

Abstract
Oxidative stress and inflammation are mediators in the development and progression of chronic kidney disease (CKD) and its complications, and they are inseparably linked as each begets and amplifies the other. CKD-associated oxidative stress is due to increased production of reactive oxygen species (ROS) and diminished antioxidant capacity. The latter is largely caused by impaired activation of Nrf2, the transcription factor that regulates genes encoding antioxidant and detoxifying molecules. Protective effects of Nrf2 are evidenced by amelioration of oxidative stress, inflammation, and kidney disease in response to natural Nrf2 activators in animal models, while Nrf2 deletion amplifies these pathogenic pathways and leads to autoimmune nephritis. Given the role of impaired Nrf2 activity in CKD-induced oxidative stress and inflammation, interventions aimed at restoring Nrf2 may be effective in retarding CKD progression. Clinical trials of the potent Nrf2 activator bardoxolone methyl showed significant improvement in renal function in CKD patients with type 2 diabetes. Results of the ongoing BEACON trial investigating the effect of this drug on time to end-stage renal disease or cardiovascular death will help further characterize the efficacy of Nrf2 pharmacological modulation in CKD. This article provides an overview of the role of impaired Nrf2 activity in the pathogenesis of CKD-associated oxidative stress and inflammation and the potential utility of targeting Nrf2 in the treatment of CKD.
Keywords: Oxidative stress, Inflammation, CKD progression, Cardiovascular disease, ESRD, Antioxidant therapy, bardoxolone methyl
OXIDATIVE STRESS AND INFLAMMATION IN CKD
Oxidative stress and inflammation are features of chronic kidney disease (CKD) and drivers of CKD progression, as well as its cardiovascular and other complications (1-4). Oxidative stress is a condition in which generation of reactive oxygen species (ROS) exceeds the capacity of the antioxidant defense system. It can occur as a result of increased ROS production, impaired antioxidant capacity, or both (5). Oxidative stress and inflammation are inseparably linked, as each begets and amplifies the other. For instance, via activation of NFκB, a redox-sensitive transcription factor that regulates expression of pro-inflammatory cytokines and chemokines, oxidative stress promotes recruitment and activation of leukocytes and resident cells, thereby eliciting inflammation (6). Likewise, via formation of pro-inflammatory oxidized lipids and advanced protein oxidation and glycation end products, oxidative stress promotes inflammation. Conversely, by generating reactive oxygen, chlorine, and nitrogen species, activated leukocytes, macrophages, and resident cells cause oxidative stress (2;6). Before proceeding with the review of the nature, mechanisms, and contribution of oxidative stress and inflammation in the pathogenesis and progression of CKD, a brief description of ROS production and metabolism, as well as the antioxidant defense system, is provided below.
Production of reactive oxygen species (ROS) and oxidative stress
Conversion of molecular oxygen to water involves its 4-electron reduction by hydrogen (O2 + 4H→ 2H2O). For the great majority of oxygen processed in mitochondria, this reaction takes place in a single step. However, for the remaining small but significant portion (1-4%) of oxygen used in the body, conversion to water occurs with the acquisition of one electron at a time, leading to formation of short-lived and highly reactive intermediary products, termed ROS (7-9). The primary ROS produced in the body are superoxide anion (O2•−) and hydrogen peroxide (H2O2), which represent the byproducts of 1 and 2 electron reductions of O2, respectively. Generation of these intermediary oxygen metabolites rises in response to reduction of O2 supply (hypoxia, ischemia), elevation of substrate supply, or both (7;9).
Besides mitochondria, a number of cytosolic enzymes, including oxygenases, oxidases, and peroxidases, generate superoxide anion and hydrogen peroxide. While uncontained hydrogen peroxide and superoxide can cause oxidative stress and cytotoxicity, when produced at a normal rate, the healthy organism is well-equipped to neutralize them (7;9). For example, superoxide anion is transformed to hydrogen peroxide (2O2•− + 2H→ H2O2) by the superoxide dismutase (SOD) family of enzymes, which are present in mitochondria (Mn-SOD), cytoplasm (Cu,Zn-SOD), and plasma membrane (EC-SOD). Similarly, hydrogen peroxide is converted to water by catalase and glutathione peroxidase. Accordingly, the ability of superoxide and hydrogen peroxide to directly cause oxidative stress and tissue injury is limited. As a matter of fact, under normal conditions they serve as signaling molecules or second messengers for various growth factors and hormones. However, under pathological conditions, they can serve as substrates for the generation of highly reactive and cytotoxic products the organism is not equipped to contain. These include production of hydroxyl radical (•OH) from hydrogen peroxide in the presence of transition metals, such as catalytically active iron (H2O2+Fe2+→•OH+OH−+ Fe3+); peroxynitrite from superoxide in the presence of nitric oxide (NO+O2• →ONOO−); and hypochlorous acid (HOCl), commonly known as bleach, from hydrogen peroxide in the presence of myeloperoxidase (H2O2+Cl−→HOCl) (Figure 1). Generation of these highly reactive and cytotoxic secondary molecules mediates the pathological changes involved in many diverse progressive and degenerative disorders (7;9).
Figure 1. Production and Metabolism of Reactive Oxygen Species.
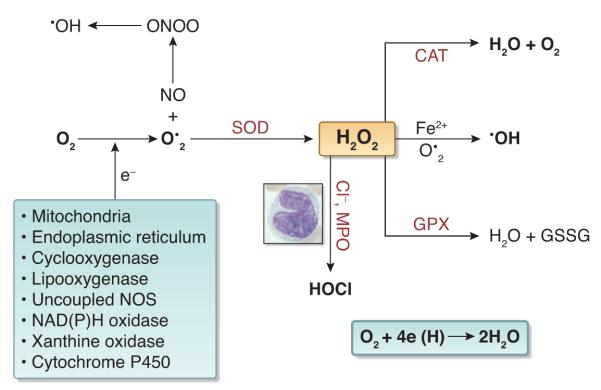
Normally over 95% of the oxygen consumed in the body is converted to water by acquisition of 2 electrons in a single step. However, for the remaining 5% this process occurs with the transfer of one electron at a time, leading to formation highly reactive and short-lived species, collectively referred to as ROS. The primary ROS produced in the body is superoxide which is formed from single electron reduction of molecular oxygen. The primary sources of superoxide include the mitochondria, endoplasmic reticulum, cyclooxygenase, lipoxygenase, uncoupled nitric oxide synthase (NOS), NAD(P)H oxidase, xanthine oxidase, and cytochrome P450. Antioxidants then act on ROS to generate less reactive species. For example, superoxide dismutase (SOD) converts superoxide into hydrogen peroxide, which is then reduced by catalase (CAT) into water and oxygen and by glutathione peroxidase (GPX) into water and oxidized glutathione. However in pathological states hydrogen peroxide serves as the substrate for formation of highly reactive and cytotoxic oxidants such as hydroxyl radical by catalytically-active iron (Fe2+) and hypochlorous acid by myeloperoxidase. An increase in ROS generation or decrease in antioxidant availability leads to oxidative stress and induction of the pro-inflammatory response, which contribute to disease pathogenesis.
Under normal conditions, ROS produced during metabolism are contained by the natural antioxidant defense system. However, when ROS production exceeds the capacity of this system, it leads to oxidative stress in which the uncontained or uncontainable ROS cause tissue damage and dysfunction by attacking, denaturing, and modifying structural and functional molecules and by activating redox-sensitive transcription factors and signal transduction pathways. These events result in necrosis, apoptosis, inflammation, fibrosis, and other disorders that participate in the disease process. Thus, oxidative stress occurs as a result of either increased ROS production and/or an impaired antioxidant defense system.
Antioxidant defense system
The natural antioxidant defense system consists of numerous ROS scavenger molecules of dietary and endogenous origin, antioxidant enzymes and substrates, and phase 2 detoxifying enzymes. Each component of this system provides a specific function and works in a highly coordinated manner with the other components to fulfill the task of protecting against tissue injury. Consequently, the components of this system are not interchangeable, and as such, supernormal quantities of one does not compensate for a deficiency in the other(s) (10).
Nuclear factor-erythroid-2-related factor 2 (Nrf2) plays a central part in basal activity and coordinated induction of over 250 genes, including those encoding antioxidant and phase 2 detoxifying enzymes and related proteins, such as catalase, SOD, UDP-glucuronosyltransferase, NAD(P)H:quinone oxidoreductase-1 (NQO1), heme oxygenase-1 (HO-1), glutamate cysteine ligase, glutathione S-transferase, glutathione peroxidase, and thioredoxin (11;12). Nrf2 is held in the cytoplasm as an inactive complex bound to Keap1 (Kelch-like ECH-associated protein 1), a repressor molecule that facilitates Nrf2 ubiquitination (Figure 2). Keap1 contains several reactive cysteine residues that serve as sensors of the intracellular redox state. Oxidative or covalent modification of thiols in some of these cysteine residues leads to conformational changes in Keap1 that result in disruption of one of the two Keap1 interactions with Nrf2 (“hinge and latch” model). By limiting proteasomal degradation of Nrf2 this process results in accumulation of the de novo synthesized Nrf2 and its translocation to the nucleus (13-15). Within the nucleus, Nrf2 binds to regulatory sequences, known as antioxidant response elements (AREs) or electrophile response elements (EpREs), in the promoter regions of genes encoding antioxidant and phase 2 detoxifying molecules. This process entails heterodimerization of Nrf2 with other transcription factors (e.g., small Maf) within the nucleus. In addition to modification of Keap1, nuclear translocation of Nrf2 may occur via phosphorylation of its threonine or serine residues by upstream kinases, such as protein kinase C, mitogen-activated protein kinases, phosphatidylinositol-3-kinase/Akt, casein kinase-2, and the endoplasmic reticulum enzyme PERK (16;17). Regulation of cellular antioxidant and anti-inflammatory machinery by Nrf2 plays a central part in defense against oxidative stress (11;12). In fact, Nrf2 disruption in mice attenuates or abrogates the induction of genes encoding antioxidants in response to oxidative stress. In addition, ablation of the Nrf2 gene causes a lupus-like autoimmune nephritis and exacerbates diabetes-induced oxidative stress, inflammation, and nephropathy in experimental animals (18;19).
Figure 2. Nrf2 inhibits reactive oxygen species and inflammatory pathways that lead to kidney dysfunction.

