 expression, but is dispensable for IL-6 production (50). Since RIG-I and MAVS were critical in the cytokine/chemokine response of BMCMC, we next wanted to examine the role of CARD9, STING, and STAT6 in the response of BMCMCs to A/WSN/33 infection. To do this, 2.5×105 BMCMC from C57BL/6, CARD9−/−, STING−/−, or STAT6−/− mice were treated with medium, A/WSN/33, or LPS for 6 hours. In agreement with our previous findings, A/WSN/33 treatment of C57BL/6-derived BMCMCs resulted in cytokine and chemokine secretion in all experiments (Figure 8a–c). The response of CARD9−/−-derived BMCMCs was similar to C57BL/6-derived BMCMCs after A/WSN/33 infection (Figure 8a). However, STING−/−- and STAT6−/−-derived BMCMCs had a ~50% reduction in their production of IL-6, CCL2, and CCL4 (Figure 8b & 8c, data not shown). Importantly, CARD9−/−, STING−/− and STAT6−/− BMCMC responded similarly to C57BL/6 BMCMC to an irrelevant stimulus, such as LPS (data not shown). Thus, while RIG-I and MAVS are absolutely essential for the cytokine response of BMCMCs in response to IAV infection (Figure 7), CARD9 is completely dispensable, while STING and STAT6 play partial roles in the inflammatory cytokine and chemokine response to IAV infection (Figure 8).
Figure 8. Mast cell activation is partially dependent on STING and STAT6.
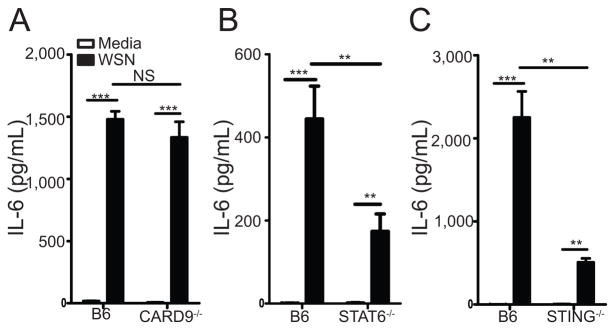
BMCMC were generated by culturing total bone marrow from either C57BL/6 or CARD9−/− mice (A), C57BL/6 or STING−/− mice (B), or C57BL/6 or STAT6−/− mice (C) with IL-3 for 5 weeks, supplementing with stem cell factor for the last 2 weeks. 2.5×105 FcRε1+ and CD117+ mast cells were treated with media or A/WSN/33 (WSN). Virus was added at an MOI of 1. Six hours after inoculation IL-6 levels were measured by Milliplex™ multiplex analysis. Similar data was observed for CCL2 and CCL4 expression (data not shown). Statistically significant differences were determined using a one-way ANOVA (***p<0.001; **p<0.01; NS=not significant). Data are representative of 2–4 independent experiments. White bars = Media treated; Black bars = A/WSN/33 treated.
Discussion
Mast cells are tissue resident hematopoietic cells. Since infectious agents enter the host through environmentally exposed barriers, such as the skin, gastrointestinal tract, and respiratory tract, mast cells are poised to be one of the first cell types to respond to invading pathogens. Furthermore, mast cells express a wide array of pattern-recognition receptors that endow them with the ability to respond to a broad range of stimuli, such as infections and pathogenic conditions (51). To date it is well established that mast cells play crucial immune surveillance roles during bacterial and parasitic infections (8, 12). In contrast, the role of mast cells in the immune surveillance of viral infections has received less attention. In the current study, we examined the role of mast cells in sensing IAV infection and initiating the subsequent inflammatory response.
A primary rationale for our work stems from the recent work by Teijaro et al that demonstrated that blunting the ‘cytokine storm’ significantly improves the health of animals infected with either the mouse-adapted IAV strain A/WSN/33 or the 2009 H1N1pdm strain A/Wisconsin/WSLH34939/2009 (6). Thus, we reasoned if mast cells are critical mediators of the inflammatory response to IAV, a further understanding of this interaction may lead to new therapeutic option(s) to improve IAV patient outcomes.
