

Abstract
There has been much recent interest in lysophosphatidic acid (LPA) signaling through one of its receptors, LPA1, in fibrotic diseases, but the mechanisms by which LPA-LPA1 signaling promotes pathological fibrosis remain to be fully elucidated. Using a mouse peritoneal fibrosis model, we demonstrate central roles for LPA and LPA1 in fibroblast proliferation. Genetic deletion or pharmacological antagonism of LPA1 protected mice from peritoneal fibrosis, blunting the increases in peritoneal collagen by 65.4 and 52.9%, respectively, compared to control animals and demonstrated that peritoneal fibroblast proliferation was highly LPA1 dependent. Activation of LPA1 on mesothelial cells induced these cells to express connective tissue growth factor (CTGF), driving fibroblast proliferation in a paracrine fashion. Activation of mesothelial cell LPA1 induced CTGF expression by inducing cytoskeleton reorganization in these cells, causing nuclear translocation of myocardin-related transcription factor (MRTF)-A and MRTF-B. Pharmacological inhibition of MRTF-induced transcription also diminished CTGF expression and fibrosis in the peritoneal fibrosis model, mitigating the increase in peritoneal collagen content by 57.9% compared to controls. LPA1-induced cytoskeleton reorganization therefore makes a previously unrecognized but critically important contribution to the profibrotic activities of LPA by driving MRTF-dependent CTGF expression, which, in turn, drives fibroblast proliferation.—Sakai, N., Chun, J., Duffield, J. S., Wada, T., Luster, A. D., Tager, A. M. LPA1-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation.
Keywords: lysophosphatidic acid, Rho GTPases, myocardin-related transcription factor-A, myocardin-related transcription factor-B, serum response factor
Fibrosis characterizes many chronic diseases that result in end-stage organ failure, causing major morbidity and mortality. Fibrosis in many of these diseases appears to result from aberrant or overexuberant wound-healing responses to injury (1), producing excessive deposition of extracellular matrix. Expansion of the matrix-producing fibroblast pool is a critical component of profibrotic injury responses (2), but the molecular mediators driving this expansion in vivo have not yet been fully identified. Better identification of these mediators will hopefully identify new therapeutic targets for fibrotic diseases, most of which are refractory to currently available therapies.
Lysophosphatidic acid (LPA) is a bioactive lipid that mediates diverse cellular responses through interactions with specific G-protein-coupled receptors (GPCRs), of which at least 5 have been definitively identified and designated LPA1–5 (3). We and others have recently demonstrated that LPA signaling specifically through LPA1 is required for the development of fibrosis in several organs (4–6), but the mechanisms through which LPA-LPA1 signaling contributes to fibrosis remain to be fully elucidated. Among its profibrotic activities, LPA-LPA1 signaling contributes to fibroblast accumulation in pulmonary fibrosis by driving fibroblast migration to sites of lung injury (4). Although fibroblast migration into the wound matrix is a central step in tissue responses to injury (7), recent evidence suggests that the proliferation of resident fibroblasts within injured tissues is central to the accumulation of these cells (8). We now demonstrate that fibroblast proliferation in vivo is dependent on LPA-LPA1 signaling, in a mouse model of fibrosis not previously shown to require LPA1, chlorhexidine gluconate (CG)-induced peritoneal fibrosis (9). Genetic deletion or pharmacological antagonism of LPA1 significantly attenuated fibroblast proliferation, as well as the development of fibrosis, in this model.
LPA itself can induce fibroblast proliferation, but this direct effect of LPA in vitro can be mediated by LPA1 or LPA2 (10). We therefore hypothesized that the specific requirement for LPA1 that we observed for fibroblast proliferation in vivo was attributable to LPA signaling specifically through LPA1 being required for the expression of fibroblast mitogens after tissue injury other than LPA. Many of the activities induced by LPA, such as cell migration and shape change, result, at least in part, from its potent ability to regulate the actin cytoskeleton through activation of the RhoA-Rho-associated coiled-coil-forming kinase (ROCK) cascade, converting globular actin (G-actin) monomers to filamentous actin (F-actin) polymers (11). The recent description of a pathway linking actin cytoskeleton reorganization to gene expression provides a potential mechanism through which LPA-LPA1 signaling could induce fibroblast mitogen expression. In this pathway, polymerization of G- into F-actin liberates myocardin-related transcription factor (MRTF)-A and MRTF-B, which then translocate to the nucleus and markedly augment serum response factor (SRF)-induced gene transcription (12–13).
SRF is a MADS box transcription factor that induces the expression of genes by binding to CArG box sequences [CC(A/T)6GG] in their promoters (14). The potent profibrotic mediator connective tissue growth factor (CTGF/CCN2) contains a CArG-like box in its promoter, and SRF has been shown to induce CTGF expression in human endothelial cells (15). CTGF is highly expressed in multiple fibrotic pathologies (16) and induces fibroblast adhesion, extracellular matrix production, and proliferation (17). Fibroblast-specific genetic deletion of CTGF was recently shown to markedly attenuate fibrosis and myofibroblast accumulation in the bleomycin model of scleroderma dermal fibrosis (18), a model that we have found to also be highly dependent on LPA-LPA1 signaling (5).
In this work, we investigated the hypotheses that LPA-LPA1 signaling induces expansion of the fibroblast pool during the development of fibrosis in vivo; that this expansion is driven by LPA-LPA1-induced CTGF expression, which then drives fibroblast proliferation; and that this CTGF expression results from LPA-LPA1-induced actin polymerization, which liberates MRTF-A and MRTF-B to translocate to the nucleus and augment SRF-dependent transcription. This proposed link between the profibrotic activities of LPA and CTGF connects 2 important mediators that are each being evaluated independently as therapeutic targets for multiple fibrotic diseases.
The disease model we investigated in these studies, peritoneal fibrosis, is an important clinical problem in multiple settings, including postsurgical peritoneal adhesions (19) and peritoneal dialysis (20), a life-sustaining therapy used by >100,000 patients with renal failure worldwide, accounting for ∼10–15% of the dialysis population (21). Dialysis solutions that are hyperosmotic, hyperglycemic, and/or acidic can cause repetitive peritoneal injury (22), producing progressive fibrosis of the submesothelial region that normally consists of a thin layer of connective tissue, with a few scattered fibroblasts, underlying a single-layered mesothelial cell barrier (23). The development of peritoneal fibrosis can markedly decrease dialysis ultrafiltration capacity and in some patients cause encapsulating peritoneal sclerosis, which is associated with bowel obstruction and mortality rates as high as 38–56% (20, 24–25). Elucidating the role of an LPA-LPA1-actin-MRTF-SRF-CTGF pathway will hopefully identify new targets for therapies to treat and/or prevent peritoneal fibrosis in all of the clinical settings in which it occurs. Characterizing this profibrotic pathway also will have the potential to identify new therapeutic targets for fibrotic diseases affecting other organs in which LPA-LPA1 signaling and/or CTGF has been implicated.
MATERIALS AND METHODS
Reagents and cells
LPA (1-oleoyl-2-hydroxy-sn-glycero-3-phosphate) was purchased from Avanti Polar Lipids (Alabaster, AL, USA) and diluted in Dulbecco's modified Eagle's medium (DMEM; Lonza, Basel, Switzerland) including 0.1% fatty acid-free bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO, USA). AM095 was the kind gift of Amira Pharmaceuticals (San Diego, CA, USA). Sodium pyruvate, NEAA mixture, and penicillin/streptomycin were obtained from Lonza. l-glutamine was from Cellgro (Manassas, VA, USA). Y27632, pertussis toxin, cycloheximide, and DMSO were from Sigma-Aldrich. Latrunculin B was from Invitrogen (Grand Island, NY, USA). CCG-1423 was from Cayman Chemical (Ann Arbor, MI, USA). Permeable C3 toxin was from Cytoskeleton (Denver, CO, USA). Recombinant TGF-β1, monoclonal anti-TGF-β1, -β2, and -β3 neutralizing antibody (1D11), and normal rabbit IgG were from R&D Systems (Minneapolis, MN, USA). NIH3T3 fibroblasts were purchased from American Type Culture Collection (Manassas, VA, USA).
Mice
We purchased C57Bl/6 mice from the National Cancer Institute (NCI)-Frederick Mouse Repository (Frederick, MD, USA). Experiments comparing LPA1-knockout (KO) and wild-type (WT) mice used offspring of mice heterozygous for the LPA1 mutant allele, which were hybrids of the C57Bl/6 and 129Sv/J genetic backgrounds (kindly provided by Dr. Jerold Chun, Scripps Research Institute, La Jolla, CA, USA; ref. 26). Experiments to identify fibroblasts used type I collagen (COLI)–green fluorescent protein (GFP) transgenic mice generated on the C57Bl/6 background (kindly provided by Dr. Jeremy Duffield, University of Washington, Seattle, WA, USA; ref. 27). All experiments used sex- and weight-matched mice at 6–10 wk of age maintained in specific pathogen-free environments and were performed in accordance with U.S. National Institutes of Health (NIH) guidelines and protocols approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee.
Peritoneal fibrosis model
Peritoneal fibrosis was induced by intraperitoneal injection of 0.1% CG (Wako Pure Chemical Industries, Osaka, Japan) dissolved in 15% ethanol/phosphate buffered saline (PBS). CG was injected every other day over a period of 21 d.
Histology and peritoneal thickness measurement
A sample of peritoneal tissue from each mouse was fixed in 10% buffered formalin (pH 7.2) and embedded in paraffin. We then stained 5-μm sections with Masson's trichrome according to our standard protocol (4). Peritoneal thickness was measured from the junction of the parietal peritoneum with the musculature of the abdominal wall to the serosal surface of the parietal peritoneum, as described previously (9), on photomicrographs (×200) of Masson's trichrome-stained sections at 5 randomly selected sites per high-power field (HPF) per section.
