

Abstract
Antigen activation of the B-cell receptor (BCR) may play a role in the pathogenesis of human follicular lymphoma (FL) and other B-cell malignancies. However, the nature of the antigen(s) recognized by tumor BCRs has not been well studied. Here, we used unbiased approaches to demonstrate that 42 (19.35%) of 217 tested FL immunoglobulins (Igs) recognized vimentin as a shared autoantigen. The epitope was localized to the N-terminal region of vimentin for all vimentin-reactive tumor Igs. We confirmed specific binding to vimentin by using recombinant vimentin and by performing competitive inhibition studies. Furthermore, using indirect immunofluorescence staining, we showed that the vimentin-reactive tumor Igs colocalized with an anti-vimentin monoclonal antibody in HEp-2 cells. The reactivity to N-terminal vimentin of IgG FL Igs was significantly higher than that of IgM FL Igs (30.4% vs. 10%; P=0.0022). However, vimentin-reactive FL Igs did not share complimentarity determining region 3 motifs and were not homologous. Vimentin was expressed in the T-cell rich regions of FL, suggesting that vimentin is available for binding with tumor BCRs within the tumor microenvironment. Vimentin was also frequently recognized by mantle cell lymphoma and multiple myeloma Igs. Our results demonstrate that vimentin is a shared autoantigen recognized by nonstereotyped FL BCRs and by the Igs of mantle cell lymphoma and multiple myeloma and suggest that vimentin may play a role in the pathogenesis of multiple B-cell malignancies. These findings may lead to better understanding of the biology and natural history of FL and other B cell malignancies.
Keywords: lymphoma, B-cell receptor, antigen, vimentin, pathogenesis
Introduction
Human B-cell lymphomas are characterized by recurrent chromosomal translocations; however, they may not be sufficient for lymphomagenesis. The t(14;18) translocation in follicular lymphoma (FL), the most common indolent B-cell lymphoma,(1, 2) results in the juxtaposition of the BCL2 gene next to the immunoglobulin (Ig) heavy (H) chain locus leading to hyperexpression of the antiapoptotic bcl-2 protein that provides a survival advantage to the tumor cells. However, the presence of t(14;18) translocation in a substantial proportion of healthy individuals suggests that it is not sufficient to initiate lymphomagenesis(3–5) and that other factors may be necessary. This suggestion is also supported by the observation that lymphoma developed in only 10%–15% of transgenic mice, in which BCL2 expression was driven by an IgH enhancer (Eμ).(6) Alternative factors that may promote lymphomagenesis include antigenic stimulation of the B-cell receptor (BCR), that may act in concert with intracellular genetic alterations by providing survival and proliferation signals.(7)
Several reports suggest a possible role for antigen activation in the pathogenesis of FL. Most FL tumors express membrane-bound Igs, and the loss of BCRs is extremely rare in FL.(8) FL tumors exhibit ongoing somatic hypermutation (SHM) during tumor growth, and the patterns of these mutations suggest their selection by antigen activation.(9, 10) Recently, cross-linking of BCRs was shown to activate mitogen-activated protein kinases Erk1/2 and p38 more rapidly and for longer duration in FL tumor B cells than in nontumor B cells, thereby suggesting that BCR signaling may promote the survival and proliferation of tumor B cells.(11) Together, these reports suggest a possible dependency of FL tumor B cells on BCRs for survival and proliferative signals and support the need for the identification of antigen(s) recognized by FL BCRs to better understand the pathogenesis and natural history of FL.
Despite the observations mentioned above, the nature of the antigen(s) recognized by FL BCRs has not been well studied. Dighiero et al. tested the reactivity of a small number of FL Igs against preselected autoantigens.(12) They observed that at least 8 of 31 FL Igs reacted to a self-antigen. The purpose of our study was to define the characteristics of FL Igs and to identify potential self-antigens recognized by them using an unbiased approach and without preconceptions about the nature of the antigenic ligands. To determine whether other tumor Igs may also recognize self-antigens, we tested tumor Igs derived from mantle cell lymphoma (MCL) and multiple myeloma (MM).
Materials and Methods
Reagents
The following reagents were obtained from commercial sources: recombinant human vimentin (GenWay), anti-vimentin V9 and H84 antibodies (Santa Cruz Biotechnology), horseradish peroxidase-conjugated mouse anti-6×His (BD Biosciences), rituximab (Genentech), cetuximab (ImClone Systems), calf thymus double-stranded DNA (Invitrogen), lipopolysaccharide and recombinant human insulin (Sigma-Aldrich), and goat antihuman Ig peroxidase conjugate (Jackson ImmunoResearch Laboratories). The human larynx epidermoid carcinoma (HEp-2) cell line was obtained from ATCC. ED38, mGO186, and mGO53 mAbs were kindly provided by Michel C. Nussenzweig.(13)
Generation of tumor Igs
The Institutional Review Board at The University of Texas MD Anderson Cancer Center approved all studies. Tumor Igs were generated from patients previously enrolled in clinical trials of idiotype vaccine after obtaining informed consent.(14–18) Tumor Igs from patients with previously untreated FL (n = 28) and MCL (n = 18) were generated by heterohybridoma fusion technology.(15, 16, 18, 19) Additional tumor Igs from patients with FL (n = 211; 180 untreated and 31 relapsed) were generated by cloning tumor-derived VH and VL sequences into a baculovirus expression vector system_containing the IgG1 and κ or λ constant region coding sequences as previously described.(14, 20) Tumor Igs from patients with previously untreated MM (n = 31) were purified from plasma by protein A affinity chromatography.(17) The monoclonal tumor IgG and IgA represented greater than 80% of the total IgG or IgA, respectively, in the plasma in all MM samples.
Sequencing and analysis of IgVH and IgVL
An Ig heavy- and light-chain gene library was constructed from fresh biopsy tissue from each patient using two different constant heavy- and light-chain cDNA primers with a commercially available 5' RACE kit (Invitrogen). Variable region gene sequences that appeared multiple times from two different cDNA reactions were considered tumor derived.(14, 20) Sequence analysis was performed using IMGT V-QUEST analysis software.(21) The usage of VH, JH, DH, Vκ, Jκ, Vλ, and Jλ genes; length of the CDR3 region; characteristics of each nucleotide mutation (replacement vs. silent) in each FWR and CDR; number of positively charged amino acids; and sites of N-glycosylation were determined as previously described.(21–24)
Analysis of amino acid frequencies
Amino acid frequencies were calculated from IMGT position 107 to 114, known as the “CDR-H3 loop” region. CDR-H3 loops derived from FL tumors were compared to a previously published compilation of normal, unique, human CDR-H3 loops obtained from the Immunogenetics database,(25) as previously described(26) CDR-H3 loops from FL tumors were also compared to normal human memory B cells; these sequences were further divided into IgM+ or IgG+ subsets.(23, 24)
Analysis of CDR-H3 hydrophobicity
The average hydrophobicity of each CDR-H3 loop was calculated with values from the normalized the Kyte-Doolittle hydrophobicity scale,(27, 28) which ranges from arginine at −1.3 to isoleucine at +1.7. The distribution of average hydrophobicities was then plotted for normal, human memory B cells and for FL tumors.
HEp-2 lysate preparation
The HEp-2 cell line (ATCC) was grown to approximately 70%–80% confluence at 37°C in 5% CO2 and Dulbecco's modified Eagle's_medium (Invitrogen) supplemented with 10% heat-inactivated ultra-low IgG fetal bovine serum (Invitrogen), 20 U/mL penicillin, and 20 μg/mL streptomycin (Invitrogen). Whole-cell lysates were prepared by lysing HEp-2 cells in cold lysate buffer (1% Triton X-100, 150 mM NaCl, 10 mM Thri [pH 7.4], 1 mM ethylenediaminetetraacetic acid, 1 mM ethylene glycol tetraacetic acid [pH 8.0], 0.2 mM sodium orthovanadate, 0.5% octylphenoxypolyethoxyethanol [Sigma-Aldrich]) in the presence of a protease inhibitor cocktail (Roche Applied Science). Hydrophilic cytoplasmic and nuclear proteins of HEp-2 cells were obtained by the use of NE-PER extraction reagents (Thermo Scientific) according to the manufacturer's protocol. After the hydrophilic proteins were extracted, the hydrophobic fraction was dissolved in 8 M urea. All protein concentrations were determined by using a 2-D Quant Kit (GE Healthcare).
