

Abstract
β-Site amyloid precursor protein convertase enzyme 1 (BACE1), a type I transmembrane aspartyl protease required to cleave amyloid precursor protein for releasing a toxic amyloid peptide, also cleaves type I and type III neuregulin-1 (Nrg-1). BACE1 deficiency in mice causes hypomyelination during development and impairs remyelination if injured. In BACE1-null mice, the abolished cleavage of neuregulin-1 by BACE1 is speculated to cause reduced myelin sheath thickness in both the central nervous system and peripheral nervous system because reduced cleavage of Nrg-1 correlates with reduced Akt phosphorylation, a downstream signaling molecule of the Nrg-1/ErbB pathway. Here we tested specifically whether increasing Akt activity alone in oligodendrocytes would be sufficient to reverse the hypomyelination phenotype in BACE1-null mice. BACE1-null mice were bred with transgenic mice expressing constitutively active Akt (Akt-DD; mutations with D308T and D473S) in oligodendrocytes. Relative to littermate BACE1-null controls, BACE1−/−/Akt-DD mice exhibited enhanced expression of myelin basic protein and promoter of proteolipid protein. The elevated expression of myelin proteins correlated with a thicker myelin sheath in optic nerves; comparison of quantified g ratios with statistic significance was used to confirm this reversion. However, it appeared that myelin sheath thickness in the sciatic nerves was not increased in BACE1−/−/Akt-DD mice, as the g ratio was not significantly different from the control. Hence, increased Akt activity in BACE1-null myelinating cells only compensates for the loss of BACE1 activity in the central nervous system, which is consistent with the observation that overexpression of Akt-DD in Schwann cells did not induce hypermyelination. Our results suggest that signaling activity other than Akt may also contribute to proper myelination in peripheral nerves.—Hu, X., Schlanger, R., He, W., Macklin, W. B., Yan, R. Reversing hypomyelination in BACE1-null mice with Akt-DD overexpression.
Keywords: BACE1, Alzheimer's β-secretase, neuregulin-1, ErbB4, dominant gain of function
β-Site amyloid precursor protein (APP) convertase enzyme 1 (BACE1) is identified as an enzyme for cleaving APP in generating the β-amyloid (Aβ) peptide (1–5); an excessive accumulation of Aβ likely causes the onset of Alzheimer's disease (6, 7). Mice with complete deficiency of BACE1 are void of Aβ production (8–10), demonstrating that BACE1 is the sole protease for processing APP at the β-secretase site. Hence, BACE1 inhibitors are expected to prevent or delay the progression of Alzheimer's disease.
On the other hand, genetic deletion of BACE1 causes hypomyelination of central nervous system (CNS) and peripheral nervous system (PNS) nerves during early development (11, 12). Sciatic nerve crush experiments also demonstrate that BACE1 is required for the optimal remyelination of peripheral nerves in adult mice (13). Initial biochemical studies suggest that the abrogated cleavage of neuregulin-1 (Nrg-1) by BACE1 contributes to hypomyelination in BACE1-null mice (11, 12). Expression of both Nrg-1 and BACE1 in neurons and the transportation of both along axons (14–16) support the above notion.
Nrg-1 is a neurotrophic factor important for mediating cell-cell signaling via interaction with cognate receptors of the ErbB protein family, and this binding to the ErbB receptor triggers a cascade of downstream signaling events, which regulate diverse functions, including cardiac and neural development (17–21). During development, binding of axonal Nrg-1 to ErbB2/ErbB3 on Schwann cells or ErbB4 on oligodendrocytes induces phosphorylation of its downstream signaling molecule Akt and regulates myelination (22, 23). Reduction of Nrg1/ErbB signaling activity reduces myelin sheath thickness (24–27).
To confirm that Nrg1 is a natural substrate of BACE1, we performed an enzymatic mapping experiment and showed that cleavage of Nrg-1 by BACE1 at the F-M site in an evolutionarily conserved ectodomain region, which is present in both type I and type III Nrg-1 isoforms (13). Reduced cleavage of Nrg-1 by BACE1 reduces binding of Nrg-1 to its cognate receptor, and the level of phosphorylated Akt is reduced in BACE1-null mouse brains and peripheral nerves. Further biochemical analysis revealed that Nrg-1 can also be cleaved by ADAM10 and ADAM17 at a site adjacent to the BACE1 cleavage site (28, 29), consistent with the reduced but not completely abolished cleavage of Nrg1. Interestingly, only inhibited cleavage of Nrg1 by BACE1, but not by ADAMs, reduces myelination (28, 29). Hence, BACE1 activity is clearly more important for proper myelin formation in mammals.