Hydroxyproline assay
Two pieces of peritoneal samples were taken by 6-mm punch biopsy apparatus (Acuderm, Fort Lauderdale, FL, USA) from each mouse to assess peritoneal collagen, measured by its hydroxyproline content as determined by the standard protocol of our laboratory (28). Assay results were expressed as micrograms of hydroxyproline per piece.
RNA analyses
Total cellular RNA was isolated from primary cells using RNeasy Mini Kits (Qiagen, Valencia, CA, USA). Total cellular RNA was isolated from peritoneal tissue by immersing the surface of peritoneum in Trizol reagent (Invitrogen) for 20 min and then extracting RNA according to the manufacturer's instructions. Quantitative real-time PCR analyses of RNA were performed using Mastercycler realplex2 (Eppendorf, Hauppauge, NY, USA).
Immunohistochemical and immunocytochemical analyses
Myofibroblasts were identified in tissue samples using a specific antibody against α smooth muscle actin (αSMA). Formalin-fixed, paraffin-embedded 5-μm sections were incubated with anti-mouse αSMA monoclonal antibody (E184; Abcam, Cambridge, MA, USA) followed by an Envision kit (Dako, Carpinteria, CA, USA). CTGF-expressing cells were identified using anti-mouse CTGF polyclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). αSMA+ and CTGF+ cells were visualized by incubating antibody-stained sections with DAB (Dako). α-SMA+ cells were then counted in all fields of the submesothelial zone and expressed as the mean ± se number per HPF. To identify proliferating fibroblasts, peritoneal sections from COLI-GFP mice were costained with anti-enhanced GFP (EGFP) monoclonal antibody (Cell Signaling, Danvers, MA, USA) and anti-mouse proliferating cell nuclear antigen (PCNA) monoclonal antibody (Abcam), using an M.O.M. kit (Vector Laboratories, Burlingame, CA, USA). Antibody-stained cells were visualized using Fluorescein avidin (Vector Laboratories) for EGFP and Texas-red avidin (Vector Laboratories) for PCNA. EGFP and PCNA single-positive cells, and EGFP-PCNA double-positive cells, were then counted in all fields of the submesothelial zone and expressed as the mean ± se number per HPF.
Nuclear vs. cytoplasmic localization of MRTF-A and MRTF-B was assessed by immunocytochemical analyses performed on primary mesothelial cells (PMCs), isolated as described below. PMCs were fixed in 4% paraformaldehyde in PBS, followed by the permeabilization with 0.2% Triton X-100 in PBS. Some PMCs were stimulated with LPA prior to fixation, and some cells were additionally treated with 5 μM Y27632 30 min before LPA stimulation. Following permeabilization, PMCs were incubated with anti-mouse MRTF-A polyclonal antibodies (Santa Cruz Biotechnology) or anti-mouse MRTF-B polyclonal antibodies (Santa Cruz Biotechnology), followed by Alexa-Fluor 488-conjugated secondary antibodies (Invitrogen). Five random fields of view were counted, and subcellular localization of MRTFs was scored as predominantly nuclear, evenly distributed, or predominantly cytoplasmic, as described previously (29).
Western blot analyses
Whole cellular lysates from primary cells were extracted with RIPA buffer (Thermo Scientific, Waltham, MA, USA) according to the manufacturer's protocol. Samples of conditioned medium (CM) were centrifuged to remove cellular debris, and concentrated (15-fold) using Amicon Ultra 10,000 (Millipore, Billerica, MA, USA). Whole-tissue lysates were extracted from peritoneal samples by immersing the surface of peritoneal tissues in RIPA buffer (Thermo Scientific) for 20 min. Cellular and tissue lysates, and concentrated CM, were then separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Invitrogen). After incubation in blocking buffer containing 5% skim milk (Bio-Rad, Hercules, CA, USA), membranes were incubated overnight at 4°C with rabbit anti-mouse CTGF polyclonal antibodies (Thermo Scientific), rabbit anti-mouse Gα12 polyclonal antibodies (Santa Cruz Biotechnology), rabbit anti-mouse Gα13 polyclonal antibodies (Santa Cruz Biotechnology), rabbit anti-mouse MRTF-A polyclonal antibodies (Santa Cruz Biotechnology), rabbit anti-mouse MRTF-B polyclonal antibodies (Santa Cruz Biotechnology), rabbit anti-mouse SRF monoclonal antibody (Cell Signaling), rabbit anti-mouse Smad3 monoclonal antibody (Cell Signaling), rabbit anti-mouse phospho-Smad3 monoclonal antibody (Cell Signaling), or rabbit anti-mouse GAPDH monoclonal antibody (Cell Signaling). Following additional incubations of the membranes with appropriate biotinylated secondary antibodies, protein bands were detected with an enhanced chemiluminescent substrate (Thermo Scientific). Quantification was performed with Image J software (NIH, Bethesda, MD, USA).
Inhibitor administration in vivo
The selective LPA1 antagonist AM095 was dissolved in sterile water, and dosages of 30 mg/kg/mouse were administered by oral gavage to COLI-GFP mice 2×/d on weekdays and 1×/d on weekends. Control COLI-GFP mice received equal volumes of sterile water alone (vehicle) on the same schedule. AM095 or vehicle control was administered from the onset of CG challenge in a preventive regimen, or beginning 7 d after the onset of CG challenge in a therapeutic regimen.
The MRTF-SRF inhibitor CCG-1423 was dissolved in vehicle (30% DMSO/15% ethanol/PBS), and a dose of 1.5 or 3.0 mg/kg/mouse, or vehicle alone, was administered by intraperitoneal injection to C57Bl/6 mice. CCG-1423 dosages, or equal volumes of vehicle alone, were administered 1×/d from the onset of CG challenge over a period of 21 d.
Isolation of primary mouse PMCs
Primary PMCs were isolated from mice by enzymatic digestion of the inner surface of the peritoneum, as described previously (30), with minor modifications. Briefly, parietal peritoneal flaps were removed and stretched on a sterile culture dish. RPMI 1640 (Lonza) containing 25 μg/ml Liberase (Roche, Basel, Switzerland) was placed on the peritoneal inner surface for 45 min at 37°C. After incubation, the surface of the digested peritoneum was gently scraped to complete the release of partially attached mesothelial cells. The cell pellet was collected by centrifugation and resuspended in growth medium composed of 10% FBS-containing DMEM. To identify the outgrowing cells as mesothelial cells, immunocytochemistry with anti-vimentin antibody (VIM13.2; Sigma-Aldrich) and anti-cytokeratin antibody (PCK-26; Sigma-Aldrich) was performed. More than 98% of these cells were positive for vimentin and cytokeratin, consistent with their being mesothelial cells (31). In vitro experiments were performed on PMCs from second to fifth passages.
RhoA activation assay
Quantification of RhoA activation in PMCs cultured in 100-mm dishes was performed using a commercially available kit (Cytoskeleton) according to the manufacturer's instructions.
siRNA transfections
In experiments using RNA interference, siRNAs targeting mouse MRTF-A, MRTF-B, SRF, and CTGF were On-Target Plus Smart Pools, siRNAs targeting Gα12 were an siGenome Smart Pool (all from Thermo Scientific), and siRNAs targeting Gα13 were from Santa Cruz Biotechnology. On-Target Plus nontargeting pool siRNAs were used as a nonspecific control. PMCs or fibroblasts were transfected with siRNAs by Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol and were incubated for 48 h prior to use in experiments.
Fibroblast proliferation assay
Cultured PMCs were transfected with either CTGF-targeting or control siRNA and then stimulated with 10 μM LPA for 24 h. The CM was then aspirated, centrifuged to remove cellular debris, and applied to serum-deprived NIH3T3 fibroblasts. Fibroblasts in these experiments were also treated with CTGF-targeting siRNA, to prevent any LPA remaining in the CM from stimulating fibroblast CTGF production. Fibroblast proliferation after incubation for 48 h with PMC-CM was determined by BrdU assay (Roche), performed according to the manufacturer's protocol.
Statistical analyses
Data are expressed as means ± se. Unpaired t tests were used for comparisons between 2 groups, and analysis of variance with post hoc Fisher's test was used for comparisons between >2 groups. Values of P < 0.05 were considered statistically significant.
RESULTS
Genetic deletion of LPA1 protects mice from CG-induced peritoneal fibrosis
LPA1-deficient (LPA1-KO) mice were dramatically protected from CG-induced peritoneal fibrosis. Masson's trichrome staining demonstrated that collagen deposition induced in WT mice by CG challenge was markedly reduced in LPA1-KO mice (Fig. 1A). The degree of protection of LPA1-KO mice was quantified by measuring peritoneal thickness, hydroxyproline content, and mRNA levels of the α1 chain of type I procollagen (COLIα1). The peritoneal thickness of LPA1-KO mice following CG challenge was significantly less than that of CG-challenged WT mice (55.3±4.7 vs. 116.0±6.0 μm; Fig. 1B). In contrast, peritoneal thickness was not different between LPA1-KO and WT mice challenged with PBS (6.3±0.3 vs. 6.6±0.2 μm; Fig. 1B). The increase in peritoneal hydroxyproline content observed in CG-challenged WT mice was blunted by 65.4% in CG-challenged LPA1-KO mice (Fig. 1C), and the increase in peritoneal expression of COLIα1 mRNA was significantly reduced as well (Fig. 1D). These data indicate that the LPA-LPA1 pathway importantly contributes to the development of peritoneal fibrosis.
Figure 1.
