

Summary
Tuberous sclerosis complex (TSC) is a genetic disease characterized by multiorgan benign tumors as well as neurological manifestations. Epilepsy and autism are two of the more prevalent neurological complications and are usually severe. TSC is caused by mutations in either the TSC1 (encodes hamartin) or TSC2 (encodes tuberin) genes with TSC2 mutations being associated with worse outcomes. Tuberin contains a highly conserved GTPase activating protein (GAP) domain that indirectly inhibits mammalian target of rapamycin complex 1 (mTORC1). mTORC1 dysregulation is currently thought to cause much of the pathogenesis in TSC but mTORC1-independent mechanisms may also contribute. We generated a novel conditional allele of Tsc2 by flanking exons 36 and 37 with loxP sites. Mice homozygous for this knock-in Tsc2 allele are viable and fertile with normal appearing growth and development. Exposure to Cre recombinase then creates an in-frame deletion involving critical residues of the GAP domain. Homozygous conditional mutant mice generated using Emx1Cre have increased cortical mTORC1 signaling, severe developmental brain anomalies, seizures and die within three weeks. We found normal levels of the mutant Tsc2 mRNA though GAP-deficient tuberin protein appears unstable and rapidly degraded. This novel animal model will allow further study of tuberin function including the requirement of the GAP domain for protein stability.
Keywords: cortical development, mTORC1, mTORC2, rapamycin, Tuberous Sclerosis Complex
Results/Discussion
TSC afflicts approximately 1 in 6,000 individuals. Patients have significant morbidity due to multi-organ tumors although neurological involvement is generally more severe. Most patients present at very young ages with epilepsy and up to 90% of patients will have seizures at some point in their life (Crino et al., 2006). Additional neurologic and psychiatric manifestations including autism, attention deficit disorder and anxiety disorders are also quite common and disabling but symptom severity may vary considerably (Joinson et al., 2003; Staley et al., 2011). Inactivating mutations in either the TSC1 or TSC2 genes cause disease although TSC2 mutations are associated with greater symptom severity (Jones et al., 1997). TSC1 and TSC2 encode hamartin and tuberin respectively, which form a heterodimeric regulatory complex. Tuberin contains a highly conserved GTPase activating (GAP) domain that inactivates the G protein Rheb (Maheshwar et al., 1997). Rheb inactivation then inhibits mammalian/mechanistic target of rapamycin complex 1 (mTORC1), a critical regulator of protein translation and cellular growth (Benvenuto et al., 2000; Inoki et al., 2003). Loss of tuberin GAP activity is known to contribute to the molecular pathogenesis of TSC (Beaumont et al., 2012; Crino, 2004; Meikle et al., 2008); however, additional mTORC1-independent and rapamycin-resistant abnormalities likely also exist in TSC (Hartman et al., 2009; Hodges et al., 2001; Momose et al., 2007; Noonan et al., 2002; Rosner et al., 2008). Multiple mouse models of TSC have been generated through conditional inactivation of the Tsc1 gene and used to study developmental brain abnormalities (Carson et al., 2012; Fu et al., 2012; Uhlmann et al., 2002; Way et al., 2012). Constitutive mutant alleles of mouse Tsc2 have been reported (Onda et al., 1999) including a variant with a deletion involving the tuberin GAP domain and amino acid substitutions in the rapabtin-5 binding motif (Chevere-Torres et al., 2012; Govindarajan et al., 2005). A conditional mutant of Tsc2 has also been generated by targeting exons 2–4 with flanking loxP sites (Hernandez et al., 2007) that is expected to have complete loss of function when exposed to Cre recombinase.