Enzyme-linked immunosorbent assay (ELISA)
All proteins were dissolved in 100 mM sodium bicarbonated buffer (pH 9.6) to a concentration of 10 μg/mL, sonicated, coated onto 96-well ELISA plates, and stored overnight at 4°C. Coated plates were blocked with 3% nonfat milk (Bio-Rad) for 1 h at room temperature, incubated with tumor Igs (12.5 μg/mL in superblock T20 blocking buffer [Thermo Scientific]) for 1 h at room temperature, and washed with 0.05% Tween 20 in phosphate-buffered saline. Bound tumor Igs were detected with goat horseradish peroxidase-conjugated antihuman Ig diluted at 1:10,000 and 2,2'-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid peroxidase substrate (Kirkegaard & Perry Laboratories) and read at 405 nm in a microplate spectrophotometer (Molecular Devices). To determine whether vimentin inhibits the binding of tumor Igs to HEp-2 cell lysates, we preincubated tumor Igs (12.5 μg/mL) for 1 h with various concentrations of recombinant human vimentin protein or control glutathione S-transferase protein and then used them in ELISA.
Western blotting and mass spectrometry
All proteins were dissolved in 8 M urea and boiled for 5 min in 0.4% SDS, 5% glycerol, 2.9 mM 2-mercaptoethanol, 0.025% bromophenol blue, and 12 mM Tris (pH 6.8). Proteins were analyzed by SDS-PAGE using 4%–20% gradient gels (Bio-Rad) and stained with Coomassie Brilliant Blue R-250 (Thermo Scientific). Alternatively, after SDS-PAGE, proteins were transferred to a nitrocellulose membrane (Bio-Rad) for Western blotting with tumor Igs or V9 and H84 antibodies. Bound tumor Igs were detected with goat alkaline phosphatase-conjugated antihuman Ig diluted at 1:10,000 and a mixture of nitro-blue tetrazolium chloride and 5-bromo-4-chloro-3'-indolyphosphate p-toluidine salt (Thermo Scientific) as alkaline phosphatase substrates. Protein bands were excised from Coomassie Blue-stained SDS-PAGE gels, digested with trypsin, and subjected to sequential mass spectrometry (liquid chromatography-mass spectrometry; Protein Chemistry Core Facility, Tufts University).
Immunofluorescence staining
HEp-2 cell-coated slides (Bion Enterprises LTD) were incubated with tumor Igs (12.5 μg/mL) and anti-vimentin V9 mAb (1:250) in SuperBlock T20 Tris buffered saline blocking buffer (Thermo Scientific) for 1 h at room temperature. The slides were washed three times in phosphate-buffered saline and 0.05% Tween 20 and incubated with antihuman IgG or IgM antibodies conjugated to Alexa Fluor 594 (Invitrogen) and antimouse IgG antibody conjugated to Alexa Fluor 488 (Invitrogen) in the blocking buffer for 1 h at room temperature. The slides were washed as before and mounted on slides with ProLong Gold antifade mounting medium (Invitrogen), and images of the stained sections were captured using a confocal laser-scanning microscope (Leica SP2 SE). The raw confocal data were converted to 8-bit red, green, and blue split mode, and green and red channels were isolated for colocalization analysis using ImageJ software and relevant plugins. A threshold of 50% was applied to exclude false signals and improve the signal-to-noise ratio. The resulting colocalized pixels were obtained as grayscale images. A Pearson correlation coefficient of >0.8 (as close to 1 as possible) was considered statistically significant. Laser intensities were compensated with single-color controls, and autofluorescence was eliminated by background correction based on secondary antibody-only controls. All experiments were performed in triplicate and repeated at least three times to ensure reproducibility.
Cloning of full-length and truncated vimentin cDNA fragments
A 6×histidine tag bacterial expression vector, pQE-30 (Qiagen), was used to express full-length, N-terminal, and C- terminal human vimentin. The full-length vimentin gene was cloned into the TA PCR 2.1_vector (Invitrogen) from total RNA from the HEp-2 cell line by RT-PCR using vimentin-specific primers Fwd-Vim (5'-AAGGATCCATGTCCACCAGGTCCGTGTCCTCG-3') and Rev-Vim-R (5'-AAAAGCTTGTTTATTCAAGGTCATCGTGATG-3') containing BamHI (underlined) and HindIII (double underlined) restriction sites, respectively. The vimentin-containing TA PCR 2.1 vector DNA was then digested with BamHI and HindIII restriction endonucleases, and the resulting fragment was subcloned into the pQE-30 vector. To generate the C-terminal vimentin fragment (aa 260–467), we used 5'-AAGGATCCGATGTTTCCAAGCCTGACCTCACGGCTG-3' as a forward primer and Rev-Vim-R as a reverse primer. To generate N-terminal vimentin fragments Vim-N1–259, Vim-5N1–224, and Vim-4N1–137, we used 5'-AAAAGCTTGTTTACACATCGATTTGGACATGCTGTTC-3', 5'-AAAAGCTTGTTTACACTTTGCGTTCAAGGTCAAGACGTGC-3', and 5'-AAAAGCTTGTTTACTGCTCGAGCTCGGCCAGCAGGATCTT-3' as reverse primers, respectively, and Fwd-Vim as a forward primer. The reverse primers were constructed with a stop codon after the HindIII site in the sense strand. The cloned full-length vimentin and truncated vimentin fragments were confirmed by sequencing.
Generation of full-length and truncated vimentin protein
Overnight cultures of BL21 (DE3) cells transformed with each plasmid were diluted to a ratio of 1:50 in fresh TB broth supplemented with 100 μg/mL ampicillin. Cells were grown at 37°C for 3 h or until a cell density of A600 = 0.5 was reached. Recombinant protein expression was induced by the addition of 1.0 mM isopropyl-β-D-thiogalactopyranoside (Invitrogen), and cultures were grown for 4 h at 37°C. Cells were harvested by centrifugation for 20 min at 10,000 × g at room temperature and lysed for 1 h in lysis buffer (100 mM NaH2PO4, 10 mM Tris-Cl, 8 M urea, and sigma protease inhibitor mixture [pH 8.0]), and the supernatant was harvested after centrifugation for 30 min at 10,000 × g at room temperature. PBS-washed Ni2+-NTA beads (Qiagen) were added to the supernatants, incubated with agitation for 1 h at room temperature, and pelleted by centrifugation for 5 min at 3000 × g at room temperature. The beads were washed with wash buffer C (100 mM NaH2PO4, 10 mM Tris-Cl, 8 M urea, and sigma protease inhibitor mixture [pH 6.3]), repelleted, washed with wash buffer D (100 mM NaH2PO4, 10 mM Tris-Cl, 8 M urea, and sigma protease inhibitor mixture [pH 5.9]), and loaded onto a column. The recombinant full-length vimentin and truncated protein fragments were eluted with elution buffer E (100 mM NaH2PO4, 10 mM Tris-Cl, 8 M urea, and sigma protease inhibitor mixture [pH 4.5]). Each recombinant protein was characterized by SDS-PAGE and stained with Coomassie Brilliant Blue, and the protein concentration was determined using a 2-D Quant kit (GE Healthcare).
Immunohistochemical staining
Formalin-fixed, paraffin-embedded tissue sections of FL and normal tonsils were deparaffinized and rehydrated, and antigen retrieval was performed according to the manufacturer's protocol (Vector Laboratories). Sections were treated for 5 min with 0.3% hydrogen peroxide solution to block endogenous peroxidase and incubated with a blocking buffer (1% BSA in PBS; Sigma) for 5 min at room temperature. Next, slides were incubated with anti-vimentin antibody (BD Biosciences) overnight at 40C at a dilution of 1:100 in blocking buffer, washed, and incubated for 30 min with appropriate secondary antibodies in blocking buffer. For CD3 and CD20 double staining, a polymer cocktail composed of anti-mouse IgG/alkaline phosphatase and anti-rabbit IgG/HRP (Thermo Scientific) was used in conjunction with primary antibodies (Thermo Scientific). The sections were stained using DAB solution (Vector Laboratories). Digital photomicrographs were acquired using DP Controller software (Olympus) mounted on a BX41 inverted microscope (Olympus).
Statistical analysis
Statistical significance of data was determined by 2 × 2 or 2 × 5 Fisher's exact test (for determining the reactivity of tumor Igs, analyzing tumor Ig gene repertoires, analyzing CDR3 positive charges, and identifying sites of N-glycosylation) or Student's t test (for determining the length of the CDR3 region and the number of mutations). Amino acid sequence analysis was performed in Microsoft Excel 2007™. Once sequences were sorted into their respective subsets, macros and formulas were created to calculate amino acid frequencies. Differences between populations were assessed by χ2 analysis or Levene test for variance using JMP® 9.0.0 (SAS, Cary, North Carolina) and Prism® 5.0 (GraphPad Software Inc., La Jolla, CA).
