

Abstract
Purpose
Recently, genomewide association analysis has revealed that the Tumor Necrosis Factor Receptor-associated factor 1-Complement 5 (TRAF1-C5) containing locus on chromosome 9 was associated with an increased risk for RA. Studies in model systems suggested that either gain- or loss-of-function TRAF1 mutations have immune effects that could plausibly lead to or exacerbate the arthritis phenotype. KRN/I-Ag7 (KxB/N) is a genetic mouse model of inflammatory arthritis. We aimed to assess the impact of TRAF1 deficiency on KRN/I-Ag7 mice.
Methods
We have bred KRN/I-Ag7 mice onto a TRAF1-deficient background and followed cohorts for the spontaneous appearance of arthritis. We have also transferred KxB/N serum to B6.I-Ag7 TRAF1KO recipients. In addition, systemic autoimmunity was induced through cGVH by injecting bm12 splenocytes into TRAF1KO recipient mice.
Results
TRAF1-deficient KRN/I-Ag7 mice spontaneously developed severe, progressive arthritis, comparable to that seen in TRAF1-intact KRN/I-Ag7 mice. However, the anti-GPI antibody titer was significantly lower in the former group. Interestingly, the TRAF1KO mice that had background levels of anti-GPI antibodies still showed severe arthritis, although with a brief delay compared to TRAF1 sufficient mice. In addition, TRAF1KO mice were fully susceptible to passive, serum transfer experiments. In another model of autoimmunity, TRAF1KO had no effect on cGVH autoantibodies production; nor was the response to an exogenous antigen impaired.
Conclusion
The pathogenesis of spontaneous KRN/I-Ag7 arthritis can largely proceed by TRAF1-independent pathways. The production of anti-GPI autoantibody, but not other autoantibody or antibody responses, was markedly impaired by TRAF1 deficiency. The spontaneous arthritis model in KRN mice appears to be much less antibody dependent than previously believed.
Keywords: Arthritis, Autoantibody, KRN, GPI, TRAF1KO
Introduction
Rheumatoid arthritis (RA) is characterized by chronic in ammation and destruction of the synovial joints leading to progressive joint damage and disability. The disease pathogenesis is influenced by environmental and genetic factors. The inheritance is multigenic [1]. Genetic studies, including genome-wide scans have mapped the human leukocyte antigen (HLA)–DRB1 [2] gene as well as several non–major histocompatibility complex (MHC) RA susceptibility loci, e.g. cytotoxic T lymphocyte-associated antigen 4 (CTLA4) [3]; protein tyrosine phosphatase (PTPN22) [4] and tumor necrosis factor alpha (TNFα) [5]. A recent genome wide association analysis revealed an additional risk-associated genetic region, the Tumor Necrosis Factor Receptor-associated factor 1-Complement 5 (TRAF1-C5) containing locus on chromosome 9 [6, 7]. TRAF1 is a unique member of the TRAF family. The TRAF1 gene encodes an intracellular protein that mediates signal transduction. It contains a single zinc finger and a TRAF domain. Members of the TNFR superfamily, which lack a death domain and include TNFR-II, CD40 and CD30, associate with members of the TRAF family of intracellular signal transduction molecules [8]. Due to its widespread expression, the potential effects of TRAF1 may be receptor and/or cell specific and may potentially provide either enhancing or suppressing activities at different stages of arthritis [9–13].
A variety of animal models of arthritis have been used in the past to gain insight into the pathogenesis of arthritis development [14]. Since 1996, the KRN/I-Ag7 (or K/BxN) model has gained a great deal of attention, given that it occurs spontaneously as a result of a well-defined T cell and B cell autoreactivity to a single antigen [15]. The KRN T cell receptor recognizes glucose-6-phosphate isomerase (GPI) in the context of the NOD-derived H-2g7class II MHC molecule [16]. This immune recognition gives rise to anti-G6PI autoantibodies, which appear to be critical to the development of joint disease, as passive transfer of anti-G6PI antibodies can result in the appearance of transient joint disease [17]. C5 has been shown to be essential for development of arthritis in both the spontaneous genetic model and the passive-serum protocol for KRN arthritis [18, 19]. Curiously, C3 is essential only for the serum transferred disease, and not for the genetic model [20].
In the current study, we have tested the role of TRAF1 in the KRN model. TRAF1-deficient mice were found to be fully susceptible to passive arthritis induced by serum transfer. KRN/I-Ag7 hybrid mice that were TRAF1 deficient also developed severe joint involvement. Interestingly, many of them made no or little detectable anti-GPI antibody. The antibody negative mice showed a slight delay in the development of arthritis compared to antibody positive TRAF1 KO or WT mice. Other immune responses in TRAF1 KO mice were equivalent to that of WT mice for both the induction of IgG antibodies after immunization with an exogenous T-dependent antigen and the production of autoantibodies in a chronic GVH. These results implicate TRAF1 in the loss of B-cell tolerance to the particular GPI autoantigen. They also indicate that the role of antibody in the spontaneous KRN model needs to be reconsidered.