Nrf2 is held in an inactive state bound to Keap1 in the cytoplasm. In response to oxidative stress signals from various sources, the Keap1-Nrf2 complex is disrupted, preventing degradation of Nrf2. As a result, newly synthesized Nrf2 translocates to the nucleus, where it activates the transcription of several antioxidant and detoxifying enzymes. Activation of Nrf2 also suppresses NFκB activity, thereby inhibiting inflammation.
Role of increased ROS production in CKD-associated oxidative stress
Oxidative stress is invariably present in all forms of CKD and caused by a combination of increased ROS production and impaired antioxidant capacity. Increased ROS production in the diseased kidney is primarily driven by activation and upregulation of ROS-producing enzymes, including NAD(P)H oxidase (NOX) isoforms, cycloxygenase-2, lipoxygenase, and uncoupled nitric oxide synthase (NOS), mitochondrial dysfunction, and endoplasmic reticulum stress (6;20). This is, in part, mediated by the pathological upregulation of the intra-renal angiotensin system, as evidenced by marked upregulation of angiotensin II receptors (AT1 and AT2) and simultaneous increases in angiotensin II-producing cells in the diseased kidney. Of note, many angiotensin II-producing cells in the diseased kidney are macrophages, which serve as ectopic sources of this hormone (21). In addition to angiotensin II and its receptors, the kidney contains angiotensinogen, angiotensin converting enzyme, and renin, making this organ ideal for the production of angiotensin II and a recipient of its effects (22;23). Interestingly, it has been recently shown that angiotensinogen detected in the kidney originates in the liver (24). Activation of the AT1 receptor by angiotensin II raises superoxide production in the kidney and vasculature via NOX, which is consistently upregulated in animals with experimental or spontaneous CKD (25;26). As described in the elegant review by Shah and associates (27), increased intra-renal ROS production and oxidative stress is found in various models of CKD. Rats with 5/6 nephrectomy-induced CKD exhibit oxidative stress, inflammation, upregulation of ROS-producing enzymes, NFκB activation, and immune cell infiltration in the remnant kidney (28;29). Similarly, oxidative stress and inflammation are prominently present in renal tissue of Imai rats with spontaneous focal segmental glomerulosclerosis (25). Oxidative stress in CKD is commonly accompanied by activation of NFκB and accumulation of inflammatory cells in the diseased kidney (25;28). Infiltrating leukocytes and resident cells are the major source of ROS in many forms of immune complex- and complement-mediated glomerulonephritis, such as anti-glomerular basement membrane antibody-mediated glomerulonephritis and membranoproliferative glomerulonephritis (30-33). However, oxidative stress is also present in experimental models of leukocyte-independent glomerulopathies, such as puromycin aminonucleoside-induced nephrotic syndrome (a widely used model of minimal change disease) (34) and in passive Heymann nephritis (a commonly employed model of membranous nephropathy) (35;36).
Role of diabetes in CKD-associated oxidative stress
Globally, diabetes is a leading cause of CKD and end-stage renal disease (ESRD) requiring dialysis or kidney transplant (37). As with other forms of CKD, diabetic kidney disease is associated with increased intra-renal ROS generation and oxidative stress. In fact, cultured mesangial cells exposed to simulated hyperglycemia (high glucose concentration in culture media) show increased ROS generation (38;39), and glomeruli isolated from diabetic rats show increased superoxide and hydrogen peroxide production (40;41). The latter observations illustrate the direct effect of hyperglycemia on ROS production. In addition, advanced glycation end products (AGEs), which accumulate in tissues of diabetic animals and humans, raise intracellular ROS generation in mesangial cells (42), a phenomenon that is mediated by binding of AGEs to their receptors on macrophages (43). One of the sources of excess ROS production in diabetic nephropathy is NOX4, which is normally expressed in vascular and renal cortical tissues and markedly upregulated in diabetic nephropathy (44). Another source of ROS in diabetic kidney disease is uncoupled NOS (45). In addition, diabetes- and hyperglycemia-induced impairment of mitochondrial metabolism can serve as major sources of ROS generation in kidney and vascular tissues (46-48).
Role of hypertension in CKD-associated oxidative stress
Hypertension is a nearly constant feature and both a cause and consequence of CKD. ROS production is invariably elevated in kidney and arterial tissues and plays a central role in the pathogenesis of hypertension in all models of genetic and acquired hypertension (49-51). Via ROS-mediated inactivation of endothelium-derived NO, depletion of the NO synthase cofactor tetrahydrobiopterin, accumulation of the potent endogenous NOS inhibitor asymmetrical dimethylarginine, formation of F2 isoprostane, and intra-renal activation of NFκB, oxidative stress increases systemic vascular resistance, renal sodium retention, and hence, arterial pressure (49-51). Conversely, hypertension promotes ROS production and oxidative stress in the arterial wall. This supposition is based on an earlier study in rats with aorta banding above the renal arteries that showed marked oxidative stress and ROS-mediated inactivation of NO in the thoracic aorta, which resides in the hypertensive zone, but not in the abdominal aorta, which resides in the normotensive zone of the arterial tree (52). By contrast, no difference was found between the corresponding segments of aorta in sham-operated control rats. Subsequent studies showed marked upregulation of the ROS-generating enzyme NOX in the aorta segment proximal (i.e., the hypertensive zone), but not distal, to the banding site in this model (53;54). Thus, associated hypertension contributes to oxidative stress in CKD.
Role of inflammation in CKD-associated oxidative stress
As noted above, inflammation is a common feature of CKD and both oxidative stress and inflammation are inseparably linked and participate in a self-perpetuating vicious circuit. Consequently, the presence and severity of systemic inflammation contribute to CKD-associated oxidative stress. Inflammation in CKD is not confined to the kidney, rather it is systemic in nature (55). Recent demonstration of disruption of the colonic epithelial tight junction in experimental CKD (56) and marked changes in the composition of the intestinal microbial flora in the CKD humans and animals (57) has unraveled the role of influx of the byproducts of microbial flora and other noxious luminal content into the internal milieu as a potential contributor to CKD-associated inflammation and oxidative stress. In fact, a number of pro-oxidant and pro-inflammatory uremic toxins, including indoxyl sulfate and p-cresol sulfate, in the uremic plasma are byproducts of colonic microbial flora (58). Consequently, disruption of the gut barrier function participates in the pathogenesis of systemic inflammation and oxidative stress in CKD.
Role of impaired antioxidant defense system in CKD-associated oxidative stress
Increased production of ROS in CKD is compounded by an impaired endogenous antioxidant defense system in the kidney (25;59;60). As noted above, Nrf2 plays a central part in basal activity and coordinated induction of several genes encoding antioxidant and phase 2 detoxifying enzymes and related proteins. Consequently, constitutive Nrf2 activity is critical for maintaining redox balance in normal conditions, and its induction in response to oxidative stress is essential for protecting against oxidative stress. However, studies conducted in animals with 5/6 nephrectomy-induced CKD have revealed that despite the presence of oxidative stress and inflammation, which should have induced Nrf2 activation and upregulation of its target genes, CKD animals exhibited a marked and time-dependent decline in nuclear Nrf2 content, pointing to its impaired activation in the remnant kidney (59). The progressive decline of Nrf2 activity was accompanied by a steady reduction of its target gene products, including antioxidant enzymes (i.e., catalase, SOD isoforms, glutathione peroxidase, and HO-1), the key enzymes responsible for glutathione synthesis (i.e., glutamate-cysteine ligase catalytic and modifier subunits), and the major detoxifying enzyme NQO1, over the course of 12 weeks post-renal ablation. These changes were associated with marked elevation of the repressor molecule Keap1, accounting for the failure of Nrf2 activation despite the prevailing oxidative stress in the remnant kidney of CKD animals.
Impaired Nrf2 activity seen in remnant kidney tissue of Sprague Dawley rats with CKD induced by renal mass reduction was subsequently confirmed in kidneys of Imai rats, which exhibit a spontaneous progressive focal and segmental glomerulosclerosis beginning at 8-10 weeks of age and reaching ESRD in several months (25;60). Failure of Nrf2 activation, oxidative stress, inflammation, progressive glomerulosclerosis, tubulo-interstitial fibrosis, upregulation of the intra-renal angiotensin system, hypertension, proteinuria, and renal insufficiency were accompanied by NFκB activation, the maladaptive endoplasmic reticulum stress response, and apoptosis in renal tissue of Imai rats employed in the latter studies. The presence of reduced Nrf2 activity in diverse CKD models (i.e., 5/6 nephrectomy-induced CKD, spontaneous CKD in Imai rats) points to its role as a common mediator in the pathogenesis and progression of CKD independent of the underlying etiologies.
The renal protective effect of Nrf2 is supported by the fact that Nrf2 gene ablation intensifies diabetes-induced inflammation, oxidative stress, and renal injury in experimental animals (19); Nrf2 knockout mice exhibit a lupus-like autoimmune nephritis (18); and ischemic and nephrotoxic insults result in a much more severe acute kidney injury and dysfunction, as well as higher mortality, in Nrf2-deficient mice than in wild-type mice (61). Conversely, weak Nrf2 activators commonly found in foods or dietary supplements have renoprotective effects in rodent models. For example sulforaphane, an organosulfur compound found in cruciferous vegetables has been shown to ameliorate nephropathy in animals with streptozotocin-induced diabetes (62), epigallocatechin-3-gallate (the bioactive polyphenol in green tea) improves renal lesions in animal models of systemic lupus erythematosus (63), mice with anti-glomerular basement membrane glomerulonephritis (64), and cisplatin-induced nephrotoxicty (65). Likewise curcumin, a polyphenolic compound isolated from turmeric, is protective in a model of chromium-induced nephrotoxicity (66). Taken together, these findings support the role of Nrf2 deficiency in the pathogenesis of oxidative stress, inflammation, and progression of CKD and the renoprotective effect of Nrf2 activators.
In addition to rendering the diseased kidney vulnerable to direct ravages of oxidative stress, Nrf2 dysfunction in CKD participates in the pathogenesis and amplification of intra-renal inflammation by accommodating tissue accumulation of hydroperoxides and lipoperoxides, which are potent activators of NFκB. This assertion is supported by several studies that have demonstrated the anti-inflammatory function of Nrf2 (11;67-69).
