

Abstract
Exposure to cyanide causes a spectrum of cardiac, neurological, and metabolic dysfunctions that can be fatal. Improved cyanide antidotes are needed, but the ideal biological pathways to target are not known. To understand better the metabolic effects of cyanide and to discover novel cyanide antidotes, we developed a zebrafish model of cyanide exposure and scaled it for high-throughput chemical screening. In a screen of 3120 small molecules, we discovered 4 novel antidotes that block cyanide toxicity. The most potent antidote was riboflavin. Metabolomic profiling of cyanide-treated zebrafish revealed changes in bile acid and purine metabolism, most notably by an increase in inosine levels. Riboflavin normalizes many of the cyanide-induced neurological and metabolic perturbations in zebrafish. The metabolic effects of cyanide observed in zebrafish were conserved in a rabbit model of cyanide toxicity. Further, humans treated with nitroprusside, a drug that releases nitric oxide and cyanide ions, display increased circulating bile acids and inosine. In summary, riboflavin may be a novel treatment for cyanide toxicity and prophylactic measure during nitroprusside treatment, inosine may serve as a biomarker of cyanide exposure, and metabolites in the bile acid and purine metabolism pathways may shed light on the pathways critical to reversing cyanide toxicity.—Nath, A. K., Roberts, L. D., Liu, Y., Mahon, S. B., Kim, S., Ryu, J. H., Werdich, A., Januzzi, J. L., Boss, G. R., Rockwood, G. A., MacRae, C. A., Brenner, M., Gerszten, R. E., Peterson, R. T. Chemical and metabolomic screens identify novel biomarkers and antidotes for cyanide exposure.
Keywords: mitochondria, inosine, zebrafish, riboflavin
The actions of cyanide are ubiquitous, poisoning body tissues by inhibiting oxidative phosphorylation, with resultant shifts of cellular metabolism from aerobic to anaerobic. As such, cyanide is most toxic to organs with high metabolic requirements, such as the brain and heart (1). Cyanide exposure can occur as a consequence of smoke inhalation, consumption of cyanide-containing foods, infusion of certain therapeutic drugs, industrial accidents, or terrorism (2). Milligram quantities of cyanide causes convulsions, seizures, cardiovascular collapse, and death in minutes, while lower doses can cause a spectrum of debilitating, long-lasting pathologies, including a Parkinson-like syndrome due to irreversible neuronal death in select brain areas (3–5).
Current therapies for cyanide exposure include combinations of inhaled amyl nitrite, infused sodium nitrite, and infused sodium thiosulfate, or infused hydroxocobalmin (1). These therapies can be effective for individual victims who have not succumbed to exposure, as the mechanisms of action involve binding, sequestration, and removal of cyanide from the blood. However, the utility of these therapies in mass exposure events or during field operations is limited by the complexities of intravenous administration and the requirement for ongoing monitoring from trained medical personnel due to the risk of dangerous decreases in blood pressure and anaphylaxis. Thus, new classes of cyanide antidotes with novel mechanisms of action are needed.
Several other mechanisms have been proposed in the literature, each with varying degrees of experimental validation (6–19). Some of the potential countermeasure mechanisms still focus on detoxification or elimination of cyanide itself, while others seek to reverse the biological sequelae of cyanide exposure (20). As it is not known which biological pathways are critical for reversing the effects of cyanide most effectively, the unbiased whole organism approach to drug discovery applied here has the potential both to identify novel antidote candidates and illuminate new biological pathways.
Cyanide is a well-established mitochondrial respiration inhibitor; however, a thorough characterization of the metabolic pathways that are disrupted is lacking. To understand better the metabolic effects of cyanide and to discover novel cyanide antidotes, we developed a zebrafish model in which exposure to cyanide causes stereotypic toxicities, including bradycardia, neuronal necrosis, metabolic dysfunction, and death. As zebrafish have been a successful model for the high-throughput identification of chemical compounds that suppress genetic defects and disease states (21–24), we next developed an in vivo zebrafish-based chemical screen to discover novel cyanide countermeasures. The incorporation of metabolomics into our platform allows for the elucidation of diagnostic biomarkers to define specific toxicants and provides a means of determining the metabolic mechanisms of toxicity. Increasingly, metabolomics studies have provided robust metabolite candidates associated with distinct toxicological end-points, such as drug-induced phospholipidosis (25), hydrazine-induced neurotoxicity (26), and nephrotoxicity (27, 28). This study represents a novel integration of high throughput chemical and metabolomics screens in the investigation of the metabolic mechanisms underlying cyanide toxicity and the search for antidotes. Determining the metabolic changes that result from cyanide exposure and countermeasure treatment may identify novel indicators of mitochondrial dysfunction, metabolites for monitoring cyanide exposure, and biochemical pathways that play a role in the diverse physiological effects induced by sublethal cyanide exposure.
MATERIALS AND METHODS
Zebrafish husbandry
Ekkwill zebrafish were maintained and embryos were obtained according to standard fish husbandry protocols in accordance with U.S. national guidelines. Zebrafish embryos were grown at 28°C in HEPES-buffered Tübingen E3 in the dark.
Heart rate assay
At 3 days postfertilization (dpf), zebrafish were exposed to various doses of potassium cyanide (KCN). After 2 h, the plate was screened using an automated assay to measure bradycardia. Wells were scored according to the change in heart rate compared with negative and positive controls in each plate (at 2 h) and on the presence and severity of necrosis or death (at 4 h). Heart rate was measured from bright-field video recordings using a method that we have previously described (22). Briefly, images were acquired at a rate of 30 s−1 using a charge-coupled device camera (Hamamatsu ORCA-ER; Hamamatsu Photonics, Hamamatsu, Japan) attached to the microscope. Image stacks of 15-s recordings were then exported and analyzed offline using custom MatLab scripts (R2012a; The Mathworks, Natick, MA, USA). Pixel intensities were measured as a function of time from regions of interest that cover the heart of each fish. Then, a fast Fourier transformation of the raw pixel intensity was performed, which yields the heart rate as the dominant frequency of temporal variation.
Glucose measurement
Zebrafish larvae at 6 dpf were exposed to freshly prepared KCN for 8 h in a plate sealed with a foil lid in place of the plastic lid. These doses were previously determined to be sublethal. Glucose was measured using Amplex Red glucose assay kit (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions.
Startle response
Using high-speed video microscopy (500 frames/s), we recorded a stereotypic startle response to high-intensity blue light. To perform the assay, ten 4-dpf larvae per well were loaded into 96-well plates. A Zeiss Axio Observer A1 microscope (Carl Zeiss, Oberkochen, Germany) was used to deliver a pulse of blue light (450 nm) for 1 s. Videos were captured using Metamorph software (Molecular Devices, Sunnyvale, CA, USA), and the latency to respond to the stimulus by moving away (startle latency) was calculated using custom scripts.