Methods
Mice
C57BL/6 (B6), B6.CgH-2g7-Tg (Ins2-CD80)3B7Flv/LwnJ (B6.I-Ag7) mice were purchased from Jackson Laboratory (Bar Harbor, ME). B6TRAF1KO mice were developed in the Choi laboratory. KRN mice were originally obtained from Diane Mathis. The strain was maintained on the B6 background (B6 KRN). Arthritis-prone mice that express both KRN and I-A were produced by crossing B6.I-Ag7 females and B6.KRN males (KRN/I-Ag7 F1).
All mice were bred and maintained in the animal facility of the School of Medicine, University of Pennsylvania. The animal breeding, maintenance, and experimental procedures were carried out by the protocols approved by the Institutional Animal Care and Use Committee of the University.
Mouse Genotyping
KRN TCR expression [21] and MHC II I-Ag7/MHC II I-Ab alleles [20] were typed by amplification of genomic DNA were from mouse tails.
Homozygous TRAF1 KO mice: PCR analysis was performed with two different primer sets. The 5′ primer sequence 5′-GGT ACA AGC GCA GGC ACA ACT TG and the 3′ primer sequence 5′-GCA TCC TTT GAT GGT ACT TTC were used to amplify a 1.2kb fragment from the WT allele. The 5′ primer sequence 5′-CGG GAG CGG CGA TAC CGT AAA GC and the 3′ primer sequence 5′-GCT TGG GTG GAG AGG CTA TTC GG were used to amplify an 800bp fragment from the KO TRAF1 allele. Each PCR was carried out in a volume of 25μl with 1.5mM MgCl2, 0.2mM dNTP, 100μM 5′ primer, 100μM 3′ primer, and 0.15 unite of Platinum Taq polymerase (Invitrogen, San Diego, CA) and 1 μl of mouse tail digestion. The conditions for the WT allele PCR were: 1 cycle of 4 minutes at 94 °C; 35 cycles of 40 seconds at 94 °C, 30 seconds at 60 °C, and 1 minute at 72 °C; followed by 7 minutes at 72 °C. The conditions for the TRAF1KO allele PCR were: 1 cycle of 4 minutes at 94 °C; 35 cycles of 40 seconds at 94 °C, 30 seconds at 66 °C, and 90 seconds at 72 °C; followed by 7 minutes at 72 °C.
Serum Transfer
Eight- to 16-week-old female B6 and B6.TRAF1KO mice received intraperitoneal injections (i.p.) of KRN/I-Ag7F1 serum 200 μL on day 1 and day 3. Ankle thickness and clinical index were evaluated as described below.
Mouse Arthritis Evaluation
Ankle thickness was measured at the malleoli with a caliper (World Precision Instrument, Sarasota, FL, USA). The clinical index of arthritis was scored on a scale from 0 to 4. Level 0, no redness and swelling of ankles or wrists; level 1, mild, but definite redness and swelling of the ankle or wrist, or apparent redness and swelling limited to individual digits regardless of the number of affected digits; level 2, moderate redness and swelling of ankles or wrists; level 3, severe redness and swelling of the entire paw including digits; level 4, maximally inflamed limb with involvement of multiple joints. The scores of all four limbs were summed.
Mouse arthritis was evaluated daily until the joint swelling reached plateau, then every other day until swelling was no longer present.
Mouse Serum Anti-GPI κ Chain Antibody Detection
Serum anti-GPI κ chain antibodies were detected by solid-phase enzyme-linked immunosorbent assays (ELISA). GPI was expressed from a his-tagged mouse GPI construct (obtained from Diane Mathis) in Protein Expression and Libraries Facility (Wistar Institute, Philadelphia, PA, USA). Polyvinylchloride flat bottom microtiter plates (Thermo Electron Corp., Milford, MA, USA) were coated with 5 μg/ml GPI in BBS (0.2 M borate buffed solution, pH 8.2) at 4°C overnight. Coated plates were blocked with BBT (0.5% bovine serum albumin and 0.4% Tween-80 in BBS) at room temperature for 1 h. The plates were washed three times with BBS after each incubation. Serum samples and controls were diluted in BBT, added to plates, and incubated at 4°C overnight. Mouse serum anti-GPI κ was detected with 25 ng/ml biotinylated rat anti-mouse κ (BD Bioscience, San Jose, CA, USA), followed by avidin-alkaline phosphatase in 1:10,000 dilution (Sigma-Aldrich, St. Louis, MO, USA). Each reagent was added and incubated at room temperature for 1 h following three washes with BBST (0.5% Tween-80 in BBS). Phosphatase substrate 4-nitrophenyl phosphate (Sigma-Aldrich) at a concentration of 1 mg/ml in 10 mM diethanolamine was added and incubated at room temperature for 4 h. Optical density at 405 nm of each well was measured using a microplate reader (E-Max, Molecular Devices Corp., Sunnyvale, CA, USA). All reagents used in this ELISA were added to the plate at a volume of 100 μl per well. A laboratory-pooled KRN/I-Ag7 F1 anti-GPI serum was used as experimental reference and positive control [22].