Role of impaired Nrf2 activity in CKD-associated lipotoxicity
Lipid accumulation in macrophages and mesangial cells is a central step in atherosclerosis and glomerulosclerosis and is mediated by uptake of oxidized lipids and lipoproteins via oxidized LDL receptor-1 and scavenger receptor class A type 1, leading to their transformation to foam cells. Normal HDL confers cardiovascular and renal protection by preventing/reversing oxidation of lipids and lipoproteins (via its potent antioxidant enzymes, paraoxonase and glutathione peroxidase) and by facilitating retrieval of surplus cholesterol and phospholipids from lipid-laden macrophages and mesangial cells. Due to the prevailing oxidative stress and inflammation, CKD results in accumulation of oxidized lipids and lipoproteins in the plasma, kidney, and vascular tissue (70-72). This is coupled with reduced plasma concentration of HDL and impaired antioxidant, anti-inflammatory, and reverse cholesterol transport capacity of HDL in CKD (73-75). CKD-induced HDL abnormalities are due to reductions in production and plasma concentration of apoA1 (the principal apolipoprotein constituent of HDL), paraoxonase, glutathione peroxidase, and lecithin-cholesterol acyltransferase (76-78).
The constellation of increased burden of oxidized lipids and lipoproteins, as well as HDL deficiency and dysfunction, results in accumulation of lipids in the arterial wall and kidney tissues in CKD (79;80). In addition, reabsorption of filtered protein-bound lipids leads to heavy accumulation of lipids and lipotoxicity in proximal tubular epithelial cells of the diseased kidney (79). Normally, oxidized lipids provoke Nrf2 activation and, hence, production of antioxidant enzymes and substrates, which mitigate the impact of oxidative stress and prevent cellular injury (81-83). However, despite the large burden of oxidized lipids (79), Nrf2 activity and its target gene products are greatly suppressed in the diseased kidney (25;59), pointing to a maladaptive response in CKD. Given the critical role of Nrf2 in regulating antioxidant enzymes, glutathione synthase, and apoA1, its impaired activity undoubtedly contributes to the accumulation of oxidized lipids and lipoproteins, HDL deficiency and dysfunction, atherosclerosis, and progression of renal disease in CKD.
Thus, via amplification of oxidative stress, inflammation, and lipotoxicity, impaired Nrf2 activity can contribute to the development and progression of CKD. As such, strategies aimed at restoring Nrf2 activity could potentially retard CKD progression. This supposition has formed the basis for randomized clinical trials to explore the efficacy of the Nrf2 activator compound, bardoxolone methyl, in patients with CKD.
BARDOXOLONE METHYL, AN NRF2 ACTIVATOR
Bardoxolone methyl (methyl 2-cyano-3,12-dioxooleana-1,9(11)dien-28-oate, CDDO-Me, or RTA 402) is a synthetic triterpenoid derived from the natural product oleanolic acid and member of a class of antioxidant inflammation modulators that are the most potent activators of Nrf2 known to-date (84-86). Bardoxolone methyl and its analogs were generated in an attempt to create compounds with significant anti-inflammatory activity that could be used as agents against chronic inflammatory diseases, namely cancer. Potent anti-inflammatory derivatives of oleanolic acid were identified through an assay measuring inhibition of the pro-inflammatory enzyme inducible nitric oxide synthase (iNOS) in mouse macrophages stimulated with IFN-γ. In this assay, bardoxolone methyl was found to be 10,000 times more potent than oleanolic acid, making this derivative an attractive development compound (85;86).
The structure and activity profile of bardoxolone methyl resembles that of 15-deoxy-delta 12,14-prostaglandin J2 (15d-PGJ2) and related cyclopentenone prostaglandins (cyPGs), which are the endogenous activators of Nrf2 that aid in the resolution of inflammation and suppress NFκB (87-92). In this regard, bardoxolone methyl provides a pharmacological means to mimic the body’s innate response to inflammation. Similar to cyPGs, bardoxolone methyl activates Nrf2 by binding to specific cysteine residues on Keap1 (84). This binding results in translocation of Nrf2 to the nucleus, where it initiates transcription of a host of enzymes involved in the antioxidant and detoxification response (93), as previously discussed. Also similar to cyPGs, bardoxolone methyl inhibits NFκB activation via binding to a cysteine residue in IκB kinase (IKKβ) (91;94). Binding of bardoxolone methyl to IKKβ prevents release of NFκB from its bound complex with IκB in the cytosol, thereby inhibiting NFκB activation and induction of downstream pro-inflammatory signaling. Therefore, bardoxolone methyl acts to upregulate the antioxidant response, while diminishing pro-inflammatory signaling.
As previously described, both oxidative stress and inflammation in their chronically activated state contribute to the pathogenesis of CKD (6;95;96). Bardoxolone methyl is proposed to counteract these pathogenic pathways via its effects on Nrf2 activation and NFκB inhibition (86), although effects on other pathways, including specific downstream targets, cannot be excluded. In mesangial cells treated with bardoxolone methyl, increased expression of the Nrf2 target genes HO-1, NQO1, and thioredoxin was observed. When these cells were exposed to albumin or TNF-α, bardoxolone methyl inhibited NFκB activation and expression of COX-2, MCP-1, and IL-1β, providing evidence that this drug attenuates inflammation induced by factors known to be upregulated in CKD (Wigley, et al. Treatment with an Antioxidant Inflammation Modulator (AIM) Increases Glomerular Filtration Rate Monitored by Inulin Clearance in Rats [Abstract]. J Am Soc Nephrol 2010; 21:170A). In proximal tubular epithelial cells, glomerular endothelial cells, and podocytes, bardoxolone methyl was also shown to induce Nrf2 targets, as well as attenuate NFκB activation and its downstream signaling (Dulubova, et al. Effects of Bardoxolone Methyl (BARD) on Renal Protein Handling and Secondary Nephropathy [Abstract]. J Am Soc Nephrol 2010; 21:280A), demonstrating that this Nrf2 activator is capable of inducing an antioxidant and anti-inflammatory response in various cells of renal origin. Additionally, bardoxolone methyl and a related triterpenoid analog (RTA 405) inhibited angiotensin II-mediated contraction in mesangial cells. Ex vivo, RTA 405 was observed to inhibit angiotensin II-induced contraction in whole glomeruli, counteracting the angiotensin-mediated morphological changes that reduce glomerular filtration. In vivo, RTA 405 attenuated the angiotensin II-mediated fall in inulin clearance, demonstrating functional improvement that accompanied the aforementioned morphological changes. Notably, the improved renal clearance was observed in the absence of changes in renal plasma flow and systemic arterial blood pressure (Wigley, et al. Treatment with an Antioxidant Inflammation Modulator (AIM) Increases Glomerular Filtration Rate Monitored by Inulin Clearance in Rats [Abstract]. J Am Soc Nephrol 2010;21:170A). These data demonstrate that bardoxolone methyl can activate the antioxidant response and reduce inflammation in cells of renal origin, resulting in both structural and functional improvement in the kidney.
Bardoxolone methyl first entered clinical trials as an anti-cancer agent in patients with an advanced solid tumor or lymphoid malignancy refractory to standard therapy (97). This first-in-man study enrolled 47 patients who received 5-1300 mg bardoxolone methyl once daily for 21 days of a 28-day cycle. In addition to the observed anti-tumor activity, significant improvement was noted in the estimated glomerular filtration rate (eGFR). Mean baseline serum creatinine levels in 36 evaluable patients (i.e., patients having both baseline and Day 21 Cycle 1 serum creatinine measurements) was 1.0 mg/dL. Bardoxolone methyl treatment resulted in a mean increase in eGFR of 26%, which occurred in 86% of patients on Day 21. In patients treated with bardoxolone methyl for six months, eGFR increases were maintained (97). Based on the significant improvements in eGFR in cancer patients, bardoxolone methyl is being evaluated for the treatment of CKD.
Clinical experience with bardoxolone methyl in CKD
Diabetes is a leading cause of CKD and ESRD, globally (37). As discussed previously, both oxidative stress and inflammation play a central role in the pathogenesis of diabetic complications, including kidney disease (95). Despite significant advances in the care of patients with diabetes, a significant number progress to later stages of CKD and many require renal replacement therapy (98). Pivotal studies performed in patients with diabetic nephropathy using the angiotensin receptor blockers (ARBs) losartan and irbesartan showed that treatment with these agents can slow progression to ESRD by only 4-6 months (99-101). No single treatment (pharmacological or behavioral) has been associated with a significant, sustained, and reproducible improvement in kidney function in patients with kidney disease and diabetes. Therapies aimed at further retarding progression or preferably reversing the almost inexorable decline in kidney function in these patients are badly needed. In light of the significant body of evidence pointing to the potential beneficial effects of Nrf2 modulation in CKD (18;19;25;59-66) and the intriguing findings in cancer patients treated with bardoxolone methyl (97), studies were designed to evaluate the effects of bardoxolone methyl in patients with CKD and type 2 diabetes.
An initial, exploratory (Phase 2a), multi-center, opens-label study was performed to evaluate the effect of bardoxolone methyl on kidney function in patients with type 2 diabetes and Stage 3b-4 CKD (102). Twenty patients received 25 mg bardoxolone methyl daily for 28 days followed by 75 mg for another 28 days. Patients were 64.4 ± 10.6 years of age, obese (101.5 ± 26.7 kg), and had adequate control of blood pressure (systolic blood pressure 142.8 ± 19.6 mmHg) and glycemia (average glycosylated hemoglobin 7.8 ± 2.2%). All participants had a history of hypertension, and 75% were on either an ARB or angiotensin converting enzyme inhibitor (ACEI). The mean baseline eGFR was 30.3 ± 8.0 mL/min/1.73m2. Treatment with bardoxolone methyl resulted in a significant increase in eGFR of +7.2 ± 5.3 mL/min/1.73m2 on Day 56. This improvement was consistent across all patients. The increase in eGFR correlated with a decrease in serum creatinine, increase in creatinine clearance, and decrease in blood urea nitrogen levels. No significant change in 24-hour urinary creatinine excretion was observed. In an exploratory analysis, bardoxolone methyl treatment resulted in a decrease in both the number and iNOS-positive stained circulating endothelial cells, suggesting reduced vascular dysfunction and decreased systemic inflammation, and no change in urinary neutrophil gelatinase-associated lipocalin (NGAL) and N-acetyl-β-D-glucosaminidase (NAG), suggesting no increase in renal injury.