To study the tissue specific requirement of the tuberin GAP domain in TSC pathogenesis, we designed a novel conditional allele of the Tsc2 gene that selectively targets exons within the GAP domain. A Tsc2 gene targeting vector was generated using recombineering techniques (Liu et al., 2003). Exons 36 and 37 were flanked with intronic loxP sites (Fig. 1a) designed to produce an in-frame deletion of the gene after exposure to Cre recombinase and expected to result in mutant tuberin protein absent 109 amino acids (aa1534-1642) within the GAP domain. A FRT flanked neomycin resistance cassette was used for positive selection of transfected mouse embryonic stem (ES) cells. We successfully targeted the Tsc2 gene in 6% (12 of 192) of screened ES clones as verified by Southern blot analyses and PCR (Fig. 1b). After confirming a normal karyotype, targeted ES cells were used for blastocyst injection. Multiple chimeric mice resulted and germline transmission of the Tsc2neo.flox allele was confirmed from two independent lines by PCR (Fig. 1c) and direct sequencing of genomic DNA (data not shown).
Fig. 1.

Generation of Tsc2neo.flox mice. (a) Schematic of targeting vector with homologous left 6.8 kb and right 5.3 kb arms. Tsc2 genomic structure and targeted allele Tsc2neo.flox containing the PGK-neo cassette flanked by Frt sites (red arrowheads) and loxP sites flanking exons 36 and 37 that encode critical residues of the GAP domain. An additional EcoRI site to aid in genotyping was also added immediately 5′ to the loxP site. Select regions have been expanded to highlight PCR primer sites. (b) Southern blot of EcoRI digested genomic DNA from mouse ES cells showing wild-type band (18.4 kb) and targeted Tsc2 allele (11.6 kb) in clone 4. C. PCR of wild-type and Tsc2neo.flox ES cells with primers P2F and P2R confirming targeting of the Tsc2 allele with 596 bp band only in wild-type but an additional 2387 bp product in Tsc2neo.flox mice.
We anticipated that the presence of the neomycin resistance cassette might interfere with Tsc2 gene transcription or translation and may result in the equivalent of a null Tsc2 allele. If so, this should lead to early embryonic lethality in homozygote mice as previously shown using conventional gene knockout approaches (Hernandez et al., 2007; Onda et al., 1999). We interbred heterozygous Tsc2neo.flox mice and in fact did not find any homozygous Tsc2neo.flox offspring from 10 separate litters resulting in 49 total offspring (data not shown). We conclude that homozygous Tsc2neo.flox mice with the neomycin selection cassette are non-viable and die during embryogenesis.
We then crossed Tsc2neo.flox mice to flippase recombinase expressing animals to remove the FRT-flanked neomycin resistance cassette in vivo, thus generating the Tsc2f36–37 conditional allele (Fig. 2a). In contrast to Tsc2neo.flox, heterozygous Tsc2f36–37 crosses produced viable homozygous offspring at Mendelian ratios (Fig. 2b), indicating restoration of Tsc2 gene function after excision of the neomycin resistance cassette. We next assayed Tsc2 gene transcript and tuberin protein levels from brain extracts of mature homozygous Tsc2f36–37 mice and found no significant difference in expression; additionally, Tsc2f36–37 homozygotes were phenotypically indistinguishable from wild-type mice with regards to long-term growth and survival (data not shown). These findings indicate no deleterious effect from the insertion of loxP sites within the Tsc2 gene and provide further evidence that the neomycin cassette accounted for the non-viability of homozygous Tsc2neo.flox mice.
Fig. 2.

Creation of Tsc2f36–37 mice with loxP sites flanking exons 36 and 37. (a) Mice heterozygous for the Tsc2neo.flox allele were crossed with flippase expressing animals to remove the PGK-neo cassette. (b) Summary of litter genotyping from ten sequential heterozygous Tsc2f36–37 crosses showing Mendelian birth proportions.