Recent studies show that increased Akt activity in myelinating cells enhances myelination and results in hypermyelination (30–35). To determine whether decreased Akt phosphorylation contributes to hypomyelination in BACE1-null mice, we took advantage of transgenic mice overexpressing Akt with mutations of D308T and D473S (HAAkt308D473D; Akt-DD) in oligodendrocytes and asked whether increased Akt activity would be sufficient to reverse hypomyelination seen in optic nerves in BACE1-null mice. In this transgenic mouse model, constitutively active Akt (Akt-DD) is expressed in oligodendrocytes driven by the promoter of proteolipid protein (PLP), and these transgenic Plp-Akt-DD mice exhibit enhanced CNS myelination (35). After crossing this transgenic mouse model with BACE1-null mice, we examined myelin sheath thickness in optic nerves and the expression of myelin proteins in hippocampi. We showed that enhancing Akt activity in BACE1-null oligodendrocytes is sufficient to normalize myelination. Noticeably, Akt activity was also elevated in Schwann cells, but this increased Akt activity appeared insufficient to reverse hypomyelination in BACE1-null mice. Together, our results suggest that BACE1 cleavage of Nrg1 indeed mediates myelination through Akt in the CNS, while Akt overexpression in the PNS neither induces hypermyelination (35) nor is sufficient to overcome the loss of BACE1 in the PNS.
MATERIALS AND METHODS
Animals
BACE1-null mice
Generation and genotyping of BACE1-null mice were described in detail in the original publication (8). The BACE1-null mice were backcrossed to C57BL/6 mice for >8 generations and maintained in this genetic background.
Plp-Akt-DD mice
Transgenic mice expressing Akt-DD driven by the Plp promoter were generated in the W.B.M. laboratory (35). The method for generating this mouse model and primers for the genotyping was described in detail in the original publication. These mice were initially generated in SJL/SWR background. To avoid mixed genetic background in our breeding, these transgenic mice were backcrossed to C57BL/6 mice for >6 generations before they were crossed with BACE1-null mice.
All experimental protocols were approved by the Institutional Animal Care and Use Committee at the Lerner Research Institute in compliance with the guidelines established by the U.S. Public Health Service Guide for the Care and Use of Laboratory Animals.
Western blotting and antibodies
Protein extraction was performed according to standard procedures. Briefly, hippocampal samples were homogenized in 0.3 ml of disrupting RIPA buffer [50 mM Tris-HCl at pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4 and a protease inhibitor cocktail (Roche, Indianapolis, IN, USA)] and centrifuged at 13,200 rpm for 90 min. Equal amounts of protein were resolved on a NuPAGE Bis-Tris Gel (Invitrogen, Carlsbad, CA, USA) and transferred onto nitrocellulose membranes (Invitrogen). Subsequently, blots were incubated with primary antibodies [PLP, 1:1000, was a gift from S. Pfeiffer, University of Connecticut, Storrs-Mansfield, CT, USA; myelin oligodendrocyte glycoprotein (MOG), 1:500, was a gift from M. Gardinier, University of Iowa, Iowa City, IA, USA; myelin basic protein (MBP), 1:1000, was from Millipore, Bedofrd, MA, USA; Akt1, 1:1000, was from Cell Signaling, Danvers, MA, USA; P0, 1:000, was from Aves Labs, Tigard, OR, USA; and actin, 1:50,000, was from Sigma, St. Louis, MO, USA] overnight at 4°C. After being washed extensively, the blots were reacted with horseradish peroxidase-conjugated secondary antibodies and visualized using enhanced chemiluminescence (Thermo Scientific, Waltham, MA, USA).
Immunofluorescent confocal microscopy
Animals were first subjected to transcardial perfusion with 4% paraformaldehyde for fixation. The mouse brain was then surgically removed and immersed in 20% sucrose overnight at 4°C. The fixed brain was sagittally cut into 16-μm-thick sections on a freezing microtome (Microm, Walldorf, Germany). Sections were permeabilized with 0.3% Triton X-100 for 30 min. After being rinsed in PBS 3 times to remove the detergent, the sections were treated by microwave in 0.05 M citrate-buffered saline (pH 6.0) for 5 min, blocked with 5% normal goat serum, and incubated with the primary antibody PLP (1:1000). After being washed with PBS 3 times, sections were incubated with the secondary antibody goat anti-rat IgG conjugated with Alexa fluor 568 (Molecular Probes).