Antibody Responses to exogenous antigen
To determine the antibody response to the T-dependent antigen ovalbumin (OVA), 4 week old mice were immunized intraperitoneally with 100 μg of ovalbumin precipitated with alum, boosted the same way at day 21 and bled at days 0, 7, 14, 21, 28, and 35. The magnitude of the anti-ovalbumin antibody response was detected by ELISA, as previously described [23]. Briefly, the OVA (100 μg/ml in BBS) was absorbed to 96-well plates overnight at 4°C. For κ chain, IgG1, IgG2c isotype determination, sera were diluted 1/500, 1/5000, and 1/1000, respectively, and added to the wells for overnight incubation at 4°C. Biotin-conjugated antibodies specific for κ chain, IgG1, IgG2c were added and incubated for 1 hr at 37°C, followed by strepavidin-alkaline conjugate according to the manufacturer’s recommendations (BD Pharmingen).
Experimental chronic GVH protocol
Chronic GVH was induced as previously described [24]. Briefly, donor bm12 splenocytes (1×108) were injected i.p. into TRAF1KO or WT recipient mice at the age of 12–16 weeks. Mice were bled serially for the following 9 weeks, and anti-chromatin antibodies were quantitated as described [24]
Statistical Analysis
The levels of serum anti-GPI were analyzed with Soft-Max v4.5. and reported as EDF (equivalent dilution factor to standards). The data were presented as geometric means from each group of samples. Error bars represent SE of each group of data. Statistical significance (p value) was determined by Student’ s t test.
Results
Mouse Arthritis in TRAF1-Deficient KRN/I-Ag7 Mice
We observed that KRN/I-Ag7 F1 that were totally TRAF1-deficient exhibited pronounced joint inflammation of all distal joints of the paws at 4 weeks, similar to that seen in KRN/I-Ag7 that had normal TRAF1 genes (Fig. 1). In order to verify TRAF1 deficiency in these mice, disrupted TRAF1 allele was reconfirmed by PCR (data not shown).
Fig. 1.

Spontaneous arthritis in KRN/I-Ag7 mice with or without TRAF1. One hundred percent of KRN/I-Ag7 mice with or without TRAF1 developed spontaneous arthritis. a–c: Representative images of right hind paws of 1.5-month-old TRAF1-deficient B6.I-Ag7(a), B6.KRN/I-Ag7(b), and TRAF1-deficient B6.KRN/I-Ag7(c) mice. d-i: Cohorts (N = 8–10) of female (d, f, h) and male (e, g, i) mice were followed for clinical arthritis between ages 3 to 10 weeks. Closed symbols represent TRAF1-sufficient mice, and open symbols represent TRAF1-deficient mice. Shown are arthritis incidence (d, e); clinical index (f, g); and ankle thickness (h, i). * Indicates that the two groups are statistically different (p < 0.05) at the given time point by Student’s T test.
In a longitudinal experiment, matched cohorts of TRAF1-deficient KRN/I-Ag7 mice and TRAF1-intact KRN/I-Ag7 were followed from weaning for clinical signs of arthritis and serum anti-GPI antibody production. The TRAF1 KO mice groups showed a modest (but statistically significant) delay in the development of arthritis between the ages of 3–6 weeks. Surprisingly, the anti-GPI κ chain antibody titers were much lower in TRAF1-deficient mice than in TRAF1-sufficient mice (Fig. 2). If we divided the TRAF1KO mice into antibody responders and non-responders, using an anti-GPI cutoff value >20 (= 2× the lower limit of detection), those mice with 0 or almost 0 titers accounted for all the observed delay in arthritis in the TRAF1 KO groups (Fig. 3a). In addition, the levels of anti-GPI antibodies in some of the TRAF1KO non-responders were completely indistinguishable from background levels in KRN− mice (data not shown). Those TRAF1 KO animals that made an anti-GPI antibody response developed arthritis equivalent to that of the WT mice (Fig. 3b). Nevertheless, the appearance of anti-GPI antibodies in this TRAF1 KO responder group was delayed in comparison with the WT mice, and remained at substantially lower levels. Importantly, clinical disease was apparent in both TRAF1-sufficient and TRAF1-deficient groups at 4 weeks, at which time serum anti-GPI antibodies were not detectable, and actually peaked in most mice before the appearance of antibody in the serum. All mice in the non-responder group also showed severe arthritis, even though many of them never had any evidence of anti-GPI antibodies in their serum.