In further support of our hypothesis regarding mast cells and IAV, recent genetic analysis of the lungs from mice infected with a model strain of the 2009 IAV pandemic virus demonstrated an enrichment of genes associated with mast cells (32). However, whether respiratory mast cells play a role during IAV infection remains elusive. Here our data demonstrate that mast cells are critical participants in local lung inflammation and systemic IAV-induced disease during A/WSN/33 infection of mice. In vivo we found that mast cells contributed to the establishment of the inflammatory milieu and lung damage during A/WSN/33 infection. This appears to not be due to dramatic differences in the lung viral burden in the absence of mast cells; rather, in the absence of mast cells (B6.Cg-KitW-sh), mice had decreased inflammatory cell infiltration into the BALF and levels of inflammatory cytokines and chemokines. Importantly, this reduction in inflammatory cells and mediators in the absence of mast cells correlated with reduced lung damage and/or vascular leakage as measured by protein levels in the BALF.
Our results fit with a recent report that the H5N1 isolate A/chicken/Henan/1/2004 was capable of activating the mastocytoma cell line P815 and inducing a significant increase in mast cell numbers within the respiratory tract over the first 5 days of IAV infection in mice (52). We extend these findings to show that IAV can activate primary mast cells (BMCMC) and importantly that H1N1, H3N2, and influenza B viruses could also activate mast cells. Additionally, Hu et al demonstrated that ketotifen, an H1 receptor inhibitor (53), could limit epithelial cell death in vivo (52). However, ketotifen can also inhibit cytokine and chemokine expression by macrophages after LPS stimulation (54) and the H1 receptor has a broad expression pattern including airway epithelial cells (55). These facts complicate the interpretation by Hu et al that mast cells are the crucial cell inhibited by ketotifen (52). In our study we specifically demonstrate that mice lacking mast cells do not develop as severe an IAV-induced inflammatory response and lung damage, which could be complemented by reconstitution of those mice with mast cells.
How mast cells are mediating their response in vivo is the focus of our future studies. In vitro Hu et al demonstrated mast cells produced high levels of IFNγ which caused significant epithelial cell death (52). However, we failed to detect any IFNγ expression by BMCMC after IAV treatment. The major differences between our two studies are the source of mast cells (primary BMCMC versus P815 cell line) and viral isolates studied (H1N1 versus H5N1 isolates), which could likely contribute to this difference. However, both our study and Hu et al. (52) suggest that human IAV isolates can activate mast cells. Interestingly, IAV infection of humans results in elevated urinary histamine levels, whose major source in vivo are mast cells and basophils (56), which peaked ~2 days after infection and its expression correlated with virus-induced illness (57). Thus, mast cells appear to be an attractive target for a host-targeted therapeutic to limit the pathological response induced by IAV infection.
Interestingly, similar to our results with IAV, mast cells have been shown to be a key role during cutaneous viral infections. Cutaneous mast cells were recently shown to play a protective role during skin vaccinia virus infection (22). Additionally, mast cells have been shown to be crucial for the in vivo immune surveillance of dengue virus after a foot-pad inoculation (19). Mast cells can also regulate endothelial cell function during dengue virus infection in vitro (58). Importantly, dengue shock syndrome in humans displays elevated levels of mast cell-derived tryptases and chymases (21). Thus, mast cells are likely crucial participants in the inflammatory response during dengue virus infection through the skin. These data, together with ours, demonstrate that mast cells may play a crucial role in the immune surveillance of viral infections in general.
A major question then is how mast cells detect viral infections and whether mast cells can serve as a suitable environment for virus replication. Again, there appears to be numerous correlations between IAV and dengue virus. We found that mast cells were directly infected with IAV in vitro as demonstrated by de novo NS1 expression, but BMCMC could not propagate A/WSN/33 in vitro. This fits with Befus and colleagues recent finding that IAV did not replicate in human mast cells (59). Similarly, the mast cells could be infected with dengue virus (19, 20); however, in contrast to our results, dengue virus could replication within mast cells (19). BMCMC could be infected with vaccinia virus at higher multiplicities of infection, but whether vaccinia virus could replicate within mast cells in vitro was not explored (22).