These provocative early findings were corroborated in a large Phase 2b trial (BEAM study) (103). In this double-blind, randomized, placebo-controlled trial, 227 adults with CKD and type 2 diabetes were assigned to one of four treatment arms (placebo or bardoxolone methyl at 25, 75, or 150 mg daily) in a 1:1:1:1 ratio for 52 weeks (primary analysis at 24 weeks); patients were followed for an additional four weeks after treatment ended. At baseline, patients were 67.7 ± 10 years of age, obese (95.2 ± 22.8 kg), and had satisfactory control of blood pressure (systolic blood pressure 130.5 ± 13.5 mmHg) and glycemia (average glycosylated hemoglobin 7.2 ± 1.2%); 98% were on an ARB, ACEI, or both. The median baseline eGFR was 31.2 ± 6.3 mL/min/1.73m2 (range for inclusion 20-45 mL/min/1.73m2). Median urinary albumin-to-creatinine ratio was 76 mg/g (equal numbers of patients with normal albuminuria, microalbuminuria, and macroalbuminuria). In the intent-to-treat (ITT) analysis, patients receiving bardoxolone methyl had significant increases in eGFR, ranging between 8.2 ± 1.5 and 11.4 ± 1.5 mL/min/1.73m2, compared to placebo at 24 weeks; these increases were maintained through Week 52. Four weeks after ending treatment with bardoxolone methyl, the eGFR remained above baseline (0.7 ± 1.6 to 2.5 ±1.6 mL/min/1.73m2). Similar to observations in the Phase 2a trial, increases in eGFR correlated with decreases in blood urea nitrogen, serum phosphorus, and uric acid. In the 75 and 150 mg dose groups, there was a small but significant increase in urinary albumin-to-creatinine ratio at 24 and 52 weeks. Adverse events tended to be mild to moderate in severity and dose-related, with the most frequent being muscle spasms, hypomagnesemia, mild increases in alanine aminotransferase levels, and gastrointestinal effects.
The exact mechanism for the eGFR increase in response to bardoxolone methyl treatment in these studies is unclear. Given the extensive evidence presented in this review for the role of oxidative stress and inflammation in the pathophysiology of kidney disease, particularly in diabetes, the leading hypotheses for the initial eGFR increase (within 2-4 weeks) are an improvement in endothelial function, reduction in vasoconstriction, and an increase in the filtration surface (i.e., decreased mesangial contraction). In the medium- to long-term (24-52 weeks), one might expect that inhibition of NFκB-mediated pro-inflammatory and pro-fibrotic effects may result in at least prevention of further mesangial expansion and fibrosis, as well as interstitial inflammation (104). It can also be speculated that chronic correction of the inflammatory and oxidative stress may result in reversal of established lesions (likely beyond the 52 weeks evaluated in the Phase 2b trial). There was no evidence of any detrimental effects associated with the increase in eGFR in bardoxolone methyl-treated patients, and as noted, this increase was sustained at 52 weeks and persisted above baseline after stopping the drug for four weeks. While these results suggest treatment with bardoxolone methyl in combination with standard of care may help retard disease progression in patients with advanced CKD and diabetes, the results require confirmation in a larger, outcomes trial.
The BEACON trial, a multi-center, international, randomized, placebo-controlled, Phase 3 study, was initiated in 2011 to evaluate the effect of treatment with either placebo or 20 mg bardoxolone methyl given orally once daily in 2000 patients with Stage 4 CKD (eGFR 15-30 mL/min/1.73m2) and type 2 diabetes (NCT01351675). Although this dose of bardoxolone methyl appears smaller than those tested previously, this is due to a change in drug formulation. While prior Phase 2 studies used the crystalline form of bardoxolone methyl, the BEACON study is using an amorphous form with increased intestinal absorption. Pharmacokinetic and pharmacodynamic studies suggested that the effects of 20 mg of the amorphous formulation are similar to 75 mg of the crystalline formulation (Pergola et al. Results from a Phase 2 Study of an Amorphous Spray-Dried Disperson (SDD) Formulation of Bardoxolone Methyl [Abstract]. Proceedings of the Interational Society of Nephrology, Vancouver, Canada, April 2011). The primary outcome measure of BEACON is the time-to-first event of the composite endpoint consisting of ESRD (need for chronic dialysis or renal transplantation) or cardiovascular death. Secondary outcome measures will evaluate the rate of change in eGFR over the study duration, time-to-first hospitalization for heart failure or death due to heart failure, and time-to-first event in the composite endpoint consisting of cardiovascular death, non-fatal myocardial infarction, non-fatal stroke, or hospitalization for heart failure. Results from this trial will provide meaningful insight into the effects of Nrf2 modulation in CKD.
NRF2 UPREGULATION: POTENTIAL USES FOR PREVENTION OF AKI AND ITS CONSEQUENCES
The above discussion clearly illustrates the potential utility of Nrf2 upregulation in mitigating progressive renal disease, in general, and diabetic kidney disease, in particular. Indeed, this is the focus of current clinical investigations, including the ongoing BEACON trial. However, it is important to note that in addition to treatment of CKD, agents that upregulate Nrf2 signaling with resultant increases in both antioxidant and anti-inflammatory defenses may have substantial application in preventing acute kidney injury (AKI) and some of its potential consequences, which include progression to CKD. This assumption is based on experimental studies illustrating oxidative stress as a key mediator of diverse forms of not only CKD but also nephrotoxic and ischemic AKI (105;106) and, once induced, intra-renal inflammation results (107-109). The latter may be triggered by: i) injury-initiated activation of renal tubular inflammatory responses, culminating in increased renal cytokine and chemokine production (e.g., TNF-α, IL-6, and MCP-1); and ii) activation of inflammatory cells (lymphocytes, macrophages, neutrophils, and dendritic cells), which enter the kidney from systemic circulation. Indeed, AKI-initiated inflammation represents an injury-sustaining “positive feedback loop” that evokes ongoing tissue damage despite withdrawal of the originating insult. The importance of this “feedback loop” is underscored by numerous experimental studies demonstrating a variety of anti-inflammatory agents directed at both humoral and cellular components of the inflammatory cascade can attenuate the course of post-ischemic or nephrotoxin-induced AKI (107-109).
For the sake of this discussion, we will focus on just one cytoprotective pathway known to be upregulated by Nrf2: HO-1. Although HO-1 activity is also controlled by its repressor Bach1, it has been demonstrated that Nrf2 has preferential binding to the Maf recognition element (MARE) relative to Bach1 (110), allowing for Nrf2-mediated upregulation of HO-1 and, thereby, making HO-1 a marker of Nrf2 activity (110-112). HO-1’s classically accepted role has been as a key enzyme involved in heme protein degradation (113). It acts by cleaving the porphyrin ring, generating free iron and biliverdin, which is subsequently converted into bilirubin. However, our view of HO-1 has expanded considerably since 1992, following Nath’s seminal observation that, in addition to functioning as a heme-degrading enzyme, HO-1 possesses dramatic cytoprotective and anti-inflammatory effects (114). For example, HO-1 upregulation in the kidney by administration of hematin or heme proteins (hemoglobin, myoglobin) provides marked functional and morphologic protection against a variety of toxic (e.g., glycerol-induced rhabdomyolysis, cisplatin, and endotoxemia) or ischemic renal insults (115-121). Alternatively, blocking HO-1 activity by administration of an enzyme inhibitor (e.g., tin protoporphyrin) increases renal susceptibility to AKI. Because virtually all pharmacologic agents can exert non-specific effects, Nath et al subsequently performed studies in mice with genetic HO-1 deletion (119;122;123). These experiments confirmed the importance of HO-1 as a cytoprotective enzyme, given that HO-1 ‘knockout’ mice were markedly sensitized to diverse forms of AKI.
Considerable progress has been made in deciphering the mechanisms by which HO-1 confers renal cytoprotective effects. Because HO-1’s protective action was first demonstrated against rhabdomyolysis-induced acute renal failure, it was initially assumed that its salutary effect was due to accelerated degradation of myoglobin’s porphyrin ring, which has known pro-oxidant effects. However, a potential paradox arises from this concept. Given that porphryin ring degradation releases “catalytic” (pro-oxidant) free iron and in light of the fact that free iron is more ‘pro-oxidant’ than iron sequestered within a porphyrin ring, if anything, an upregulation of HO-1 might be assumed to worsen, not protect against, heme protein-induced acute renal failure. Thus, alternative explanations were sought. This led to the recognition that as HO-1 releases free iron from porphyrin rings, the iron scavenging protein ferritin is also upregulated, and thus, prevents iron-mediated damage (114). Therefore, pro-oxidant heme protein effects are abrogated without induction of free iron toxicity. A second downstream consequence of HO-1 activity is generation of biliverdin and, ultimately, bilirubin, both of which possess potent antioxidant effects (124). Finally, it is now recognized that a byproduct of the HO-1 pathway is the production of carbon monoxide, which can serve as a potent vasodilator and anti-inflammatory agent (125-129). By mitigating renal vasoconstriction during the induction phase of AKI and by exerting anti-inflammatory effects, carbon monoxide production likely contributes to HO-1-mediated protection. Finally, it is important to note that HO-1’s cytoprotective properties can be expressed against injury in extra-renal tissues (e.g., heart, lung, brain, and liver). Indeed, given that severe acute renal failure frequently exists in the setting of multi-organ failure, a strong theoretical rationale exists for upregulating HO-1 within extra-renal tissues, as well as in the kidney.
Bardoxolone methyl confers protection against ischemic and nephrotoxic AKI
In light of the above information, it is logical to postulate that agents that upregulate Nrf2 signaling might increase HO-1 expression in the kidney, and thus, confer protection against ischemic or nephrotoxic AKI. To test this hypothesis, Wu and colleagues pre-treated mice with bardoxolone methyl twice a day for two days and then subjected the mice to an ischemia model of AKI (130). Marked protection resulted, as denoted by an almost complete prevention of post-ischemic azotemia and a corresponding reduction in renal histologic damage. Furthermore, bardoxolone methyl evoked a significant increase in both Nrf2 mRNA and protein levels, culminating in increased HO-1 gene transcription and translation (assessed by mRNA and protein levels, respectively). Of considerable interest was while bardoxolone methyl increased Nrf2 and HO-1 levels in sham-operated (control) mice, even greater increases were observed in post-AKI kidneys (130), suggesting preferential activity in injured tissues. In order to determine the intra-renal site of bardoxolone methyl-mediated HO-1 increases, immunohistochemistry studies were performed. Increased HO-1 localization was observed predominantly within renal tubular cells, as well as in interstitial leukocytes (130), providing an anatomic–functional correlate as to why decreased renal tubular damage and interstitial inflammation resulted. Equally noteworthy was that bardoxolone methyl’s effects were not restricted to Nrf2-mediated HO-1 increases, as a surprising upregulation of the renal cytoprotective PPARγ signaling pathway was observed within glomerular and peritubular capillary endothelial cells (130). This underscores that the Nrf2 pathway, in general, and bardoxolone methyl, in particular, may exert protective effects in the kidney, the net result being potential cytoprotection against ischemic AKI.