Results
Characteristics of FL Igs
The isotype of the FL Igs used in this study was determined by flow cytometry of tumor samples derived from 239 patients (208 untreated and 31 relapsed). Consistent with previous reports,(8, 29) we observed that IgM was expressed more frequently than IgG in FL (119 vs. 98; 50% vs. 41%) (Fig. 1A). A subset of the tumors expressed IgD, IgA, or dual Igs (4.3% IgD+IgM, 2.6% IgA, 1.7% IgD, and 0.41% IgG+IgM). Interestingly, the kappa light chain was more frequently expressed in the IgM+ tumors, whereas the lambda light chain was more commonly expressed in the IgG+ tumors (Fig. 1B; P=0.0121). Sequencing of the variable regions of the heavy and light chains was performed on 198 FL tumors (167 untreated and 31 relapsed). The usage of VH, JH, DH, Vκ, Jκ, Vλ, and Jλ genes did not differ significantly between IgM+ and IgG+ FL tumors except the usage of VH3, which was more frequent in IgM+ FL than in IgG+ FL (76% vs. 56%; P=0.012) (Supplementary Fig. 1A, Supplementary Table 1).
Figure 1. Heavy and light chain usage, mutational status, and amino acid usage of tumor Igs in FL.

(A) The percentage of tumors (n = 239) expressing different heavy chains as determined by flow cytometry. (B) The percentage of IgM+ and IgG+ FL tumors (n = 239) expressing Vκ and Vλ light chains as determined by flow cytometry. P was calculated using a 2 × 2 Fisher's exact test. (C) The total number of mutations in VH, Vκ, and Vλ genes from FWR1 to FWR3 in individual IgM+ (n = 112) and IgG+ (n = 86) FL tumors. Each dot represents data from one tumor, and the horizontal lines represent the average number of mutations for the group. P values were calculated using a two-tailed Student's t-test. (D) The frequency of replacement (R; black bar) and silent (S; gray) mutations in VH, Vκ, and Vλ were calculated and expressed as follows in the bar graphs: (number of nucleotide exchanges in each FWR and CDR / total number of nucleotides in the respective FWR and CDR) × 100. The R:S mutation ratio for each FWR and CDR is shown on the x-axis. (E) The frequency of amino acid usage in CDR-H3 loops of all FL Igs (middle) was compared with normal human B cells (top). Amino acids are listed in order of hydrophobicity as per a normalized Kyte-Doolittle hydrophobicity scale. Differences in amino acid frequencies between the two groups (bottom) were assessed by χ2 analysis (**p<0.001, ***p<0.0001).
FL Igs are highly mutated and have altered amino acid usage
To determine whether somatic mutations may correlate with antigen reactivity, we analyzed the variable region sequences of FL Igs. We found that FL Igs had an average of 34.2, 19.7, and 20.4 somatic mutations in IgH, Igκ, and Igλ variable regions, respectively (Fig. 1C, Supplementary Table 1). The number of somatic mutations was significantly higher in IgG variable heavy (P=0.0195) and κ light chains (P=0.006) than in IgM+ FL tumors, but did not differ in the λ light chain (P=0.628). As expected, the mutations were more frequent in the complementarity determining regions (CDRs) than in the framework regions (FWRs) of both IgM+ and IgG+ FL tumors (Fig. 1D). Furthermore, the ratio of replacement mutations (nucleotide changes yielding amino acid replacements) to silent mutations (nucleotide changes not leading to amino acid replacements) was consistently higher in the CDRs than in the FWRs of both tumor types. The length of the CDR3 region and the number of positively charged amino acid (aa) residues did not differ significantly between the variable regions of IgM+ and IgG+ tumors (Supplementary Figs. 1 B, C; Supplementary Table 1). N-glycosylation sites were more commonly observed in IgM+ than in IgG+ tumors (Supplementary Figs. 1 D, E; P=0.0032). Comparison of tumor Igs from untreated and relapsed FL patients did not show significant differences in variable gene usage and the rate and pattern of somatic mutation. However, the high number of mutations in the tumor Ig variable regions and, in particular, the high ratio of replacement to silent mutations in the CDRs suggests an antigen-driven process in FL pathogenesis. Analysis of aa frequencies in the CDR3 regions of heavy chains (CDR-H3) showed that aa usage differed between FL and normal human CDR-H3 loops (Fig. 1E). Amino acid usage was also different between IgM+ and IgG+ FL and their respective normal memory B cells (Supplementary Figs. 2 A, B). But, the average CDR-H3 loop hydrophobicity was not significantly different between FL and normal memory B cells (Supplementary Fig. 2C).
Tumor Igs are frequently self-reactive
To determine whether tumor Igs are self-reactive, we tested their reactivity in an ELISA(30) against the HEp-2 cell line, which is commonly used to identify antigens recognized by autoantibodies.(13) We observed that 24 (11.06%) of 217 FL Igs were self-reactive and that IgG+ FL Igs were more commonly self-reactive than those derived from IgM+ FL (20.4% vs. 3.4%; P=0.0001) (Figs. 2A–C). Our results also showed that 6 (33.33%) of 18 MCL Igs and 14 (45.16%) of 31 MM Igs were self-reactive (Figs. 2 D, E). We did not observe any light-chain bias in HEp-2 reactivity in FL, MCL, or MM (Figs. 2B–D) (P>0.05 for FL, MCL, and MM). Together, these data suggest that tumor Igs from FL, MCL, and MM are frequently reactive against a self-antigen.
Figure 2. Tumor Igs are frequently self-reactive.

FL Igs were tested in triplicate for reactivity against HEp-2 whole-cell lysates using ELISA as described in Materials and Methods. (A) The percentage of IgM+ and IgG+ FL Igs reacting to HEp-2 whole-cell lysates. P was calculated using a 2 × 2 Fisher's exact test. (B–E) Reactivity of individual tumor Igs from (B) IgM+ FL (n = 119), (C) IgG+ FL (n = 98), (D) MCL (n = 18), and (E) MM (n = 31) against HEp-2 whole-cell lysates is shown by optical density (O.D.) determined at 405 nm. Each tumor Ig is indicated by a unique number, and a polyreactive ED38 antibody was used as a positive control (P). Nonreactive mG053 antibody (N), and rituximab (R) were used as negative controls. Tumor Igs were considered to be HEp-2-reactive if the O.D. was greater than 5 times the O.D. of the nonreactive mG053 antibody (indicated by red line). Error bars represent the standard deviation for triplicate samples, and the stars indicate HEp-2-reactive tumor Igs. Data were reproducible in three independent experiments.
Tumor Igs recognized vimentin
To identify the self-antigen recognized by the HEp-2-reactive tumor Igs, we extracted hydrophilic nuclear and cytoplasmic protein fractions and a hydrophobic fraction from Hep-2 cells and used them as antigenic material in separate ELISAs. We tested five randomly selected HEp-2-reactive tumor Igs (FL15, MCL29, MCL32, MCL43, and MM63) and found that they specifically recognized the hydrophobic fraction but not the hydrophilic nuclear or cytoplasmic fractions of HEp-2 cells (Fig. 3A). The hydrophobic fraction was dissolved in urea and subjected to SDS-PAGE and Western blotting. The five tumor Igs recognized a single protein band of approximately 56 kDa (Fig. 3B), which was then excised and analyzed by mass spectrometry. Using the Basic Local Alignment Search Tool to find regions of similarity and homology, we determined that seven peptide sequences matched and encompassed 23% of the vimentin protein (Fig. 3C). By performing ELISA (Fig. 3D) and Western blotting (Fig. 3E), we confirmed that the five tumor Igs specifically bound to recombinant human vimentin protein but not bovine serum albumin (Figs. 3 D, E). We did not observe any significant reactivity to vimentin by a non-HEp-2-reactive tumor Ig (FL1). To further validate the specificity, we preincubated the tumor Igs with recombinant human vimentin and observed that it significantly inhibited reactivity to the HEp-2 hydrophobic fraction in a dose-dependent manner, whereas preincubation with control glutathione S-transferase protein did not (Fig. 3F). In addition, using indirect immunofluorescence staining, we showed that the vimentin-reactive tumor Igs FL15, MCL32, and MM63 colocalized with a mouse anti-vimentin V9 mAb in HEp-2 cells but the non-HEp-2-reactive tumor Ig FL26 did not (Fig. 4). Taken together, these results suggest that the self-antigen recognized by the five HEp-2-reactive tumor Igs tested was vimentin.
Figure 3. Tumor Igs recognize vimentin.