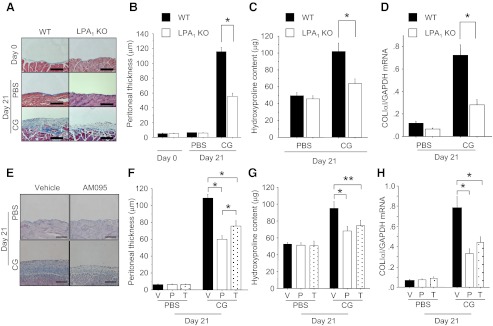
Genetic deletion or pharmacological inhibition of LPA1 protects mice from CG-induced peritoneal fibrosis. A–D) Protection by genetic deletion of LPA1. Data are expressed as means ± se. A) Masson's trichrome-stained peritoneal sections of WT (left) and LPA1-KO mice (right). Representative sections are shown from untreated mice (d 0) and mice at d 21 of PBS or CG injections (×200). B) Peritoneal thickness of WT and LPA1-KO mice following PBS or CG challenges (d 0, n=3 mice/genotype; d 21 PBS, n=8 mice/genotype; d 21 CG, n=8 mice/genotype). C) Biochemical analysis of CG-induced peritoneal fibrosis. Hydroxyproline content was measured in the peritoneum of WT and LPA1-KO mice following CG or PBS challenges (d 21 PBS, n=6 mice/genotype; d 21 CG, n=6 mice/genotype). D) Peritoneal expression of COL1α1 mRNA in WT and LPA1-KO mice following CG or PBS challenges mice (d 21 PBS, n=5 mice/genotype; d 21 CG, n=5 mice/genotype). E–H) Protection by pharmacologic inhibition of LPA1. Data are expressed as means ± se. E) Representative Masson's trichrome-stained peritoneal sections of vehicle-treated (left) and preventive AM095-treated mice (right, ×200). F) Peritoneal thickness following PBS or CG challenge (d 21 PBS, n=5 mice/treatment group; d 21 CG, n=5 mice/treatment group). V, vehicle treatment; P, preventive regimen of AM095; T, therapeutic regimen of AM095. G) Hydroxyproline content in the peritoneum following CG or PBS challenges (d 21 PBS, n=5 mice/treatment group; d 21 CG, n=5 mice/treatment group). H) Peritoneal expression of COLIα1 mRNA following CG or PBS challenges (d 21 PBS, n=6 mice/treatment group; d 21 CG, n=6 mice/treatment group). Scale bars = 100 μm. *P < 0.01, **P < 0.05.
Pharmacological inhibition of LPA1 protects mice from CG-induced peritoneal fibrosis
We next determined whether CG-induced peritoneal fibrosis could be suppressed by administration of the LPA1-selective oral small molecule antagonist AM095 (kind gift of Amira Pharmaceuticals; sodium{4′-[3-methyl-4-((R)-1-phenyl-ethoxycarbonylamino)-isoxazol-5-yl]-biphenyl-4-yl}-acetate; ref. 5). AM095 was administered from CG challenge onset in a preventive regimen to LPA1-sufficient mice. Since fibrosis is already established by d 7 in the CG model (32), we examined the potential of LPA1 inhibition to treat peritoneal fibrosis by administering AM095 beginning 7 d after CG challenge onset in a therapeutic regimen. To enable the analyses of fibroblast accumulation and proliferation described below, the LPA1-sufficient mice used in these experiments were COLI-GFP mice, in which fibroblasts can be identified by their transgenic expression of EGFP driven by the fibroblast-specific collagen type I, α1 promoter (27). Mice treated with AM095 in the preventive regimen were dramatically protected from peritoneal fibrosis, as indicated by Masson's trichrome staining of peritoneal collagen (Fig. 1E), measurements of peritoneal thickness (60.0±4.6 μm in preventive AM095-treated mice vs. 108.5±4.8 μm in vehicle-treated CG-challenged mice; Fig. 1F), peritoneal hydroxyproline content (Fig. 1G), and peritoneal COLIα1 mRNA expression levels (Fig. 1H). The increase in peritoneal hydroxyproline content observed in vehicle-treated CG-challenged mice was reduced by 52.9% in preventive AM095-treated mice. Delayed administration of AM095 in the therapeutic regimen also attenuated CG-induced peritoneal thickness, hydroxyproline content, and COLIα1 expression (Fig. 1F–H). Therapeutic AM095 treatment mitigated the increase in peritoneal hydroxyproline content of vehicle-treated CG-challenged mice by 41.2%. These data indicate an ongoing requirement for the LPA-LPA1 pathway in the maintenance of pathological peritoneal fibrosis, suggesting that targeting this pathway may be an effective therapeutic strategy for peritoneal fibrosis.
CG-induced peritoneal αSMA+ myofibroblast accumulation is dependent on LPA1
Myofibroblasts are prominent in areas of increased collagen deposition in fibrotic tissues and are thought to be key effector cells producing pathological extracellular matrix in fibrotic diseases (33). We assessed the effect of LPA-LPA1 signaling on the peritoneal accumulation of myofibroblasts, identified as αSMA-expressing cells by immunostaining. As demonstrated by the representative peritoneal sections in Fig. 2A, CG challenges induced a robust, mainly submesothelial accumulation of αSMA+ myofibroblasts. The increase in myofibroblasts induced in WT mice by CG challenge was markedly attenuated in LPA1-KO mice (97.8±3.7 vs. 38.2±3.4 cells/HPF, respectively; Fig. 2A, B). The increase in peritoneal expression of αSMA mRNA induced by CG challenge was similarly reduced in LPA1-KO mice (Fig. 2C), as well as in mice treated with AM095 in either preventive or therapeutic regimens. These findings indicate that LPA signaling through LPA1 is required for αSMA+ myofibroblast accumulation during peritoneal fibrogenesis.
Figure 2.
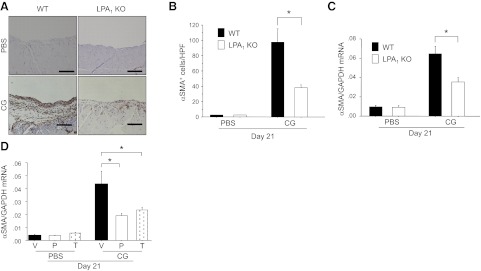
CG-induced peritoneal αSMA+ myofibroblast accumulation is dependent on LPA1. A) Peritoneal accumulation of αSMA+ myofibroblasts after CG challenges. Representative peritoneal sections of WT (left) and LPA1-KO mice (right) stained with anti-αSMA antibody/peroxidase are shown from mice after 21 d of PBS or CG injections (×200). Scale bars = 100 μm. B) Numbers of αSMA+ cells in the submesothelial zone, expressed as mean number per HPF (d 21 PBS, n=5 mice/genotype; d 21 CG, n=5 mice/genotype). C) Peritoneal expression of αSMA mRNA in WT and LPA1-KO mice following CG or PBS challenges, expressed as mean copies of αSMA mRNA relative to copies of GAPDH mRNA (d 21 PBS, n=5 mice/genotype; d 21 CG, n=5 mice/genotype). D) Peritoneal expression of αSMA mRNA following CG or PBS challenges in AM095-treated mice, expressed as mean copies of αSMA mRNA relative to copies of GAPDH mRNA (d 21 PBS, n=6 mice/treatment group; d 21 CG, n=6 mice/treatment group). V, vehicle; P, preventive AM095; T, therapeutic AM095. Data are expressed as means ± se. *P < 0.01.
CG-induced peritoneal fibroblast accumulation and proliferation is dependent on LPA-LPA1
Although recruitment of bone marrow-derived progenitors and transdifferentiation of other cells types may play important roles in increasing myofibroblast numbers during the development of fibrosis, recent evidence suggests that the proliferation of fibroblasts within injured tissues makes an essential contribution to myofibroblast accumulation (8). We compared fibroblast proliferation induced by CG challenges in AM095- vs. vehicle-treated COLI-GFP mice. As demonstrated in the representative sections in Fig. 3A, CG challenges induced a marked peritoneal accumulation of GFP+ fibroblasts that was significantly reduced by LPA1 inhibition with AM095. The increased number of GFP+ fibroblasts present in CG-challenged, vehicle-treated mice was significantly reduced in CG-challenged, AM095-treated mice (283.7±23.9 cells/HPF in CG-challenged, vehicle-treated mice vs. 73.5±13.4/HPF in CG-challenged, AM095-treated mice; Fig. 3B). To specifically identify proliferating fibroblasts, we double-stained peritoneal sections with anti-PCNA antibody and anti-GFP antibody. As demonstrated in the representative sections in Fig. 3A, fibroblast proliferation induced by CG challenges was also dependent on LPA-LPA1 signaling. The number of proliferating (PCNA+) fibroblasts (GFP+) in the peritoneum after CG challenges was significantly higher in vehicle- vs. AM095-treated mice (158.3±17.2 vs. 23.0±4.6 cells/HPF; Fig. 3C), as was the percentage of proliferating fibroblasts among total fibroblasts (percentage of PCNA+GFP+ cells among total GFP+ cells): 56.0 ± 4.4 vs. 31.4 ± 2.6% in vehicle- vs. AM095-treated mice (Fig. 3D). These data suggest that reduced fibroblast proliferation in the absence of LPA1 signaling contributes to reduced peritoneal myofibroblast accumulation in LPA1-KO mice following CG challenges.
Figure 3.
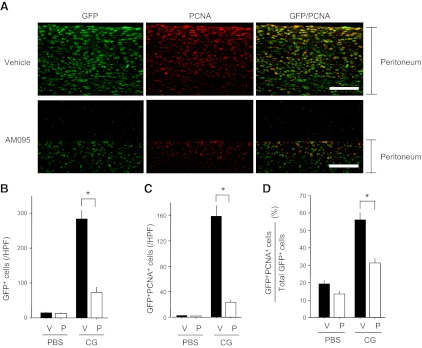
CG-induced peritoneal fibroblast accumulation and proliferation is dependent on LPA-LPA1. A) Peritoneal accumulation of fibroblasts, proliferating cells, and proliferating fibroblasts after CG challenges. Representative peritoneal sections of vehicle-treated (top panel) and preventive AM095-treated COLI-GFP mice (bottom panel) stained with anti-GFP antibody (green) and anti-PCNA antibody (red) are shown (×400). Scale bars = 50 μm. B) Numbers of submesothelial GFP+ cells (fibroblasts), expressed as mean number per HPF (d 21 PBS, n=6 mice/treatment group; d 21 CG, n=6 mice/treatment group). C) Numbers of submesothelial GFP+PCNA+ cells (proliferating fibroblasts), expressed as mean number per HPF (d 21 PBS, n=6 mice/treatment group; d 21 CG, n=6 mice/treatment group). D) Percentages of submesothelial fibroblasts that are proliferating (GFP+PCNA+ cells/total GFP+ cells; d 21 PBS, n=6 mice/treatment group; d 21 CG, n=6 mice/treatment group). V, vehicle; P, preventive AM095. Data are expressed as means ± se. *P < 0.01.