To verify the functional integrity of the loxP sites and address the impact of this conditional allele upon neurodevelopment, we generated conditional knockout mice using Emx1Cre expressing mice (Tsc2f36–37 Emx1 CKO). Emx1Cre mice are well characterized for Cre expression in dorsal neural progenitor cells. These cells give rise to excitatory neurons of the cerebral cortex as well as most astrocytes and a subset of oligodendrocytes (Gorski et al., 2002). We also chose Emx1Cre to compare these results to what we have previously seen from the conditional loss of the Tsc1 gene in dorsal neural progenitors (Carson et al., 2012; Magri et al., 2011). We first determined levels of Tsc2 mRNA using quantitative PCR using RNA extracted from P5 dorsal cortex of Tsc2f36–37 Emx1 CKO. There was no significant difference in Tsc2 mRNA expression from Tsc2f36–37 Emx1 CKO RNA extracts compared to control littermates (Fig. 3a). Despite equivalent amounts of mRNA, we found much reduced levels of tuberin protein from P5 dorsal cortex protein extracts (Fig. 3b). We sequenced a portion of the Tsc2del36–37 cDNA product surrounding the deleted region and confirmed that loss of tuberin protein did not result from use of an ectopic splice site or a frame shift after Cre recombination (data not shown). It is possible that the small amount of tuberin observed from Tsc2f36–37 Emx1 CKO brain is the mutant protein; however, we have not been able to resolve a distinct band corresponding to the predicted molecular weight of the mutant protein to support this possibility. Much more likely, rather, we think that the observed tuberin band from CKO extracts is due the minority of cells in the neocortex that are not expected to express Emx1Cre.
Fig. 3.
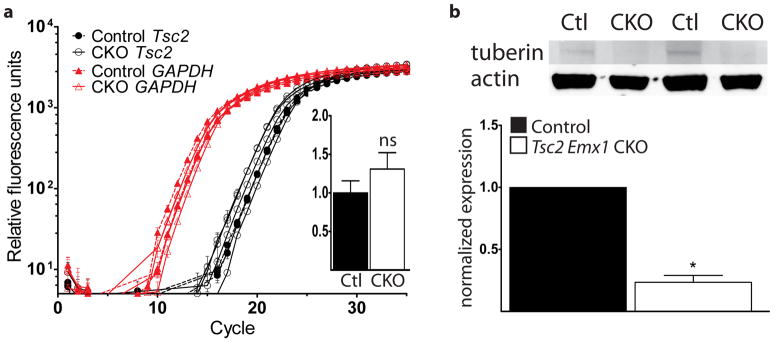
Normal levels of Tsc2 mRNA but decreased tuberin protein in Tsc2f36–37 Emx1 CKO mice. (a) We used quantitative PCR to measure Tsc2 mRNA extracted from the dorsal cortex of P5 control littermate and Tsc2f36–37 Emx1 CKO mice. N=3 control and 3 Tsc2f36–37 Emx1 CKO mice. Inset: normalized Tsc2 expression. ns = not significant (b) Immunoblotting using an anti-tuberin antibody that recognizes an N-terminal epitope shows much decreased protein in the dorsal cortical extracts from Tsc2f36–37 Emx1 CKO mice compared to control littermates. *p<0.05 by Student’s t-test. N=4 control and 4 Tsc2f36–37 Emx1 CKO mice.