Fig. 2.

Anti-GPI antibodies in individual TRAF1-sufficient and TRAF1-deficient KRN/I-Ag7 mice. Anti-GPI antibodies levels were tested weekly. Solid diamonds represent TRAF1-sufficient male mice, while open diamonds represent TRAF1-deficient male mice (Fig. 2a, N=8 in TRAF1-sufficient group; N=10 in TRAF1-deficient group); solid circles represent TRAF1-sufficient female mice while open circles represent TRAF1-deficient female mice (Fig. 2b, N=10 in each group).
Fig. 3.
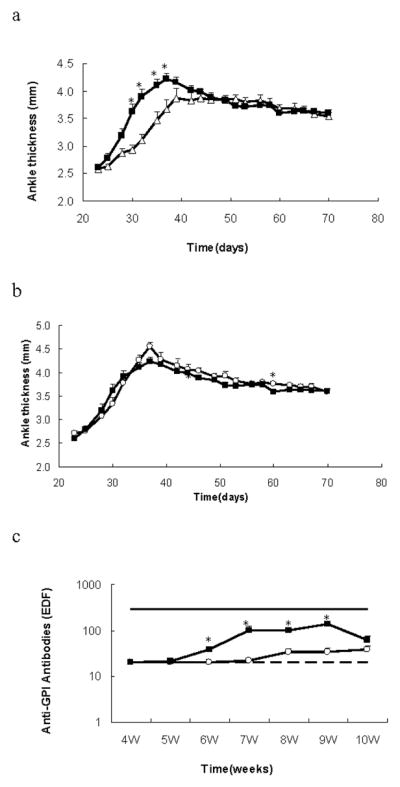
The role of the anti-GPI response on the spontaneous development of arthritis in TRAF1-sufficient and TRAF1-deficient KRN/I-Ag7 mice. TRAF1KO mice were divided into serological responders and non-responders, using an anti-GPI cutoff value >20. The ankle thickness and clinical score (not shown) of mice were quantitated from age 3 to 10 weeks. Anti-GPI antibodies levels were tested weekly. Shown are arithmetic means and SE. Open triangles represent non-responder TRAF1-deficient mice (N=11), open circles represent responder TRAF1-deficient mice (N=9), and solid squares represent TRAF1-sufficient mice (N=18). The mice without antibody had delayed arthritis (Fig 3a), while those animals that had antibodies had arthritis equivalent to that of the WT mice (Fig 3b). However the antibody titers were relatively lower even in the TRAF1KO responders compared to WT (Fig 3c; solid and dotted lines represent EDF value of positive and negative controls, respectively). *p<0.05 for comparison of two groups.
Passive Arthritis in TRAF1-Deficient KRN/I-Ag7 Mice
Since TRAF1KO KRN/I-Ag7 F1 mice had pronounced joint inflammation with significantly lower amount of serum anti-GPI antibodies, we tested the role of TRAF1 in the distal mechanisms of the KRN model by transferring 200 μL anti-GPI serum to B6.TRAF1KO.I-Ag7 and B6.I-Ag7 mice. The responses in both groups of mice were identical: inflammation was visible 24 h after first serum injection and attained maximal ankle thickness at nine days (Fig 4). The clinical index measurements were also consistent (data not shown).
Fig. 4.
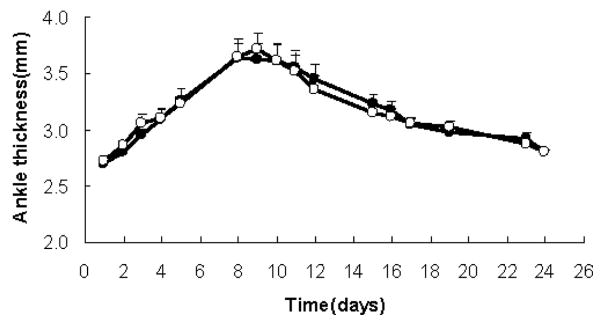
KRN/I-Ag7 serum-induced arthritis. Two-hundred μl of KRN/I-Ag7 serum was administrated I.P. on day 1 and day 3 to TRAF1-intact female mice (black circles N=15) or to TRAF1-deficient female mice (open squares N=13). Shown are arithmetic means and SE.