CG-induced CTGF expression requires LPA1, and is generated by peritoneal mesothelial cells
We hypothesized that LPA-LPA1 signaling drives fibroblast proliferation after tissue injury in vivo at least in part by driving the expression of important fibroblast mitogens other than LPA, such as CTGF. We focused on CTGF for several reasons: CTGF stimulates fibroblast proliferation (17, 34); fibroblast-specific deletion of CTGF markedly attenuates myofibroblast accumulation in the bleomycin model of dermal fibrosis (18); CTGF has been implicated in the pathogenesis of peritoneal fibrosis in peritoneal dialysis patients (35); and CTGF has a CArG-like box in its promoter (15), which would allow it to be induced by an LPA-LPA1-actin-MRTF-SRF pathway. CG challenges markedly induced peritoneal CTGF mRNA (Fig. 4A) and protein (Fig. 4B) expression, both of which were significantly attenuated in LPA1-KO mice. Peritoneal CTGF mRNA expression was also significantly suppressed by the LPA1 antagonist AM095 administered in either preventive or therapeutic regimens (Fig. 4C). These data indicate that LPA-LPA1 signaling is required for the induction of CTGF expression during the development of peritoneal fibrosis. To determine the cellular sources of CTGF in this model, we identified CTGF+ cells by immunostaining. Robust CTGF staining was detected in the PMCs of CG-challenged WT mice, with reduced staining being detected in the PMCs of LPA1-KO mice (Fig. 4D). Although some CTGF staining was also detected in cells in the expanded peritoneal interstitium of CG-challenged mice, the robust expression of CTGF by PMCs is consistent with accumulating evidence that these cells are a critically important source of profibrotic molecules, including cytokines, growth factors, and matrix proteins, in the pathogenesis of peritoneal fibrosis (36).
Figure 4.
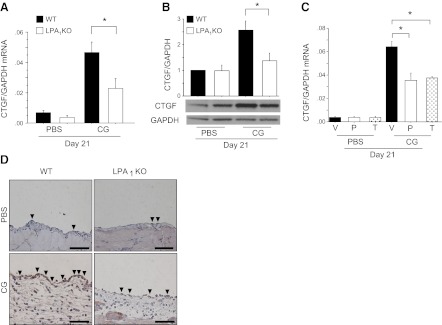
CG-induced CTGF expression is dependent on LPA1 and is predominantly attributable to peritoneal mesothelial cells. A) Peritoneal expression of CTGF mRNA in WT and LPA1-KO mice following CG or PBS challenges (d 21 PBS, n=5 mice/genotype; d 21 CG, n=5 mice/genotype). Data are expressed as mean copies of CTGF mRNA relative to copies of GAPDH mRNA. B) Peritoneal expression of CTGF protein in WT and LPA1-KO mice following CG or PBS challenges (d 21 PBS, n=4 mice/genotype; d 21 CG, n=4 mice/genotype. Quantification was performed with ImageJ software; data are expressed as mean density of CTGF bands relative to GAPDH bands. C) Peritoneal expression of CTGF mRNA in vehicle and AM095-treated mice following CG or PBS challenges (d 21 PBS, n=6 mice/treatment group; d 21 CG, n=6 mice/treatment group). V, vehicle; P, preventive AM095; T, therapeutic AM095. Data are expressed as mean copies of CTGF mRNA relative to copies of GAPDH mRNA. D) Peritoneal location of CTGF protein in representative sections from WT and LPA1-KO mice following PBS and CG challenges, stained with anti-CTGF antibody/peroxidase (×400). Arrowheads indicate CTGF+ mesothelial cells. Scale bars = 50 μm. Data are expressed as means ± se. *P < 0.01.
LPA-LPA1 signaling induces PMC CTGF expression, which induces fibroblast proliferation
To investigate the ability of LPA-LPA1 signaling to directly induce PMC CTGF expression, we isolated primary mouse PMCs for in vitro studies. LPA induced robust CTGF mRNA expression in primary PMCs isolated from WT mice (WT-PMCs) in a time- and dose-dependent manner (Fig. 5A, B). Maximal CTGF mRNA induction was observed 2 h after stimulation with 10 μM LPA. LPA similarly induced CTGF protein production by WT-PMCs, also in a time- and dose-dependent manner (Fig. 5C, D). To determine which LPA receptors are responsible for LPA-induced CTGF expression by WT-PMCs, we first determined the LPA receptor expression profile of these cells. Of the 5 definitively identified LPA receptors, LPA1 was the most highly expressed by WT-PMCs (LPA1>LPA2>LPA3≅LPA4≅LPA5; Fig. 5E). Primary mouse PMCs isolated from LPA1-KO mice (LPA1-KO PMCs) did not express LPA1 as expected, and their LPA1 deficiency did not cause compensatory changes in their expression of other LPA receptors (Fig. 5E). LPA-induced CTGF mRNA expression was almost completely abrogated in LPA1-KO PMCs (Fig. 5F), indicating that signaling through LPA1 is predominantly responsible for CTGF induction by LPA.
Figure 5.
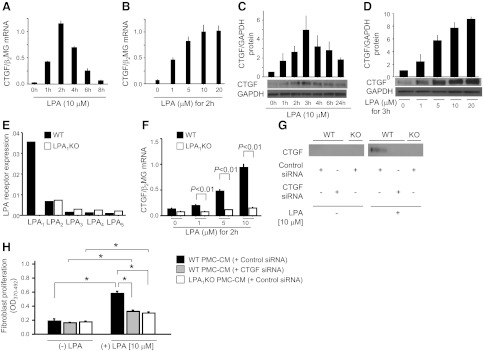
Mesothelial cell-derived CTGF expression is dependent on LPA-LPA1, and regulates fibroblast proliferation. A, B) LPA induces CTGF mRNA expression time dependently (A) and dose dependently (B) in PMCs (n=3 cell preparations/group). C, D) LPA induces CTGF protein expression time dependently (C) and dose dependently (D) in PMCs (n=2 cell preparations/group). E) LPA receptor expression of PMCs. Data are presented as copies of receptor mRNA relative to copies of β2 microglobulin mRNA. F) LPA-induced CTGF mRNA expression was abrogated in LPA1-KO PMCs (n=3 cell preparations/group). G) Identification of CTGF protein in CM from PMCs by Western blot. WT PMCs were transfected with control or CTGF siRNA, and LPA1-KO PMCs with control siRNA. All PMCs were then stimulated with control medium or LPA (10 μM) for 24 h, and their CM was assayed for CTGF. H) NIH3T3 fibroblasts were transfected with CTGF siRNA, and then incubated with CM from the same groups of PMCs as in G. BrdU proliferation assays were performed after incubation with CM for 48 h (n=3 cell preparations/group), and expressed as mean OD value (OD370–492). Data are expressed as means ± se. *P < 0.01.
To investigate the potential role of CTGF as a downstream mediator of LPA's ability to drive peritoneal fibroblast proliferation, we investigated the ability of medium conditioned by LPA-stimulated PMCs to induce fibroblast proliferation, and whether any of the proliferation induced was attributable to CTGF produced by PMCs in response to LPA-LPA1 signaling. Medium conditioned by LPA-stimulated PMCs contained increased CTGF (Fig. 5G) and stimulated increased fibroblast proliferation (Fig. 5H), compared with medium conditioned by unstimulated PMCs. CTGF content and fibroblast proliferative activity of medium conditioned by LPA-stimulated PMCs were simultaneously reduced when PMCs were transfected with siRNAs targeting CTGF, or when PMCs were isolated from LPA1-KO mice (Fig. 5G, H). To prevent any LPA remaining in the CM from directly stimulating CTGF expression by the responding fibroblasts, the responding fibroblasts in these experiments were also transfected with siRNA targeting CTGF. Taken together, these data support the hypothesis that LPA-LPA1 signaling promotes fibroblast proliferation during the development of peritoneal fibrosis by inducing CTGF expression by PMCs.
LPA-induced CTGF expression is independent of de novo protein synthesis and TGF-β activation
CTGF expression is highly induced by TGF-β (37), and LPA has been demonstrated to induce activation of latent TGF-β by lung epithelial cells (38) and smooth muscle cells (39). To investigate whether LPA-induced CTGF expression required de novo production of TGF-β, or any other proteins in an autocrine fashion, we investigated whether this CTGF expression was sensitive to inhibition of protein synthesis with cycloheximide. As shown in Fig. 6A, cycloheximide treatment did not block LPA-induced CTGF mRNA expression by PMCs, actually resulting in CTGF “superinduction,” i.e., augmented mRNA induction following agonist stimulation in the presence of translational blockers, such as cycloheximide (40). LPA-induced CTGF expression in PMCs was therefore independent of new TGF-β or other protein production. Since TGF-β activity in vivo is primarily regulated by the post-translational conversion of latent TGF-β complexes to active TGF-β (41), we investigated whether LPA induced CTGF expression by activating latent TGF-β. Treatment with pan-specific TGF-β-neutralizing antibody had no effect on LPA-induced CTGF expression (Fig. 6B), and LPA stimulation did not induce phosphorylation of Smad3 (Fig. 6C). Taken together, these results suggest that PMC CTGF expression induced by the LPA-LPA1 pathway is independent of TGF-β production, activation, or canonical Smad signaling.