Further evidence for the potential importance of the Nrf2 pathway in AKI comes from two recent studies of cisplatin-induced acute renal failure. Aleksunes et al demonstrated that Nrf2-null mice were markedly sensitized to cisplatin nephrotoxicity (131). Conversely, when the Nrf2 pathway was upregulated in wild-type mice using the bardoxolone methyl congener CDDO-Im, protection against cisplatin nephrotoxicity resulted. Again, this protection could not be directly ascribed to increased HO-1 expression, per se, as the authors suggested that a number of other Nrf2-associated cytoprotective and anti-inflammatory pathways might also have been involved (131). In the study of Wu et al, bardoxolone methyl-mediated protection against cisplatin-induced acute renal failure was also observed (130). In sum, the above results and fact that bardoxolone methyl therapy appears to be clinically well tolerated suggest potential application for preventing human AKI. Indeed, future clinical trials to assess this possibility (e.g., in high-risk patients receiving radiocontrast agents, cisplatin chemotherapy, or individuals undergoing extracorporeal circulation during cardiovascular surgery) seem warranted given the available experimental and clinical data.
Potential role of Nrf2 activators in established acute renal failure
A growing body of experimental and clinical information suggests that acute renal failure is not only an independent risk for mortality, but may also trigger chronic progressive renal disease. Underscoring the latter possibility is a study from Kaiser Permanente of California that documented a 28-fold increased risk of CKD in patients who sustained a bout of severe acute renal failure (requiring temporary dialysis/CRRT), compared to a hospitalized-matched non-AKI patient cohort (132). In a recent experimental study (133), we observed that a unilateral mouse model of ischemic AKI led to a two-thirds loss of renal mass over a 3-week period (equivalent in human life to ~3 years). As depicted in Figure 3, a fundamental feature of this model is progressive tubular injury (increased NGAL expression), increasing renal inflammation (denoted by rising intra-renal cytokine levels, e.g., TNF-α and MCP-1), and stepwise reductions in levels of HO-1 and IL-10 (an anti-inflammatory cytokine). Thus, the balance between pro-inflammatory vs. anti-inflammatory influences appeared to be “tipped” towards a pro-inflammatory state. We also demonstrated that one potential reason for this ‘tipping of the balance’ towards inflammation was injury-induced ‘gene activating’ epigenetic remodeling at transcribed regions of pro-inflammatory genes (133-137). This likely facilitates unbridled gene transcription, inflammation, and hence, progressive renal damage. Furthermore, we have demonstrated that the post-AKI kidney hyper-responds to superimposed pro-inflammatory influences (e.g., endotoxemia) that signal through the innate immune pathway (i.e., Toll-like receptor) (138). Again, this could reflect epigenetic re-programming, with the consequence of predisposing to further renal damage.
Figure 3. Acute kidney injury leads to progressive renal disease and decline in anti-inflammatory HO-1 and IL-10.

(A) Following the induction of unilateral ischemic injury, a progressive loss of renal mass occurs over a three week period, culminating in a 2/3rds reduction of renal weight (left panel). Progressive tubular injury during this period is underscored by a progressive increase in the mRNA for the biomarker protein NGAL. A role for inflammation in this process is indicated by a progressive increase in pro-inflammatory cytokines, with stepwise increases in TNF-α mRNA expression. Similar results were obtained for a pro-inflammatory chemokine, MCP-1, as well as for a pro-fibrotic cytokine, TGF-β1. (Data presented in modified form from studies presented in reference 117 and from additional unpublished data; RZ). BL, baseline values; 1 d, one day, 1 and 3 weeks (wks) post ischemia. The mRNA values were quantified by RT-PCR and were divided by simultaneously obtained GAPDH levels, used as a “housekeeping” gene. (B) Whereas progressive renal disease and inflammation were noted over 3 weeks post ischemia (as shown in Panel A), there was a relative failure of HO-1 expression. While HO-1 protein levels rose at 1 day post ischemia, the levels then fell over the ensuing 3 weeks. Even more dramatic reductions in anti-inflammatory IL-10 protein levels were observed. Thus, the falling anti-inflammatory protein levels, with rising expression of pro-inflammatory genes (as depicted in Panel A), appear to represent reciprocal changes that tip the balance towards a pro-inflammatory / injury promoting state. These results are from ref. 117 and additional unpublished data from one of the authors; RZ). The HO-1 and IL-10 values are presented after factoring by total protein in the tissue extract.
Given this experimental backdrop, it is tempting to postulate that AKI-induced epigenetic remodeling and ongoing inflammation set the stage for progressive renal disease, as noted in the clinical literature. Thus, an appealing concept would be to ‘re-set’ the balance from a pro-inflammatory to anti-inflammatory phenotype, thereby slowing or preventing post-AKI disease progression to CKD. For example, reconstitution of diminished IL-10 levels might contribute to such a goal. However, the lack of clinically available IL-10 makes this an untestable hypothesis at present. Alternatively, given the clinical availability of upregulators of the Nrf2 signaling pathway, this approach might have utility. Indeed, if the BEACON trial, designed to test bardoxolone methyl as an agent to slow CKD progression, shows promising results, then trials of its effects on both AKI prevention, as well as post-AKI disease progression to CKD, may represent an additional promising step for further clinical testing.
CONCLUSION
Oxidative stress and its constant companion inflammation are constant features and major mediators of CKD progression and the associated complications. CKD-associated oxidative stress is caused by increased ROS production and impaired Nrf2 activity. The latter renders the organism defenseless against ravages of oxidative stress. This phenomenon lends support for the use of Nrf2-targeted therapies to combat the prevailing oxidative stress and inflammation and hence progression of kidney disease. Bardoxolone methyl is a potent Nrf2 activator being used for the treatment of CKD. The results of the Phase 2 trials of bardoxolone methyl have demonstrated improvement in kidney function. The ongoing longer-term Phase 3 trial (BEACON) seeks to determine the potential efficacy of Nrf2 activation with bardoxolone in delaying the progression of late-stage CKD in patients with type 2 diabetes to ESRD. These results will be especially insightful as similar pathogenic mechanisms are involved in AKI, which may contribute to the onset of CKD.
Acknowledgments
This work was supported by grants from the National Institutes of Health: DK38432; DK-083310 (RA Zager).
Footnotes
DISCLOSURE
S Ruiz is an employee of Reata Pharmaceuticals, Inc.; PE Pergola is an investigator in the BEACON study sponsored by Reata Pharmaceuticals, Inc.; RA Zager has served as a consultant for and received research support from Reata Pharmaceuticals, Inc.; and ND Vaziri is a member of the BEACON steering committee and receives research support from Reata Pharmaceuticals, Inc.
REFERENCES
- (1).Himmelfarb J, Hakim RM. Oxidative stress in uremia. Curr Opin Nephrol Hypertens. 2003 Nov;12(6):593–8. doi: 10.1097/00041552-200311000-00004. [DOI] [PubMed] [Google Scholar]
- (2).Himmelfarb J, Stenvinkel P, Ikizler TA, Hakim RM. The elephant in uremia: oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002 Nov;62(5):1524–38. doi: 10.1046/j.1523-1755.2002.00600.x. [DOI] [PubMed] [Google Scholar]
- (3).Vaziri ND. Roles of oxidative stress and antioxidant therapy in chronic kidney disease and hypertension. Curr Opin Nephrol Hypertens. 2004 Jan;13(1):93–9. doi: 10.1097/00041552-200401000-00013. [DOI] [PubMed] [Google Scholar]
- (4).Vaziri ND. Oxidative stress in uremia: nature, mechanisms, and potential consequences. Semin Nephrol. 2004 Sep;24(5):469–73. doi: 10.1016/j.semnephrol.2004.06.026. [DOI] [PubMed] [Google Scholar]
- (5).Halliwell B. Biochemistry of oxidative stress. Biochem Soc Trans. 2007 Nov;35(Pt 5):1147–50. doi: 10.1042/BST0351147. [DOI] [PubMed] [Google Scholar]
- (6).Cachofeiro V, Goicochea M, de Vinuesa SG, Oubina P, Lahera V, Luno J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney Int Suppl. 2008 Dec;(111):S4–S9. doi: 10.1038/ki.2008.516. [DOI] [PubMed] [Google Scholar]
- (7).Rosenthal J, Nocera DG. Role of proton-coupled electron transfer in O-O bond activation. Acc Chem Res. 2007 Jul;40(7):543–53. doi: 10.1021/ar7000638. [DOI] [PubMed] [Google Scholar]
- (8).Gennis RB. Coupled proton and electron transfer reactions in cytochrome oxidase. Front Biosci. 2004 Jan 1;9:581–91. doi: 10.2741/1237. [DOI] [PubMed] [Google Scholar]
- (9).Bartz RR, Piantadosi CA. Clinical review: oxygen as a signaling molecule. Crit Care. 2010;14(5):234. doi: 10.1186/cc9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gutteridge JM, Halliwell B. Antioxidants: Molecules, medicines, and myths. Biochem Biophys Res Commun. 2010 Mar 19;393(4):561–4. doi: 10.1016/j.bbrc.2010.02.071. [DOI] [PubMed] [Google Scholar]