Quantification of g ratio
The g ratio of axons from the optic nerve and the sciatic nerve was determined as described previously (11) and ImageJ software (NIH Image; U.S. National Institutes of Health, Bethesda, MD, USA) was used for digital tracing. The myelinated axon diameter and circumference were measured by digitally tracing the inner and outer layers of the myelinated fiber. The g ratio was calculated by dividing the inner circumference of the axon (without myelin) by the outer circumference of the total fiber (including myelin). The g-ratio data are displayed as a scatter plot against axon diameter.
Statistical analysis
Statistical analysis was performed using Microsoft Excel software (Microsoft, Redmond, WA, USA). All data were analyzed for statistical significance using an F test for equal variance, followed by a 2-tailed Student's t test. Values of P < 0.01 were considered significant. All data values in this article are expressed as means ± se.
RESULTS
In this study, we investigated whether increasing Akt activity alone in oligodendrocytes would be sufficient to rescue the hypomyelination phenotype in BACE1-null mice. We bred transgenic Plp-Akt-DD mice with BACE1-null mice, and offspring were examined. Characterization of transgenic Plp-Akt-DD mice has been previously described (35). To avoid the effect of different genetic backgrounds on optimal myelination, we first bred transgenic Plp-Akt-DD mice with wild-type C57BL/6 mice for 6 generations to ensure a relatively pure congenic background; BACE1-null mice were routinely maintained in the C57BL/6 background. Similar to a previous report (35), the expression of transgene Akt-DD (D308 replaced by T and D473 replaced by S) was readily detected by an antibody specific to Akt1 in brains of P10 offspring, and its level increased during the first 2 mo and reached a plateau between P60 and P120 (data not shown). Therefore, BACE1−/−/Akt-DD mice were analyzed at the age of 2 mo to quantify myelin proteins and myelin sheath thickness relative to control mice.
In our previous report (11), we showed that BACE1-null dentate gyrus exhibited weaker staining by MBP antibody compared with wild-type mice. We also found that BACE1-null dentate gyrus showed similar weakened staining compared with the wild-type control when analyzed for PLP expression (Fig. 1, top panels). Notably, matched brain sections from transgenic Plp-Akt-DD mice (BACE1+/+/Akt-DD) showed significantly enhanced PLP staining relative to normal wild-type mice (Fig. 1, left panels). When BACE1 was knocked out in these transgenic mice (BACE1−/−/Akt-DD), PLP staining was only slightly reduced and was still significantly stronger that in BACE1-null dentate gyrus.
Figure 1.
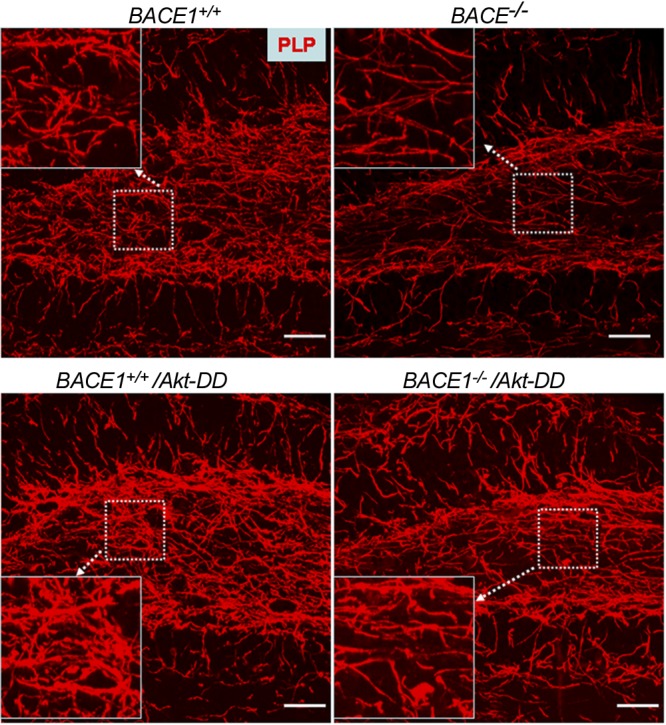
PLP expression was increased in BACE1-null mice with expression of Akt-DD. Fixed brain sections were stained with primary antibody specific to PLP and Alexa-568-labeled second antibody. Images were captured from similar areas in the dentate gyrus. Dashed box was further enlarged for the purpose of a clearer view. Scale bar = 30 μm.