Tumor Igs from FL (15), MCL (29, 32, 63), and MM (63) were tested in various assays to characterize the antigens they recognized. Polyreactive ED38 antibody was used as a positive control (P), and nonreactive mG053 antibody was used as a negative control (N) where appropriate. Error bars represent the standard deviation for triplicate samples. (A) Tumor Igs were tested in triplicate for reactivity against hydrophilic cytoplasmic and nuclear proteins fractions and a hydrophobic fraction from HEp-2 cells using ELISA as described in Materials and Methods. (B) The hydrophobic fraction from HEp-2 cells was dissolved in urea, subjected to SDS-PAGE, and transferred to nitrocellulose membrane. Immunoblotting was performed with tumor Igs (5 μg/ml), and bound tumor Igs were detected by goat alkaline phosphatase-conjugated antihuman Ig. (C) The corresponding bands (arrows in panel B) were excised from Coomassie Blue-stained gels and subjected to sequential mass spectrometry. Seven peptides (marked in red) that matched the vimentin protein sequence were observed. (D) Tumor Igs were tested in triplicate for reactivity against the HEp-2 hydrophobic fraction, commercially obtained recombinant human vimentin, and BSA (50 μl of 10 μg/ml) using ELISA. (E) Tumor Igs were tested for reactivity against commercially obtained recombinant human vimentin using Western blotting. (F) Tumor Igs were preincubated with various concentrations of recombinant human vimentin or glutathione S-transferase and then tested for reactivity against the HEp-2 hydrophobic fraction.
Figure 4. Vimentin-reactive tumor Igs colocalize with anti-vimentin V9 mAb.

Immunofluorescence staining of HEp-2 cells was performed with tumor Igs from FL15, FL26, MCL32, and MM63, and colocalization with anti-vimentin V9 mAb was determined. Each column represents staining with one tumor Ig. Top to bottom: V9 mAb alone (green), tumor Ig (red), V9 mAb and tumor Ig overlay, and colocalization pixels.
Tumor Igs recognized N-terminal region of vimentin
To determine the location of the epitope recognized by the vimentin-reactive tumor Igs, we generated recombinant N-terminal (aa 1–259) and C-terminal (aa 260–466) 6×His-tagged vimentin fusion proteins (Fig. 5A). We found that all five vimentin-reactive tumor Igs (FL15, MCL29, MCL32, MCL43, and MM63) bound to N- but not C-terminal vimentin fragments when tested by ELISA (Fig. 5B) and Western blotting (Fig. 5C). As expected, the C-terminal-specific mouse anti-vimentin V9 mAb recognized C-terminal vimentin, the N-terminal-specific rabbit anti-vimentin H84 polyclonal antibody recognized N-terminal vimentin, and the non-HEp-2-reactive FL1 Ig did not recognize either. To further define the precise location of the epitope, we generated additional truncated N-terminal vimentin protein fragments (Fig. 5A). Remarkably, four of the five HEp-2-reactive tumor Igs tested (FL15, MCL29, MCL32, and MCL43) bound to Vim-N (aa 1–259) protein fragments but not to Vim-5N (aa 1–224) or Vim-4N (aa 1–137) protein fragments (Fig. 5D). The MM63 Ig bound to Vim-N and Vim-5N protein fragments but not to Vim-4N protein fragment. These results suggest that the epitope recognized by four of the five vimentin-reactive tumor Igs derived from FL and MCL is located between aa residues 224 and 259 of the vimentin protein.
Figure 5. Tumor Igs recognize N-terminal region of vimentin.

(A) Structure of full-length and truncated vimentin generated with the head, tail, and subdomains 1A, 1B, 2A, and 2B connected by linkers. The aa numbers for each protein fragment are indicated. (B and D) Tumor Igs from FL (1, 15), MCL (29, 32, 43), and MM (63) were tested in triplicate for reactivity against full-length vimentin (Vim-W) and C-terminal (Vim-C) and N-terminal (Vim-N, Vim-5N, Vim-4N) vimentin fragments using ELISA. Mouse anti-vimentin V9 mAb specific for Vim-C and rabbit anti-vimentin H84 polyclonal antibody specific for Vim-N were used as positive controls. Anti-epidermal growth factor receptor antibody, cetuximab (Cx) was used as negative control. Error bars represent the standard deviation for triplicate samples. Results were reproducible in three independent experiments. (C) Tumor Igs were tested for reactivity against Vim-C and Vim-N fragments using Western blotting. Mouse anti-vimentin V9 mAb and mouse anti-6×His mAb were used as positive controls.
Nonstereotyped tumor Igs recognized N-terminal vimentin
Since the reactivity of some of the tumor Igs (MCL29, MCL32, and MCL43) was markedly higher to N-terminal-truncated vimentin than to full-length vimentin (Fig. 5B), we screened all available tumor Igs for reactivity against N-terminal vimentin (aa 1–259). We observed that 42 (19.35%) of 217 FL Igs, 9 (50%) of 18 MCL Igs, and 12 (38.7%) of 31 MM Igs were reactive with N-terminal-truncated vimentin (Figs. 6A–E). However, only a fraction of the tumor Igs that bound to N-terminal vimentin bound to full-length vimentin (26 (61.9%) of 42 FL Igs, 3 (33.3%) of 9 MCL Igs, and 6 (50%) of 12 MM Igs) (Supplementary Figs. 3A–D) and none of them bound to C-terminal vimentin (aa 260–466). Similar to the results of our experiments with HEp-2 cell reactivity (Figs. 2A–C), the N-terminal vimentin reactivity of IgG FL Igs was significantly higher than that of IgM FL Igs (30.4% vs. 10%; P=0.0022) (Figs. 6A–C). However, we did not observe any significant correlation between HEp-2 or vimentin reactivity and VH, JH, Vκ, Jκ, Vλ, and Jλ gene usage; CDR3 length; the number of positively charged aa residues in CDR3; the rate and pattern of mutations; or the rate and pattern of N-glycosylation (Table 1; Supplementary Table 1). Furthermore, the N-terminal vimentin-reactive FL Igs did not share CDR3 motifs and were not homologous (Table 1). Similarly, the vimentin-nonreactive FL Igs were not homologous (Supplementary Table 1). Interestingly, among the MM Igs, 11 (78.57%) of 14 IgGκ tumor Igs bound to N-terminal vimentin as opposed to 1 (10%) of 10 IgGλ tumor Igs and 0 (0%) of 7 IgA tumor Igs (Fig. 6E). However, we did not see this pattern of light chain bias in FL and MCL. The reactivity with vimentin was variable between samples and suggests differences in the binding affinity of the tumor Igs.
Figure 6. Reactivity of tumor Igs with N-terminal vimentin.

Tumor Igs were tested in triplicate for reactivity against N-terminal vimentin (Vim-N; aa 1–259) using ELISA as described in Materials and Methods. (A) The percentage of IgM+ and IgG+ FL Igs reacting to Vim-N. P was calculated using a 2 × 2 Fisher's exact test. (B–E) Reactivity of individual tumor Igs from (B) IgM+ FL (n = 119), (C) IgG+ FL (n = 98), (D) MCL (n = 18), and (E) MM (n = 31) against Vim-N is shown by optical density (O.D.) determined at 405 nm. Each tumor Ig is indicated by a unique number, and vimentin-reactive MM tumor Ig 63 was used as a positive control (P). Nonreactive mG053 mAb (N) and cetuximab (Cx) were used as negative controls. Tumor Igs were considered to be Vim-N-reactive if the O.D. was greater than 5 times the O.D. of the negative control cetuximab (indicated by line). Error bars represent the standard deviation for triplicate samples. The black stars indicate Vim-N-specific tumor Igs, and the gray stars indicate polyreactive tumor Igs (Fig. 7). Results were reproducible in three independent experiments.
Table 1.