Tsc2f36–37 Emx1 CKO mice were born at expected Mendelian proportions but were obviously impaired with poor growth evident during the first few days of life (Fig. 4a). They started dying at P4 with complete mortality by P21 (Fig. 4b). Spontaneous seizures were witnessed just prior to death in multiple Tsc2f36–37 Emx1 CKO mice during routine handling, suggesting that recurrent seizures contributed to their early demise. These phenotypes of seizures and increased mortality were similar but more severe relative to Tsc1 Emx1 CKO mice (Carson et al., 2012) and is also consistent with other conditional knockout models of Tsc2 relative to their Tsc1 counterparts (Zeng et al., 2011). To see if these mice had altered mTOR signaling we determined levels of phosphorylated S6 (pS6), a well established downstream marker of mTORC1 signaling (Hay and Sonenberg, 2004; Ruvinsky et al., 2005). We found diffuse expression of pS6 throughout the dorsal cortex in both control and CKO sections though there was a much wider expression seen throughout the depth of the cerebral cortex in Tsc2f36–37 Emx1 CKO (Fig. 5a). To more closely quantitate mTORC1 signaling in the brain of P5 Tsc2f36–37 Emx1 CKO mice, we used dorsal cortical protein extracts for immunoblotting pS6 levels as well as phosphorylated 4E-BP1 (p4E-BP1), a direct target of mTORC1 (Schalm et al., 2003). p4E-BP1 was significantly increased in Tsc2f36–37 Emx1 CKO dorsal cortex; however, levels of pS6 did not differ significantly between control and CKO (Fig. 5b). The discrepancy between these two readouts may possibly reflect mTORC1 independent activation of S6-kinase (Balendran et al., 1999; Pullen et al., 1998) resulting in high basal levels of S6 phosphorylation even in control cerebral cortex at this early stage of brain development. Finally, given the well documented response of TSC mouse models to mTORC1 inhibitors (Carson et al., 2012; Meikle et al., 2008; Way et al., 2012), we treated several mice with rapamycin. We found that postnatal rapamycin treatment significantly improved growth and prolonged survival in Tsc2f36–37 Emx1 CKO mice (Fig. 5c and d) confirming aberrant mTORC1 signaling in Tsc2f36–37 Emx1 CKO mice.
Fig. 4.
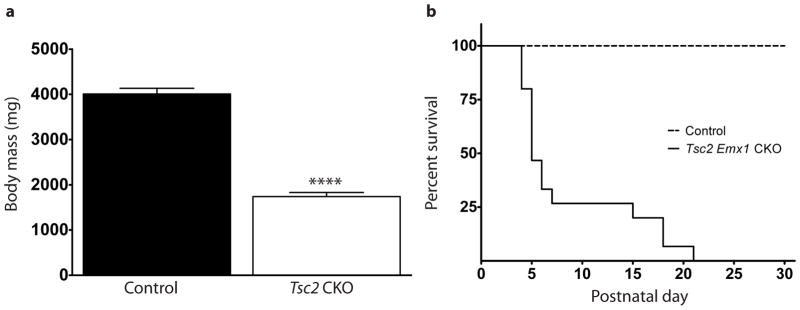
Tsc2f36–37 Emx1 CKO mice are smaller than control littermates and die by three weeks of life. (a) Mice were weighed at postnatal day 5, Tsc2f36–37 Emx1 CKO were much smaller and runted appearing. N=7 control and 7 Tsc2f36–37 Emx1 CKO mice. (b) They started dying by the 4th postnatal day and all Tsc2f36–37 Emx1 CKO mice were dead by postnatal day 21. N=14 control and 11 Tsc2f36–37 Emx1 CKO mice.****p<0.001, Student’s t-test for decreased body mass. The increased mortality of Tsc2f36–37 Emx1 CKO mice was statistically significant with a p<0.001 using a Log-Rank test.
Fig. 5.

Tsc2f36–37 Emx1 CKO mice have increased mTORC1 signaling in the brain and respond to rapamycin treatment. (a) Increased expression of phospho-S6 throughout the cerebral cortex of P5 Tsc2f36–37 Emx1 CKO mice. n=4 control and 4 Tsc2f36–37 Emx1 CKO mice. Size bar equals 100 μm. CL I=cortical layer I, ML=midline, WM=white matter/corpus callosum, LV=lateral ventricle. (b) Immunoblotting of dorsal extracts from P5 Tsc2f36–37 Emx1 CKO mice shows no change in phospho-S6 but increased overall levels of phospho-4E-BP1. Two representative control and Tsc2f36–37 Emx1 CKO extracts are shown. Quantitation of immunoblotting confirms increased levels of phospho-4E-BP1. *p<0.05 by Student’s t-test. (c) Rapamycin treatment rescues small size of Tsc2f36–37 Emx1 CKO mice, *p<0.05 by Student’s t-test. (d) Rapamycin treatment of Tsc2f36–37 Emx1 CKO mice prevented early death as compared to the previous cohort of untreated CKO mice. Rescue was statistically significant using a Log-Rank test, p<0.001. N=5 Tsc2f36–37 Emx1 CKO mice.