Figure 6.
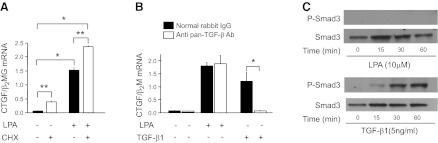
Mesothelial CTGF expression induced by LPA-LPA1 signaling is independent of de novo protein synthesis, latent TGF-β activation, and Smad signaling. A) Effect of cycloheximide (CHX) on LPA-induced CTGF expression. PMCs were preincubated with CHX (20 μg/ml) or control medium for 2 h, and then stimulated with control medium or LPA (10 μM) for an additional 2 h (n=3 cell preparations/group). B) Effect of TGF-β neutralization on LPA-induced CTGF expression. PMCs were exposed to LPA (10 μM, for 2 h) or TGF-β1 (1 ng/ml, for 4 h), with or without a pan-specific TGF-β neutralizing antibody (Ab, 10 μg/ml; n=3 cell preparations/group). C) Ability of LPA vs. TGF-β to induce Smad phosphorylation. PMCs were incubated with LPA (10 μM) or TGF-β1 (5 ng/ml), and their lysates were assayed for phosphorylated Smad3 by Western blot. The experiment was performed in 2 independent series of PMC preparations. Data are expressed as mean ± sem copies of CTGF mRNA relative to copies of β2 microglobulin mRNA. *P < 0.01, **P < 0.05.
LPA-induced CTGF expression is dependent on Gα12/13, RhoA, ROCK, and actin polymerization
LPA receptors couple to different classes of G proteins, including those containing Gα12/13, Gαi/o, or Gαq α subunits, to mediate the diverse activities of LPA (11). Transfection of PMCs with siRNAs targeting either Gα12 or Gα13 demonstrated the involvement of Gα12/13-containing G proteins in LPA-induced CTGF expression. The extent of siRNA-induced suppression of Gα12 or Gα13 expression in PMCs is shown Fig. 7A, B, and the ability of these siRNAs to significantly suppress LPA-induced PMC CTGF expression is shown in Fig. 7C. In contrast, pretreatment of PMCs with pertussis toxin, which inhibits Gαi/o–containing G proteins, had no significant effect on LPA-induced CTGF expression (Fig. 7D).
Figure 7.
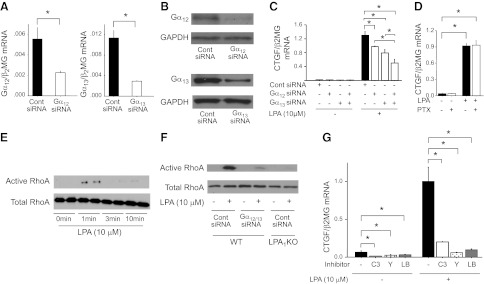
Mesothelial CTGF expression induced by LPA-LPA1 signaling is dependent on Gα12/13 signaling, RhoA and ROCK activation, and actin polymerization. A, B) Validation of siRNA inhibition of Gα12 and Gα13 expression. PMCs were transfected with Gα12, Gα13, or control siRNA, and targeted Gα subunit inhibition was determined at mRNA (A) and protein (B) levels. mRNA data are expressed as copies of Gα12 or Gα13 mRNA relative to copies of β2 microglobulin mRNA. C) Effects of Gα12 and/or Gα13 knockdown on LPA-induced CTGF expression, expressed as copies of CTGF mRNA relative to copies of β2 microglobulin mRNA. PMCs were transfected with control siRNA or Gα12 and/or Gα13 siRNA, and then stimulated with control medium or LPA (10 μM; n=3 cell preparations/group). D) Effect of pertussis toxin (PTX) on LPA-induced CTGF expression, expressed as copies of CTGF mRNA relative to copies of β2 microglobulin mRNA. PMCs were preincubated with PTX (100 ng/ml) or control medium for 18 h, and then stimulated for 2 h with control medium or LPA (10 μM). E) RhoA activation induced by LPA. Time course of active and total RhoA in PMCs following stimulation with LPA (10 μM). F) Dependence of LPA-induced RhoA activation on LPA1 and Gα12/13. WT PMCs were transfected with siRNAs targeting Gα12 and Gα13 or with control siRNA, and LPA1-KO PMCs were transfected with control siRNA. Levels of active and total RhoA were then assayed 1 min after stimulation with control medium or LPA (10 μM; n=2 cell preparations/group). G) Dependence of LPA-induced CTGF expression on RhoA, ROCK, and actin polymerization. PMCs were preincubated with no inhibitor, or with C3 toxin (C3; 2.0 μg/ml for 10 h), Y27632 (Y; 5 μM for 30 min) or latrunculin B (LB; 1 μg/ml for 30 min). PMCs were then stimulated with LPA (10 μM) or control medium for an additional 2 h. Data are expressed as copies of CTGF mRNA relative to copies of β2 microglobulin mRNA (n=3 cell preparations/group). Data are expressed as means ± se. *P < 0.01.
The effects of LPA receptor G-protein-coupled signaling on the actin cytoskeleton are mediated by Rho family of GTPases, including RhoA and Rac (11). Whereas Gαi/o G proteins typically mediate LPA-induced Rac activation, Gα12/13 G proteins are typically responsible for LPA-induced activation of RhoA, and we consequently hypothesized that LPA-induced CTGF expression would depend on RhoA. We found that LPA stimulation of WT-PMCs activated RhoA, with the greatest activation at early times (1 min) poststimulation (Fig. 7E). LPA-induced RhoA activation was suppressed in LPA1-KO PMCs, and in WT-PMCs transfected with Gα12 and Gα13 siRNAs (Fig. 7F), indicating that LPA-induced RhoA activation in PMCs is mediated by LPA1 and Gα12/13 signaling. LPA-induced CTGF expression was significantly attenuated by treatment of PMCs with the RhoA inhibitor C3 toxin (Fig. 7G), which reduced basal levels of CTGF mRNA in PMCs as well, indicating that LPA's ability to activate RhoA is required for its ability to induce CTGF expression.
RhoA signaling induces focal adhesion formation, actin polymerization, and stress fiber formation by activating ROCKs. LPA-induced CTGF expression was significantly attenuated by treatment of PMCs with the nonselective ROCK inhibitor Y-27632, as well as by the actin polymerization inhibitor latrunculin B (Fig. 7G). Both Y-27632 and latrunculin B reduced basal levels of CTGF mRNA in PMCs as well. Both of these inhibitors abrogated actin polymerization induced by LPA. Using phalloidin to visualize polymeric F-actin, LPA stimulation induced F-actin formation in PMCs as expected (Fig. 8A, B), and this actin polymerization was completely inhibited by pretreatment with Y27632 or latrunculin B (Fig. 8C, D). Taken together, these data indicate that LPA-LPA1-induced CTGF expression is mediated by a signaling pathway involving Gα12/13, RhoA, ROCK, and actin polymerization.
Figure 8.

Y27632 (Y) and latrunculin B (LB) abrogate LPA-induced actin polymerization. A, B) Actin polymerization was visualized by immunocytochemical staining for phalloidin in PMCs that had been incubated in serum-free medium for 24 h and then stimulated for 30 min with control medium (A) or LPA (10 μM; B). C, D) PMCs were treated with Y27632 (Y; 5 μM; C) or latrunculin B (LB; 1 μg/ml; D) for 30 min prior to LPA stimulation. All images were captured using identical exposure settings. Scale bars = 50 μm.
LPA promotes the nuclear translocation of MRTF-A and MRTF-B
MRTF-A and MRTF-B have recently been identified as novel links between actin dynamics and gene expression (13). The MRTFs form stable complexes with monomeric G-actin, resulting in their sequestration in the cytoplasm. Polymerization of G-actin into the F-actin filaments liberates MRTFs, which then translocate to the nucleus. In the nucleus, the MRTFs act as cofactors for SRF, augmenting the activity of this nuclear transcription factor (13). We therefore hypothesized that the LPA-LPA1-Gα12/13-RhoA-ROCK-actin signaling pathway induces gene expression by promoting the nuclear translocation of MRTF-A and MRTF-B. LPA stimulation of PMCs significantly decreased the percentage of cells with predominantly cytoplasmic localization of MRTF-A from 28.5 to 6.4%, and significantly increased the percentage of cells with predominantly nuclear MRTF-A from 44.3 to 86.6% (Fig. 9A, B). Similarly, LPA stimulation of PMCs significantly decreased the percentage of cells with predominantly cytoplasmic MRTF-B from 60.4 to 33.5%, and significantly increased the number of cells with predominantly nuclear MRTF-B from 18.0 to 41.2% (Fig. 9C, D). Consistent with LPA-induced MRTF nuclear translocation being mediated through the LPA1-Gα12/13-RhoA-ROCK-actin pathway, PMC pretreatment with Y-27632 markedly blocked LPA-induced MRTF nuclear translocation, reducing the percentages of cells with predominantly nuclear MRTF-A and MRTF-B to below control levels (Fig. 9).
Figure 9.

LPA promotes the nuclear translocation of MRTF-A and MRTF-B in a ROCK-dependent manner. A, C) Subcellular distributions of MRTF-A (A) and MRTF-B (C) were visualized by immunocytochemistry in PMCs that had been incubated in serum-free medium for 24 h and then stimulated with medium containing LPA (10 μM for 30 min) or control medium. Green represents MRTF-A or MRTF-B staining; blue represents DAPI staining. PMCs were additionally pretreated with Y27632 (Y; 5 μM) or control medium for 30 min before LPA stimulation. All images were captured using identical exposure settings. Scale bars = 100 μm. B, D) Quantification of the subcellular distribution of MRTF-A (B) and MRTF-B (D). Five random fields of view were counted per slide. Subcellular distributions were classified as nuclear (N; nuclear staining > cytoplasmic staining); equal (E; nuclear staining = cytoplasmic staining); or cytoplasmic (C; nuclear staining < cytoplasmic staining). Two independent series of PMCs were analyzed. All data are expressed as mean ± se distribution. Significant (P<0.01) comparisons for both MRTF-A and MRTF-B were percentages of cells with nuclear distributions, and percentages of cells with cytoplasmic distributions, in LPA alone vs. control, and LPA alone vs. LPA + Y27632.