Sera of all groups of mice were collected at 24 h after the second anti-GPI serum injection (day 4) and when ankle redness and swelling generally started to decrease (day 12). Figure 5 shows that the serum levels of anti-GPI κ chain were comparable in mice with or without TRAF1. This suggests that the half-life of the anti-GPI antibody is comparable in the presence and in the absence of TRAF1.
Fig. 5.
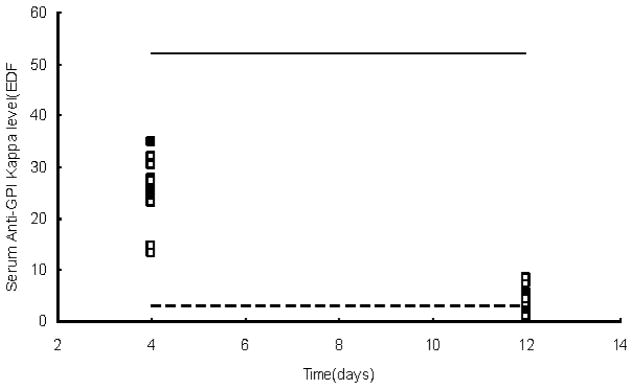
Ant-GPI levels in KRN/I-Ag7 serum transferred mice. Blood samples were collected at day 4 and day 12 respectively. Solid squares represent individual TRAF1-intact B6.I-Ag7 female mice, and open squares represent individual TRAF1 deficient B6.I-Ag7 female mice. For comparison, titers are shown from 10-week-old KRN/I-Ag7 F1 female mice with or without TRAF1 (solid and dotted lines, respectively). Values shown are EDF in reference to a pooled KRN/I-Ag7 F1 anti-GPI serum.
To determine the role of TRAF1 in antibody immune responses to a foreign antigen, we immunized TRAF1KO.I-Ag7 mice and I-Ag7 mice with the T-dependent antigen ovalbumin. In addition, cGVH was induced by injecting bm12 splenocytes into TRAF1KO recipient mice. Figure 6 shows that TRAF1KO mice have normal κ chain, IgG1, and IgG2c anti-ovalbumin responses (Fig. 6a–c), suggesting intact T cell help and intact immunoglobulin isotype switching in B cells. Figure 7 shows that TRAF1KO had no significant effect on cGVH autoantibodies production. Thus, the markedly decreased production of anti-GPI autoantibody in TRAF1 deficient KRN/ I-Ag7 F1 mice was not part of a general defect in antibody responses.
Fig. 6.
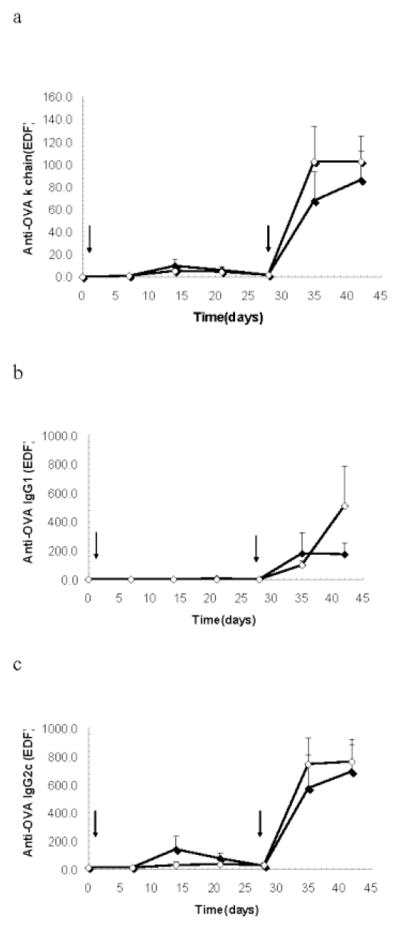
Response to the T-dependent antigen ovalbumin. TRAF1KO.I-Ag7 (open diamonds) and I-Ag7 (closed diamonds) male mice were intraperitoneally immunized with 100μg ovalbumin at day 0 and boosted with the same dose at day 28 (N=5 in each group). Arrows represent times of immunization. Ovalbumin-specific κ chain (a), IgG1 (b), IgG2c (c) were measured once a week until day 42. Shown are arithmetic means and SE.
Fig. 7.