In conclusion, we generated a novel conditional allele of Tsc2 generating an in-frame deletion of exons 36 and 37 encoding a critical portion of the tuberin GAP domain. The conditional nature of our knock-in mutation allows study of tissue specific disease mechanisms under endogenous gene regulatory conditions making it distinct from the constitutively active, transgenic allele used by Chevere-Torres et al. despite sharing a similar mutation. Also, in contrast the conditional Tsc2 knock-in allele reported by Hernandez et al., we have confirmed stability of the mutant transcript. The absence of demonstrable tuberin, increased mTORC1 signaling and response to rapamycin does, however, indicate a loss of function allele that appears to be due to an unexpected requirement of the GAP domain for tuberin protein stability. Our novel Tsc2 allele may then better model the pathogenesis seen in patients with TSC due to GAP domain mutations. Additional investigations will be required to determine the precise mechanism of protein loss from this allele, but it is likely that the tuberin GAP domain is required for overall protein stability and that this allele can be used to study other aspects of developmental disorders in TSC.
METHODS
Knock-in vector construction
A Tsc2 gene targeting vector was generated using BAC recombineering (Lee et al., 2001; Liu et al., 2003). A BAC clone (bMQ 249d21) was used containing 3′ sequence of Tsc2 generated from 129S7/SvEv mice (Wellcome Trust Sanger Institute). Recombineering was used to isolate a region of BAC DNA encompassing Tsc2 GAP encoding exons into the PL253 retrieval vector for subsequent modifications. We used the neomycin selectable loxP cassette from PL452 to place a 5′ loxP site within intron 35. An EcoRI restriction site was inserted adjacent to the 5′ loxP site to permit screening by Southern blotting. A selectable cassette from PL451 with a FRT flanked neomycin resistance gene and another loxP site was targeted to intron 37 (Fig. 1a). All modifications were confirmed by DNA sequencing.
Generation of knock-in and conditional knockout mice
The targeting construct included left 6.8 kb homology arms and right 5.3 kb homology arms. The knock-in vector was linearized with NotI and transfected into 129S6 mouse ES cells. G418-resistant ES clones were screened by Southern blot using a 3′ external probe (Fig. 1b) with additional confirmation by PCR (Fig. 1c) using primers P1F (5′-TGG TGA GGA CTT CAA ACT GGG CAC C) and P1R (5′-AAG GCC ACG CTG CTT CCC CAC TT) that generate products of 596 base pairs (wildtype) and 2387 base pairs (knock-in). 12 of 192 (6.3%) of the drug resistant clones were successfully targeted. After confirming a normal karyotype, targeted ES cells were injected into C57BL/6 blastocysts to generate chimeric mice. Male chimeras were bred to 129S1/SvImJ mice and their offspring were genotyped by PCR to validate germline transmission. The FRT-flanked neomycin resistance gene was then deleted in vivo by breeding Tsc2neo.flox/+ mice with FLPe expressing mice (129S4/SvJaeSor-Gt(ROSA)26Sortm1(FLP1)Dym/J, #3946 Jackson Laboratory, Bar Harbor, ME) (Farley et al., 2000). Tsc2 Emx1 conditional knockout mice were generated by breeding homozygous Tsc2f36–37 mice to Tsc2f36–37/+; Emx1Cre mice. Tsc2f36–37 mice were on a 129 mixed substrain background and Emx1Cre mice (B6.129S2-Emx1tm1(cre)Krj/J, #5628, Jackson Laboratory, Bar Harbor, ME) were maintained on a C57BL/6J background. All procedures involving mice were approved by the Vanderbilt University IACUC. Tsc2f36–37 mice will be made available to the research community upon request.