LPA-induced CTGF expression in PMCs is dependent on MRTF-A, MRTF-B, and SRF
Transfection of PMCs with siRNAs targeting MRTF-A, MRTF-B, or SRF demonstrated the involvement of these transcription factors in LPA-induced CTGF expression. The extent of siRNA-induced suppression of MRTF-A, MRTF-B, and SRF in PMCs is shown at the mRNA and protein levels in Fig. 10A, B, respectively, and the ability of these siRNAs to significantly suppress LPA-induced PMC CTGF expression is shown in Fig. 10C. To confirm that LPA-induced CTGF expression requires MRTF-SRF signaling, we treated PMCs with CCG-1423 (N-[2-[4(4-chlorophenyl)amino]-1-methyl-2-oxoethoxy]-3,5-bis-(trifluoromethyl)-benzamide), a small molecule inhibitor of the MRTF-activating pathway of SRF-dependent transcription (42). Treatment of PMCs with CCG-1423 abrogated LPA-induced CTGF expression, and suppressed basal levels of CTGF mRNA as well (Fig. 10D). Taken together, these results suggest that the MRTF-SRF circuit of transcriptional regulation is also part of the signaling pathway through which LPA induces CTGF expression in PMCs.
Figure 10.
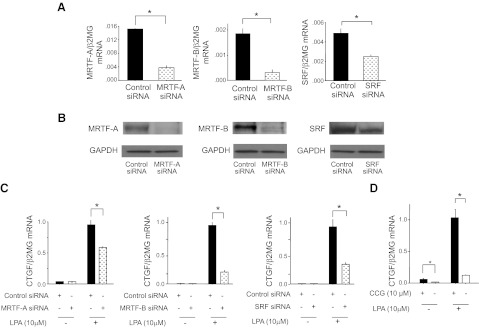
LPA-induced CTGF expression is dependent on MRTF-A, MRTF-B, and SRF signaling. A, B) Validation of siRNA inhibition of MRTF-A, MRTF-B, and SRF expression. PMCs were transfected with MRTF-A, MRTF-B, SRF, or control siRNA, and targeted transcription factor inhibition was determined at mRNA (A) and protein (B) levels. mRNA data are expressed as copies of MRTF-A, MRTF-B, or SRF mRNA relative to copies of β2 microglobulin mRNA. C) Effects of MRTF-A, MRTF-B, or SRF knockdown on LPA-induced CTGF expression by PMCs. PMCs were transfected with control siRNA or siRNA targeting MRTF-A, MRTF-B, or SRF, and then stimulated with control medium or LPA (10 μM). mRNA data are expressed as copies of CTGF mRNA relative to copies of β2 microglobulin mRNA. (n=3 cell preparations/group). D) Effect of MRTF-SRF inhibition on LPA-induced CTGF expression. PMCs were preincubated with CCG-1423 (CCG; 10 μM) or control medium for 16 h, and then stimulated with LPA (10 μM) or control media for an additional 2 h. Data are expressed as copies of CTGF mRNA relative to copies of β2 microglobulin mRNA (n=3 cell preparations/group). Data are expressed as means ± se. *P < 0.01.
Pharmacological inhibition of the MRTF-activating pathway of SRF-dependent transcription attenuates peritoneal fibrosis
To determine the relevance of the MRTF-SRF-activating pathway to peritoneal fibrosis in vivo, we evaluated CCG-1423 in the CG-induced peritoneal fibrosis model. CCG-1423 treatment significantly suppressed CG-induced increases in peritoneal collagen staining, thickness, and hydroxyproline content observed in vehicle-treated control mice (Fig. 11A–C). The increases in peritoneal hydroxyproline content observed in vehicle-treated CG-challenged mice were reduced by 52.6 and 57.9% in mice treated with CCG-1423 at 1.5 and 3.0 mg/kg dosages, respectively. CG-induced increases in peritoneal expression of CTGF and αSMA were also significantly reduced in CCG-1423-treated mice (Fig. 11D, E). We also demonstrated inhibition of peritoneal expression of other known SRF target genes, including zyxin and vinculin, in CCG-1423-treated mice, providing evidence that CCG-1423 inhibits SRF-induced transcription in these in vivo experiments (Fig. 11F). Taken together, these data implicate the MRTF-SRF-activating pathway in the pathogenesis of peritoneal fibrosis in vivo.
Figure 11.
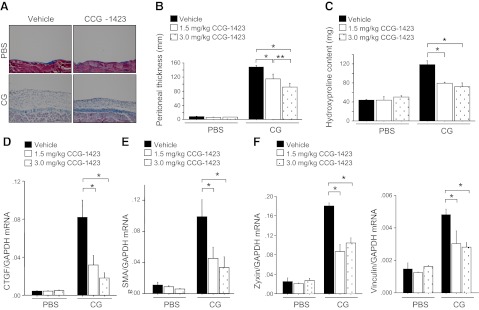
Pharmacological inhibition of the MRTF SRF-activating pathway attenuates peritoneal fibrosis. A) Representative Masson's trichrome-stained peritoneal sections of vehicle-treated (left panel) and CCG-1423-treated (3.0 mg/kg) mice (right panel) at d 21 of PBS or CG infections (×200). Scale bars = 100 μm. B) Peritoneal thickness following PBS or CG challenge (d 21 PBS, n=5 mice/treatment group; d 21 CG, n=5 mice/treatment group). Treatment groups are as indicated. C) Biochemical analysis of CG-induced peritoneal fibrosis. Peritoneal hydroxyproline content following CG or PBS challenges (d 21 PBS, n=5 mice/treatment group; d 21 CG, n=5 mice/treatment group). D–F) Peritoneal expression of CTGF, (D) αSMA (E), and zyxin and vinculin mRNA (F) following CG or PBS challenges (d 21 PBS, n=5 mice/treatment group; d 21 CG, n=5 mice/treatment group). Data are expressed as means ± se. *P < 0.01, **P < 0.05.
DISCUSSION
We found that LPA-LPA1 signaling is required for the development of peritoneal fibrosis. Genetic deletion or pharmacological antagonism of LPA1 protected mice from fibrosis produced by CG peritoneal injury. Disrupting the LPA-LPA1 pathway dramatically reduced peritoneal CTGF expression, fibroblast proliferation, and peritoneal myofibroblast accumulation. LPA engagement of PMC LPA1 induced CTGF expression, which accounted for the majority of the fibroblast proliferative activity produced by LPA-treated PMCs. LPA engagement of PMC LPA1 induced CTGF expression through Gα12/13 signaling, RhoA and ROCK activation, actin polymerization, MRTF-A and MRTF-B nuclear translocation, and SRF-induced transcription. Pharmacological inhibition of MRTF-SRF-induced transcription reduced CTGF expression and peritoneal fibrosis in the CG model. We believe these data place LPA-LPA1 signaling at the center of a profibrotic collaboration between mesothelial cells and fibroblasts in the development of peritoneal fibrosis (Fig. 12), in which LPA drives mesothelial cell CTGF expression, and this mesothelial CTGF in turn drives fibroblast proliferation.
Figure 12.

Proposed profibrotic collaboration between mesothelial cells and fibroblasts in peritoneal fibrosis. LPA-LPA1 signaling drives mesothelial cell CTGF expression through a Gα12/13-RhoA-ROCK-MRTF-SRF pathway, and this mesothelial CTGF, in turn, drives fibroblast proliferation.
We had previously found evidence that LPA1 expressed by fibroblasts contributes to the recruitment of these cells during the development of pulmonary fibrosis (4). In contrast, we found that fibroblast LPA1 expression is not required for the proliferation of these cells in response to lung injury. We found that most of the fibroblast migration induced by bronchoalveolar lavage (BAL) from mice in the bleomycin model of pulmonary fibrosis was lost when the responding fibroblasts were LPA1 deficient. In contrast, we found no significant differences between the proliferative responses of WT and LPA1-deficient fibroblasts to BAL from bleomycin-challenged mice (4). Our current data in the CG model of peritoneal fibrosis suggests a noncell autonomous requirement of LPA1 for fibroblast proliferation in vivo: LPA1 expression by PMCs is required for their expression of CTGF, which induces fibroblast proliferation in a paracrine fashion. This proposed mesothelial cell-fibroblast profibrotic crosstalk is consistent with accumulating evidence that paracrine interactions between multiple cell types are central to the development of fibrosis (1).
There have been prior indications that LPA may be an important regulator of CTGF expression. Similar to our findings in peritoneal fibrosis, CTGF expression has previously been demonstrated to be LPA dependent in a mouse model of pulmonary fibrosis induced by radiation (43), and similar to our findings with PMCs, LPA has been shown to induce CTGF expression in mouse skeletal muscle cells (44). The molecular pathways through which LPA induces CTGF may be cell type dependent, however. LPA-induced skeletal muscle CTGF expression required TGF-β receptor signaling and Smad 2/3 expression (44), whereas we found that treatment of PMCs with a pan-TGF-β neutralizing antibody had no effect on their CTGF expression induced by LPA. As we have described, this TGF-β-independent pathway through which LPA induces CTGF in PMCs sequentially involves LPA1 and Gα12/13 signaling, RhoA and ROCK activation, actin polymerization, MRTF-A and MRTF-B nuclear translocation, and SRF-induced transcription. Although the ability of LPA and RhoA to induce SRF-dependent transcriptional activation has also been reported (45), to our knowledge the molecular connections between LPA-induced RhoA-mediated cytoskeletal rearrangements and SRF activation have not been previously described. Similarly, the importance of LPA-induced SRF activation to CTGF-induced fibroblast proliferation and the development of pathological fibrosis have not previously been appreciated. Our demonstration that pharmacological inhibition of the MRTF-SRF transcriptional activating pathway attenuated peritoneal CTGF expression and fibrosis in the CG model implicates this pathway in peritoneal fibrogenesis. The therapeutic potential of targeting the MRTF-SRF activating pathway for fibrotic diseases more broadly is underscored by recent studies with MRTF-A-deficient mice. These mice were dramatically protected from cardiac fibrosis produced by either myocardial infarction or angiotensin II infusion (46). Of note, LPA signaling through LPA1 has recently been reported to induce nuclear translocation of Yes-associated protein (YAP), a major downstream transcription factor of the Hippo pathway, through a signaling pathway that also involves Gα12/13, RhoA, and the actin cytoskeleton (47). It will be of great interest to describe the relative roles of SRF- vs. YAP-induced transcription in mediating the multiple effects of LPA-LPA1 signaling.