Heavy and light chain CDR3 sequences of vimentin-reactive FL Igs
| PtID # | Isotype | VH | D | JH | VH-CDR3 | VL | JL | VL-CDR3 |
|---|---|---|---|---|---|---|---|---|
| 259 | IgMκ | 3-11 | 2-21 | 3 | AKNRSHT VMVFNN ETRRNDA TRHDVFD I | Vκ1-33 | 2 | QQCDNF PYT |
| 195 | IgMκ | 3-23 | 5-12 | 6 | ATSRGYD NYMDV | Vκ3-20 | 1 | HQYGRS PQT |
| 185 | IgMλ | 3-33 | 3-10 | 3 | ARNATST SGTYSVG YAFDF | Vλ2-23 | 3 | CSYAGTS TLV |
| 212 | IgMκ | 3-48 | 2-21 | 5 | AKGSGLL SNAFDP | Vκ1-39 | 1 | QQSYSG PWT |
| 286 | IgMκ | 3-48 | 6-19 | 5 | ARVGGA VRHWFD P | Vκ3-11 | 3 | QQRTNW PPT |
| 186 | IgMκ | 3-7 | 3-10 | 4 | ARNRSEF DY | Vκ2-30 | 4 | MQGTHW PPFT |
| 314 | IgMκ | 4-34 | 2-2 | 4 | VRGEIVIV PASRRSY MDV | Vκ3-20 | 5 | QQYGNS YPVT |
| 209 | IgMκ | 4-39 | 4-17 | 4 | ARRPLNG VTTVTHE DY | Vκ3-11 | 4 | QQRSNW PLT |
| 305 | IgMk | 4-59 | 3-16 | 5 | ARDRGG DRFDH | Vκ4-1 | 1 | HQYYNSP AT |
| 310 | IgMλ | 4-59 | 5-24 | 6 | ARHREA NIQVTNT SPLSDG MDV | Vλ3-25 | 3 | QSADST GTCV |
| 337 | IgMλ | 4-59 | 6-19 | 3 | ARGAVAG SLDGFDV | Vλ2-14 | 3 | TSFTSST TLV |
| 224 | IgGk | 1-18 | 4-17 | 6 | ATADGAY NTMDV | Vκ1-5 | 5 | QSYKVY GVS |
| 231 | IgGk | 1-18 | 1-1 | 5 | VNASGTV GDWLDP | Vκ1-39 | 2 | QQTYSTL RET |
| 294 | IgGk | 1-18 | 2-21 | 6 | ARNSTSV SYSDTD WHGMDF | Vκ1-16 | 1 | QQYYVY PPT |
| 200 | IgGλ | 1-46 | 4 | 4 | ARDDYIK TRLYFLD S | Vλ1-47 | 3 | TTWDDSL PGYWV |
| 228 | IgGλ | 1-69 | 6-6 | 6 | ARGLFYS TSSRYYH YYAMDV | Vλ1-44 | 3 | AAWDDS LNGWV |
| 301 | IgGk | 1-8 | 6-19 | 4 | AVNYTSG SLDY | Vκ4-1 | 1 | QQYYNT PRT |
| 266 | IgGλ | 3-15 | 4-4 | 5 | VNSTNN WAGNNL VDP | Vλ1-44 | 7 | AAWDDS LSGLV |
| 13 | IgGλ | 3-21 | 6-6 | 6 | VRNSSSS AGSDYC GLDV | Vλ1-40 | 7 | QSYDSR VSGSVV |
| 313 | IgGλ | 3-21 | ARIAGGA TVLVHSY VMDV | Vλ2-18 | ALFTNNL KWV | |||
| 316 | IgGλ | 3-21 | 3-3 | 6 | TRDLRPS FWSAFQ RLAYYYG MDV | Vλ3-19 | 2 | NSRDTN DNHVL |
| 351 | igGk | 3-23 | 6-13 | 4 | SAAVGLP LET | Vκ3-20 | 1 | RQYGHS PQT |
| 15 | IgGk | 3-30 | 3-10 | 4 | AKPPTP WKGTNY YLDF | Vκ3-11 | 3 | QQRRV |
| 253 | IgGA | 3-30 | 1-1 | 3 | VNKTGTH GFDI | Vλ2-8 | 3 | SSYAGG DNFLWV |
| 211 | IgGk | 3-48 | 4-17 | 4 | ARVKKS WGRLGG LDL | Vκ1-39 | 1 | QQTYSG PQT |
| 384 | IgGk | 3-48 | 3-9 | 5 | ARTTACN TSDCSVK AFDC | Vκ3-15 | 2 | QQYSNW PPYT |
| 323 | IgGλ | 3-48 | 3-3 | 3 | VRNESW AFDM | Vλ2-11 | 3 | CSYAGSF TWV |
| 325 | IgGλ | 3-48 | 3-3 | 1 | ARHRVR SGNYAKT LEK | Vλ1-44 | 3 | AAWDDS LNGWV |
| 280 | IgGλ | 3-49 | 3-16 | 3 | ARSPQM NLLVDLPI IFGNNVL DL | Vλ1-40 | 2 | QSYDKTR RSLL |
| 339 | IgGk | 3-53 | 3-16 | 6 | ARESLAG EEIPNGM DV | Vκ4-1 | 2 | QQFYSSS RT |
| 189 | IgGλ | 3-53 | 4-23 | 6 | AIGTSTT VGGMDV | Vλ1-47 | 2 | AAWDDT LSGPV |
| 265 | IgGλ | 3-74 | 3-10 | 4 | TRNASGP LMDFDK | Vλ4-69 | 3 | QTWGTGI DWV |
| 25 | InGλ | 3-h | 1-26 | 5 | TRDLYNG AYTLHP | Vλ1-44 | 2 | ASWDDR LNGLL |
| 21 | IgGk | 4-34 | 1-7 | 6 | AEGGPK SYYGLDV | Vκ3-15 | 1 | QQSSHG PPKT |
| 245 | IgGk | 4-39 | 1-1 | 5 | ATNFSRN SNDKPFV Y | Vκ1-5 | 4 | QQYNFS ST |
| 299 | IgGk | 4-39 | 2-2 | 5 | VKRSTSG RNWFDP | Vκ1-39 | 4 | QQTYSIP LT |
| 285 | IgGλ | 4-39 | 4-17 | 6 | ARNMTLV TGGFYGL DV | Vλ1-47 | 3 | AIWDDRL SGWV |
| 317 | IgGλ | 4-39 | 2-2 | 4 | VRLNSTT FFDN | Vλ1-51 | 3 | GTWNSS LTGNTSL SVHWV |
| 303 | IgGλ | 4-59 | 3-10 | 4 | ARGPQIR LHFGESL LFDL | Vλ2-23 | 2 | CSYASSN TFI |
Pt 9,23, and 27: Data not available; Pt 23: polyreactive tumor Ig; Gray boxes: polyreactive tumor Igs
To determine whether the N-terminal vimentin-reactive tumor Igs were polyreactive, we tested their reactivity against a panel of antigens, including double-stranded DNA, insulin, and lipopolysaccharide, by ELISA as previously described.(13) Tumor Igs that bound to at least one of these antigens were considered polyreactive. We observed that 14 (33.33%) of 42 FL, 0 (0%) of 7 MCL, and 2 (16.67%) of 12 MM N-terminal vimentin-reactive tumor Igs were polyreactive (Figs. 7A–D). The IgG FL Igs were more commonly polyreactive than the IgM FL Igs but did not share recurrent CDR3 motifs (Table 1). Interestingly, we did not observe polyreactivity in any of the non-vimentin-reactive or non-HEp-2-reactive FL Igs (Supplementary Table 1). Taken together, these results suggest that N-terminal vimentin is frequently recognized by nonstereotyped FL BCRs as well as MCL and MM Igs.
Figure 7. Polyreactive N-terminal vimentin-reactive tumor Igs.

N-terminal vimentin-reactive tumor Igs (Fig. 6) were tested in triplicate for reactivity against insulin (inverted triangle), dsDNA (black circle), and LPS (gray circle) using ELISA as described in Materials and Methods. Reactivity of individual tumor Igs from (A) IgM+ FL (n = 12), (B) IgG+ FL (n = 30), (C) MCL (n = 7), and (D) MM (n = 12) against dsDNA, LPS, and insulin is shown by optical density (O.D.) determined at 405 nm. Each tumor Ig is indicated by a unique number, and ED38 mAb was used as a positive control (P). Cetuximab (C) and rituximab (R) were used as negative controls. Tumor Igs were considered to be polyreactive if the O.D. at 405 nm was > 5-fold compared with the negative control, cetuximab (indicated by line). Error bars represent the standard deviation for triplicate samples. The results were reproducible in three independent experiments.
We also examined whether the method of tumor Ig generation impacted their reactivity as glycosylation can be different in insect cells compared with mammalian cells. We found that 7/28 (25%) heterohybridoma-derived FL Igs were vimentin-reactive of which 4/28 (14%) were vimentin-specific and 3/28 (11%) were polyreactive (Supplementary Table 1). Of the 189 baculovirus system-derived FL Igs, 35 (19%) were vimentin-reactive of which 22/189 (12%) were vimentin-specific and 13/189 (7%) were polyreactive (Supplementary Table 1). Furthermore, 9/18 (50%) MCL Igs all of which were generated by heterohybridoma technology were vimentin-specific. Together, these results suggest that the expression system for the FL Igs did not appear to impact their reactivity.