Photomotor response (PMR) assay
An automated behavioral assay, PMR, was performed on 10 embryos/well [30–42 hours postfertilization (hpf)] as previously described (21). Briefly, a series of stereotypic movements in response to a light stimulus was captured on digital videos and analyzed using custom scripts in Metamorph (Molecular Devices). Ultimately, a motion index that correlates to the overall movement in each well over time was generated.
Chemical screen
Approximately 400 nl of small-molecule stock solution was pin transferred from 96-well format library plates to 96-well plates containing three 6-dpf larval zebrafish per well. The final small-molecule concentration in each well was ∼8 μg/ml. Subsequently, 50 μM of KCN was added, and the plate was sealed with a foil lid. Viability was assessed the following morning. We screened the Spectrum library (2000 compounds; Microsource Discovery Systems, Gaylordsville, CT, USA) and the Prestwick library (1120 compounds; Prestwick Chemical, Illkirch, France). For follow-up assays, cobinamide and nonoxidized cobinamide were provided by Gerry Boss (University of California–San Diego).
Zebrafish metabolite extraction
LC-MS-grade water (400 μl; Sigma-Aldrich, St. Louis, MO, USA) was added to 8-dpf zebrafish (n=15). The zebrafish were homogenized (1 min) using a tissue homogenizer (Power Gen 125; Fisher Scientific, Pittsburgh, PA, USA). Extracts were sonicated for 5 min and spun, and the supernatants were then snap-frozen.
Rabbit cyanide treatment
Rabbits were housed in accordance with U.S. national guidelines. All animals were weight and age matched. Rabbits were anesthetized with ketamine/xylazine, intubated, and ventilated with air; then central lines were placed. Rabbits were treated with a sublethal dose of cyanide (10 mg NaCN in 60 cc 0.9% NaCl given intravenously at 0.166 mg/min for 1 h). Treatment was then stopped, and the rabbits were allowed to recover for 1 h. Blood plasma was collected at baseline, at 5 min prior to the end of cyanide treatment, and after 1 h of recovery, and snap-frozen (29).
Nitroprusside-treated patients
From a cohort study of patients admitted to an intensive care unit for management of severe end-stage heart failure (30), 5 were treated with intravenous nitroprusside as a vasodilator. From these subjects, blood samples were collected in serum separator tubes at 2 time points (0 and 48 h). The first blood draw was the baseline prenitroprusside sample. The samples were centrifuged at 4000 rpm, portioned into aliquots, and frozen at −80°C until the first thaw for the present analysis. The study protocols were approved by the Institutional Review Boards of Massachusetts General Hospital.
Metabolic profiling
The metabolomics platform included the hydrophilic interaction liquid chromatography (HILIC) method for amines, amino acids, and amino acid conjugates, and LC-MS methods described in the literature (31) for intermediary metabolites (organic acids, bile acids, purines, and pyrimidines). Plasma samples were prepared for LC-MS analyses by biphasic protein precipitation and metabolite extraction. Methanol:chloroform (2:1; 300 μl) was added to 200 μl of plasma or fish homogenate. Chloroform:water (1:1; 400 μl) was then added. Samples were centrifuged (16,100 g, 20 min), and the organic and aqueous phases were separated. Prior to further analysis, the aqueous fraction was dried in an evacuated centrifuge. Samples were then reconstituted in methanol containing stable isotope-labeled internal standard for phenylalanine-d8 (Cambridge Isotope Laboratories, Andover, MA, USA), transferred, and injected directly.
Statistical analyses
All results are expressed as means ± se. A 2-tailed Student's t test, paired t test, or ANOVA was used to determine P values.
RESULTS
Cyanide exposure causes reproducible and representative effects in zebrafish
To establish zebrafish as a tractable model system for studying cyanide toxicity, we exposed zebrafish larvae to various doses of cyanide and measured both lethality and physiological endpoints. Survival curves indicate that the dose that induces 100% lethality is dependent on both the age of zebrafish and the length of cyanide exposure (Fig. 1A). A survival curve for overnight cyanide exposure in 6-dpf zebrafish demonstrates 100% lethality at 40 μM (Fig. 1A). In 3-dpf embryos exposed to cyanide for 4 h, 100% lethality is induced at 200 μM.
Figure 1.
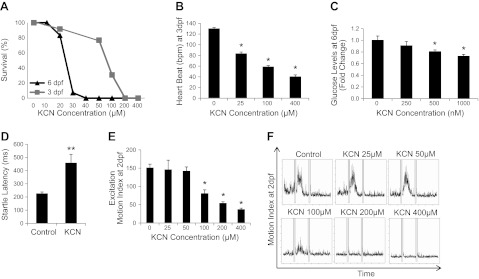
Cyanide exposure causes reproducible and representative physiological changes in zebrafish. A) Dose-response effect of cyanide exposure on survival of 3-dpf fish exposed to cyanide for 4 h (gray line) and 6-dpf zebrafish exposed to cyanide overnight (black line) (n=3). B) Dose-response effect of sublethal and lethal doses of cyanide on heart rate in 3-dpf embryos after a 1-h exposure (n=3). C) Dose-response effect of sublethal doses of cyanide on glucose levels in 6-dpf zebrafish after an 8-h exposure (n=5). D) Effect of cyanide on startle latency in 4-dpf zebrafish. E) Dose-response effect of sublethal and lethal doses of cyanide on the excitation phase of the photomotor response in 30-hpf embryos after a 1-h exposure (n=4). F) Graphs depict the series of phases of the photometer response: a period of basal movement (0–7 s), an intense light stimulus (double lines; 7 s), a brief latency period (7–8 s), an excitation phase in which the embryos move vigorously (9–13 s), and a refractory period in which a second light pulse does not elicit movement (18–32 s). Each graph displays the mean motion index of 10 wells/condition over time. All data are represented as means ± se. *P ≤ 0.01, **P < 0.05.
Heart rate was assessed in 3-dpf embryos exposed to cyanide for 2 h using an automated high-throughput imaging platform (22). Doses of 25–400 μM cyanide significantly decreased heart rate (Fig. 1B). The dose to achieve 50% reduction in heart rate was ∼100 μM. At the highest dose tested (400 μM), heart rate decreased from 130 ± 2 to 40 ± 3 beats/min (P<0.001). Changes in heart rate could be detected within 15 min of exposure and remained steady for a period of almost 2 h. After 4 h, all embryos in the 200 and 400 μM treatment groups were dead. These data demonstrate changes in cardiac physiology that are consistent with mammalian responses to cyanide (1, 6, 7).