Interestingly, it appears each phase of the mast cell response, immediate degranulation and de novo mediators synthesis, is regulated by a distinct innate immune sensing receptor and signaling network during the course of IAV infection. Expression of de novo synthesized cytokines and chemokines following IAV inoculation required RIG-I and MAVS. Similar to our results with IAV, chemokine expression by mast cells in response to dengue virus infection was dependent on sensing by both the cytosolic RNA sensors, RIG-I and Mda5 (19, 20). Moreover, IL-6, CXCL10, and type I interferon expression by BMCMC in response to VSV treatment was dependent on RIG-I and Mda5 (60). Upon interacting with RNA, RIG-I and Mda5 undergo a conformational change allowing it bind with the scaffolding protein MAVS. Originally, MAVS was described as a mitochondrial localized protein (44), but recently MAVS has been found to also localize to the peroxisome (61). Interestingly, the downstream signaling events of mitochondrial or peroxisome localized RIG-I/MAVS complexes differs (61). Specifically, peroxisome localized RIG-I/MAVS results in the activation of early interferon-regulated proteins in the absence IFNα/β expression (61). In contrast, mitochondrial localized RIG-I/MAVS results in the production of IFNα/β with delayed kinetics (61). RIG-I appears to interact with MAVS at the mitochondria-associated endoplasmic reticulum membrane (MAM) (62). STING is an endoplasmic reticulum protein which is important for the scaffolding protein for signaling of receptors detecting cytosolic nucleic acids (49). STING-RIG-I interactions were recently demonstrated to be important for the anti-viral response to Japanese encephalitis virus (JEV) (63). Moreover, STING recruits Stat6-dependent anti-viral immunity through the production of CCL2, CCL20 and CCL26, but not IL-6 and IFNα/β (50). Our data are consistent with a more central role of peroxisome-mediated RIG-I/MAVS mediated signaling during IAV infection of BMCMC for two reasons: 1) we observed nominal expression of IFNα/β, CXCL9, and CXCL10 and 2) we observed only a partial STING- and Stat6-dependency for the inflammatory response of BMCMC. However, formal testing of whether peroxisome or mitochondrial MAVS is more important during IAV infection of mast cells remains to be explored. Additionally, the PB1-F2 protein of IAV was shown to interfere with MAVS-mediated signaling through disruption of the mitochondrial membrane potential (64–66). Potentially, PB1-F2’s expression in mast cells could strongly impair mitochondria localized MAVS, thus making peroxisome localized MAVS the dominant signaling pathway in mast cells. Thus, it will be intriguing to test the anti-viral cellular response of BMCMC in the absence of these IAV proteins.
In addition to viral detection mechanisms, how mast cells degranulate in response to IAV, VSV, or dengue virus infection remains an open question. Histamine production from BMCMC occurred rapidly after IAV inoculation and appears to occur prior to endosomal escape of the virus (data not shown). Furthermore, histamine production by BMCMC after IAV infection was normal in the absence of RIG-I. Analogously, BMCMC degranulation in response to VSV treatment occurred normally following RIG-I or Mda5 knock-down by siRNA (60). Other pattern-recognition receptors detecting early stages of the IAV infectious cycle could be important, such as TLR3 or TLR7 (34–36). However, triggering of synthetic polyI:C could not induce the degranulation of human mast cells (67). Alternatively, STING was recently shown to ‘recognize’ viral membrane fusion (68), but it is role in mast cell degranulation has not be explored. The one viral infection we have at least a partial understanding of how the virus is inducing mast cell degranulation is vaccinia virus infection. During vaccinia virus infection BMCMC degranulation occurred in a membrane fusion-dependent and S1PR2-dependent manner (22). S1P signaling is important inducing the cytokine storm during IAV infection (6) and in vitro can regulate IAV growth (69); however, whether S1P signaling plays a role in regulating mast cell function during IAV infection remains unknown.
In conclusion, we demonstrate that mast cells can be infected by IAV resulting in their activation. Importantly, mast cells were critical participants in inducing IAV-induced cytokine storm and systemic disease during A/WSN/33 infection of mice. Our results suggest that the mast cell response to IAV infection is regulated by multiple receptors controlling either degranulation or de novo mediator synthesis. Importantly, based on our findings it is important to consider the role mast cells are playing in regulating the excessive inflammatory response during infections when trying to control such adverse events. It remains to be answered whether degranulation, de novo mediator synthesis, or both are required for mast cells to enhance the inflammatory response and pathology during IAV infection. Understanding these events is imperative for developing a host-targeted therapeutic which is aim at curtailing mast cell function to limit IAV-induced morbidity and mortality.