Genotyping
Genomic DNA was isolated from tail or ear samples and analyzed using PCR. Mice carrying the wild-type and Tsc2f36–37 alleles were genotyped in a single PCR reaction using forward primer P2F (5′-TGGCAGGACAGAGGGTCATCATGG) and reverse primer P2R (5′-TTCAGAGTCACCTGGCAGGCTCG) with product lengths of 388 base pairs (wildtype) and 312 base pairs (Tsc2f36–37 allele). PCR conditions: 94° C for 3 minutes, (94° C for 30 seconds, 68° C for 30 seconds, 72° C for 1 minutes) × 31 cycles, and 72° C for 10 minutes.
Immunohistochemistry
Tissues for immunohistochemistry were processed as previously described (Carson et al., 2012; Fu et al., 2012). Briefly, P5–6 pup brains were rapidly dissected and fixed overnight with 4% paraformaldehyde then cryoprotected with 30% sucrose solution for 2 days. Coronal sections at 10–20 μm were mounted on glass slides. Primary antibody: rabbit phospho-S6 (S240/244) (1:200, Cell Signaling). Secondary antibody: Alexa Fluor 568 goat anti-rabbit IgG (1:500, Life Technologies). Negative controls for each experiment were performed by omission of primary antibody. Photomicrographs were obtained using a Zeiss epifluorescence microscope.
Real time RT-PCR
Total RNA was isolated from fresh frozen dorsal cortical brain tissue using an RNeasy Mini kit (Qiagen). cDNA for use in quantitative PCR was generated using an oligo(dT) primer (Life Technologies). Age matched wildtype mouse brain tissue was used as the control. Tsc2 specific primers were used to amplify a 5′ region of the cDNA outside of the targeted region. Relative expression of Tsc2 mRNA in control and Tsc2 Emx1 CKO brain was normalized with levels of GAPDH mRNA expression. Each cDNA sample was assayed in triplicate and relative quantification was determined using the ΔΔC(t) method. qPCR reactions were quantified using a SYBR green based kit (Life Technologies) on a Bio-Rad CFX96 Real-Time PCR system using the following conditions: 95° C for 10 minutes, (95° C for 15 seconds, 60° C for 1 minute) × 40 cycles.
Immunoblotting
Immunoblots were done as previously reported (Carson et al., 2012; Fu et al., 2012). Tissues for immunoblotting were dissected and flash frozen in liquid nitrogen prior to protein extraction. Primary antibodies: rabbit tuberin N-terminus (1:500, abcam); phospho-S6 (Serine 240/244), S6, phospho-4E-BP1, (Threonine 37/46) all rabbit and used at 1:1000 (Cell Signaling). Primary antibody against β-actin was from mouse (1:2000, Sigma). Secondary antibodies: Alexa Fluor 680 goat anti-rabbit IgG (Life Technologies) and IRDye 800 goat anti-mouse IgG (Li-Cor). Fluorescence was detected using the Odyssey fluorescence imaging system (Li-Cor). Digital band density was quantified using ImageJ software (NIH, Bethesda, MD). Absolute density for each band was determined using an area-under-the curve method. Data from each sample was normalized to internal loading control proteins as indicated and expressed as fraction of the experimental control.
Rapamycin Treatment
Tsc2 Emx1 CKO mice (n=5) were treated with intraperitoneal injections of rapamycin at a dose of 0.1 mg/kg/day. Rapamycin (LC Laboratories, Woburn, MA) was dissolved as a stock at 30 mg/mL in ethanol and then diluted with vehicle consisting of 0.25% Tween-20/0.25% polyethylene glycol in PBS.