- (11).Li W, Khor TO, Xu C, Shen G, Jeong WS, Yu S, et al. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol. 2008 Dec 1;76(11):148–59. doi: 10.1016/j.bcp.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW. When NRF2 talks, who’s listening? Antioxid Redox Signal. 2010 Dec 1;13(11):1649–63. doi: 10.1089/ars.2010.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, et al. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006 Jan;26(1):221–9. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Sykiotis GP, Bohmann D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci Signal. 2010;3(112):re3. doi: 10.1126/scisignal.3112re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Uruno A, Motohashi H. The Keap1-Nrf2 system as an in vivo sensor for electrophiles. Nitric Oxide. 2011 Aug 1;25(2):153–60. doi: 10.1016/j.niox.2011.02.007. [DOI] [PubMed] [Google Scholar]
- (16).Surh YJ, Kundu JK, Na HK. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med. 2008 Oct;74(13):1526–39. doi: 10.1055/s-0028-1088302. [DOI] [PubMed] [Google Scholar]
- (17).Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004 May 7;279(19):20108–17. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- (18).Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, et al. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001 Oct;60(4):1343–53. doi: 10.1046/j.1523-1755.2001.00939.x. [DOI] [PubMed] [Google Scholar]
- (19).Yoh K, Hirayama A, Ishizaki K, Yamada A, Takeuchi M, Yamagishi S, et al. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells. 2008 Nov;13(11):1159–70. doi: 10.1111/j.1365-2443.2008.01234.x. [DOI] [PubMed] [Google Scholar]
- (20).Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007 Dec;9(12):2277–93. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- (21).Vaziri ND, Bai Y, Ni Z, Quiroz Y, Pandian R, Rodriguez-Iturbe B. Intra-renal angiotensin II/AT1 receptor, oxidative stress, inflammation, and progressive injury in renal mass reduction. J Pharmacol Exp Ther. 2007 Oct;323(1):85–93. doi: 10.1124/jpet.107.123638. [DOI] [PubMed] [Google Scholar]
- (22).Vio CP, Jeanneret VA. Local induction of angiotensin-converting enzyme in the kidney as a mechanism of progressive renal diseases. Kidney Int Suppl. 2003 Oct;(86):S57–S63. doi: 10.1046/j.1523-1755.64.s86.11.x. [DOI] [PubMed] [Google Scholar]
- (23).Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007 Sep;59(3):251–87. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- (24).Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H, et al. Liver Angiotensinogen Is the Primary Source of Renal Angiotensin II. J Am Soc Nephrol. 2012 Apr 19; doi: 10.1681/ASN.2011121159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kim HJ, Sato T, Rodriguez-Iturbe B, Vaziri ND. Role of intrarenal angiotensin system activation, oxidative stress, inflammation, and impaired nuclear factor-erythroid-2-related factor 2 activity in the progression of focal glomerulosclerosis. J Pharmacol Exp Ther. 2011 Jun;337(3):583–90. doi: 10.1124/jpet.110.175828. [DOI] [PubMed] [Google Scholar]
- (26).Vaziri ND, Dicus M, Ho ND, Boroujerdi-Rad L, Sindhu RK. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int. 2003 Jan;63(1):179–85. doi: 10.1046/j.1523-1755.2003.00702.x. [DOI] [PubMed] [Google Scholar]
- (27).Shah SV, Baliga R, Rajapurkar M, Fonseca VA. Oxidants in chronic kidney disease. J Am Soc Nephrol. 2007 Jan;18(1):16–28. doi: 10.1681/ASN.2006050500. [DOI] [PubMed] [Google Scholar]
- (28).Fujihara CK, Antunes GR, Mattar AL, Malheiros DM, Vieira JM, Jr., Zatz R. Chronic inhibition of nuclear factor-kappaB attenuates renal injury in the 5/6 renal ablation model. Am J Physiol Renal Physiol. 2007 Jan;292(1):F92–F99. doi: 10.1152/ajprenal.00184.2006. [DOI] [PubMed] [Google Scholar]
- (29).Cho KH, Kim HJ, Rodriguez-Iturbe B, Vaziri ND. Niacin ameliorates oxidative stress, inflammation, proteinuria, and hypertension in rats with chronic renal failure. Am J Physiol Renal Physiol. 2009 Jul;297(1):F106–F113. doi: 10.1152/ajprenal.00126.2009. [DOI] [PubMed] [Google Scholar]
- (30).Poelstra K, Hardonk MJ, Koudstaal J, Bakker WW. Intraglomerular platelet aggregation and experimental glomerulonephritis. Kidney Int. 1990 Jun;37(6):1500–8. doi: 10.1038/ki.1990.141. [DOI] [PubMed] [Google Scholar]
- (31).Gaertner SA, Janssen U, Ostendorf T, Koch KM, Floege J, Gwinner W. Glomerular oxidative and antioxidative systems in experimental mesangioproliferative glomerulonephritis. J Am Soc Nephrol. 2002 Dec;13(12):2930–7. doi: 10.1097/01.asn.0000034908.43113.5d. [DOI] [PubMed] [Google Scholar]
- (32).Boyce NW, Tipping PG, Holdsworth SR. Glomerular macrophages produce reactive oxygen species in experimental glomerulonephritis. Kidney Int. 1989 Mar;35(3):778–82. doi: 10.1038/ki.1989.52. [DOI] [PubMed] [Google Scholar]
- (33).Oberle GP, Niemeyer J, Thaiss F, Schoeppe W, Stahl RA. Increased oxygen radical and eicosanoid formation in immune-mediated mesangial cell injury. Kidney Int. 1992 Jul;42(1):69–74. doi: 10.1038/ki.1992.262. [DOI] [PubMed] [Google Scholar]
- (34).Kawaguchi M, Yamada M, Wada H, Okigaki T. Roles of active oxygen species in glomerular epithelial cell injury in vitro caused by puromycin aminonucleoside. Toxicology. 1992;72(3):329–40. doi: 10.1016/0300-483x(92)90183-f. [DOI] [PubMed] [Google Scholar]
- (35).Neale TJ, Ullrich R, Ojha P, Poczewski H, Verhoeven AJ, Kerjaschki D. Reactive oxygen species and neutrophil respiratory burst cytochrome b558 are produced by kidney glomerular cells in passive Heymann nephritis. Proc Natl Acad Sci U S A. 1993 Apr 15;90(8):3645–9. doi: 10.1073/pnas.90.8.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Shah SV. Evidence suggesting a role for hydroxyl radical in passive Heymann nephritis in rats. Am J Physiol. 1988 Mar;254(3 Pt 2):F337–F344. doi: 10.1152/ajprenal.1988.254.3.F337. [DOI] [PubMed] [Google Scholar]
- (37).U.S. Renal Data System . USRDS 2011 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; Bethesda, MD: 2011. [Google Scholar]
- (38).Ha H, Yu MR, Choi YJ, Kitamura M, Lee HB. Role of high glucose-induced nuclear factor-kappaB activation in monocyte chemoattractant protein-1 expression by mesangial cells. J Am Soc Nephrol. 2002 Apr;13(4):894–902. doi: 10.1681/ASN.V134894. [DOI] [PubMed] [Google Scholar]
- (39).Ha H, Kim KH. Pathogenesis of diabetic nephropathy: the role of oxidative stress and protein kinase C. Diabetes Res Clin Pract. 1999 Sep;45(2-3):147–51. doi: 10.1016/s0168-8227(99)00044-3. [DOI] [PubMed] [Google Scholar]
- (40).Chen HC, Guh JY, Shin SJ, Tsai JH, Lai YH. Reactive oxygen species enhances endothelin-1 production of diabetic rat glomeruli in vitro and in vivo. J Lab Clin Med. 2000 Apr;135(4):309–15. doi: 10.1067/mlc.2000.105616. [DOI] [PubMed] [Google Scholar]
- (41).Koya D, Hayashi K, Kitada M, Kashiwagi A, Kikkawa R, Haneda M. Effects of antioxidants in diabetes-induced oxidative stress in the glomeruli of diabetic rats. J Am Soc Nephrol. 2003 Aug;14(8 Suppl 3):S250–S253. doi: 10.1097/01.asn.0000077412.07578.44. [DOI] [PubMed] [Google Scholar]
- (42).Scivittaro V, Ganz MB, Weiss MF. AGEs induce oxidative stress and activate protein kinase C-beta(II) in neonatal mesangial cells. Am J Physiol Renal Physiol. 2000 Apr;278(4):F676–F683. doi: 10.1152/ajprenal.2000.278.4.F676. [DOI] [PubMed] [Google Scholar]
- (43).Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, et al. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994 Apr 1;269(13):9889–97. [PubMed] [Google Scholar]
- (44).Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, et al. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem. 2005 Nov 25;280(47):39616–26. doi: 10.1074/jbc.M502412200. [DOI] [PubMed] [Google Scholar]
- (45).Satoh M, Fujimoto S, Haruna Y, Arakawa S, Horike H, Komai N, et al. NAD(P)H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am J Physiol Renal Physiol. 2005 Jun;288(6):F1144–F1152. doi: 10.1152/ajprenal.00221.2004. [DOI] [PubMed] [Google Scholar]
- (46).Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001 Dec 13;414(6865):813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- (47).Lee HB, Yu MR, Yang Y, Jiang Z, Ha H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J Am Soc Nephrol. 2003 Aug;14(8 Suppl 3):S241–S245. doi: 10.1097/01.asn.0000077410.66390.0f. [DOI] [PubMed] [Google Scholar]
- (48).Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000 Apr 13;404(6779):787–90. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- (49).Vaziri ND, Rodriguez-Iturbe B. Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2006 Oct;2(10):582–93. doi: 10.1038/ncpneph0283. [DOI] [PubMed] [Google Scholar]
- (50).Rodriguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ. Oxidative stress, renal infiltration of immune cells, and salt-sensitive hypertension: all for one and one for all. Am J Physiol Renal Physiol. 2004 Apr;286(4):F606–F616. doi: 10.1152/ajprenal.00269.2003. [DOI] [PubMed] [Google Scholar]
- (51).Wilcox CS. Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? Am J Physiol Regul Integr Comp Physiol. 2005 Oct;289(4):R913–R935. doi: 10.1152/ajpregu.00250.2005. [DOI] [PubMed] [Google Scholar]
- (52).Barton CH, Ni Z, Vaziri ND. Enhanced nitric oxide inactivation in aortic coarctation-induced hypertension. Kidney Int. 2001 Sep;60(3):1083–7. doi: 10.1046/j.1523-1755.2001.0600031083.x. [DOI] [PubMed] [Google Scholar]
- (53).Sindhu RK, Roberts CK, Ehdaie A, Zhan CD, Vaziri ND. Effects of aortic coarctation on aortic antioxidant enzymes and NADPH oxidase protein expression. Life Sci. 2005 Jan 7;76(8):945–53. doi: 10.1016/j.lfs.2004.10.014. [DOI] [PubMed] [Google Scholar]
- (54).Vaziri ND, Ni Z. Expression of NOX-I, gp91phox, p47phox and P67phox in the aorta segments above and below coarctation. Biochim Biophys Acta. 2005 May 25;1723(1-3):321–7. doi: 10.1016/j.bbagen.2005.03.003. [DOI] [PubMed] [Google Scholar]