Supplementary Material
Acknowledgments
The authors wish to thank all the investigators who provided knock-out mice and/or bone marrow for our in vitro BMCMC activation studies: Dr. Michael Gale (University of Washington), Dr. Mathias Schnell (Thomas Jefferson University), Dr. John Cambier (National Jewish Health), Dr. Tobias Hohl (Fred Hutchinson Cancer Center), and Dr. Daniel Campbell (Benoyra Institute). Also, we thank Dr. David Topham (University of Rochester) for providing the initial A/WSN/33 viral stock. Dr. Leo Lefrançois (University of Connecticut Health Center), Dr. Jonathan Yewdell (NIH/NIAID), Dr. Allen Harmsen (Montana State University), and Dr. Robert Cramer (Geisel School of Medicine at Dartmouth College) provided many helpful suggestions about this project.
Footnotes
Work in this study was supported by NIH grants P20 RR-020185 (JJO), P20 GM-103500 (JJO), and K22 AI-091647 (JJO), the Montana State University Agricultural Experimental Station, and equipment grant from the M.J. Murdock Charitable Trust.
Abbreviations: IAV = influenza A virus; RIG-I = Retinoic acid inducible gene I; RLR = RIG-I like receptors; NLR = Nod-like recepotors; CLR = C-type lectin-like receptors; BALF = broncheoalveolar fluid; BMCMC = bone marrow cultured mast cells; LTB4 = leukotriene B4
Author Contributions
Conceived and designed the experiments: ACG and JJO
Performed the experiments: ACG, KMH, JMZ, and JJO
Analyzed the data: ACG, KMH, JMZ, and JJO
Wrote the paper: ACG and JJO
References
- 1.Taubenberger JK, Morens DM. The pathology of influenza virus infections. Annu Rev Pathol. 2008;3:499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baskin CR, Bielefeldt-Ohmann H, Tumpey TM, Sabourin PJ, Long JP, Garcia-Sastre A, Tolnay AE, Albrecht R, Pyles JA, Olson PH, Aicher LD, Rosenzweig ER, Murali-Krishna K, Clark EA, Kotur MS, Fornek JL, Proll S, Palermo RE, Sabourin CL, Katze MG. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc Natl Acad Sci U S A. 2009;106:3455–3460. doi: 10.1073/pnas.0813234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008;4:e1000115. doi: 10.1371/journal.ppat.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, Garcia-Sastre A, Swayne DE, Katze MG. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443:578–581. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baas T, Baskin CR, Diamond DL, Garcia-Sastre A, Bielefeldt-Ohmann H, Tumpey TM, Thomas MJ, Carter VS, Teal TH, Van Hoeven N, Proll S, Jacobs JM, Caldwell ZR, Gritsenko MA, Hukkanen RR, Camp DG, 2nd, Smith RD, Katze MG. Integrated molecular signature of disease: analysis of influenza virus-infected macaques through functional genomics and proteomics. J Virol. 2006;80:10813–10828. doi: 10.1128/JVI.00851-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146:980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moiseeva EP, Bradding P. Mast cells in lung inflammation. Adv Exp Med Biol. 2011;716:235–269. doi: 10.1007/978-1-4419-9533-9_13. [DOI] [PubMed] [Google Scholar]
- 8.Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol. 2010;10:440–452. doi: 10.1038/nri2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shelburne CP, Abraham SN. The mast cell in innate and adaptive immunity. Adv Exp Med Biol. 2011;716:162–185. doi: 10.1007/978-1-4419-9533-9_10. [DOI] [PubMed] [Google Scholar]
- 10.Klemm S, Gutermuth J, Hultner L, Sparwasser T, Behrendt H, Peschel C, Mak TW, Jakob T, Ruland J. The Bcl10-Malt1 complex segregates Fc epsilon RI-mediated nuclear factor kappa B activation and cytokine production from mast cell degranulation. J Exp Med. 2006;203:337–347. doi: 10.1084/jem.20051982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Supajatura V, Ushio H, Nakao A, Akira S, Okumura K, Ra C, Ogawa H. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J Clin Invest. 2002;109:1351–1359. doi: 10.1172/JCI14704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marshall JS. Mast-cell responses to pathogens. Nat Rev Immunol. 2004;4:787–799. doi: 10.1038/nri1460. [DOI] [PubMed] [Google Scholar]