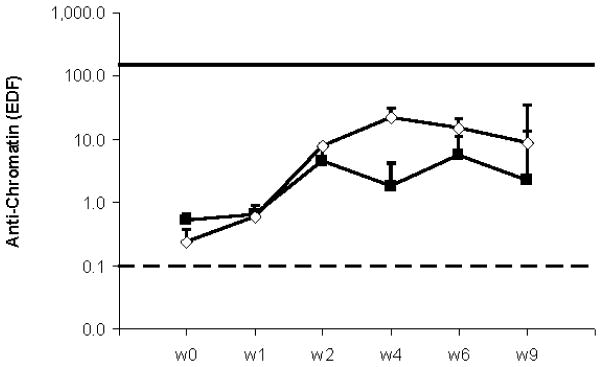
Induction of cGVH model in TRAF1KO mice. TRAF1KO (open diamonds) and WT (closed squares) male mice were intravenously immunized with donor bm12 splenocytes (1×108) at day 1 (N=4 in each group). Anti-chromatin and anti-dsDNA (not shown) were measured once a week until week 9. Shown are arithmetic means and SE. Solid and dotted lines represent EDF value of positive and negative control respectively.
Discussion
TRAF1 is an intriguing candidate as an arthritis risk gene [6, 7]. Studies have shown immune modulatory effects of TRAF1 on the innate and adaptive arms of the immune response that might potentially regulate onset or severity of arthritis [25]. In tg mice that overexpress TRAF1 in T and B cells, TRAF1 limits signaling via TRAF2. In TRAF1 KO, the hyper-responsiveness of T cells to TNF or antigen also suggests that TRAF1 negatively regulates signaling through TNFR2 and TCR [26]. Conversely, it has been reported that TRAF1 plays a key role in survival of activated and memory CD8 T cells via modulation of pro-and anti-apoptotic Bcl-2 family members [12]. Other recent data suggested that TRAF1 enhances TNFR2 induced NF-κB activation, thus acting as a positive regulator of TNF-signaling [11]. Thus, the various effects of TRAF1 may be receptor and/or cell specific and could potentially provide either enhancing or suppressing activities at different stages of arthritis. This complexity must be well understood if we are to realize the potential of TRAF1 as a therapeutic target. Our study tested the effect of lack of expression of TRAF1 in animal models of inflammatory arthritis. The present findings indicated that the role of TRAF1 gene in KRN arthritis model is indeed complicated. In the spontaneous genetic model in KRN/I-Ag7 mice, the anti-GPI κ chain antibodies level was markedly lower in TRAF1-deficient mice than in TRAF1-sufficient mice. In fact 8/20 TRAF1 KO mice showed no detectable anti-GPI antibody response up until 10 weeks of age, when the experiment was terminated. If we divide the TRAF1KO mice into responders and non-responders, using an anti-GPI cutoff EDF value >20, those mice with no or minimal titers had delayed arthritis compared to WT mice, while those animals that had significant levels of antibodies had arthritis equivalent to that of the WT mice. Eventually, even the mice that never produced anti-GPI antibodies detectable above background also showed severe clinical arthritis. Even the antibody positive TRAF1 KO mice always had anti-GPI antibody titers much lower than those of the WT KRN/I-Ag7 mice. The isotype of these responding TRAF1 KO mice was largely IgG1, as it was in the WT mice, and as has been reported by others [27] (data not shown).
It is possible that the antibody made a modest contribution to the arthritis and the delayed disease onset in those TRAF1KO that apparently failed to make antibody was a consequence of this absence. Accordingly, this interpretation would imply that the non-antibody part of the response was not at all affected by TRAF1KO. On the other hand, since the antibody titers were relatively low even in the responder TRAF1KO mice (Fig. 3c), the situation may be more complicated.
In any case, it appears that much if not most of the clinical arthritis we observed could occur in the apparent complete absence of serum antibody in some mice. In many other KRN/I-Ag7 F1 mice, including some of the TRAF1 wild type, severe clinical arthritis appeared weeks before autoantibody was detectable (data not shown). In individual KRN/I-Ag7 F1 mice, we saw no overall correlation between anti-GPI levels and clinical arthritis, in either the TRAF1KO or TRAF1 WT mice. The persistence of autoantibodies in the intact genetic model may be more critical than peak levels. Unfortunately, this is very difficult to test in a transfer model, as it would require impractical amounts of KRN serum. We have not directly tested the effectiveness of the anti-GPI autoantibodies produced in TRAF1-deficient KRN/I-Ag7 F1 mice to transfer arthritis to either TRAF1KO or TRAF1 WT recipients, although they were largely of the isotype (IgG1) that is reported to be most arthritogenic.