Cyanide has well-characterized effects on the electron transport chain and glucose homeostasis. To evaluate whether zebrafish larvae have similar metabolic responses to cyanide exposure, 6-dpf zebrafish were treated with cyanide for 8 h. Glucose concentrations were measured in fish treated with a sublethal dose of cyanide to avoid confounding due to the generalized toxicity and cell death induced at lethal doses of cyanide. There was a dose-dependent decrease in glucose concentrations (Fig. 1C); treatment with 1 μM KCN resulted in a 27 ± 3% (P=0.009) decrease in glucose concentration. These results are consistent with increased glucose utilization, as flux is increased through the glycolytic pathway due to inhibition of the electron transport chain by cyanide.
In addition to these fundamental physiological and metabolic effects described above, it is possible to detect subtle perturbations of higher function in zebrafish using neurobehavioral assays. A well-characterized behavior in zebrafish is the rapid escape reflex (the startle response), which is controlled by a defined neural circuit in the brain stem and spinal cord (32, 33). Videos of a stereotypic startle response to high-intensity blue light (450 nm) were captured and analyzed. Cyanide exposure in 4-dpf larvae resulted in a significant prolongation of the startle latency. Control fish demonstrated a startle latency time of 227 ± 11 ms, while fish exposed for 30 min to cyanide exhibited a significantly longer startle latency time of 459 ±65 ms (n=10/condition, P=0.03; Fig. 1D). These data suggest that cyanide interferes with the function of the neural circuitry that underlies this reflex in zebrafish. As response to a visual stimulus requires multistep processing and integration (34), this assay represents a rapid method to assess the effects of cyanide on neurobehavior in the whole organism. Data from this assay demonstrate that, as in humans, cyanide is a neurotoxicant.
To evaluate further the neurological effects of cyanide in zebrafish, we utilized an automated behavioral assay, the photomotor response assay, which measures the complete series of stages of a specific neurobehavior (21). In these studies, a quantitative readout of a reproducible, stereotypic movement in response to photostimulation is acquired. Control embryos at 30 hpf (a stage when they are still within their chorions) display low basal movement but respond to an intense light stimulus with a brief latency period, an excitation phase in which the embryos move vigorously, and a refractory period in which a second light pulse does not elicit movement. To obtain the motion index, the average motor activity in 10 wells was plotted over time (Fig. 1E). The graphs in Fig. 1F depict a period of basal movement (t=0–7 s), an intense light stimulus (double lines; t=7 s), a brief latency period (t=8–9 s), an excitation phase in which the embryos move vigorously (t=9–13 s), and a refractory period in which a second light pulse (double lines) does not elicit movement (t=18–32 s). Embryos were exposed to cyanide for 2 h prior to the assay. In response to cyanide doses ≥ 100 μM, basal activity was decreased, the latency period before motion was increased, and the response to the light stimulus was suppressed (Fig. 1E, F). The motion index for the excitation phase significantly decreased at doses of 100–400 μM (control 151±9, 100 μM 81±9, 200 μM 54±3, and 400 μM 37±3, P<0.001; Fig. 1E). At the highest dose of cyanide, the zebrafish remained viable throughout the duration of the assay, as documented by a constant heartbeat. The photomotor response is an integrated readout of the nervous system, and the behaviors that underlie the PMR are affected by many neurotransmitter pathways (21, 34). These data demonstrate that cyanide affects complex neural behaviors in zebrafish. Collectively, these data on cardiac, neuronal, and metabolic physiology and on survival parallel known human responses to cyanide, suggesting that the zebrafish is a useful in vivo model of cyanide intoxication (3–5).
Zebrafish screening identifies novel cyanide antidotes
To identify antidotes of cyanide with novel mechanisms of action, we developed a high-throughput screen for cyanide toxicity. Three 6-dpf larvae were plated into each well of 96-well plates, small molecules were arrayed into wells, and a lethal dose of cyanide (50 μM) was added. The wells were assessed for viability the following morning. Cyanide consistently induced 100% lethality in control wells, which received only cyanide. In a screen of 3120 bioactive small molecules, we identified 4 hits that have been confirmed in ≥3 independent experiments across a range of doses. These include cisplatin, carboplatin, methotrexate, and riboflavin (Fig. 2A). Cisplatin, carboplatin, and methotrexate are all chemotherapeutic agents, while riboflavin is a vitamin. Two of the molecules, cisplatin and carboplatin, contain a heavy metal (platinum), suggesting that they may function by scavenging cyanide.
Figure 2.

HTS chemical screening in zebrafish identifies novel cyanide antidotes. A) Chemical structures of novel small-molecule antidotes identified in this screen. B) Survival curve of 6-dpf zebrafish treated with cyanide plus either novel antidotes identified in this screen or known antidotes. C) Effects of the combination of riboflavin plus a known cyanide antidote, hydroxocobalamin, on survival. D) Zebrafish embryos (30 hpf) were treated with a sublethal dose of cyanide and increasing doses of riboflavin for 1 h prior to the PMR assay. Each graph displays the mean motion index of 10 wells/condition over time. E) Excitation motion index for each condition. All data are represented as means ± se. *P ≤ 0.01.
Cisplatin and riboflavin were compared to known cyanide antidotes cobinamide and nonoxidized cobinamide (NOC) that act via scavenging and induced 100% survival at 20 μM (Fig. 2B). Cisplatin was less potent than the existing scavengers, but riboflavin was slightly more potent than cobinamide or NOC (Fig. 2B). Notably, unlike cisplatin, no overt toxicities were observed with riboflavin treatment in zebrafish.
Next, we tested whether riboflavin can be used in conjunction with hydroxocobalamin, a known FDA-approved cyanide scavenger (Fig. 2C). In this experiment, we treated cyanide-exposed zebrafish with increasing doses of riboflavin in the presence of fixed, subeffective doses of hydroxocobalamin. We found that combining subeffective doses of riboflavin and hydroxocobalamin could completely protect the animals from cyanide killing. For example, 6.4 μM hydroxocobalamin or 3.2 μM riboflavin alone provided no survival benefit in cyanide-treated animals, but combination of the two increased survival from 0 to 90% (Fig. 2C). These results suggest a positive interaction (additive or synergistic) between riboflavin and hydroxocobalamin. Collectively, the chemical screening data suggest that riboflavin is a potential antidote to cyanide and can be used in conjunction with known cyanide scavengers, and that antidotes of novel structural classes can be identified by in vivo chemical screening in zebrafish.