- 13.Dietrich N, Rohde M, Geffers R, Kroger A, Hauser H, Weiss S, Gekara NO. Mast cells elicit proinflammatory but not type I interferon responses upon activation of TLRs by bacteria. Proc Natl Acad Sci U S A. 2010;107:8748–8753. doi: 10.1073/pnas.0912551107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guhl S, Franke R, Schielke A, Johne R, Kruger DH, Babina M, Rang A. Infection of in vivo differentiated human mast cells with hantaviruses. J Gen Virol. 2010;91:1256–1261. doi: 10.1099/vir.0.019505-0. [DOI] [PubMed] [Google Scholar]
- 15.King CA, Anderson R, Marshall JS. Dengue virus selectively induces human mast cell chemokine production. J Virol. 2002;76:8408–8419. doi: 10.1128/JVI.76.16.8408-8419.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugiyama K. Histamine release from rat mast cells induced by Sendai virus. Nature. 1977;270:614–615. doi: 10.1038/270614a0. [DOI] [PubMed] [Google Scholar]
- 17.Burke SM, Issekutz TB, Mohan K, Lee PW, Shmulevitz M, Marshall JS. Human mast cell activation with virus-associated stimuli leads to the selective chemotaxis of natural killer cells by a CXCL8-dependent mechanism. Blood. 2008;111:5467–5476. doi: 10.1182/blood-2007-10-118547. [DOI] [PubMed] [Google Scholar]
- 18.Orinska Z, Bulanova E, Budagian V, Metz M, Maurer M, Bulfone-Paus S. TLR3-induced activation of mast cells modulates CD8+ T-cell recruitment. Blood. 2005;106:978–987. doi: 10.1182/blood-2004-07-2656. [DOI] [PubMed] [Google Scholar]
- 19.St John AL, Rathore AP, Yap H, Ng ML, Metcalfe DD, Vasudevan SG, Abraham SN. Immune surveillance by mast cells during dengue infection promotes natural killer (NK) and NKT-cell recruitment and viral clearance. Proc Natl Acad Sci U S A. 2011;108:9190–9195. doi: 10.1073/pnas.1105079108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown MG, McAlpine SM, Huang YY, Haidl ID, Al-Afif A, Marshall JS, Anderson R. RNA Sensors Enable Human Mast Cell Anti-Viral Chemokine Production and IFN-Mediated Protection in Response to Antibody-Enhanced Dengue Virus Infection. PLoS One. 2012;7:e34055. doi: 10.1371/journal.pone.0034055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furuta T, Murao LA, Lan NT, Huy NT, Huong VT, Thuy TT, Tham VD, Nga CT, Ha TT, Ohmoto Y, Kikuchi M, Morita K, Yasunami M, Hirayama K, Watanabe N. Association of mast cell-derived VEGF and proteases in dengue shock syndrome. PLoS Negl Trop Dis. 2012;6:e1505. doi: 10.1371/journal.pntd.0001505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Lai Y, Bernard JJ, Macleod DT, Cogen AL, Moss B, Di Nardo A. Skin Mast Cells Protect Mice against Vaccinia Virus by Triggering Mast Cell Receptor S1PR2 and Releasing Antimicrobial Peptides. J Immunol. 2011;188:345–357. doi: 10.4049/jimmunol.1101703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clementsen P, Jensen CB, Hannoun C, Soborg M, Norn S. Influenza A virus potentiates basophil histamine release caused by endotoxin-induced complement activation. Examination of normal individuals and patients with intrinsic asthma. Allergy. 1988;43:93–99. doi: 10.1111/j.1398-9995.1988.tb00400.x. [DOI] [PubMed] [Google Scholar]
- 24.Grunewald SM, Hahn C, Wohlleben G, Teufel M, Major T, Moll H, Brocker EB, Erb KJ. Infection with influenza a virus leads to flu antigen-induced cutaneous anaphylaxis in mice. J Invest Dermatol. 2002;118:645–651. doi: 10.1046/j.1523-1747.2002.01732.x. [DOI] [PubMed] [Google Scholar]
- 25.de Vries VC, Pino-Lagos K, Nowak EC, Bennett KA, Oliva C, Noelle RJ. Mast cells condition dendritic cells to mediate allograft tolerance. Immunity. 2011;35:550–561. doi: 10.1016/j.immuni.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol. 2005;167:835–848. doi: 10.1016/S0002-9440(10)62055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakano T, Sonoda T, Hayashi C, Yamatodani A, Kanayama Y, Yamamura T, Asai H, Yonezawa T, Kitamura Y, Galli SJ. Fate of bone marrow-derived cultured mast cells after intracutaneous, intraperitoneal, and intravenous transfer into genetically mast cell-deficient W/Wv mice. Evidence that cultured mast cells can give rise to both connective tissue type and mucosal mast cells. J Exp Med. 1985;162:1025–1043. doi: 10.1084/jem.162.3.