We did not detect differences in the serum transfer model, either in clinical symptoms or levels of anti-GPI antibodies, between WT and TRAF1 KO recipients. TRAF1 deficiency did not affect anti-GPI autoantibody catabolism. The serum levels of anti-GPI at day 4 were about 50% of those seen in sera for 10-week-old KRN/I-Ag7 mice; and clearly significantly higher than WT mice. Whether there are levels or isotypes of anti-GPI antibodies that are undetectable in our anti-GPI assays and yet still pathogenic in vivo is a matter of speculation.
Korganow et al have previous demonstrated the requirement for B cells in the KRN model, and these B cells cannot have a repertoire restricted to a single specificity by a homozygous Ig transgene [17]. They demonstrated a role of B cells in activating GPI specific T cells, but concluded that the main pathological mechanism in the model ultimately required the production of GPI specific antibodies through the participation of the activated T cells. Jacobs later demonstrated that this antibody-centric view may not fully capture the role of T cells in the K/BxN arthritis model, as T cells can augment antibody-induced arthritis independently of their influence on antibody production [28]. The autoreactive T cells can promote arthritis through both the generation of arthritogenic autoantibodies and an IL-17-mediated effector response that amplifies inflammation. Recently, gut-residing segmented filamentous bacteria have been shown to affect kinetics of K/BxN arthritis by inducing Th17 cells in the lamina propria [29]. Our current data further support the potential importance of antibody-independent mechanisms in the severe spontaneous arthritis in the KRN/I-Ag7 hybrid mice. We do not know why TRAF1 deficiency has such a profound effect on production of anti-GPI antibodies in this model, but not on other T dependent responses. Nevertheless, the strong dissociation of this effect on anti-GPI antibody production and the development of severe arthritis are striking.
The implications for RA are not clear at this time. It is likely that the role of TRAF1 in RA may be more complicated. In addition, as the spontaneous arthritis model in KRN mice may be much less antibody dependent than previously believed, the likely pathogenic autoantibodies in RA (anti-CCP [30]) may be but one of several specific effector mechanisms.
Conclusion
The pathogenesis of spontaneous KRN/I-Ag7 arthritis can largely proceed by TRAF1-independent pathways. The production of anti-GPI autoantibody, but not other serological responses, was markedly impaired by TRAF1 deficiency. The spontaneous arthritis model in KRN mice appears to be much less antibody dependent than previously believed.
Acknowledgments
We thank Xin Tian for helpful mouse bleeding.
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 2.Newton JL, Harney SM, Wordsworth BP, Brown MA. A review of the MHC genetics of rheumatoid arthritis. Genes Immun. 2004;5:151. doi: 10.1038/sj.gene.6364045. [DOI] [PubMed] [Google Scholar]
- 3.Plenge RM, Padyukov L, Remmers EF, Purcell S, Lee AT, Karlson EW, et al. Replication of putative candidate-gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: association of susceptibility with PTPN22, CTLA4, and PADI4. Am J Hum Genet. 2005;77:1044–60. doi: 10.1086/498651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee AT, Li W, Liew A, Bombardier C, Weisman M, Massarotti EM, et al. The PTPN22 R620W polymorphism associates with RF positive rheumatoid arthritis in a dose-dependent manner but not with HLA-SE status. Genes Immun. 2005;6:129–33. doi: 10.1038/sj.gene.6364159. [DOI] [PubMed] [Google Scholar]
- 5.Maini RN. Current and new antitumor necrosis factor agents in perspective. Arthritis Res Ther. 2004;6:S1–S2. doi: 10.1186/ar994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurreeman, Fina AS, Padyukov, Leonid, Marques, Rute B, et al. A candidate gene approach identifies the TRAF1/C5 region as a risk factor for rheumatoid arthritis. PLoS Med. 2007;4:e278. doi: 10.1371/journal.pmed.0040278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis- a genomewide study. N Engl J Med. 2007;357:1199. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arch RH, Gedrich RW, Thompson CB. Tumor necrosis factor receptor-associated factors (TRAFs)-α family of adapter proteins that regulates life and death. Gene Dev. 1998;12:2821–30. doi: 10.1101/gad.12.18.2821. [DOI] [PubMed] [Google Scholar]
- 9.Fotin-Mleczek M, Henkler F, Hausser A, Glauner H, Samel D, Graness A, et al. TRAF1 regulates CD40-induced TRAF2-mediated NF-κB activation. J Biol Chem. 2004;279:677–85. doi: 10.1074/jbc.M310969200. [DOI] [PubMed] [Google Scholar]