Riboflavin rescues cyanide-induced suppression of neurobehavioral activity
We next determined whether riboflavin could rescue the neurobehavioral defects induced by sublethal cyanide exposure using the PMR assay. Embryos (30 hpf) were exposed to a sublethal dose of cyanide (100 μM) for 2 h. This dose of cyanide resulted in a significant suppression in the excitation motion index (control 235±14 vs. cyanide 159±15, P<0.001; Fig. 2D, E). However, the addition of riboflavin at doses > 5 μM rescued the excitation response (10 μM 198±21, 20 μM 250±21, P=0.1 and P=0.5, respectively; Fig. 2D, E). Riboflavin (20 μM) alone does not affect the excitation response. These data demonstrate that riboflavin reverses subtle neurobehavioral defects induced by cyanide exposure in zebrafish.
Riboflavin rescues cyanide-induced metabolic perturbation
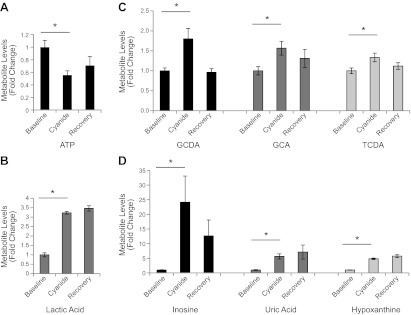
To profile the metabolic changes induced by cyanide, we used an LC-MS metabolomics platform (∼200 metabolites) to analyze the metabolic phenotype of zebrafish treated with a sublethal dose of cyanide. Cyanide treatment had a significant effect on carbohydrate and energy metabolism. As expected, cyanide decreased the concentration of ATP (−35.6±5%, P=0.02). Concomitantly, glycolytic activity was increased as indicated by a decrease in several carbohydrate species (fructose −14.6±2%, P=0.02; lactose −49.3±6%, P=0.03), and an increase in the concentration of lactic acid (+51±22%, P=0.04). These changes in carbohydrate and energy metabolism are consistent with the known effects of cyanide. Interestingly, cyanide treatment caused modest, but highly significant, increases in the concentration of several bile acids in the zebrafish [taurochenodeoxycholic acid (TCDA), +21±4%, P=0.00007; taurocholic acid (TCA), +15±2%, P=0.002]. Furthermore, cyanide perturbed purine metabolism, resulting in a significant increase in the concentration of inosine (+12±3%, P=0.004). These data indicate both expected changes in carbohydrate/energy metabolism and novel potential biomarkers in bile acids and purine metabolism.
Because riboflavin increased survival of zebrafish exposed to cyanide, we investigated whether riboflavin acted to correct the perturbed metabolic profile induced by cyanide. Using our LC-MS metabolomic platform we focused on carbohydrate, energy, bile acid, and purine metabolism, as these were identified as being altered by cyanide treatment in the original phenotyping study. Riboflavin rescued ATP concentrations following their depletion with cyanide treatment (ANOVA, P=0.01; Fig. 3). Treatment with riboflavin also normalized bile acid concentrations, reducing the increase in TCDA (ANOVA, P<0.0001) and TCA (ANOVA, P<0.0001) to control levels (Fig. 3). The effect of cyanide on purine metabolism was also corrected by riboflavin treatment. Inosine is increased with cyanide exposure; treatment with riboflavin restored inosine to control concentrations (Fig. 3). These data demonstrate that riboflavin treatment after cyanide exposure reestablishes the baseline metabolic profile.
Figure 3.

Riboflavin rescues the metabolic perturbations induced by cyanide in zebrafish. Metabolomic analysis of energy, bile acid, and purine metabolism in zebrafish exposed to cyanide, or cyanide plus riboflavin (control, n=7; cyanide, n=7; and riboflavin plus cyanide, n=7). Riboflavin restores control concentrations of ATP, TCDA, TCA, and inosine. All data are represented as means ± se. *P ≤ 0.01.
Cyanide-induced metabolic perturbations are conserved in a rabbit model
To determine whether the metabolic changes induced by sublethal cyanide exposure in zebrafish were conserved in a mammalian system, we used the LC-MS metabolomics platform to analyze the metabolic phenotype of rabbits treated with a sublethal dose of cyanide for 1 h, followed by a 1 h recovery. Cyanide treatment had a significant effect on carbohydrate and energy metabolism in the treated rabbits (Fig. 4A). As in the zebrafish, cyanide decreased the plasma concentration of ATP in the rabbits (−44±6%, P=0.02). After 60 min of recovery, the concentration of ATP in the plasma of cyanide-treated rabbits began to return to baseline concentrations. Cyanide's effects on glycolytic activity were also conserved in the rabbits with a detected increase in the concentration of lactic acid (+223±46%, P=0.002; Fig. 4B). Cyanide-induced perturbation of bile acid metabolism was also preserved across species. The concentrations of several bile acids were increased in the plasma of rabbits [glycochenodeoxycholic acid (GCDA), +80±25%, P=0.03; glycoholic acid (GCA), +56.6±17%, P=0.01; TCDA, +33.4±10%, P=0.02; Fig. 4C]. The concentrations of bile acids in the plasma of treated rabbits rapidly returned to baseline levels after 60 min of recovery. Notably, exposure of rabbits to cyanide also caused a perturbation of purine metabolism with a significant increase in the plasma concentrations of hypoxanthine (+382±3%, P=0.01), uric acid (+463±8%, P=0.002) and most prominently inosine (+2315±895%, P=0.03) (Fig. 4D). Inosine concentrations decreased following 60 min of recovery. These changes in carbohydrate, energy, bile acid, and oxypurine metabolism demonstrate conservation of the effects of cyanide across the zebrafish and rabbit model systems used in this study.
Figure 4.
Cyanide-induced metabolic perturbations are conserved in a rabbit model of cyanide toxicity. A, B) Metabolic profiling analysis of carbohydrate and energy metabolism in cyanide-treated rabbits (n=8). Changes in the circulating plasma concentration of ATP (A) and lactic acid (B) were measured using an LC-MS metabolomics platform in rabbits treated with a sublethal dose of cyanide for 1 h, followed by a 1-h recovery period. C) Metabolic profiling analysis of bile acids in cyanide-treated rabbits: GCDA, TCDA, and GCA. D) Metabolic profiling analysis of purine metabolites in cyanide-treated rabbits. All data are represented as means ± se. *P < 0.05.