To verify the immunofluorescent data, we performed a Western blot analysis to measure the levels of myelin proteins in the hippocampi of these mice. As expected, the protein levels of myelin proteins, such as PLP, MBP, and MOG, were all reduced in BACE1-null mice compared with wild-type controls (Fig. 2). Notably, this reduction was completely reversed when Akt-DD was expressed in these oligodendrocytes. Clearly, our Western blot results were in line with the immunofluorescence results and confirmed that elevated Akt activity appeared to be both dominant in the production of myelin proteins and capable of compensating for the loss of BACE1.
Figure 2.

Myelin protein levels were increased in BACE1-null mice with overexpression of Akt-DD transgene. Protein lysates were prepared from the indicated genotypes of mouse brains, and equal amounts of proteins were examined by Western blot analysis. Akt-DD transgene was detectable by antibody specific to Akt1. Protein level relative to the loading control actin was quantified and is presented as bar graphs (n=4 animals/group). *P < 0.01.
To further confirm the above Western blot and staining results, we examined myelin sheath thickness of optic nerves by electron microscopy. The myelin sheath was visibly thinner in BACE1-null optic nerves than that in wild-type optic nerves (Fig. 3). The calculated g ratios measured from these two genotypes of optic nerves were 0.693 ± 0.067 and 0.778 ± 0.064, respectively (n=200 axons in wild-type and n=200 in BACE−/− mice; P<0.001, Student's t test). Overexpressing gain of function Akt-DD mutant clearly enhanced myelin sheath thickness, consistent with a prior report (35). This increased Akt activity was sufficient to reverse the hypomyelination in BACE1-null mice (Fig. 3), and the g ratio in BACE1+/+/Akt-DD optic nerves was 0.599 ± 0.058 compared with 0.607 ± 0.056 in BACE1−/−/Akt-DD optic nerves (n=200 axons in BACE1−/−/Akt-DD vs. n=200 in BACE1+/+/Akt-DD optic nerves; P=0.23, Student's t test). It was noted that thick myelinated axons in optic nerves appeared to have distorted axon shape, and the reason for this distortion was not fully understood.
Figure 3.

Myelin sheath was thickened in BACE1-null mice if Akt-DD was overexpressed. Fixed optic nerves were examined by electron microscopy; a representative electrograph from each genotype of mice is shown. Visibly, mice expressing Akt-DD have a much thicker myelin sheath and hypomyelination in BACE1-null optic nerves was clearly reversed. Relative thickness of the myelin sheath can be compared based on the g-ratio calculation. A scatter plot of the g ratios of myelinated axons shows quantitative evidence of hypomyelination in the optic nerves of BACE1–null mice compared with wild-type controls. The g ratio in BACE1−/−/Akt-DD was similar to that in BACE1+/+/Akt-DD (n=3 animals, P=0.23). Scale bars = 1 μm.
Since the transgene was expressed under the control of PLP promoter, Akt-DD was not only expressed by oligodendrocytes but also by Schwann cells (35). Consistent with this prior report, we also found expression of transgene Akt-DD in sciatic nerves (Fig. 4). To determine whether such an increase of Akt activity in sciatic nerves would affect myelination, peripheral sciatic nerves were examined by electron microscopy. We noticed that the hypomyelination in BACE1-null sciatic nerves was essentially unaffected, as the g ratio in BACE1-null and BACE1−/−/Akt-DD sciatic nerves did not differ significantly (Fig. 5; 0.793±0.060 in BACE1−/− compared with 0.786±0.064 in BACE1−/−/Akt-DD sciatic nerves; n=200 axons in BACE1−/− vs. n=200 in BACE1−/−/Akt-DD sciatic nerves; P=0.22, Student's t test). Hence, increased Akt activity in Schwann cells appears to be insufficient to enhance myelination of sciatic nerves.
Figure 4.