- 10.Kato T, Jr, Gotoh Y, Hoffmann A, Ono Y. Negative regulation of constitutive NF-κB and JNK signaling by PKN1-mediated phosphorylation of TRAF1. Genes to Cells. 2008;13:509–20. doi: 10.1111/j.1365-2443.2008.01182.x. [DOI] [PubMed] [Google Scholar]
- 11.Lavorgna A, De Filippi R, Formisano S, Leonardi A. TNF receptor-associated factor 1 is a positive regulator of the NF-κB alternative pathway. Mol Immunol. 2009;46:3278–82. doi: 10.1016/j.molimm.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 12.Sabbagh L, Srokowski CC, Pulle G, Snell LM, Sedgmen BJ, Liu Y, et al. A critical role for TNF receptor-associated factor 1 and Bimdown-regulation in CD8 memory T cell survival. Proc Natl Acad Sci US A. 2006;103:18703–8. doi: 10.1073/pnas.0602919103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wicovsky A, Henkler F, Salzmann S, Scheurich P, Kneitz C, Wajant H. Tumor necrosis factor receptor-associated factor-1 enhances proinflammatory TNF receptor-2 signaling and modifies TNFR1-TNFR2 cooperation. Oncogene. 2009;28:1769–81. doi: 10.1038/onc.2009.29. [DOI] [PubMed] [Google Scholar]
- 14.Asquith DL, Miller AM, McInnes IB, Liew FY. Animal models of rheumatoid arthritis. Eur J Immunol. 2009;39:2040–4. doi: 10.1002/eji.200939578. [DOI] [PubMed] [Google Scholar]
- 15.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–22. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 16.Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286:1732–5. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- 17.Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–61. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 18.Ji Hong, Ohmura Koichiro, Mahmood Umar, Lee David M, Hofhuis Frans MA, Boackle Susan A, et al. Arthritis critically dependent on innate immune system players. Immunity. 2002;16:157–68. doi: 10.1016/s1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- 19.Binstadt BA, Hebert JL, Ortiz-Lopez A, Bronson R, Benoist C, Mathis D. The same systemic autoimmune disease provokes arthritis and endocarditis via distinct mechanisms. Proc Natl Acad Sci US A. 2009;106:16758–63. doi: 10.1073/pnas.0909132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsao PY, Arora V, Ji MQ, Wright AC, Eisenberg RA. KRN/I-Ag7 mouse arthritis is independent of complement C3. J Clin Immunol. 2011;31:857–63. doi: 10.1007/s10875-011-9562-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monach PA, Mathis D, Benoist C. The K/BxN arthritis model. In: Coligan JE, et al., editors. Current protocols in immunology. Unit 15. Chapter 15. New York: Wiley; 2008. p. 22. [DOI] [PubMed] [Google Scholar]
- 22.Tsao PY, Jiao J, Ji MQ, Cohen PL, Eisenberg RA. T cell-independent spontaneous loss of tolerance by anti-double-stranded DNA B cells in C57BL/6 mice. J Immunol. 2008;181:7770–7. doi: 10.4049/jimmunol.181.11.7770. [DOI] [PubMed] [Google Scholar]
- 23.Spergel JM, Mizoguchi E, Brewer JP, Martin TR, Bhan AK, Geha RS. Epicutaneous sensitization with protein anti-gen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J Clin Investig. 1998;101:1614–22. doi: 10.1172/JCI1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feuerstein N, Chen F, Madaio M, Maldonado M, Eisenberg RA. Induction of autoimmunity in a transgenic model of B cell receptor peripheral tolerance: changes in coreceptors and B cell receptor-induced tyrosine-phosphoproteins. J Immunol. 1999;163:5287–97. [PubMed] [Google Scholar]
- 25.Xavier RJ, Rioux JD. Genome-wide association studies: a new window into immune-mediated diseases. Nat Rev Immunol. 2008;8:631–43. doi: 10.1038/nri2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsitsikov EN, Laouini D, Dunn IF, Sannikova TY, Davidson L, Alt FW, et al. TRAF1 Is a Negative Regulator of TNF Signaling: Enhanced TNF Signaling in TRAF1-Deficient Mice. Immunity. 2001;15:647–57. doi: 10.1016/s1074-7613(01)00207-2. [DOI] [PubMed] [Google Scholar]
- 27.Kyburz, Diego, Corr, Maripat The KRN mouse model of inflammatory arthritis. Springer Semin Immun. 2003;25:79–90. doi: 10.1007/s00281-003-0131-5. [DOI] [PubMed] [Google Scholar]
- 28.Jacobs JP, Wu HJ, Benoist C, Mathis D. IL-17-producing T cells can augment autoantibody-induced arthritis. Proc Natl Acad Sci US A. 2009;106:21789–94. doi: 10.1073/pnas.0912152106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815–27. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schellekens GA, Visser H, De Jong BAW, van den HFHJ, Hazes JMW, Breedveld FC, et al. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000;43:155–63. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]