Vimentin is expressed in lymphoma tissues
To determine whether vimentin is expressed in lymphoma tissues, we performed immunohistochemical staining of formalin-fixed, paraffin-embedded tissue sections. We found that vimentin expression was present predominantly in the T cell-rich interfollicular regions of FL (Fig. 8). Vimentin expression was cytoplasmic and possibly membranous on T cells, and this result suggested that vimentin may be available for binding to the BCR of tumor cells within the tumor microenvironment.
Figure 8. Vimentin is expressed in FL tissues.
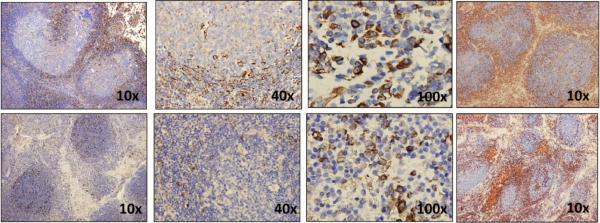
Immunohistochemical staining for vimentin (left three columns) was performed on formalin-fixed, paraffin-embedded FL (top row) and normal tonsil (bottom row) tissues. Original magnification, ×10, ×40, and ×100 are shown. Immunohistochemical staining was also performed for CD3 (red) and CD20 (blue) in the same tissues (farthest right column) and shown at original magnification ×10. The data represent one of two samples tested for FL and normal tonsil.
Discussion
Our results showed that a significant proportion of FL Igs recognized vimentin as a shared autoantigen suggesting a possible role for self antigens in the pathogenesis of FL. A recent report by Sachen and colleagues that was published as this manuscript was under preparation showed that 26% of FL Igs were Hep-2-reactive and the self-antigen was identified as myoferlin for one tumor Ig.(31) Using very stringent criteria and a different assay, we found that 11% of FL Igs were Hep-2-reactive and 19.35% were vimentin-reactive when tested using purified N-terminal vimentin protein. The higher reactivity seen with purified protein as compared with Hep-2 cell lysate may be due to better availability of antigenic epitopes. Under normal conditions, naïve mature B cells proliferate vigorously when their BCR is exposed to the cognate self or non-self antigens in secondary lymphoid organs. If the antigen-activated B cells already contain t(14;18) chromosomal translocation, they are likely to have a survival advantage because of the hyperexpressed antiapoptotic bcl-2 protein. If such antigen-activated t(14;18)-containing B cells are specific to abundantly expressed self antigens like vimentin, they are likely to undergo chronic stimulation, proliferate indefinitely, and develop into FL (Fig. 9). However, if such antigen-activated t(14;18)-containing B cells are specific to non-self antigens, they may differentiate normally into memory B cells and plasma cells and may undergo limited proliferation due to brief antigen exposure. The presence of multiple memory B-cell clones with t(14;18) in up to 60% of healthy individuals without apparent development of lymphoma(3–5) suggests that in most individuals, the t(14;18)-containing B cells are probably specific to non-self antigens. Thus, we speculate that for FL to develop, the t(14;18)-containing B cells need to be specific for self antigen. FL may be initially dependent on the self antigen for survival and proliferation but may eventually become antigen independent as additional genetic alterations are acquired, especially those leading to the transformation to diffuse large B-cell lymphoma.(32)
Figure 9. Hypothetical model for the role of antigens in the pathogenesis of FL.

First and second row. During normal B-cell development, naïve mature B cells arise from B-cell progenitors in the bone marrow and are released into the periphery. Mature B cells activated by a self or non-self antigen undergo affinity maturation by a process of somatic hypermutation (SHM) in the germinal centers of secondary lymphoid organs. A subset of these B cells with improved binding to the antigen may undergo clonal expansion and class switch recombination (CSR) and eventually differentiate into memory B cells and plasma cells. Chronic exposure to self-antigen may lead to autoimmunity depending on whether the self antigen-specific B cells are subjected to mechanism of self-tolerance or not. Third row. Because of errors during the V(D)J recombination in early B-cell development, chromosomal translocations such as t(14;18) may be introduced in the B-cell progenitors in the bone marrow. Mature B cells containing t(14;18) hyperexpress the antiapoptotic bcl-2 protein and have a survival advantage following antigen activation in the periphery. If the antigen-activated B cells containing t(14;18) are specific to self antigen, they undergo chronic stimulation and proliferation and may develop into FL. The FL may initially undergo antigen-dependent proliferation but may eventually become antigen independent as they acquire additional genetic alterations. Fourth row. Antigen-activated B cells containing t(14;18) that are specific to non-self antigens may differentiate normally into memory B cells and into plasma cells and may not develop into FL due to brief antigen exposure.
Despite the shared reactivity, IgVH- or IgVL-CDR3 motifs were not recurrent among the vimentin-reactive FL Igs (Table 1). In contrast, a restricted Ig repertoire with a shared CDR3 homology and shared antigen reactivity has been reported in CLL and MALT lymphomas.(33–36) However, the recognition of vimentin as a shared antigen by nonstereotyped FL BCRs was an unexpected finding in our study and supports our notion that vimentin may play a role in the pathogenesis of FL. Interestingly, we also observed that vimentin was recognized by tumor Igs from MCL and MM. Whereas FL and MCL tumors express Ig on their cell surface, MM is a disorder of plasma cells that do not express surface Ig but secrete the Ig.(7) Therefore, the role of antigen activation is less clear in the propagation of MM although the myeloma stem cell is presumed to be a postgerminal center B cell and may express surface Ig.(37) The reactivity to an abundantly expressed self-antigen such as vimentin was unexpected since B cells with self-reactive BCRs normally undergo clonal deletion. However, the presence of anti-vimentin IgG antibodies in the serum of healthy individuals and various pathological conditions(38, 39) suggested that vimentin-specific B cells are not centrally deleted in most individuals. The presence of anti-vimentin antibodies in healthy individuals(38) also suggests that they are not necessarily associated with autoimmune phenomena. However, anti-vimentin antibodies targeting the vimentin/cardiolipin complex occur in patients with systemic lupus erythematosus, antiphospholipid syndrome, and other autoimmune conditions, and may play a pathological role in initiating thrombosis.(39) Since lymphoma and myeloma patients are known to be at increased risk of thrombosis, it would be of interest to evaluate whether there is a correlation between vimentin-reactivity of the tumor Ig and thrombosis risk.
Vimentin is a class III intermediate filament protein that is expressed intracellularly and is an important cytoskeletal protein in many cell types. However, recent studies have demonstrated that vimentin may also be secreted by endothelial cells(40) and activated macrophages(41) in response to proinflammatory stimuli. Indeed, vimentin was shown to be present in the serum of healthy individuals.(40) Furthermore, vimentin is expressed on the cell surface of activated and apoptotic T cells, macrophages, neutrophils, platelets, viable tumor cells, and endothelial cells.(33, 41–45) Cell surface vimentin may mediate the binding of Streptococcus pyogenes(46) and may also function as a signal for the phagocytosis of apoptotic cells.(47) We found that all vimentin-reactive tumor Igs recognized the N-terminal fragment of vimentin but not the C-terminal fragment. The N-terminal fragment of vimentin was more commonly recognized by tumor Igs than full-length vimentin, presumably because of better exposure of the epitope in the N-terminal fragment (Fig. 6; Supplementary Fig. 3). The N-terminal fragment of vimentin is likely to be generated during apoptotic cell death since Asp259 in its L1-2 linker domain has been shown to be a cleavage site for caspase-6 and corresponds to similar cleavage sites in other intermediate filament proteins such as lamins and cytokeratins, which are also substrates for caspases during apoptosis.(48) Additionally, the 1B domain from the N-terminal region of vimentin is rapidly expressed on the cell surface after activation of normal T cells.(42) Taken together with our observation that vimentin is present in the T cell-rich interfollicular regions of FL (Fig. 8), these reports suggest that full-length vimentin or its N-terminal fragment may be secreted or expressed on the surface of nonmalignant cells under physiological conditions. Furthermore, they suggest that vimentin is potentially available for antigenic stimulation of the tumor BCRs in the tumor microenvironment.