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsai M, Grimbaldeston MA, Yu M, Tam SY, Galli SJ. Using mast cell knock-in mice to analyze the roles of mast cells in allergic responses in vivo. Chem Immunol Allergy. 2005;87:179–197. doi: 10.1159/000087644. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez AR, Yu JJ, Murthy AK, Guentzel MN, Klose KE, Forsthuber TG, Chambers JP, Berton MT, Arulanandam BP. Mast cell/IL-4 control of Francisella tularensis replication and host cell death is associated with increased ATP production and phagosomal acidification. Mucosal Immunol. 2011;4:217–226. doi: 10.1038/mi.2010.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ketavarapu JM, Rodriguez AR, Yu JJ, Cong Y, Murthy AK, Forsthuber TG, Guentzel MN, Klose KE, Berton MT, Arulanandam BP. Mast cells inhibit intramacrophage Francisella tularensis replication via contact and secreted products including IL-4. Proc Natl Acad Sci U S A. 2008;105:9313–9318. doi: 10.1073/pnas.0707636105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu X, Zhang D, Lyubynska N, Wolters PJ, Killeen NP, Baluk P, McDonald DM, Hawgood S, Caughey GH. Mast cells protect mice from Mycoplasma pneumonia. Am J Respir Crit Care Med. 2006;173:219–225. doi: 10.1164/rccm.200507-1034OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Josset L, Belser JA, Pantin-Jackwood MJ, Chang JH, Chang ST, Belisle SE, Tumpey TM, Katze MG. Implication of Inflammatory Macrophages, Nuclear Receptors, and Interferon Regulatory Factors in Increased Virulence of Pandemic 2009 H1N1 Influenza A Virus after Host Adaptation. J Virol. 2012;86:7192–7206. doi: 10.1128/JVI.00563-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nigrovic PA, Gray DH, Jones T, Hallgren J, Kuo FC, Chaletzky B, Gurish M, Mathis D, Benoist C, Lee DM. Genetic inversion in mast cell-deficient (W(sh)) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am J Pathol. 2008;173:1693–1701. doi: 10.2353/ajpath.2008.080407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, Si-Tahar M. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol. 2007;178:3368–3372. doi: 10.4049/jimmunol.178.6.3368. [DOI] [PubMed] [Google Scholar]
- 35.Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 37.Ichinohe T, I, Pang K, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol. 2010;11:404–410. doi: 10.1038/ni.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas PG, Dash P, Aldridge JR, Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, Doherty PC, Kanneganti TD. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M., Jr Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 43.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 44.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 45.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 46.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 47.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 48.Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschlager N, Schlee M, Rothenfusser S, Barchet W, Kato H, Akira S, Inoue S, Endres S, Peschel C, Hartmann G, Hornung V, Ruland J. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11:63–69. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 49.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen H, Sun H, You F, Sun W, Zhou X, Chen L, Yang J, Wang Y, Tang H, Guan Y, Xia W, Gu J, Ishikawa H, Gutman D, Barber G, Qin Z, Jiang Z. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell. 2011;147:436–446. doi: 10.1016/j.cell.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 51.Rao KN, Brown MA. Mast cells: multifaceted immune cells with diverse roles in health and disease. Ann N Y Acad Sci. 2008;1143:83–104. doi: 10.1196/annals.1443.023. [DOI] [PubMed] [Google Scholar]