Involvement of G proteins containing Gα12/13 subunits in LPA-induced CTGF expression was demonstrated by significant inhibition of both LPA-induced RhoA activation and CTGF expression by siRNA knockdown of Gα12 and/or Gα13. Neither the inhibition of RhoA activation nor CTGF expression by these siRNA knockdowns was complete, however, suggesting that other G proteins may be involved in the signaling pathway through which LPA induces CTGF expression. In addition to previously recognized RhoA activation by Gα12/13-containing G proteins, Gαq-containing G proteins have recently been demonstrated to activate RhoA through a phospholipase C-Ca2+ pathway (48–49), raising the possibility that such G proteins could also contribute to LPA-induced CTGF expression. Involvement of both MRTF-A and MRTF-B in LPA-induced CTGF expression was also demonstrated by significant inhibition of this CTGF expression by siRNA knockdown of either MRTF-A or MRTF-B. Of note, inhibition of this CTGF expression by MRTF-B knockdown was greater than the inhibition produced by MRTF-A knockdown, despite MRTF-A knockdown being more efficient than that of MRTF-B. These data suggest that LPA-induced CTGF expression may be more dependent on MRTF-B than MRTF-A.
In addition to the CTGF expression that is induced by LPA in PMCs being able to drive fibroblast proliferation, fibroblasts themselves have also been reported to produce CTGF in response to LPA (50), and this CTGF has been reported to drive fibroblast proliferation in an autocrine fashion (51). Fibroblast proliferation initially driven by CTGF produced by PMCs in response to LPA therefore could be amplified by LPA-induced fibroblast CTGF expression. In addition to its capacity to induce fibroblast proliferation, CTGF directs other important profibrotic fibroblast behaviors such as myofibroblast differentiation and collagen synthesis (51). We would expect LPA-induced CTGF to contribute to these behaviors as well, and decreased CTGF-induced myofibroblast differentiation may also have contributed to the dramatic reduction in peritoneal myofibroblast accumulation we observed in CG-challenged LPA1-KO mice.
Finally, our results add yet another tissue type to the growing list of tissues and organs in which fibrogenesis critically depends on the LPA-LPA1 pathway. Including this study, data now implicate the LPA-LPA1 pathway in the development of lung (4), skin (5), kidney (6, 52), liver (53), and peritoneal fibrosis, suggesting that this pathway is of fundamental importance in the pathogenesis of fibrotic diseases associated with tissue injury. The accumulating number of tissues for which fibrosis requires LPA-LPA1 signaling suggests that inhibiting this pathway may be broadly beneficial in pathological fibrosis. Additionally, by defining a mechanistic link between the profibrotic activities of LPA and CTGF, we connect 2 important mediators that are currently being evaluated independently as therapeutic targets in patients with fibrotic diseases.
Acknowledgments
The authors gratefully acknowledge support from U.S. National Institutes of Health (NIH) grants R01-HL095732 and R01-HL108975, and Scleroderma Research Foundation, Pulmonary Fibrosis Foundation, Coalition for Pulmonary Fibrosis, and Nirenberg Center for Advanced Lung Disease grants to A.M.T, the Japan Society for the Promotion of Science (JSPS) Excellent Young Researcher Overseas Visit Program to N.S., and NIH grants R01-MH051699 and R01-DA019674 to J.C.
The authors also thank B. A. Fontaine for his expert assistance, and Amira Pharmaceuticals (San Diego, CA, USA) for the kind gift of AM095. The authors declare no conflicts of interest.
Footnotes
- αSMA
- α smooth muscle actin
- BAL
- bronchoalveolar lavage
- CG
- chlorhexidine gluconate
- CM
- conditioned medium
- COLI
- type I collagen
- COLIα1
- α1 chain of type I procollagen
- CTGF
- connective tissue growth factor
- DMEM
- Dulbecco's modified Eagle's medium
- EGFP
- enhanced green fluorescent protein
- F-actin
- filamentous actin
- G-actin
- globular actin
- GFP
- green fluorescent protein
- GPCR
- G-protein-coupled receptor
- HPF
- high-power field
- KO
- knockout
- LPA
- lysophosphatidic acid
- MRTF
- myocardin-related transcription factor
- PBS
- phosphate-buffered saline
- PCNA
- proliferating cell nuclear antigen
- PMC
- peritoneal mesothelial cell
- ROCK
- Rho-associated coiled-coil-forming kinase
- SRF
- serum response factor
- WT
- wild-type
REFERENCES
- 1. Selman M., King T. E., Pardo A. (2001) Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 134, 136–151 [DOI] [PubMed] [Google Scholar]
- 2. Scotton C. J., Chambers R. C. (2007) Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest 132, 1311–1321 [DOI] [PubMed] [Google Scholar]
- 3. Choi J. W., Herr D. R., Noguchi K., Yung Y. C., Lee C. W., Mutoh T., Lin M. E., Teo S. T., Park K. E., Mosley A. N., Chun J. (2010) LPA receptors: subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 50, 157–186 [DOI] [PubMed] [Google Scholar]
- 4. Tager A. M., LaCamera P., Shea B. S., Campanella G. S., Selman M., Zhao Z., Polosukhin V., Wain J., Karimi-Shah B. A., Kim N. D., Hart W. K., Pardo A., Blackwell T. S., Xu Y., Chun J., Luster A. D. (2008) The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 14, 45–54 [DOI] [PubMed] [Google Scholar]
- 5. Castelino F. V., Seiders J., Bain G., Brooks S. F., King C. D., Swaney J. S., Lorrain D. S., Chun J., Luster A. D., Tager A. M. (2011) Amelioration of dermal fibrosis by genetic deletion or pharmacologic antagonism of lysophosphatidic acid receptor 1 in a mouse model of scleroderma. Arthritis Rheum. 63, 1405–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pradere J. P., Klein J., Gres S., Guigne C., Neau E., Valet P., Calise D., Chun J., Bascands J. L., Saulnier-Blache J. S., Schanstra J. P. (2007) LPA1 receptor activation promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 18, 3110–3118 [DOI] [PubMed] [Google Scholar]
- 7. Martin P. (1997) Wound healing: aiming for perfect skin regeneration. Science 276, 75–81 [DOI] [PubMed] [Google Scholar]
- 8. Hoyles R. K., Derrett-Smith E. C., Khan K., Shiwen X., Howat S. L., Wells A. U., Abraham D. J., Denton C. P. (2011) An essential role for resident fibroblasts in experimental lung fibrosis is defined by lineage-specific deletion of high-affinity type II transforming growth factor beta receptor. Am. J. Respir. Crit. Care Med. 183, 249–261 [DOI] [PubMed] [Google Scholar]
- 9. Yoshio Y., Miyazaki M., Abe K., Nishino T., Furusu A., Mizuta Y., Harada T., Ozono Y., Koji T., Kohno S. (2004) TNP-470, an angiogenesis inhibitor, suppresses the progression of peritoneal fibrosis in mouse experimental model. Kidney Int. 66, 1677–1685 [DOI] [PubMed] [Google Scholar]
- 10. Contos J. J., Ishii I., Fukushima N., Kingsbury M. A., Ye X., Kawamura S., Brown J. H., Chun J. (2002) Characterization of lpa(2) (Edg4) and lpa(1)/lpa(2) (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: signaling deficits without obvious phenotypic abnormality attributable to lpa(2). Mol. Cell. Biol. 22, 6921–6929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moolenaar W. H., van Meeteren L. A., Giepmans B. N. (2004) The ins and outs of lysophosphatidic acid signaling. Bioessays 26, 870–881 [DOI] [PubMed] [Google Scholar]
- 12. Miralles F., Posern G., Zaromytidou A. I., Treisman R. (2003) Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113, 329–342 [DOI] [PubMed] [Google Scholar]
- 13. Olson E. N., Nordheim A. (2010) Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 11, 353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miano J. M. (2003) Serum response factor: toggling between disparate programs of gene expression. J. Mol. Cell. Cardiol. 35, 577–593 [DOI] [PubMed] [Google Scholar]
- 15. Muehlich S., Cicha I., Garlichs C. D., Krueger B., Posern G., Goppelt-Struebe M. (2007) Actin-dependent regulation of connective tissue growth factor. Am. J. Physiol. Cell Physiol. 292, C1732–C1738 [DOI] [PubMed] [Google Scholar]
- 16. Leask A., Parapuram S. K., Shi-Wen X., Abraham D. J. (2009) Connective tissue growth factor (CTGF, CCN2) gene regulation: a potent clinical bio-marker of fibroproliferative disease? J. Cell. Commun. Signal. 3, 89–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leask A., Abraham D. J. (2003) The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem. Cell Biol. 81, 355–363 [DOI] [PubMed] [Google Scholar]
- 18. Liu S., Shi-wen X., Abraham D. J., Leask A. (2011) CCN2 is required for bleomycin-induced skin fibrosis in mice. Arthritis Rheum. 63, 239–246 [DOI] [PubMed] [Google Scholar]