Statistical Analysis
Results were assessed for statistical significance using a Student’s t-test or Log-Rank test as appropriate with a p-value less than 0.05.
Acknowledgments
Funding: This work was supported by awards from the Tuberous Sclerosis Alliance (KCE and CF) and 1R01 NS078289 from NINDS, NIH (KCE)
We would like to thank Peggy Winzenberger and Sherry Syed for excellent technical assistance. Mouse ES cell gene targeting was performed by the Vanderbilt Transgenic Mouse/ESC shared resource that is in part supported by the Vanderbilt Center for Molecular Neuroscience. This work was supported by awards from the Tuberous Sclerosis Alliance (KCE and CF) and 1R01 NS078289 from NINDS, NIH (KCE).
LITERATURE CITED
- Balendran A, Currie R, Armstrong CG, Avruch J, Alessi DR. Evidence that 3-phosphoinositide-dependent protein kinase-1 mediates phosphorylation of p70 S6 kinase in vivo at Thr-412 as well as Thr-252. J Biol Chem. 1999;274:37400–37406. doi: 10.1074/jbc.274.52.37400. [DOI] [PubMed] [Google Scholar]
- Beaumont TL, Limbrick DD, Smyth MD. Advances in the management of subependymal giant cell astrocytoma. Childs Nerv Syst. 2012 doi: 10.1007/s00381-012-1785-x. [DOI] [PubMed] [Google Scholar]
- Benvenuto G, Li SW, Brown SJ, Braverman R, Vass WC, Cheadle JP, Halley DJJ, Sampson JR, Wienecke R, DeClue JE. The tuberous sclerosis-1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene. 2000;19:6306–6316. doi: 10.1038/sj.onc.1204009. [DOI] [PubMed] [Google Scholar]
- Carson RP, Van Nielen DL, Winzenburger PA, Ess KC. Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol Dis. 2012;45:369–380. doi: 10.1016/j.nbd.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevere-Torres I, Maki JM, Santini E, Klann E. Impaired social interactions and motor learning skills in tuberous sclerosis complex model mice expressing a dominant/negative form of tuberin. Neurobiol Dis. 2012;45:156–164. doi: 10.1016/j.nbd.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB. Molecular pathogenesis of tuber formation in tuberous sclerosis complex. J Child Neurol. 2004;19:716–725. doi: 10.1177/08830738040190091301. [DOI] [PubMed] [Google Scholar]
- Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- Farley FW, Soriano P, Steffen LS, Dymecki SM. Widespread recombinase expression using FLPeR (flipper) mice. Genesis. 2000;28:106–110. [PubMed] [Google Scholar]
- Fu C, Cawthon B, Clinkscales W, Bruce A, Winzenburger P, Ess KC. GABAergic interneuron development and function is modulated by the Tsc1 gene. Cereb Cortex. 2012;22:2111–2119. doi: 10.1093/cercor/bhr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan B, Brat DJ, Csete M, Martin WD, Murad E, Litani K, Cohen C, Cerimele F, Nunnelley M, Lefkove B, Yamamoto T, Lee C, Arbiser JL. Transgenic expression of dominant negative tuberin through a strong constitutive promoter results in a tissue-specific tuberous sclerosis phenotype in the skin and brain. J Biol Chem. 2005;280:5870–5874. doi: 10.1074/jbc.M411768200. [DOI] [PubMed] [Google Scholar]
- Hartman TR, Liu D, Zilfou JT, Robb V, Morrison T, Watnick T, Henske EP. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet. 2009;18:151–163. doi: 10.1093/hmg/ddn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hernandez O, Way S, McKenna J, 3rd, Gambello MJ. Generation of a conditional disruption of the Tsc2 gene. Genesis. 2007;45:101–106. doi: 10.1002/dvg.20271. [DOI] [PubMed] [Google Scholar]
- Hodges AK, Li S, Maynard J, Parry L, Braverman R, Cheadle JP, DeClue JE, Sampson JR. Pathological mutations in TSC1 and TSC2 disrupt the interaction between hamartin and tuberin. Hum Mol Genet. 2001;10:2899–2905. doi: 10.1093/hmg/10.25.2899. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joinson C, O’Callaghan FJ, Osborne JP, Martyn C, Harris T, Bolton PF. Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol Med. 2003;33:335–344. doi: 10.1017/s0033291702007092. [DOI] [PubMed] [Google Scholar]