Elevated Akt activity in sciatic nerves. Sciatic nerve segments were dissected out from the indicated genotypes of mice for preparation of protein lysates. Equal amounts of protein from each sample were loaded for Western blot analysis. Akt1 antibody was used to detect Akt-DD mutant, P0 antibody was used to detect peripheral myelin protein P0, and β-actin served as loading control.
Figure 5.

Hypomyelination in BACE1-null sciatic nerves was not affected by the overexpression of Akt-DD. Fixed sections of sciatic nerves from the indicated genotype of mice (2 mo old) were examined by electron microscopy. Hypomyelination is evident in the nerves of BACE1-null mice (top right image) compared with wild-type controls (top left image). Slightly thicker myelin sheaths were seen in BACE1−/−/TgAktDD (bottom right image) and BACE1+/+/TgAktDD (bottom left image) sciatic nerves. However, g ratios, calculated from the indicated genotype of sciatic nerves shown in the scatter plot (right panel), showed no significant reversion of hypomyelination (n=3 animals). Scale bars = 1 μm.
DISCUSSION
Nrg-1 is an important signaling molecule that exerts its signaling function via interaction with the ErbB receptor. In humans, mutation of Nrg-1 is genetically linked to the pathogenesis of schizophrenia (36) and this has been further validated in various animal models with altered Nrg1/ErbB functions (37). As demonstrated by biochemical studies, BACE1 as a type I transmembrane aspartyl protease that can cleave Nrg-1. Reduced Nrg-1 signaling activity has been shown in BACE1-null mice by both reduced myelination (11, 12) and schizophrenia-like behaviors (38). The reduced Nrg-1 signaling activity in BACE1-null mice correlates with reduced Akt signaling activity. Presumably, Nrg-1 on the axonal surface binds its cognate receptor ErbB2/3 in Schwann cells or ErbB4 in oligodendrocytes to provide instructive myelination signals. However, prior studies failed to establish whether the reduced Akt phosphorylation in BACE1-null mice is truly due to reduced Nrg-1 signaling in myelinating glial cells.
To address this question, we took advantage of transgenic mice that express the gain-of-function Akt-DD transgene in both oligodendrocytes and Schwann cells (35). Increased Akt activity alone has been shown to increase myelination in the CNS (30, 35). By introducing higher Akt activity in BACE1-null oligodendrocytes, we could answer whether reduced myelination on BACE1 deficiency can be reversed. In this study, we demonstrate that an increase in Akt activity in BACE1-null oligodendrocytes can clearly increase the expression of myelin proteins and myelination. It appears that a high level of active Akt plays a dominant role in the control of myelin sheath thickness, as the thickness of the myelin sheath in BACE1−/−/Akt-DD optic nerves was comparable to that of BACE1+/+/Akt-DD optic nerves.
Interestingly, transgenic mice expressing Akt-DD under the control of Plp promoter also produce a high level of Akt in Schwann cells (35) and BACE1-null mice have reduced Akt activity in sciatic nerves (13). Surprisingly, this increase of active Akt in Schwann cells failed to reverse the hypomyelination seen in BACE1-null sciatic nerves. A recent study suggests that the presence of abundant Dlg1 in Schwann cells could provide a stop signal for preventing hypermyelination due to abnormally active Akt (39, 40). Although it is not known whether the expression of Dlg1 is altered on BACE1 deficiency, our results indicate that an increase of active Akt in Schwann cells is not sufficient to compensate for the reduced Nrg-1 signaling activity. Our study suggests that signaling molecules other than Akt are also required for proper myelination in peripheral nerves (41–43).
The reduced myelin sheath thickness due to BACE1 deficiency has provided an important tool for monitoring signaling control of myelination in vivo. By utilizing mouse genetic approaches, we have demonstrated that Akt appears to play a differential role in the control of myelin sheath thickness. The knowledge gained from this study suggests that additional signaling molecules other than Akt are required for peripheral myelination. Further studies utilizing BACE1-null mice will be helpful for deciphering whether reduced Nrg1 signaling activity alone is sufficient to contribute to the impaired myelination.
Acknowledgments
The authors thank M. Yin (Electron Microscopy Facility, Cleveland Clinic Foundation, Cleveland, OH, USA) for assistance in electron microscopic analysis.