Cyanide-induced metabolic perturbations are conserved in humans
To further assess the translational potential of the zebrafish cyanide model, we used metabolic profiling to analyze plasma from human subjects treated with the vasodilator nitroprusside. Nitroprusside is a commonly administered arterial vasodilator used in cases of hypertensive crisis and advanced heart failure (35). On administration, nitroprusside degrades, releasing cyanide ions, mandating that patients treated with nitroprusside are closely monitored for signs of cyanide toxicity, particularly in the context of renal dysfunction (36, 37). We obtained baseline and postnitroprusside treatment blood samples in a pilot cohort of 5 individuals given nitroprusside, with samples obtained a mean of 48 h after inception. Our analyses focused on the key metabolic pathways highlighted by the cyanide and riboflavin rescue experiments in zebrafish. As in the zebrafish and rabbit cyanide models, bile acids were elevated in nitroprusside-treated humans. GCDA was increased by the greatest magnitude within the bile acid class of metabolites in the plasma of cyanide-treated rabbits (+80%). The plasma concentration of the bile acid modestly but significantly increased ∼10% in nitroprusside-treated humans (paired t test, P=0.02; Fig. 5A). The perturbation in purine metabolism observed in both zebrafish and rabbits was also conserved in the nitroprusside-treated patients. Inosine was increased in zebrafish and produced the largest percentage change of any single metabolite in the plasma of the cyanide-treated rabbits. After 2 d of nitroprusside treatment in humans, inosine plasma concentrations doubled (paired t test, P=0.04; Fig. 5B). The increased inosine in both animal and human data sets is suggestive of a conserved effect of cyanide and raises the possibility that this metabolite may prove to be a useful marker for cyanide toxicity, including that associated with nitroprusside treatment.
Figure 5.
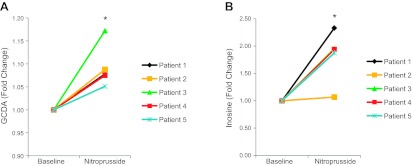
Metabolic perturbations in humans treated with nitroprusside. A) LC-MS metabolomic analysis of GCDA in the plasma of nitroprusside treated humans (n=5). B) Metabolic profiling of inosine in the plasma of nitroprusside-treated humans (n=5). Data are represented as metabolite concentrations prenitroprusside and postnitroprusside treatment for each patient. *P < 0.05; paired t test.
DISCUSSION
Our investigation of the effects of cyanide on zebrafish indicates that cyanide toxicity in the zebrafish parallels the cardiac, neurological, and metabolic effects observed in mammals. These data demonstrate that increasing doses of cyanide shift metabolism to anaerobic glycolysis, decrease heart rate, and suppress complex neural behaviors in zebrafish. Collectively, these data on cardiac, neuronal, and metabolic physiology, as well as on survival, parallel known human responses to cyanide.
Motivated by the potent toxicity of cyanide, the risk of exposure, and the difficulty of administering any of the current treatments to mass casualty exposure victims, we sought to identify effective countermeasure candidates by using unbiased whole-organism chemical screening. We established a first-in-field high-throughput assay and screened bioactive small molecules to identify those that reverse cyanide's diverse effects. From this screen, we identified four FDA-approved molecules that demonstrate potent protective effects against cyanide toxicity, the most efficacious being riboflavin. We showed that riboflavin improves survival of zebrafish exposed to doses of cyanide established to be in the lethal range, and using a metabolomics screen, we determined that it normalizes metabolic defects in animals treated with sublethal doses of cyanide. Many of the effects of cyanide on the metabolic profile in the zebrafish model were conserved in a mammalian system (rabbit) and in humans treated with the vasodilator nitroprusside.
Unlike the highly cytotoxic molecules cisplatin, carboplatin, and methotrexate, riboflavin does not appear to have significant toxic side effects in zebrafish or humans, where doses of up to 400 mg/d over a 3-mo period are well tolerated (38). Riboflavin is also orally available, unlike current therapies that are limited by the complexities of intravenous administration, the requirement for ongoing monitoring from trained medical personnel due to the risk of dangerous decreases in blood pressure, and the risk of anaphylaxis. Therefore, riboflavin has potential as a safe and inexpensive antidote in a number of relevant human exposure scenarios. Of course, testing of riboflavin in mammalian models will be necessary to determine whether its efficacy is conserved in mammals. Furthermore, additional work may be required to optimize formulation and delivery methods, an important consideration for countermeasure development and particularly relevant for riboflavin, which has exhibited limited water solubility in prior studies (39). Encouragingly, derivatives of riboflavin, such as riboflavin-5-phosphate, exhibit increased solubility.
Existing cyanide countermeasures function by sequestering cyanide, either by direct binding to a metal atom (e.g., hydroxocobalamin, cobinamide, methemoglobin) or by catalyzed reaction of cyanide with the sulfur-containing countermeasures (e.g., thiosulfate, 3-mercaptopyruvate). Riboflavin, lacking both a metal atom and sulfur, is not likely to function in either of these manners. Therefore, it is likely that riboflavin functions by a mechanism distinct from that of the currently available therapies. It is noteworthy that riboflavin was found to act positively with the FDA-approved cyanide antidote hydroxocobalamin, which functions by scavenging the cyanide anion and could, therefore, theoretically be used in conjunction with existing antidotes.
Riboflavin is essential in the production of the cofactors flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN). The cofactors FAD and FMN in flavoproteins impart substantial redox potential and are required for mitochondrial electron transport chain function, acting as electron acceptors from NADH and succinate in complexes I and II, respectively. In addition, FAD is required for the conversion of pyruvate to α-ketoglutarate, retinol to retinoic acid, tryptophan to niacin, and oxidized glutathione to reduced glutathione. Flavoproteins participate in these and a wide variety of biochemical reactions that underlie fat, carbohydrate, and protein metabolism, and therefore play a central role in energy metabolism. Although we cannot preclude the possibility of other mechanisms of action, the role of riboflavin in electron transport chain and intermediary metabolism may explain the normalization of the metabolic phenotype by riboflavin in cyanide-treated zebrafish.
Interestingly, examination of the metabolic phenotype of cyanide-treated zebrafish and rabbits revealed elevated bile acids. Bile acids, the end products of cholesterol metabolism, not only facilitate lipid digestion but also act as signaling molecules (40, 41). Through activation of the G-protein-coupled receptor TGR5, nuclear hormone receptors, such as farnesoid X, and cell signaling pathways (MAPK, AKT, and ERK), these metabolite-signaling molecules alter the expression of genes involved in energy metabolism (42). Thus, bile acid-controlled signaling pathways are promising drug targets to improve the metabolic phenotype induced by toxicants, obesity, or disease. Although, chronic cyanide exposure in rabbits is known to decrease total serum cholesterol, to our knowledge, alterations in bile acid species has not been reported (43).
Our study discovered increased GCDA, GCA, TCDA, and TCA. The majority of primary bile acids in humans are cholic and chenodeoxycholic acids, which are conjugated with one of two amino acids, glycine or taurine, in the liver. The ratio of glycine to taurine bile acids is species specific and altered in some disease states. Very low concentrations of bile acids trigger mitochondrial depolarization in pancreatic acinar cells and may be part of the mechanism of bile-induced pancreatitis (40). Further studies are required to determine whether cyanide induces bile acid-mediated toxicity or if bile acid-mediated signaling plays a role in the reversal of cyanide toxicity.