- (55).Yilmaz MI, Carrero JJ, Axelsson J, Lindholm B, Stenvinkel P. Low-grade inflammation in chronic kidney disease patients before the start of renal replacement therapy: sources and consequences. Clin Nephrol. 2007 Jul;68(1):1–9. doi: 10.5414/cnp68001. [DOI] [PubMed] [Google Scholar]
- (56).Vaziri ND, Yuan J, Rahimi A, Ni Z, Said H, Subramanian VS. Disintegration of colonic epithelial tight junction in uremia: a likely cause of CKD-associated inflammation. Nephrol Dial Transplant. 2011 Nov 29; doi: 10.1093/ndt/gfr624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Vaziri ND, Wong J, Pahl MV, Piceno YM, Yuan J, DeSantis TZ, et al. Chronic kidney disease alters the composition of intestinal microbial flora. Kidney Int. 2012 doi: 10.1038/ki.2012.345. (In press) [DOI] [PubMed] [Google Scholar]
- (58).Aronov PA, Luo FJ, Plummer NS, Quan Z, Holmes S, Hostetter TH, et al. Colonic contribution to uremic solutes. J Am Soc Nephrol. 2011 Sep;22(9):1769–76. doi: 10.1681/ASN.2010121220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol. 2009 Dec 9; doi: 10.1152/ajprenal.00421.2009. [DOI] [PubMed] [Google Scholar]
- (60).Aminzadeh AM, Sato T, Vaziri ND. Participation of endoplasmic reticulum stress in the pathogenesis of spontaneous glomerulosclerosis - Role of intra-renal angiotensin system. Transl Res. 2012 doi: 10.1016/j.trsl.2012.03.003. (In press) [DOI] [PubMed] [Google Scholar]
- (61).Liu M, Grigoryev DN, Crow MT, Haas M, Yamamoto M, Reddy SP, et al. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009 May 13; doi: 10.1038/ki.2009.157. [DOI] [PubMed] [Google Scholar]
- (62).Zheng H, Whitman SA, Wu W, Wondrak GT, Wong PK, Fang D, et al. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes. 2011 Nov;60(11):3055–66. doi: 10.2337/db11-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Tsai PY, Ka SM, Chang JM, Chen HC, Shui HA, Li CY, et al. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic Biol Med. 2011 Aug 1;51(3):744–54. doi: 10.1016/j.freeradbiomed.2011.05.016. [DOI] [PubMed] [Google Scholar]
- (64).Peng A, Ye T, Rakheja D, Tu Y, Wang T, Du Y, et al. The green tea polyphenol (-)-epigallocatechin-3-gallate ameliorates experimental immune-mediated glomerulonephritis. Kidney Int. 2011 Sep;80(6):601–11. doi: 10.1038/ki.2011.121. [DOI] [PubMed] [Google Scholar]
- (65).Sahin K, Tuzcu M, Gencoglu H, Dogukan A, Timurkan M, Sahin N, et al. Epigallocatechin-3-gallate activates Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in rats. Life Sci. 2010 Aug 14;87(7-8):240–5. doi: 10.1016/j.lfs.2010.06.014. [DOI] [PubMed] [Google Scholar]
- (66).Molina-Jijon E, Tapia E, Zazueta C, El HM, Zatarain-Barron ZL, Hernandez-Pando R, et al. Curcumin prevents Cr(VI)-induced renal oxidant damage by a mitochondrial pathway. Free Radic Biol Med. 2011 Oct 15;51(8):1543–57. doi: 10.1016/j.freeradbiomed.2011.07.018. [DOI] [PubMed] [Google Scholar]
- (67).Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, et al. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Physiol Heart Circ Physiol. 2006 May;290(5):H1862–H1870. doi: 10.1152/ajpheart.00651.2005. [DOI] [PubMed] [Google Scholar]
- (68).Morimitsu Y, Nakagawa Y, Hayashi K, Fujii H, Kumagai T, Nakamura Y, et al. A sulforaphane analogue that potently activates the Nrf2-dependent detoxification pathway. J Biol Chem. 2002 Feb 1;277(5):3456–63. doi: 10.1074/jbc.M110244200. [DOI] [PubMed] [Google Scholar]
- (69).Surh YJ, Kundu JK, Na HK, Lee JS. Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J Nutr. 2005 Dec;135(12 Suppl):2993S–3001S. doi: 10.1093/jn/135.12.2993S. [DOI] [PubMed] [Google Scholar]
- (70).Vaziri ND. Lipotoxicity and impaired high density lipoprotein-mediated reverse cholesterol transport in chronic kidney disease. J Ren Nutr. 2010 Sep;20(5 Suppl):S35–S43. doi: 10.1053/j.jrn.2010.05.010. [DOI] [PubMed] [Google Scholar]
- (71).Vaziri ND. Dyslipidemia of chronic renal failure: the nature, mechanisms, and potential consequences. Am J Physiol Renal Physiol. 2006 Feb;290(2):F262–F272. doi: 10.1152/ajprenal.00099.2005. [DOI] [PubMed] [Google Scholar]
- (72).Vaziri ND. Causes of dysregulation of lipid metabolism in chronic renal failure. Semin Dial. 2009 Nov;22(6):644–51. doi: 10.1111/j.1525-139X.2009.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Moradi H, Pahl MV, Elahimehr R, Vaziri ND. Impaired antioxidant activity of high-density lipoprotein in chronic kidney disease. Transl Res. 2009 Feb;153(2):77–85. doi: 10.1016/j.trsl.2008.11.007. [DOI] [PubMed] [Google Scholar]
- (74).Vaziri ND, Navab M, Fogelman AM. HDL metabolism and activity in chronic kidney disease. Nat Rev Nephrol. 2010 May;6(5):287–96. doi: 10.1038/nrneph.2010.36. [DOI] [PubMed] [Google Scholar]
- (75).Vaziri ND, Moradi H, Pahl MV, Fogelman AM, Navab M. In vitro stimulation of HDL anti-inflammatory activity and inhibition of LDL pro-inflammatory activity in the plasma of patients with end-stage renal disease by an apoA-1 mimetic peptide. Kidney Int. 2009 Aug;76(4):437–44. doi: 10.1038/ki.2009.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Vaziri ND, Deng G, Liang K. Hepatic HDL receptor, SR-B1 and Apo A-I expression in chronic renal failure. Nephrol Dial Transplant. 1999 Jun;14(6):1462–6. doi: 10.1093/ndt/14.6.1462. [DOI] [PubMed] [Google Scholar]
- (77).Vaziri ND, Liang K, Parks JS. Down-regulation of hepatic lecithin:cholesterol acyltransferase gene expression in chronic renal failure. Kidney Int. 2001 Jun;59(6):2192–6. doi: 10.1046/j.1523-1755.2001.00734.x. [DOI] [PubMed] [Google Scholar]
- (78).Vaziri ND, Norris K. Lipid disorders and their relevance to outcomes in chronic kidney disease. Blood Purif. 2011;31(1-3):189–96. doi: 10.1159/000321845. [DOI] [PubMed] [Google Scholar]
- (79).Kim HJ, Moradi H, Yuan J, Norris K, Vaziri ND. Renal mass reduction results in accumulation of lipids and dysregulation of lipid regulatory proteins in the remnant kidney. Am J Physiol Renal Physiol. 2009 Jun;296(6):F1297–F1306. doi: 10.1152/ajprenal.90761.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Moradi H, Yuan J, Ni Z, Norris K, Vaziri ND. Reverse cholesterol transport pathway in experimental chronic renal failure. Am J Nephrol. 2009;30(2):147–54. doi: 10.1159/000210020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Bea F, Hudson FN, Chait A, Kavanagh TJ, Rosenfeld ME. Induction of glutathione synthesis in macrophages by oxidized low-density lipoproteins is mediated by consensus antioxidant response elements. Circ Res. 2003 Mar 7;92(4):386–93. doi: 10.1161/01.RES.0000059561.65545.16. [DOI] [PubMed] [Google Scholar]
- (82).Cho S, Hazama M, Urata Y, Goto S, Horiuchi S, Sumikawa K, et al. Protective role of glutathione synthesis in response to oxidized low density lipoprotein in human vascular endothelial cells. Free Radic Biol Med. 1999 Mar;26(5-6):589–602. doi: 10.1016/s0891-5849(98)00232-9. [DOI] [PubMed] [Google Scholar]
- (83).Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, et al. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res. 2004 Mar 19;94(5):609–16. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- (84).Dinkova-Kostova AT, Liby KT, Stephenson KK, Holtzclaw WD, Gao X, Suh N, et al. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci U S A. 2005 Mar 22;102(12):4584–9. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Honda T, Rounds BV, Bore L, Finlay HJ, Favaloro FG, Jr., Suh N, et al. Synthetic oleanane and ursane triterpenoids with modified rings A and C: a series of highly active inhibitors of nitric oxide production in mouse macrophages. J Med Chem. 2000 Nov 2;43(22):4233–46. doi: 10.1021/jm0002230. [DOI] [PubMed] [Google Scholar]
- (86).Sporn MB, Liby KT, Yore MM, Fu L, Lopchuk JM, Gribble GW. New synthetic triterpenoids: potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J Nat Prod. 2011 Mar 25;74(3):537–45. doi: 10.1021/np100826q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, et al. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J. 2004 Mar 1;378(Pt 2):373–82. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Cernuda-Morollon E, Pineda-Molina E, Canada FJ, Perez-Sala D. 15-Deoxy-Delta 12,14-prostaglandin J2 inhibition of NF-kappaB-DNA binding through covalent modification of the p50 subunit. J Biol Chem. 2001 Sep 21;276(38):35530–6. doi: 10.1074/jbc.M104518200. [DOI] [PubMed] [Google Scholar]
- (89).Cuzzocrea S, Wayman NS, Mazzon E, Dugo L, Di PR, Serraino I, et al. The cyclopentenone prostaglandin 15-deoxy-Delta(12,14)-prostaglandin J(2) attenuates the development of acute and chronic inflammation. Mol Pharmacol. 2002 May;61(5):997–1007. doi: 10.1124/mol.61.5.997. [DOI] [PubMed] [Google Scholar]
- (90).Kawamoto Y, Nakamura Y, Naito Y, Torii Y, Kumagai T, Osawa T, et al. Cyclopentenone prostaglandins as potential inducers of phase II detoxification enzymes. 15-deoxy-delta(12,14)-prostaglandin j2-induced expression of glutathione S-transferases. J Biol Chem. 2000 Apr 14;275(15):11291–9. doi: 10.1074/jbc.275.15.11291. [DOI] [PubMed] [Google Scholar]
- (91).Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000 Jan 6;403(6765):103–8. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- (92).Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, et al. 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci U S A. 2000 Apr 25;97(9):4844–9. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- (94).Ahmad R, Raina D, Meyer C, Kharbanda S, Kufe D. Triterpenoid CDDO-Me Blocks the NF-{kappa}B Pathway by Direct Inhibition of IKKbeta on Cys-179. J Biol Chem. 2006 Nov 24;281(47):35764–9. doi: 10.1074/jbc.M607160200. [DOI] [PubMed] [Google Scholar]