This study was supported by U.S. National Institutes of Health grant NS-074256 (to R.Y.) and National Multiple Sclerosis Society grant RG-4012A1/1 (to R.Y.).
The authors declare no conflicts of interests.
Footnotes
- Aβ
- β-amyloid
- Akt-DD
- Akt with mutations of D308T and D473S (HAAkt308D473D)
- APP
- amyloid precursor protein
- BACE1
- β-site amyloid precursor protein convertases enzyme 1
- Nrg1
- neuregulin-1
- CNS
- central nervous system
- MBP
- myelin basic protein
- MOG
- myelin oligodendrocyte glycoprotein
- PLP
- promoter of proteolipid protein
- PNS
- peripheral nervous system
REFERENCES
- 1. Vassar R., Bennett B. D., Babu-Khan S., Kahn S., Mendiaz E. A., Denis P., Teplow D. B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M. A., Biere A. L., Curran E., Burgess T., Louis J. C., Collins F., Treanor J., Rogers G., Citron M. (1999) Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 [DOI] [PubMed] [Google Scholar]
- 2. Yan R., Bienkowski M. J., Shuck M. E., Miao H., Tory M. C., Pauley A. M., Brashier J. R., Stratman N. C., Mathews W. R., Buhl A. E., Carter D. B., Tomasselli A. G., Parodi L. A., Heinrikson R. L., Gurney M. E. (1999) Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature 402, 533–537 [DOI] [PubMed] [Google Scholar]
- 3. Hussain I., Powell D., Howlett D. R., Tew D. G., Meek T. D., Chapman C., Gloger I. S., Murphy K. E., Southan C. D., Ryan D. M., Smith T. S., Simmons D. L., Walsh F. S., Dingwall C., Christie G. (1999) Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol. Cell. Neurosci. 14, 419–427 [DOI] [PubMed] [Google Scholar]
- 4. Sinha S., Anderson J. P., Barbour R., Basi G. S., Caccavello R., Davis D., Doan M., Dovey H. F., Frigon N., Hong J., Jacobson-Croak K., Jewett N., Keim P., Knops J., Lieberburg I., Power M., Tan H., Tatsuno G., Tung J., Schenk D., Seubert P., Suomensaari S. M., Wang S., Walker D., Zhao J., McConlogue L., John V. (1999) Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402, 537–540 [DOI] [PubMed] [Google Scholar]
- 5. Lin X., Koelsch G., Wu S., Downs D., Dashti A., Tang J. (2000) Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc. Natl. Acad. Sci. U. S. A. 97, 1456–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sisodia S. S., Price D. L. (1995) Role of the beta-amyloid protein in Alzheimer's disease. FASEB J. 9, 366–370 [DOI] [PubMed] [Google Scholar]
- 7. Haas C., Hung A. Y., Citron M., Teplow D. B., Selkoe D. J. (1995) beta-Amyloid, protein processing and Alzheimer's disease. Arzneimittelforschung 45, 398–402 [PubMed] [Google Scholar]
- 8. Cai H., Wang Y., McCarthy D., Wen H., Borchelt D. R., Price D. L., Wong P. C. (2001) BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 4, 233–234 [DOI] [PubMed] [Google Scholar]
- 9. Luo Y., Bolon B., Kahn S., Bennett B. D., Babu-Khan S., Denis P., Fan W., Kha H., Zhang J., Gong Y., Martin L., Louis J. C., Yan Q., Richards W. G., Citron M., Vassar R. (2001) Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 4, 231–232 [DOI] [PubMed] [Google Scholar]
- 10. Roberds S. L., Anderson J., Basi G., Bienkowski M. J., Branstetter D. G., Chen K. S., Freedman S. B., Frigon N. L., Games D., Hu K., Johnson-Wood K., Kappenman K. E., Kawabe T. T., Kola I., Kuehn R., Lee M., Liu W., Motter R., Nichols N. F., Power M., Robertson D. W., Schenk D., Schoor M., Shopp G. M., Shuck M. E., Sinha S., Svensson K. A., Tatsuno G., Tintrup H., Wijsman J., Wright S., McConlogue L. (2001) BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum. Mol. Genet. 10, 1317–1324 [DOI] [PubMed] [Google Scholar]
- 11. Hu X., Hicks C. W., He W., Wong P., Macklin W. B., Trapp B. D., Yan R. (2006) Bace1 modulates myelination in the central and peripheral nervous system. Nat. Neurosci. 9, 1520–1525 [DOI] [PubMed] [Google Scholar]