The findings of this study suggest that the metabolite inosine may serve as a candidate biomarker for cyanide toxicity. Inosine is elevated in cyanide-exposed zebrafish and rabbits and is also elevated in humans in response to treatment with nitroprusside, suggesting that it could be a useful biomarker for monitoring cyanide toxicity in patients. A potential model explaining why inosine is elevated by cyanide exposure is the following: cyanide decreases tissue and, consequently, plasma ATP concentrations, which activates AMP deaminase. AMP deamination produces IMP, which is readily dephosphorylated to inosine, and, in turn, can be catalyzed to the oxypurines hypoxanthine and uric acid (44, 45).
In addition to inosine's role as a candidate biomarker, increases in this metabolite may have important biological consequences in the nervous system. In a rat model of stroke and in patients with multiple sclerosis, inosine treatment is neuroprotective by promoting axonal rewiring and inhibiting axon degeneration (46, 47). In addition, inosine improved cell viability in glial cells exposed to glucose deprivation and mitochondrial respiratory chain inhibition (48). Notably, inosine is in phase II trials for Parkinson's disease and is hypothesized to improve symptoms via its metabolism to the antioxidant urate (49). One attractive hypothesis about the biological consequences of increased inosine is that it confers neural protection against the mitochondrial and metabolic dysfunction induced by cyanide.
The clinical features of nitroprusside poisoning are central nervous system and cardiovascular system dysfunction. Diagnosis is made on the basis of clinical presentation and nonspecific laboratory tests (serum lactate, basic blood chemistry, and arterial blood gas). Blood cyanide and thiocyanate concentrations may be obtained to confirm the diagnosis, but the assays are generally sent to specialized laboratories hampering clinical utility. Interestingly, in rabbits, the concentration of inosine increased prior to any indication of acidosis. Furthermore, during cyanide treatment of rabbits, the plasma inosine levels returned to baseline after 60 min of recovery, whereas lactate concentrations remained elevated. These findings may indicate greater sensitivity and responsiveness of inosine compared to lactate as a biomarker of cyanide toxicity. One limitation of the human studies presented is the small cohort size and clinical complexity of these patients. However, the conservation of the inosine elevation in zebrafish and rabbits adds additional support for the idea that inosine levels are associated with cyanide toxicity. Interestingly, inosine is also a candidate biomarker for other situations in which cellular hypoxia occurs, such as cardiac ischemia or stroke (50). Therefore, this marker may be useful in identifying people in need of intervention due to mitochondrial dysfunction and/or defects in oxidation phosphorylation.
Cellular hypoxia after cyanide exposure is a major pathway leading to dysfunction in the central nervous system and cardiovascular system. The long-term effects of hypoxia and other cyanide-induced sequelae are observed in survivors of cyanide exposure. The central nervous system is particularly sensitive to fluctuations in metabolism and oxygen. Sublethal cyanide exposure has been reported to cause cognitive deficits and neuroanatomical changes (3–5, 51). In humans, sublethal doses of cyanide are associated with Parkinson-like syndrome (52, 53). Simple, robust, and reliable assays are required to decipher and treat the pathological mechanisms that underlie these changes in the central nervous system. The zebrafish, as a vertebrate, shares many similarities with the mammalian nervous system. However, there are several relatively simple neural circuits that underlie stereotypic behaviors in zebrafish, which can be experimentally exploited in a way that is impossible in mammals. A strength of our physiology-based platform is the ability to identify subtle changes in the neurobehavior of zebrafish at sublethal doses. Our data reveal that riboflavin rescues cyanide effects on multiple forms of stimulus-induced excitation. Compiling and analyzing the effects of potential antidotes on various neurological behaviors and metabolic phenotypes, combined with chemical structure-function analysis, has the potential to yield a wealth of novel avenues for the study of sublethal cyanide exposure, which may be beneficial to survivors of cyanide poisoning.
In summary, this combined high-throughput chemical and metabolomics screen establishes the first large-scale, phenotype-based in vivo screen for cyanide countermeasures. We have demonstrated that this platform has the potential to discover diverse compounds with divergent mechanisms of chemical and metabolic action. Riboflavin shows promise as a candidate to provide safe, efficacious treatment for cyanide toxicity, while metabolites in the bile acid and purine metabolism pathways may shed light on the pathways critical to reversing cyanide toxicity. In addition, riboflavin may have promise as a prophylactic measure for cyanide poisoning during nitroprusside treatment. Finally, plasma inosine concentrations may have a role as a biomarker of cyanide exposure, allowing for the diagnosis and monitoring of at-risk patients. While further studies will be required to address these questions, we believe our findings highlight new approaches to the discovery of countermeasures that may have a significant effect on the future treatment and survival of humans at risk from cyanide exposure.
Acknowledgments
Funding for this project has been provided by U.S. National Institutes of Health (NIH) grant T32 HL-007208 (A.K.N. and Y.L.) and NIH Countermeasures Against Chemical Threats (Counter ACT) grants U54NS063718 and U54NS079201 (S.B.M. and M.B.) and U54NS079201 (R.T.P). The zebrafish research was supported, in part, by the CounterACT Program, NIH Office of the Director, and the National Institute of Allergy and Infectious Diseases, NIH/U.S. Army Medical Research and Materiel Command interagency agreement no. Y1-OD-9611-01/A120-B.P2009-1 (G.A.R.).