- Jones AC, Daniells CE, Snell RG, Tachataki M, Idziaszczyk SA, Krawczak M, Sampson JR, Cheadle JP. Molecular genetic and phenotypic analysis reveals differences between TSC1 and TSC2 associated familial and sporadic tuberous sclerosis. Hum Mol Genet. 1997;6:2155–2161. doi: 10.1093/hmg/6.12.2155. [DOI] [PubMed] [Google Scholar]
- Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73:56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magri L, Cambiaghi M, Cominelli M, Alfaro-Cervello C, Cursi M, Pala M, Bulfone A, Garcia-Verdugo JM, Leocani L, Minicucci F, Poliani PL, Galli R. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell. 2011;9:447–462. doi: 10.1016/j.stem.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Maheshwar MM, Cheadle JP, Jones AC, Myring J, Fryer AE, Harris PC, Sampson JR. The GAP-related domain of tuberin, the product of the TSC2 gene, is a target for missense mutations in tuberous sclerosis. Hum Mol Genet. 1997;6:1991–1996. doi: 10.1093/hmg/6.11.1991. [DOI] [PubMed] [Google Scholar]
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28:5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momose S, Kobayashi T, Tada N, Itoyama S, Hino O. N-terminal hamartin-binding and C-terminal GAP domain of tuberin can separate in vivo. Biochem Biophys Res Commun. 2007;356:693–698. doi: 10.1016/j.bbrc.2007.03.036. [DOI] [PubMed] [Google Scholar]
- Noonan DJ, Lou D, Griffith N, Vanaman TC. A calmodulin binding site in the tuberous sclerosis 2 gene product is essential for regulation of transcription events and is altered by mutations linked to tuberous sclerosis and lymphangioleiomyomatosis. Arch Biochem Biophys. 2002;398:132–140. doi: 10.1006/abbi.2001.2682. [DOI] [PubMed] [Google Scholar]
- Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ. Tsc2(+/−) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest. 1999;104:687–695. doi: 10.1172/JCI7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G. Phosphorylation and activation of p70s6k by PDK1. Science. 1998;279:707–710. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- Rosner M, Hanneder M, Siegel N, Valli A, Hengstschlager M. The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partners. Mutat Res. 2008;658:234–246. doi: 10.1016/j.mrrev.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, Dor Y, Zisman P, Meyuhas O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005;19:2199–2211. doi: 10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalm SS, Fingar DC, Sabatini DM, Blenis J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr Biol. 2003;13:797–806. doi: 10.1016/s0960-9822(03)00329-4. [DOI] [PubMed] [Google Scholar]
- Staley BA, Vail EA, Thiele EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics. 2011;127:e117–125. doi: 10.1542/peds.2010-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, Yamada K, Gutmann DH. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52:285–296. doi: 10.1002/ana.10283. [DOI] [PubMed] [Google Scholar]
- Way SW, Rozas NS, Wu HC, McKenna J, 3rd, Reith RM, Hashmi SS, Dash PK, Gambello MJ. The differential effects of prenatal and/or postnatal rapamycin on neurodevelopmental defects and cognition in a neuroglial mouse model of tuberous sclerosis complex. Hum Mol Genet. 2012 doi: 10.1093/hmg/dds156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum Mol Genet. 2011;20:445–454. doi: 10.1093/hmg/ddq491. [DOI] [PMC free article] [PubMed] [Google Scholar]