- 12. Willem M., Garratt A. N., Novak B., Citron M., Kaufmann S., Rittger A., DeStrooper B., Saftig P., Birchmeier C., Haass C. (2006) Control of peripheral nerve myelination by the beta-secretase BACE1. Science 314, 664–666 [DOI] [PubMed] [Google Scholar]
- 13. Hu X., He W., Diaconu C., Tang X., Kidd G. J., Macklin W. B., Trapp B. D., Yan R. (2008) Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J. 22, 2970–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park S. K., Solomon D., Vartanian T. (2001) Growth factor control of CNS myelination. Dev. Neurosci. 23, 327–337 [DOI] [PubMed] [Google Scholar]
- 15. Kamal A., Almenar-Queralt A., LeBlanc J. F., Roberts E. A., Goldstein L. S. (2001) Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 414, 643–648 [DOI] [PubMed] [Google Scholar]
- 16. Lazarov O., Morfini G. A., Lee E. B., Farah M. H., Szodorai A., DeBoer S. R., Koliatsos V. E., Kins S., Lee V. M., Wong P. C., Price D. L., Brady S. T., Sisodia S. S. (2005) Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J. Neurosci. 25, 2386–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burden S., Yarden Y. (1997) Neuregulins and their receptors: a versatile signaling module in organogenesis and oncogenesis. Neuron 18, 847–855 [DOI] [PubMed] [Google Scholar]
- 18. Buonanno A., Fischbach G. D. (2001) Neuregulin and ErbB receptor signaling pathways in the nervous system. Curr. Opin. Neurobiol. 11, 287–296 [DOI] [PubMed] [Google Scholar]
- 19. Mei L., Xiong W. C. (2008) Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 9, 437–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Falls D. L. (2003) Neuregulins: functions, forms, and signaling strategies. Exp. Cell Res. 284, 14–30 [DOI] [PubMed] [Google Scholar]
- 21. Altiok N., Altiok S., Changeux J. P. (1997) Heregulin-stimulated acetylcholine receptor gene expression in muscle: requirement for MAP kinase and evidence for a parallel inhibitory pathway independent of electrical activity. EMBO J. 16, 717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lemke G. (2006) Neuregulin-1 and myelination. Sci. STKE 2006, e11. [DOI] [PubMed] [Google Scholar]
- 23. Nave K. A., Salzer J. L. (2006) Axonal regulation of myelination by neuregulin 1. Curr. Opin. Neurobiol. 16, 492–500 [DOI] [PubMed] [Google Scholar]
- 24. Michailov G. V., Sereda M. W., Brinkmann B. G., Fischer T. M., Haug B., Birchmeier C., Role L., Lai C., Schwab M. H., Nave K. A. (2004) Axonal neuregulin-1 regulates myelin sheath thickness. Science 304, 700–703 [DOI] [PubMed] [Google Scholar]
- 25. Taveggia C., Zanazzi G., Petrylak A., Yano H., Rosenbluth J., Einheber S., Xu X., Esper R. M., Loeb J. A., Shrager P., Chao M. V., Falls D. L., Role L., Salzer J. L. (2005) Neuregulin-1 type III determines the ensheathment fate of axons. Neuron 47, 681–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tao Y., Dai P., Liu Y., Marchetto S., Xiong W. C., Borg J. P., Mei L. (2009) Erbin regulates NRG1 signaling and myelination. Proc. Natl. Acad. Sci. U. S. A. 106, 9477–9482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taveggia C., Thaker P., Petrylak A., Caporaso G. L., Toews A., Falls D. L., Einheber S., Salzer J. L. (2008) Type III neuregulin-1 promotes oligodendrocyte myelination. Glia 56, 284–293 [DOI] [PubMed] [Google Scholar]
- 28. La M. R., Cerri F., Horiuchi K., Bachi A., Feltri M. L., Wrabetz L., Blobel C. P., Quattrini A., Salzer J. L., Taveggia C. (2011) TACE (ADAM17) inhibits Schwann cell myelination. Nat. Neurosci. 14, 857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo X., Prior M., He W., Hu X., Tang X., Sheng W., Yadav S., Kiryu-Seo S., Miller R., Trapp B. D., Yan R. (2011) Cleavage of neuregulin-1 by BACE1 or ADAM10 produces differential effects on myelination. J. Biol. Chem. 286, 23967–23974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Narayanan S. P., Flores A. I., Wang F., Macklin W. B. (2009) Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J. Neurosci. 