- 19. Van der Wal J. B., Jeekel J. (2007) Biology of the peritoneum in normal homeostasis and after surgical trauma. Colorectal Dis. 9(Suppl. 2), 9–13 [DOI] [PubMed] [Google Scholar]
- 20. Kawaguchi Y., Kawanishi H., Mujais S., Topley N., Oreopoulos D. G. (2000) Encapsulating peritoneal sclerosis: definition, etiology, diagnosis, and treatment. International Society for Peritoneal Dialysis Ad Hoc Committee on Ultrafiltration Management in Peritoneal Dialysis. Perit. Dial. Int. 20(Suppl. 4), S43–55 [PubMed] [Google Scholar]
- 21. Grassmann A., Gioberge S., Moeller S., Brown G. (2005) ESRD patients in 2004: global overview of patient numbers, treatment modalities and associated trends. Nephrol. Dial. Transplant. 20, 2587–2593 [DOI] [PubMed] [Google Scholar]
- 22. Higuchi C., Nishimura H., Sanaka T. (2006) Biocompatibility of peritoneal dialysis fluid and influence of compositions on peritoneal fibrosis. Ther. Apher. Dial. 10, 372–379 [DOI] [PubMed] [Google Scholar]
- 23. Aroeira L. S., Aguilera A., Sanchez-Tomero J. A., Bajo M. A., del Peso G., Jimenez-Heffernan J. A., Selgas R., Lopez-Cabrera M. (2007) Epithelial to mesenchymal transition and peritoneal membrane failure in peritoneal dialysis patients: pathologic significance and potential therapeutic interventions. J. Am. Soc. Nephrol. 18, 2004–2013 [DOI] [PubMed] [Google Scholar]
- 24. Rigby R. J., Hawley C. M. (1998) Sclerosing peritonitis: the experience in Australia. Nephrol. Dial. Transplant. 13, 154–159 [DOI] [PubMed] [Google Scholar]
- 25. Bansal S., Sheth H., Siddiqui N., Bender F. H., Johnston J. R., Piraino B. (2010) Incidence of encapsulating peritoneal sclerosis at a single U.S. university center. Adv. Perit. Dial. 26, 75–81 [PubMed] [Google Scholar]
- 26. Contos J. J., Fukushima N., Weiner J. A., Kaushal D., Chun J. (2000) Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc. Natl. Acad. Sci. U. S. A. 97, 13384–13389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin S. L., Kisseleva T., Brenner D. A., Duffield J. S. (2008) Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 173, 1617–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tager A. M., Kradin R. L., LaCamera P., Bercury S. D., Campanella G. S., Leary C. P., Polosukhin V., Zhao L. H., Sakamoto H., Blackwell T. S., Luster A. D. (2004) Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am. J. Respir. Cell Mol. Biol. 31, 395–404 [DOI] [PubMed] [Google Scholar]
- 29. Vartiainen M. K., Guettler S., Larijani B., Treisman R. (2007) Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 316, 1749–1752 [DOI] [PubMed] [Google Scholar]
- 30. Hjelle J. T., Golinska B. T., Waters D. C., Steidley K. R., McCarroll D. R., Dobbie J. W. (1989) Isolation and propagation in vitro of peritoneal mesothelial cells. Perit. Dial. Int. 9, 341–347 [PubMed] [Google Scholar]
- 31. Stylianou E., Jenner L. A., Davies M., Coles G. A., Williams J. D. (1990) Isolation, culture and characterization of human peritoneal mesothelial cells. Kidney Int. 37, 1563–1570 [DOI] [PubMed] [Google Scholar]
- 32. Sawada T., Ishii Y., Tojimbara T., Nakajima I., Fuchinoue S., Teraoka S. (2002) The ACE inhibitor, quinapril, ameliorates peritoneal fibrosis in an encapsulating peritoneal sclerosis model in mice. Pharmacol. Res. 46, 505–510 [DOI] [PubMed] [Google Scholar]
- 33. Hinz B., Phan S. H., Thannickal V. J., Prunotto M., Desmouliere A., Varga J., De Wever O., Mareel M., Gabbiani G. (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol. 180, 1340–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grotendorst G. R. (1997) Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. 8, 171–179 [DOI] [PubMed] [Google Scholar]
- 35. Mizutani M., Ito Y., Mizuno M., Nishimura H., Suzuki Y., Hattori R., Matsukawa Y., Imai M., Oliver N., Goldschmeding R., Aten J., Krediet R. T., Yuzawa Y., Matsuo S. (2010) Connective tissue growth factor (CTGF/CCN2) is increased in peritoneal dialysis patients with high peritoneal solute transport rate. Am. J. Physiol. Renal Physiol. 298, F721–F733 [DOI] [PubMed] [Google Scholar]
- 36. Yung S., Li F. K., Chan T. M. (2006) Peritoneal mesothelial cell culture and biology. Perit. Dial. Int. 26, 162–173 [PubMed] [Google Scholar]
- 37. Igarashi A., Okochi H., Bradham D. M., Grotendorst G. R. (1993) Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol. Biol. Cell 4, 637–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu M. Y., Porte J., Knox A. J., Weinreb P. H., Maher T. M., Violette S. M., McAnulty R. J., Sheppard D., Jenkins G. (2009) Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q). Am. J. Pathol. 174, 1264–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tatler A. L., John A. E., Jolly L., Habgood A., Porte J., Brightling C., Knox A. J., Pang L., Sheppard D., Huang X., Jenkins G. (2011) Integrin alphavbeta5-mediated TGF-beta activation by airway smooth muscle cells in asthma. J. Immunol. 187, 6094–6107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lau L. F., Nathans D. (1987) Expression of a set of growth-related immediate early genes in BALB/c 3T3 cells: coordinate regulation with c-fos or c-myc. Proc. Natl. Acad. Sci. U. S. A. 84, 1182–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Annes J. P., Munger J. S., Rifkin D. B. (2003) Making sense of latent TGF-beta activation. J. Cell Sci. 116, 217–224 [DOI] [PubMed] [Google Scholar]
- 42. Evelyn C. R., Wade S. M., Wang Q., Wu M., Iniguez-Lluhi J. A., Merajver S. D., Neubig R. R. (2007) CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol. Cancer Ther. 6, 2249–2260 [DOI] [PubMed] [Google Scholar]
- 43. Gan L., Xue J. X., Li X., Liu D. S., Ge Y., Ni P. Y., Deng L., Lu Y., Jiang W. (2011) Blockade of lysophosphatidic acid receptors LPAR1/3 ameliorates lung fibrosis induced by irradiation. Biochem. Biophys. Res. Commun. 409, 7–13 [DOI] [PubMed] [Google Scholar]
- 44. Cabello-Verrugio C., Cordova G., Vial C., Zuniga L. M., Brandan E. (2011) Connective tissue growth factor induction by lysophosphatidic acid requires transactivation of transforming growth factor type beta receptors and the JNK pathway. Cell. Signal. 23, 449–457 [DOI] [PubMed] [Google Scholar]
- 45. Price M. A., Hill C., Treisman R. (1996) Integration of growth factor signals at the c-fos serum response element. Philos. Trans. R. Soc. Lond. B Biol. Sci. 351, 551–559 [DOI] [PubMed] [Google Scholar]
- 46. Small E. M., Thatcher J. E., Sutherland L. B., Kinoshita H., Gerard R. D., Richardson J. A., Dimaio J. M., Sadek H., Kuwahara K., Olson E. N. (2010) Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res. 107, 294–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yu F. X., Zhao B., Panupinthu N., Jewell J. L., Lian I., Wang L. H., Zhao J., Yuan H., Tumaneng K., Li H., Fu X. D., Mills G. B., Guan K. L. (2012) Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Deng X., Mercer P. F., Scotton C. J., Gilchrist A., Chambers R. C. (2008) Thrombin induces fibroblast CCL2/JE production and release via coupling of PAR1 to Galphaq and cooperation between ERK1/2 and Rho kinase signaling pathways. Mol. Biol. Cell 19, 2520–2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Singh I., Knezevic N., Ahmmed G. U., Kini V., Malik A. B., Mehta D. (2007) Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J. Biol. Chem. 282, 7833–7843 [DOI] [PubMed] [Google Scholar]
- 50. Heusinger-Ribeiro J., Eberlein M., Wahab N. A., Goppelt-Struebe M. (2001) Expression of connective tissue growth factor in human renal fibroblasts: regulatory roles of RhoA and cAMP. J. Am. Soc. Nephrol. 12, 1853–1861 [DOI] [PubMed] [Google Scholar]
- 51. Grotendorst G. R., Duncan M. R. (2005) Individual domains of connective tissue growth factor regulate fibroblast proliferation and myofibroblast differentiation. FASEB J. 19, 729–738 [DOI] [PubMed] [Google Scholar]
- 52. Swaney J. S., Chapman C., Correa L. D., Stebbins K. J., Broadhead A. R., Bain G., Santini A. M., Darlington J., King C. D., Baccei C. S., Lee C., Parr T. A., Roppe J. R., Seiders T. J., Ziff J., Prasit P., Hutchinson J. H., Evans J. F., Lorrain D. S. (2011) Pharmacokinetic and pharmacodynamic characterization of an oral lysophosphatidic acid type 1 receptor-selective antagonist. J. Pharmacol. Exp. Ther. 336, 693–700 [DOI] [PubMed] [Google Scholar]
- 53. Watanabe N., Ikeda H., Nakamura K., Ohkawa R., Kume Y., Tomiya T., Tejima K., Nishikawa T., Arai M., Yanase M., Aoki J., Arai H., Omata M., Fujiwara K., Yatomi Y. (2007) Plasma lysophosphatidic acid level and serum autotaxin activity are increased in liver injury in rats in relation to its severity. Life Sci. 81, 1009–1015 [DOI] [PubMed] [Google Scholar]