- 52.Hu Y, Jin Y, Han D, Zhang G, Cao S, Xie J, Xue J, Li Y, Meng D, Fan X, Sun LQ, Wang M. Mast cell-induced lung injury in mice infected with H5N1 influenza virus. J Virol. 2012 doi: 10.1128/JVI.06053-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grant SM, Goa KL, Fitton A, Sorkin EM. Ketotifen. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in asthma and allergic disorders. Drugs. 1990;40:412–448. doi: 10.2165/00003495-199040030-00006. [DOI] [PubMed] [Google Scholar]
- 54.Hung CH, Suen JL, Hua YM, Chiang W, Chang HC, Chen CN, Jong YJ. Suppressive effects of ketotifen on Th1- and Th2-related chemokines of monocytes. Pediatr Allergy Immunol. 2007;18:378–384. doi: 10.1111/j.1399-3038.2007.00535.x. [DOI] [PubMed] [Google Scholar]
- 55.Hill SJ, Ganellin CR, Timmerman H, Schwartz JC, Shankley NP, Young JM, Schunack W, Levi R, Haas HL. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol Rev. 1997;49:253–278. [PubMed] [Google Scholar]
- 56.Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol. 2005;23:749–786. doi: 10.1146/annurev.immunol.21.120601.141025. [DOI] [PubMed] [Google Scholar]
- 57.Skoner DP, Gentile DA, Fireman P, Cordoro K, Doyle WJ. Urinary histamine metabolite elevations during experimental influenza infection. Ann Allergy Asthma Immunol. 2001;87:303–306. doi: 10.1016/s1081-1206(10)62244-2. [DOI] [PubMed] [Google Scholar]
- 58.Brown MG, Hermann LL, Issekutz AC, Marshall JS, Rowter D, Al-Afif A, Anderson R. Dengue virus infection of mast cells triggers endothelial cell activation. J Virol. 2011;85:1145–1150. doi: 10.1128/JVI.01630-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marcet CW, St Laurent CD, Moon TC, Singh N, Befus AD. Limited replication of influenza A virus in human mast cells. Immunol Res. 2012 doi: 10.1007/s12026-012-8377-4. [DOI] [PubMed] [Google Scholar]
- 60.Fukuda M, Ushio H, Kawasaki J, Niyonsaba F, Takeuchi M, Baba T, Hiramatsu K, Okumura K, Ogawa H. Expression and Functional Characterization of Retinoic Acid-Inducible Gene-I-Like Receptors of Mast Cells in Response to Viral Infection. J Innate Immun. 2012 doi: 10.1159/000343895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Horner SM, Liu HM, Park HS, Briley J, Gale M., Jr Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A. 2011;108:14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nazmi A, Mukhopadhyay R, Dutta K, Basu A. STING Mediates Neuronal Innate Immune Response Following Japanese Encephalitis Virus Infection. Sci Rep. 2012;2:347. doi: 10.1038/srep00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Varga ZT, Ramos I, Hai R, Schmolke M, Garcia-Sastre A, Fernandez-Sesma A, Palese P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011;7:e1002067. doi: 10.1371/journal.ppat.1002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Varga ZT, Grant A, Manicassamy B, Palese P. The Influenza Virus Protein PB1-F2 Inhibits the Induction of Type I Interferon by Binding to MAVS and Decreasing the Mitochondrial Membrane Potential. J Virol. 2012 doi: 10.1128/JVI.01122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dudek SE, Wixler L, Nordhoff C, Nordmann A, Anhlan D, Wixler V, Ludwig S. The influenza virus PB1-F2 protein has interferon antagonistic activity. Biol Chem. 2012;392:1135–1144. doi: 10.1515/BC.2011.174. [DOI] [PubMed] [Google Scholar]
- 67.Kulka M, Alexopoulou L, Flavell RA, Metcalfe DD. Activation of mast cells by double-stranded RNA: evidence for activation through Toll-like receptor 3. J Allergy Clin Immunol. 2004;114:174–182. doi: 10.1016/j.jaci.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 68.Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, Gonzalez-Dosal R, Rasmussen SB, Christensen MH, Yarovinsky TO, Rixon FJ, Herold BC, Fitzgerald KA, Paludan SR. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol. 2012 doi: 10.1038/ni.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seo YJ, Blake C, Alexander S, Hahm B. Sphingosine 1-phosphate-metabolizing enzymes control influenza virus propagation and viral cytopathogenicity. J Virol. 2010;84:8124–8131. doi: 10.1128/JVI.00510-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.