29, 6860–6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barros C. S., Nguyen T., Spencer K. S., Nishiyama A., Colognato H., Muller U. (2009) Beta1 integrins are required for normal CNS myelination and promote AKT-dependent myelin outgrowth. Development 136, 2717–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goebbels S., Oltrogge J. H., Wolfer S., Wieser G. L., Nientiedt T., Pieper A., Ruhwedel T., Groszer M., Sereda M. W., Nave K. A. (2012) Genetic disruption of Pten in a novel mouse model of tomaculous neuropathy. EMBO Mol. Med. 4, 486–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen S., Velardez M. O., Warot X., Yu Z. X., Miller S. J., Cros D., Corfas G. (2006) Neuregulin 1-erbB signaling is necessary for normal myelination and sensory function. J. Neurosci. 26, 3079–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brinkmann B. G., Agarwal A., Sereda M. W., Garratt A. N., Muller T., Wende H., Stassart R. M., Nawaz S., Humml C., Velanac V., Radyushkin K., Goebbels S., Fischer T. M., Franklin R. J., Lai C., Ehrenreich H., Birchmeier C., Schwab M. H., Nave K. A. (2008) Neuregulin-1/ErbB signaling serves distinct functions in myelination of the peripheral and central nervous system. Neuron 59, 581–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Flores A. I., Narayanan S. P., Morse E. N., Shick H. E., Yin X., Kidd G., Avila R. L., Kirschner D. A., Macklin W. B. (2008) Constitutively active Akt induces enhanced myelination in the CNS. J. Neurosci. 28, 7174–7183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stefansson H., Sigurdsson E., Steinthorsdottir V., Bjornsdottir S., Sigmundsson T., Ghosh S., Brynjolfsson J., Gunnarsdottir S., Ivarsson O., Chou T. T., Hjaltason O., Birgisdottir B., Jonsson H., Gudnadottir V. G., Gudmundsdottir E., Bjornsson A., Ingvarsson B., Ingason A., Sigfusson S., Hardardottir H., Harvey R. P., Lai D., Zhou M., Brunner D., Mutel V., Gonzalo A., Lemke G., Sainz J., Johannesson G., Andresson T., Gudbjartsson D., Manolescu A., Frigge M. L., Gurney M. E., Kong A., Gulcher J. R., Petursson H., Stefansson K. (2002) Neuregulin 1 and susceptibility to schizophrenia. Am. J. Hum. Genet. 71, 877–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Corfas G., Roy K., Buxbaum J. D. (2004) Neuregulin 1-erbB signaling and the molecular/cellular basis of schizophrenia. Nat. Neurosci. 7, 575–580 [DOI] [PubMed] [Google Scholar]
- 38. Savonenko A. V., Melnikova T., Laird F. M., Stewart K. A., Price D. L., Wong P. C. (2008) Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc. Natl. Acad. Sci. U. S. A. 105, 5585–5590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Macklin W. B. (2010) The myelin brake: when enough is enough. Sci. Signal. 3, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cotter L., Ozcelik M., Jacob C., Pereira J. A., Locher V., Baumann R., Relvas J. B., Suter U., Tricaud N. (2010) Dlg1-PTEN interaction regulates myelin thickness to prevent damaging peripheral nerve overmyelination. Science 328, 1415–1418 [DOI] [PubMed] [Google Scholar]
- 41. Newbern J., Birchmeier C. (2010) Nrg1/ErbB signaling networks in Schwann cell development and myelination. Semin. Cell Dev. Biol. 21, 922–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Syed N., Kim H. A. (2010) Soluble neuregulin and Schwann cell myelination: a therapeutic potential for improving remyelination of adult axons. Mol. Cell. Pharmacol. 2, 161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goebbels S., Oltrogge J. H., Kemper R., Heilmann I., Bormuth I., Wolfer S., Wichert S. P., Mobius W., Liu X., Lappe-Siefke C., Rossner M. J., Groszer M., Suter U., Frahm J., Boretius S., Nave K. A. (2010) Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J. Neurosci. 30, 8953–8964 [DOI] [PMC free article] [PubMed] [Google Scholar]