Footnotes
- dpf
- days postfertilization
- FAD
- flavin adenine dinucleotide
- FMN
- flavin mononucleotide
- GCA
- glycolic acid
- GCDA
- glycochenodeoxycholic acid
- hpf
- hours postfertilization
- KCN
- potassium cyanide
- PMR
- photomotor response
- TCA
- taurocholic acid
- TCDA
- taurochenodeoxycholic acid
REFERENCES
- 1. Gracia R., Shepherd G. (2004) Cyanide poisoning and its treatment. Pharmacotherapy 24, 1358–1365 [DOI] [PubMed] [Google Scholar]
- 2. Alcorta R. (2004) Smoke inhalation & acute cyanide poisoning. Hydrogen cyanide poisoning proves increasingly common in smoke-inhalation victims. JEMS 29(Suppl.), 6–15; quiz 16–17 [PubMed] [Google Scholar]
- 3. Borgohain R., Singh A. K., Radhakrishna H., Rao V. C., Mohandas S. (1995) Delayed onset generalised dystonia after cyanide poisoning. Clin. Neurol. Neurosurg. 97, 213–215 [DOI] [PubMed] [Google Scholar]
- 4. Grandas F., Artieda J., Obeso J. A. (1989) Clinical and CT scan findings in a case of cyanide intoxication. Mov. Disord. 4, 188–193 [DOI] [PubMed] [Google Scholar]
- 5. Rosenow F., Herholz K., Lanfermann H., Weuthen G., Ebner R., Kessler J., Ghaemi M., Heiss W. D. (1995) Neurological sequelae of cyanide intoxication—the patterns of clinical, magnetic resonance imaging, and positron emission tomography findings. Ann. Neurol. 38, 825–828 [DOI] [PubMed] [Google Scholar]
- 6. Baskin S. I., Porter D. W., Rockwood G. A., Romano J.A., Jr., Patel H. C., Kiser R. C., Cook C. M., Ternay A.L., Jr. (1999) In vitro and in vivo comparison of sulfur donors as antidotes to acute cyanide intoxication. J. Appl. Toxicol. 19, 173–183 [DOI] [PubMed] [Google Scholar]
- 7. Hall A. H., Kulig K. W., Rumack B. H. (1989) Suspected cyanide poisoning in smoke inhalation: complications of sodium nitrite therapy. J. Toxicol. Clin. Exp. 9, 3–9 [PubMed] [Google Scholar]
- 8. Sauer S. W., Keim M. E. (2001) Hydroxocobalamin: improved public health readiness for cyanide disasters. Ann. Emerg. Med. 37, 635–641 [DOI] [PubMed] [Google Scholar]
- 9. Hall A. H., Rumack B. H. (1987) Hydroxycobalamin/sodium thiosulfate as a cyanide antidote. J. Emerg. Med. 5, 115–121 [DOI] [PubMed] [Google Scholar]
- 10. Marrs T. C., Swanston D. W., Bright J. E. (1985) 4-Dimethylaminophenol and dicobalt edetate (Kelocyanor) in the treatment of experimental cyanide poisoning. Hum. Toxicol. 4, 591–600 [DOI] [PubMed] [Google Scholar]
- 11. Nagahara N., Li Q., Sawada N. (2003) Do antidotes for acute cyanide poisoning act on mercaptopyruvate sulfurtransferase to facilitate detoxification? Curr. Drug Targets Immune. Endocr. Metabol. Disord. 3, 198–204 [DOI] [PubMed] [Google Scholar]
- 12. Muller U., Krieglstein J. (1995) Inhibitors of lipid peroxidation protect cultured neurons against cyanide-induced injury. Brain Res. 678, 265–268 [DOI] [PubMed] [Google Scholar]
- 13. Pearce L. L., Bominaar E. L., Hill B. C., Peterson J. (2003) Reversal of cyanide inhibition of cytochrome c oxidase by the auxiliary substrate nitric oxide: an endogenous antidote to cyanide poisoning? J. Biol. Chem. 278, 52139–52145 [DOI] [PubMed] [Google Scholar]
- 14. Ardelt B. K., Borowitz J. L., Isom G. E. (1989) Brain lipid peroxidation and antioxidant protectant mechanisms following acute cyanide intoxication. Toxicology 56, 147–154 [DOI] [PubMed] [Google Scholar]
- 15. Lee Q. P., Park H. W., Thayer J., Mirkes P. E., Juchau M. R. (1996) Apoptosis induced in cultured rat embryos by intra-amniotically microinjected sodium nitroprusside. Teratology 53, 21–30 [DOI] [PubMed] [Google Scholar]
- 16. Yamada M., Momose K., Richelson E. (1996) Sodium nitroprusside-induced apoptotic cellular death via production of hydrogen peroxide in murine neuroblastoma N1E-115 cells. J. Pharmacol. Toxicol. Methods 35, 11–17 [DOI] [PubMed] [Google Scholar]
- 17. Shimizu S., Eguchi Y., Kamiike W., Waguri S., Uchiyama Y., Matsuda H., Tsujimoto Y. (1996) Retardation of chemical hypoxia-induced necrotic cell death by Bcl-2 and ICE inhibitors: possible involvement of common mediators in apoptotic and necrotic signal transductions. Oncogene 12, 2045–2050 [PubMed] [Google Scholar]
- 18. Mills E. M., Gunasekar P. G., Pavlakovic G., Isom G. E. (1996) Cyanide-induced apoptosis and oxidative stress in differentiated PC12 cells. J. Neurochem. 67, 1039–1046 [DOI] [PubMed] [Google Scholar]
- 19. Leavesley H. B., Li L., Mukhopadhyay S., Borowitz J. L., Isom G. E. (2010) Nitrite-mediated antagonism of cyanide inhibition of cytochrome c oxidase in dopamine neurons. Toxicol. Sci. 115, 569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Isom G. E., Borowitz J. L. (1995) Modification of cyanide toxicodynamics: mechanistic based antidote development. Toxicol. Lett. 82–83, 795–799 [DOI] [PubMed] [Google Scholar]
- 21. Kokel D., Bryan J., Laggner C., White R., Cheung C. Y., Mateus R., Healey D., Kim S., Werdich A. A., Haggarty S. J., Macrae C. A., Shoichet B., Peterson R. T. (2010) Rapid behavior-based identification of neuroactive small molecules in the zebrafish. Nat. Chem. Biol. 6, 231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peal D. S., Mills R. W., Lynch S. N., Mosley J. M., Lim E., Ellinor P. T., January C. T., Peterson R. T., Milan D. J. (2011) Novel chemical suppressors of long QT syndrome identified by an in vivo functional screen. Circulation 123, 23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peterson R. T., Fishman M. C. (2004) Discovery and use of small molecules for probing biological processes in zebrafish. Methods Cell Biol. 76, 569–591 [DOI] [PubMed] [Google Scholar]
- 24. Sachidanandan C., Yeh J. R., Peterson Q. P., Peterson R. T. (2008) Identification of a novel retinoid by small molecule screening with zebrafish embryos. PLoS One 3, e1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nicholls A. W., Nicholson J. K., Haselden J. K., Waterfield. C.J. (2000) A metabonomic approach to the investigation of drug-induced phospholipidosis: an NMR spectroscopy and pattern recognition study. Biomarkers 5, 410–423 [DOI] [PubMed] [Google Scholar]
- 26. Nicholls A. W., Holmes E., Lindon J. C., Shockor J. P., Farrant R. D., Hasleden J. N., Damment S. J., Waterfield C. J., Nicholson J. K. (2001) Metabonomic investigations into hydrazine toxicity in the rat. Chem. Res. Toxicol. 14, 975–987 [DOI] [PubMed] [Google Scholar]