- (95).Rivero A, Mora C, Muros M, Garcia J, Herrera H, Navarro-Gonzalez JF. Pathogenic perspectives for the role of inflammation in diabetic nephropathy. Clin Sci (Lond) 2009 Mar;116(6):479–92. doi: 10.1042/CS20080394. [DOI] [PubMed] [Google Scholar]
- (96).Navarro-Gonzalez JF, Mora-Fernandez C. Inflammatory pathways. Contrib Nephrol. 2011;170:113–23. doi: 10.1159/000325646. [DOI] [PubMed] [Google Scholar]
- (97).Hong DS, Kurzrock R, Supko JG, He X, Naing A, Wheler J, et al. A Phase I First-in-Human Trial of Bardoxolone Methyl in Patients with Advanced Solid Tumors and Lymphomas. Clin Cancer Res. 2012 May 25; doi: 10.1158/1078-0432.CCR-11-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Rossing P. Diabetic nephropathy: worldwide epidemic and effects of current treatment on natural history. Curr Diab Rep. 2006 Dec;6(6):479–83. doi: 10.1007/s11892-006-0083-y. [DOI] [PubMed] [Google Scholar]
- (99).Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001 Sep 20;345(12):861–9. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- (100).Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001 Sep 20;345(12):851–60. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- (101).Vilayur E, Harris DC. Emerging therapies for chronic kidney disease: what is their role? Nat Rev Nephrol. 2009 Jul;5(7):375–83. doi: 10.1038/nrneph.2009.76. [DOI] [PubMed] [Google Scholar]
- (102).Pergola P, Krauth M, Huff W, Ferguson D, Ruiz S, Meyer C, et al. Effect of Bardoxolone on Kidney Function in Patients with T2D and Stage 3b - 4 CKD. Am J Nephrol. 2011;33:469–76. doi: 10.1159/000327599. [DOI] [PubMed] [Google Scholar]
- (103).Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011 Jul 28;365(4):327–36. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- (104).Navarro-Gonzalez JF, Mora-Fernandez C, Muros de FM, Garcia-Perez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011 Jun;7(6):327–40. doi: 10.1038/nrneph.2011.51. [DOI] [PubMed] [Google Scholar]
- (105).Shah SV, Rajapurkar MM, Baliga R. The role of catalytic iron in acute kidney injury. Clin J Am Soc Nephrol. 2011 Oct;6(10):2329–31. doi: 10.2215/CJN.08340811. [DOI] [PubMed] [Google Scholar]
- (106).Aksu U, Demirci C, Ince C. The pathogenesis of acute kidney injury and the toxic triangle of oxygen, reactive oxygen species and nitric oxide. Contrib Nephrol. 2011;174:119–28. doi: 10.1159/000329249. [DOI] [PubMed] [Google Scholar]
- (107).Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011 Nov;121(11):4210–21. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Lee DW, Faubel S, Edelstein CL. Cytokines in acute kidney injury (AKI) Clin Nephrol. 2011 Sep;76(3):165–73. doi: 10.5414/cn106921. [DOI] [PubMed] [Google Scholar]
- (109).Kinsey GR, Okusa MD. Pathogenesis of acute kidney injury: foundation for clinical practice. Am J Kidney Dis. 2011 Aug;58(2):291–301. doi: 10.1053/j.ajkd.2011.02.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Miyazaki T, Kirino Y, Takeno M, Samukawa S, Hama M, Tanaka M, et al. Expression of heme oxygenase-1 in human leukemic cells and its regulation by transcriptional repressor Bach1. Cancer Sci. 2010 Jun;101(6):1409–16. doi: 10.1111/j.1349-7006.2010.01550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Igarashi K, Sun J. The heme-Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid Redox Signal. 2006 Jan;8(1-2):107–18. doi: 10.1089/ars.2006.8.107. [DOI] [PubMed] [Google Scholar]
- (112).Keum YS, Han YH, Liew C, Kim JH, Xu C, Yuan X, et al. Induction of heme oxygenase-1 (HO-1) and NAD[P]H: quinone oxidoreductase 1 (NQO1) by a phenolic antioxidant, butylated hydroxyanisole (BHA) and its metabolite, tert-butylhydroquinone (tBHQ) in primary-cultured human and rat hepatocytes. Pharm Res. 2006 Nov;23(11):2586–94. doi: 10.1007/s11095-006-9094-2. [DOI] [PubMed] [Google Scholar]
- (113).Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci U S A. 1968 Oct;61(2):748–55. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, et al. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest. 1992 Jul;90(1):267–70. doi: 10.1172/JCI115847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006 Aug;70(3):432–43. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- (116).Inguaggiato P, Gonzalez-Michaca L, Croatt AJ, Haggard JJ, Alam J, Nath KA. Cellular overexpression of heme oxygenase-1 up-regulates p21 and confers resistance to apoptosis. Kidney Int. 2001 Dec;60(6):2181–91. doi: 10.1046/j.1523-1755.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- (117).Agarwal A, Balla J, Alam J, Croatt AJ, Nath KA. Induction of heme oxygenase in toxic renal injury: a protective role in cisplatin nephrotoxicity in the rat. Kidney Int. 1995 Oct;48(4):1298–307. doi: 10.1038/ki.1995.414. [DOI] [PubMed] [Google Scholar]
- (118).Vogt BA, Alam J, Croatt AJ, Vercellotti GM, Nath KA. Acquired resistance to acute oxidative stress. Possible role of heme oxygenase and ferritin. Lab Invest. 1995 Apr;72(4):474–83. [PubMed] [Google Scholar]
- (119).Agarwal A, Nick HS. Renal response to tissue injury: lessons from heme oxygenase-1 GeneAblation and expression. J Am Soc Nephrol. 2000 May;11(5):965–73. doi: 10.1681/ASN.V115965. [DOI] [PubMed] [Google Scholar]
- (120).Nath KA. Heme oxygenase-1: a redoubtable response that limits reperfusion injury in the transplanted adipose liver. J Clin Invest. 1999 Dec;104(11):1485–6. doi: 10.1172/JCI8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Zarjou A, Kim J, Traylor AM, Sanders PW, Balla J, Agarwal A, et al. Paracrine effects of mesenchymal stem cells in cisplatin-induced renal injury require heme oxygenase-1. Am J Physiol Renal Physiol. 2011 Jan;300(1):F254–F262. doi: 10.1152/ajprenal.00594.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Pittock ST, Norby SM, Grande JP, Croatt AJ, Bren GD, Badley AD, et al. MCP-1 is up-regulated in unstressed and stressed HO-1 knockout mice: Pathophysiologic correlates. Kidney Int. 2005 Aug;68(2):611–22. doi: 10.1111/j.1523-1755.2005.00439.x. [DOI] [PubMed] [Google Scholar]
- (123).Tracz MJ, Juncos JP, Croatt AJ, Ackerman AW, Grande JP, Knutson KL, et al. Deficiency of heme oxygenase-1 impairs renal hemodynamics and exaggerates systemic inflammatory responses to renal ischemia. Kidney Int. 2007 Nov;72(9):10735–80. doi: 10.1038/sj.ki.5002471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987 Feb 27;235(4792):1043–6. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- (125).Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med. 2006 Oct 2;203(10):2377–89. doi: 10.1084/jem.20060845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med. 2007 Jun;13(6):703–10. doi: 10.1038/nm1586. [DOI] [PubMed] [Google Scholar]
- (127).Tracz MJ, Alam J, Nath KA. Physiology and pathophysiology of heme: implications for kidney disease. J Am Soc Nephrol. 2007 Feb;18(2):414–20. doi: 10.1681/ASN.2006080894. [DOI] [PubMed] [Google Scholar]
- (128).Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation. 2008 Jan 15;117(2):231–41. doi: 10.1161/CIRCULATIONAHA.107.698316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008 Mar;60(1):79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- (130).Wu QQ, Wang Y, Senitko M, Meyer C, Wigley WC, Ferguson DA, et al. Bardoxolone Methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes - Nrf2, PPAR{gamma}, and HO-1. Am J Physiol Renal Physiol. 2011 Feb 2; doi: 10.1152/ajprenal.00353.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Aleksunes LM, Goedken MJ, Rockwell CE, Thomale J, Manautou JE, Klaassen CD. Transcriptional regulation of renal cytoprotective genes by Nrf2 and its potential use as a therapeutic target to mitigate cisplatin-induced nephrotoxicity. J Pharmacol Exp Ther. 2010 Oct;335(1):2–12. doi: 10.1124/jpet.110.170084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Lo LJ, Go AS, Chertow GM, McCulloch CE, Fan D, Ordonez JD, et al. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int. 2009 Oct;76(8):893–9. doi: 10.1038/ki.2009.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Zager RA, Johnson AC, Becker K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am J Physiol Renal Physiol. 2011 Dec;301(6):F1334–F1345. doi: 10.1152/ajprenal.00431.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Zager RA, Johnson AC. Progressive histone alterations and proinflammatory gene activation: consequences of heme protein/iron-mediated proximal tubule injury. Am J Physiol Renal Physiol. 2010 Mar;298(3):F827–F837. doi: 10.1152/ajprenal.00683.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Naito M, Zager RA, Bomsztyk K. BRG1 increases transcription of proinflammatory genes in renal ischemia. J Am Soc Nephrol. 2009 Aug;20(8):1787–96. doi: 10.1681/ASN.2009010118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Zager RA, Johnson AC. Renal ischemia-reperfusion injury upregulates histone-modifying enzyme systems and alters histone expression at proinflammatory/profibrotic genes. Am J Physiol Renal Physiol. 2009 May;296(5):F1032–F1041. doi: 10.1152/ajprenal.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Naito M, Bomsztyk K, Zager RA. Endotoxin mediates recruitment of RNA polymerase II to target genes in acute renal failure. J Am Soc Nephrol. 2008 Jul;19(7):1321–30. doi: 10.1681/ASN.2007121368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Zager RA, Johnson AC, Hanson SY, Lund S. Ischemic proximal tubular injury primes mice to endotoxin-induced TNF-alpha generation and systemic release. Am J Physiol Renal Physiol. 2005 Aug;289(2):F289–F297. doi: 10.1152/ajprenal.00023.2005. [DOI] [PubMed] [Google Scholar]