The pattern of distribution of replacement and silent mutations in the CDRs and FWRs of FL (Fig. 1D) that we observed was similar to that reported in memory B cells in healthy individuals.(23, 24) However, the average somatic mutation frequency in FL tumors was significantly higher than the average somatic mutation frequency in healthy donor memory B cells (33.0 vs. 9.6 for IgH, 18.0 vs. 4.0 for Igκ, and 18.96 vs. 5.4 for Igλ for IgM+ FL tumors vs. IgM+ memory B cells(24) and 36.8 vs. 18.0 for IgH, 25.4 vs. 10.0 for Igκ, and 20.3 vs. 7.9 for Igλ for IgG+ FL tumors vs. IgG+ memory B cells).(23) Consistent with previous reports,(22) the number of N-glycosylation sites was significantly higher in FL Igs than in normal Igs. The usage of V and J genes in IgH, Igκ, and Igλ was comparable between FL tumors and healthy donor memory B cells except for Jλ-7, which was more common in FL.(23, 24) We also found that the variation in CDR3 length, the average hydrophobicity, and the number of positively charged aa residues in FL was comparable to that in healthy donor memory B cells.(23, 24) Nonetheless, we observed that the aa usage in FL is altered (Fig. 1E; Supplementary Fig. 2) suggesting that the FL repertoire may be distinct. But, without direct comparison with normal germinal center B cells, we cannot state with certainty whether the FL repertoire represents transformation of a distinct repertoire or antigen-driven selection from a normal B-cell repertoire. The likelihood, however, is the latter; that this repertoire represents antigen-driven selection for a specific type of reactivity, possibly self-reactivity like CLL.
In conclusion, our results show that vimentin is a shared autoantigen recognized by nonstereotyped FL BCRs as well as tumor Igs from MCL and MM. These findings suggest that vimentin may play a role in the pathogenesis of these malignancies and may lead to better understanding of their biology and natural history. It is likely that other self antigens are also recognized by tumor Igs and additional studies are needed to identify them.
Supplementary Material
Acknowledgments
The tissue samples were provided by the University of Texas MD Anderson Cancer Center Lymphoma Tissue Bank. We thank Markeda Wade for editorial assistance.
Grant support: This work was supported by an American Society of Clinical Oncology Career Development Award (SSN), an American Society of Hematology Junior Faculty Scholar Award in Clinical/Translational Research (SSN), Leukemia & Lymphoma Society of Translational Research Program (LWK),National Institutes of Health grants K23CA123149 (SSN), R21AI088498 (LSW, HWS), R01AI090742 (HWS) and U01AI090902 (HWS), and The University of Texas MD Anderson Cancer Center Institutional Research Grant Program (SSN). The Lymphoma Tissue Bank and the DNA Analysis Core Facility are supported by the MD Anderson Cancer Center Support Grant CA16672 (National Institutes of Health).
Footnotes
SCC and HQ contributed equally to this work.
Author Contributions The authors have made the following declarations about their contributions: Designed the study: SCC, HQ, LWK, SSN. Performed the experiments: SCC, SK, SR, WW, JO, JW, BK, SK, MSP, DSP, FEB, KK. Analyzed the data: SCC, HQ, SK, SR, LSW, WW, JO, JW, BK, SK, MSP, DSP, FEB, RED, HWS, LWK, SSN. Provided the samples: AL. Wrote the paper: SCC, RED, SSN. Supervised the experiments: HQ Directed the work: LWK, SSN. All authors read and approved the manuscript.
Disclosure of Conflicts of Interest The authors declare no competing financial interests.
References
- 1.Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 2.Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science. 1985;229:1390–1393. doi: 10.1126/science.3929382. [DOI] [PubMed] [Google Scholar]
- 3.Limpens J, Stad R, Vos C, de Vlaam C, de Jong D, van Ommen GJ, Schuuring E, Kluin PM. Lymphoma-associated translocation t(14;18) in blood B cells of normal individuals. Blood. 1995;85:2528–2536. [PubMed] [Google Scholar]
- 4.Roulland S, Navarro JM, Grenot P, Milili M, Agopian J, Montpellier B, Gauduchon P, Lebailly P, Schiff C, Nadel B. Follicular lymphoma-like B cells in healthy individuals: a novel intermediate step in early lymphomagenesis. J Exp Med. 2006;203:2425–2431. doi: 10.1084/jem.20061292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dolken G, Dolken L, Hirt C, Fusch C, Rabkin CS, Schuler F. Age-dependent prevalence and frequency of circulating t(14;18)-positive cells in the peripheral blood of healthy individuals. J Natl Cancer Inst Monogr. 2008:44–47. doi: 10.1093/jncimonographs/lgn005. [DOI] [PubMed] [Google Scholar]
- 6.McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18) Nature. 1991;349:254–256. doi: 10.1038/349254a0. [DOI] [PubMed] [Google Scholar]
- 7.Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5:251–262. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- 8.Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, Delsol G, De Wolf-Peeters C, Falini B, Gatter KC, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994;84:1361–1392. [PubMed] [Google Scholar]
- 9.Aarts WM, Bende RJ, Steenbergen EJ, Kluin PM, Ooms EC, Pals ST, van Noesel CJ. Variable heavy chain gene analysis of follicular lymphomas: correlation between heavy chain isotype expression and somatic mutation load. Blood. 2000;95:2922–2929. [PubMed] [Google Scholar]
- 10.Bahler DW, Levy R. Clonal evolution of a follicular lymphoma: evidence for antigen selection. Proc Natl Acad Sci U S A. 1992;89:6770–6774. doi: 10.1073/pnas.89.15.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irish JM, Czerwinski DK, Nolan GP, Levy R. Altered B-cell receptor signaling kinetics distinguish human follicular lymphoma B cells from tumor-infiltrating nonmalignant B cells. Blood. 2006;108:3135–3142. doi: 10.1182/blood-2006-02-003921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dighiero G, Hart S, Lim A, Borche L, Levy R, Miller RA. Autoantibody activity of immunoglobulins isolated from B-cell follicular lymphomas. Blood. 1991;78:581–585. [PubMed] [Google Scholar]
- 13.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 14.Freedman A, Neelapu SS, Nichols C, Robertson MJ, Djulbegovic B, Winter JN, Bender JF, Gold DP, Ghalie RG, Stewart ME, Esquibel V, Hamlin P. Placebo-controlled phase III trial of patient-specific immunotherapy with mitumprotimut-T and granulocyte-macrophage colony-stimulating factor after rituximab in patients with follicular lymphoma. J Clin Oncol. 2009;27:3036–3043. doi: 10.1200/JCO.2008.19.8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neelapu SS, Baskar S, Gause BL, Kobrin CB, Watson TM, Frye AR, Pennington R, Harvey L, Jaffe ES, Robb RJ, Popescu MC, Kwak LW. Human autologous tumor-specific T-cell responses induced by liposomal delivery of a lymphoma antigen. Clin Cancer Res. 2004;10:8309–8317. doi: 10.1158/1078-0432.CCR-04-1071. [DOI] [PubMed] [Google Scholar]
- 16.Neelapu SS, Kwak LW, Kobrin CB, Reynolds CW, Janik JE, Dunleavy K, White T, Harvey L, Pennington R, Stetler-Stevenson M, Jaffe ES, Steinberg SM, Gress R, Hakim F, Wilson WH. Vaccine-induced tumor-specific immunity despite severe B-cell depletion in mantle cell lymphoma. Nat Med. 2005;11:986–991. doi: 10.1038/nm1290. [DOI] [PubMed] [Google Scholar]
- 17.Neelapu SS, Munshi NC, Jagannath S, Watson TM, Pennington R, Reynolds C, Barlogie B, Kwak LW. Tumor antigen immunization of sibling stem cell transplant donors in multiple myeloma. Bone Marrow Transplant. 2005;36:315–323. doi: 10.1038/sj.bmt.1705057. [DOI] [PubMed] [Google Scholar]
- 18.Bendandi M, Gocke CD, Kobrin CB, Benko FA, Sternas LA, Pennington R, Watson TM, Reynolds CW, Gause BL, Duffey PL, Jaffe ES, Creekmore SP, Longo DL, Kwak LW. Complete molecular remissions induced by patient-specific vaccination plus granulocyte-monocyte colony-stimulating factor against lymphoma. Nat Med. 1999;5:1171–1177. doi: 10.1038/13928. [DOI] [PubMed] [Google Scholar]
- 19.Carroll WL, Thielemans K, Dilley J, Levy R. Mouse × human heterohybridomas as fusion partners with human B cell tumors. J Immunol Methods. 1986;89:61–72. doi: 10.1016/0022-1759(86)90032-3. [DOI] [PubMed] [Google Scholar]