- 27. Gartland K. P., Bonner F. W., Nicholson J. K. (1989) Investigations into the biochemical effects of region-specific nephrotoxins. Mol. Pharmacol. 35, 242–250 [PubMed] [Google Scholar]
- 28. Anthony M. L., Gartland K. P., Beddell C. R., Lindon J. C., Nicholson J. K. (1994) Studies of the biochemical toxicology of uranyl nitrate in the rat. Arch. Toxicol. 68, 43–53 [PubMed] [Google Scholar]
- 29. Brenner M., Kim J. G., Lee J., Mahon S. B., Lemor D., Ahdout R., Boss G. R., Blackledge W., Jann L., Nagasawa H. T., Patterson S. E. (2010) Sulfanegen sodium treatment in a rabbit model of sub-lethal cyanide toxicity. Toxicol. Appl. Pharmacol. 248, 269–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zilinski J. L., Shah R. V., Gaggin H. K., Gantzer M. L., Wang T. J., Januzzi J. L., Jr. (2012) Measurement of multiple biomarkers in advanced stage heart failure patients treated with pulmonary artery catheter-guided therapy. Crit. Care 16, R135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shaham O., Wei R., Wang T. J., Ricciardi C., Lewis G. D., Vasan R. S., Carr S. A., Thadhani R., Gerszten R. E., Mootha V. K. (2008) Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Mol. Syst. Biol. 4, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kimmel C. B., Patterson J., Kimmel R. O. (1974) The development and behavioral characteristics of the startle response in the zebra fish. Dev. Psychobiol. 7, 47–60 [DOI] [PubMed] [Google Scholar]
- 33. Kohashi T., Oda Y. (2008) Initiation of Mauthner- or non-Mauthner-mediated fast escape evoked by different modes of sensory input. J. Neurosci. 28, 10641–10653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Portugues R., Engert F. (2009) The neural basis of visual behaviors in the larval zebrafish. Curr. Opin. Neurobiol. 19, 644–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chobanian A. V., Bakris G. L., Black H. R., Cushman W. C., Green L. A., Izzo J. L., Jr., Jones D. W., Materson B. J., Oparil S., Wright J. T., Jr., Roccella E. J. (2003) The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: The JNC 7 Report. JAMA 289, 2560–2572 [DOI] [PubMed] [Google Scholar]
- 36. Robin E. D., McCauley R. (1992) Nitroprusside-related cyanide poisoning. Time (long past due) for urgent, effective interventions. Chest 102, 1842–1845 [DOI] [PubMed] [Google Scholar]
- 37. Vesey C. J., Cole P. V., Simpson P. J. (1976) Cyanide and thiocyanate concentrations following sodium nitroprusside infusion in man. Br. J. Anaesth. 48, 651–660 [DOI] [PubMed] [Google Scholar]
- 38. Boehnke C., Reuter U., Flach U., Schuh-Hofer S., Einhäupl K. M., Arnold G. (2004) High-dose riboflavin treatment is efficacious in migraine prophylaxis: an open study in a tertiary care centre. Eur. J. Neurol. 11, 475–477 [DOI] [PubMed] [Google Scholar]
- 39. Unna K., Greslin J. G. (1942) Studies on the toxicity and pharmacology of riboflavin. J. Pharmacol. Exp. Ther. 76, 75–80 [Google Scholar]
- 40. Voronina S. G., Barrow S. L., Gerasimenko O. V., Petersen O. H., Tepikin A. V. (2004) Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. J. Biol. Chem. 279, 27327–27338 [DOI] [PubMed] [Google Scholar]
- 41. Venglovecz V., Rakonczay Z., Hegyi P. (2012) The effects of bile acids on pancreatic ductal cells. Pancreapedia doi: 10.3998/panc.2012.8 [DOI] [PubMed] [Google Scholar]
- 42. Houten S. M., Watanabe M., Auwerx J. (2006) Endocrine functions of bile acids. EMBO J. 25, 1419–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Okolie N. P. (2002) Hypocholesterolemic and hypertriglycerolemic effects of chronic cyanide intoxication in rabbits. Global J. Pure Appl. Sci. 8, 491–496 [Google Scholar]
- 44. Woods H. F., Eggleston L. V., Krebs H. A. (1970) The cause of hepatic accumulation of fructose 1-phosphate on fructose loading. Biochem. J. 119, 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Katz A., Andersson D. C., Yu J., Norman B., Sandstrom M. E., Wieringa B., Westerblad H. (2003) Contraction-mediated glycogenolysis in mouse skeletal muscle lacking creatine kinase: the role of phosphorylase b activation. J. Physiol. 553, 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen P., Goldberg D. E., Kolb B., Lanser M., Benowitz L. I. (2002) Inosine induces axonal rewiring and improves behavioral outcome after stroke. Proc. Natl. Acad. Sci. 99, 9031–9036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Markowitz C. E., Spitsin S., Zimmerman V., Jacobs D., Udupa J. K., Hooper D. C., Koprowski H. (2009) The treatment of multiple sclerosis with inosine. J. Altern. Complement. Med. 15, 619–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jurkowitz M. S., Litsky M. L., Browning M. J., Hohl C. M. (1998) Adenosine, inosine, and guanosine protect glial cells during glucose deprivation and mitochondrial inhibition: correlation between protection and ATP preservation. J. Neurochem. 71, 535–548 [DOI] [PubMed] [Google Scholar]
- 49. Ascherio A., LeWitt P. A., Xu K., Eberly S., Watts A., Matson W. R., Marras C., Kieburtz K., Rudolph A., Bogdanov M. B., Schwid S. R., Tennis M., Tanner C. M., Beal M. F., Lang A. E., Oakes D., Fahn S., Shoulson I., Schwarzschild M. A. (2009) Urate as a predictor of the rate of clinical decline in Parkinson disease. Arch. Neurology. 66, 1460–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Farthing D., Xi L., Gehr L., Sica D., Larus T., Karnes H. T. (2006) High-performance liquid chromatography (HPLC) determination of inosine, a potential biomarker for initial cardiac ischaemia, using isolated mouse hearts. Biomarkers 11, 449–459 [DOI] [PubMed] [Google Scholar]
- 51. Di Filippo M., Tambasco N., Muzi G., Balucani C., Saggese E., Parnetti L., Calabresi P., Rossi A. (2008) Parkinsonism and cognitive impairment following chronic exposure to potassium cyanide. Mov. Disord. 23, 468–470 [DOI] [PubMed] [Google Scholar]
- 52. Rosenberg N. L., Myers J. A., Martin W. R. (1989) Cyanide-induced parkinsonism: clinical, MRI, and 6-fluorodopa PET studies. Neurology 39, 142–144 [DOI] [PubMed] [Google Scholar]
- 53. Top H., Sarikaya A., Aygit A. C., Benlier E., Kiyak M. (2006) Review of monitoring free muscle flap transfers in reconstructive surgery: role of 99mTc sestamibi scintigraphy. Nucl. Med. Commun. 27, 91–98 [DOI] [PubMed] [Google Scholar]