- 20.Redfern CH, Guthrie TH, Bessudo A, Densmore JJ, Holman PR, Janakiraman N, Leonard JP, Levy RL, Just RG, Smith MR, Rosenfelt FP, Wiernik PH, Carter WD, Gold DP, Melink TJ, Gutheil JC, Bender JF. Phase II trial of idiotype vaccination in previously treated patients with indolent non-Hodgkin's lymphoma resulting in durable clinical responses. J Clin Oncol. 2006;24:3107–3112. doi: 10.1200/JCO.2005.04.4289. [DOI] [PubMed] [Google Scholar]
- 21.Giudicelli V, Chaume D, Lefranc MP. IMGT/V-QUEST, an integrated software program for immunoglobulin and T cell receptor V-J and V-D-J rearrangement analysis. Nucleic Acids Res. 2004;32:W435–440. doi: 10.1093/nar/gkh412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu D, McCarthy H, Ottensmeier CH, Johnson P, Hamblin TJ, Stevenson FK. Acquisition of potential N-glycosylation sites in the immunoglobulin variable region by somatic mutation is a distinctive feature of follicular lymphoma. Blood. 2002;99:2562–2568. doi: 10.1182/blood.v99.7.2562. [DOI] [PubMed] [Google Scholar]
- 23.Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, Wardemann H. Autoreactivity in human IgG+ memory B cells. Immunity. 2007;26:205–213. doi: 10.1016/j.immuni.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsuiji M, Yurasov S, Velinzon K, Thomas S, Nussenzweig MC, Wardemann H. A checkpoint for autoreactivity in human IgM+ memory B cell development. J Exp Med. 2006;203:393–400. doi: 10.1084/jem.20052033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zemlin M, Klinger M, Link J, Zemlin C, Bauer K, Engler JA, Schroeder HW, Jr., Kirkham PM. Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. Journal of molecular biology. 2003;334:733–749. doi: 10.1016/j.jmb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 26.Ivanov, Schelonka RL, Zhuang Y, Gartland GL, Zemlin M, Schroeder HW., Jr Development of the expressed Ig CDR-H3 repertoire is marked by focusing of constraints in length, amino acid use, and charge that are first established in early B cell progenitors. Journal of immunology. 2005;174:7773–7780. doi: 10.4049/jimmunol.174.12.7773. [DOI] [PubMed] [Google Scholar]
- 27.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. Journal of molecular biology. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 28.Eisenberg D. Three-dimensional structure of membrane and surface proteins. Annual review of biochemistry. 1984;53:595–623. doi: 10.1146/annurev.bi.53.070184.003115. [DOI] [PubMed] [Google Scholar]
- 29.Bende RJ, Smit LA, van Noesel CJ. Molecular pathways in follicular lymphoma. Leukemia. 2007;21:18–29. doi: 10.1038/sj.leu.2404426. [DOI] [PubMed] [Google Scholar]
- 30.Herve M, Xu K, Ng YS, Wardemann H, Albesiano E, Messmer BT, Chiorazzi N, Meffre E. Unmutated and mutated chronic lymphocytic leukemias derive from self-reactive B cell precursors despite expressing different antibody reactivity. J Clin Invest. 2005;115:1636–1643. doi: 10.1172/JCI24387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sachen KL, Strohman MJ, Singletary J, Alizadeh AA, Kattah NH, Lossos C, Mellins ED, Levy S, Levy R. Self-antigen recognition by follicular lymphoma B-cell receptors. Blood. 2012;120:4182–4190. doi: 10.1182/blood-2012-05-427534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Irish JM, Myklebust JH, Alizadeh AA, Houot R, Sharman JP, Czerwinski DK, Nolan GP, Levy R. Inaugural Article: B-cell signaling networks reveal a negative prognostic human lymphoma cell subset that emerges during tumor progression. Proc Natl Acad Sci U S A. doi: 10.1073/pnas.1002057107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanemo Myhrinder A, Hellqvist E, Sidorova E, Soderberg A, Baxendale H, Dahle C, Willander K, Tobin G, Backman E, Soderberg O, Rosenquist R, Horkko S, Rosen A. A new perspective: molecular motifs on oxidized LDL, apoptotic cells, and bacteria are targets for chronic lymphocytic leukemia antibodies. Blood. 2008;111:3838–3848. doi: 10.1182/blood-2007-11-125450. [DOI] [PubMed] [Google Scholar]
- 34.Ghiotto F, Fais F, Valetto A, Albesiano E, Hashimoto S, Dono M, Ikematsu H, Allen SL, Kolitz J, Rai KR, Nardini M, Tramontano A, Ferrarini M, Chiorazzi N. Remarkably similar antigen receptors among a subset of patients with chronic lymphocytic leukemia. J Clin Invest. 2004;113:1008–1016. doi: 10.1172/JCI19399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bende RJ, Aarts WM, Riedl RG, de Jong D, Pals ST, van Noesel CJ. Among B cell non-Hodgkin's lymphomas, MALT lymphomas express a unique antibody repertoire with frequent rheumatoid factor reactivity. J Exp Med. 2005;201:1229–1241. doi: 10.1084/jem.20050068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu CC, Catera R, Hatzi K, Yan XJ, Zhang L, Wang XB, Fales HM, Allen SL, Kolitz JE, Rai KR, Chiorazzi N. Chronic lymphocytic leukemia antibodies with a common stereotypic rearrangement recognize nonmuscle myosin heavy chain IIA. Blood. 2008;112:5122–5129. doi: 10.1182/blood-2008-06-162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y, Smith BD, Civin CI, Jones RJ. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103:2332–2336. doi: 10.1182/blood-2003-09-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blaschek MA, Boehme M, Jouquan J, Simitzis AM, Fifas S, Le Goff P, Youinou P. Relation of antivimentin antibodies to anticardiolipin antibodies in systemic lupus erythematosus. Ann Rheum Dis. 1988;47:708–716. doi: 10.1136/ard.47.9.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ortona E, Capozzi A, Colasanti T, Conti F, Alessandri C, Longo A, Garofalo T, Margutti P, Misasi R, Khamashta MA, Hughes GR, Valesini G, Sorice M. Vimentin/cardiolipin complex as a new antigenic target of the antiphospholipid syndrome. Blood. 116:2960–2967. doi: 10.1182/blood-2010-04-279208. [DOI] [PubMed] [Google Scholar]
- 40.Xu B, deWaal RM, Mor-Vaknin N, Hibbard C, Markovitz DM, Kahn ML. The endothelial cell-specific antibody PAL-E identifies a secreted form of vimentin in the blood vasculature. Mol Cell Biol. 2004;24:9198–9206. doi: 10.1128/MCB.24.20.9198-9206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. Vimentin is secreted by activated macrophages. Nat Cell Biol. 2003;5:59–63. doi: 10.1038/ncb898. [DOI] [PubMed] [Google Scholar]
- 42.Huet D, Bagot M, Loyaux D, Capdevielle J, Conraux L, Ferrara P, Bensussan A, Marie-Cardine A. SC5 mAb represents a unique tool for the detection of extracellular vimentin as a specific marker of Sezary cells. J Immunol. 2006;176:652–659. doi: 10.4049/jimmunol.176.1.652. [DOI] [PubMed] [Google Scholar]
- 43.Garg A, Barnes PF, Porgador A, Roy S, Wu S, Nanda JS, Griffith DE, Girard WM, Rawal N, Shetty S, Vankayalapati R. Vimentin expressed on Mycobacterium tuberculosis-infected human monocytes is involved in binding to the NKp46 receptor. J Immunol. 2006;177:6192–6198. doi: 10.4049/jimmunol.177.9.6192. [DOI] [PubMed] [Google Scholar]
- 44.Moisan E, Girard D. Cell surface expression of intermediate filament proteins vimentin and lamin B1 in human neutrophil spontaneous apoptosis. J Leukoc Biol. 2006;79:489–498. doi: 10.1189/jlb.0405190. [DOI] [PubMed] [Google Scholar]
- 45.Leong HS, Mahesh BM, Day JR, Smith JD, McCormack AD, Ghimire G, Podor TJ, Rose ML. Vimentin autoantibodies induce platelet activation and formation of platelet-leukocyte conjugates via platelet-activating factor. J Leukoc Biol. 2008;83:263–271. doi: 10.1189/jlb.0607339. [DOI] [PubMed] [Google Scholar]
- 46.Bryant AE, Bayer CR, Huntington JD, Stevens DL. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis. 2006;193:1685–1692. doi: 10.1086/504261. [DOI] [PubMed] [Google Scholar]
- 47.Nijenhuis S, Zendman AJ, Vossenaar ER, Pruijn GJ, vanVenrooij WJ. Autoantibodies to citrullinated proteins in rheumatoid arthritis: clinical performance and biochemical aspects of an RA-specific marker. Clin Chim Acta. 2004;350:17–34. doi: 10.1016/j.cccn.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 48.Marceau N, Schutte B, Gilbert S, Loranger A, Henfling ME, Broers JL, Mathew J, Ramaekers FC. Dual roles of intermediate filaments in apoptosis. Exp Cell Res. 2007;313:2265–2281. doi: 10.1016/j.yexcr.2007.03.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.