

Abstract
Histone deacetylase (HDAC) inhibitors have emerged as effective antineoplastic agents in the clinic. Studies from our lab and others have reported that magnetic resonance spectroscopy (MRS)-detectable phosphocholine (PC) is elevated following SAHA treatment, providing a potential noninvasive biomarker of response. Typically, elevated PC is associated with cancer while a decrease in PC accompanies response to antineoplastic treatment. The goal of this study was therefore to elucidate the underlying biochemical mechanism by which HDAC inhibition leads to elevated PC. We investigated the effect of SAHA on MCF-7 breast cancer cells using 13C MRS to monitor [1,2-13C] choline uptake and phosphorylation to PC. We found that PC synthesis was significantly higher in treated cells, representing 154±19% of control. This was within standard deviation of the increase in total PC levels detected by 31P MRS (129±7% of control). Furthermore, cellular choline kinase activity was elevated (177±31%), while cytidylyltransferase activity was unchanged. Expression of the intermediate-affinity choline transporter SLC44A1 and choline kinase α increased (144% and 161%, respectively) relative to control, as determined by mRNA microarray analysis with protein-level confirmation by Western blotting. Taken together, our findings indicate that the increase in PC levels following SAHA treatment results from its elevated synthesis. Additionally, the concentration of glycerophosphocholine (GPC) increased significantly with treatment to 210±45%. This is likely due to the upregulated expression of several phospholipase A2 (PLA2) isoforms, resulting in increased PLA2 activity (162±18%) in SAHA-treated cells. Importantly, the levels of total choline (tCho)-containing metabolites, comprised of choline, PC and GPC, are readily detectable clinically using 1H MRS. Our findings thus provide an important step in validating clinically translatable non-invasive imaging methods for follow-up diagnostics of HDAC inhibitor treatment.
Introduction
The histone deacetylase (HDAC) enzymes catalyze the removal of acetyl groups from the epsilon-amine of lysine residues of histone tails. The opposing actions of HDACs and histone acetyltransferases (HATs) dictate the acetylation status of histone lysine residues, regulating gene transcription through chromatin packaging [1], [2]. HDAC enzymes also modify at least 50 non-histone proteins of known biological function, including transcription factors, transcription regulators, signal transduction mediators, chaperone proteins and inflammation mediators [2], resulting in altered stability and activity of the target proteins. Interestingly, epigenetic modulation of genes involved in tumor initiation and progression has been linked to altered expression or mutation of genes encoding HAT, HDAC or their binding partners [3], [4]. Additionally, aberrant recruitment of HDAC enzymes has been associated with the development of certain human cancers. Consistent with these findings, HDAC inhibitors (HDACi), a recently developed class of anticancer drugs, have been shown to induce differentiation, growth arrest and apoptosis in treated cells and tumors, while normal cells are relatively resistant to HDACi effects [2], [5]. Vorinostat (suberoylanilide hydroxamic acid; SAHA) has been approved by the FDA for treatment of cutaneous T-cell lymphoma, while newly developed HDAC inhibitors are having success in clinical trials in various tumor types, including breast cancer, acute myeloid leukemia, and Hodgkin lymphoma [3], [6]–[8].
Clinical use of therapeutic drugs is significantly enhanced when a non-invasive means of longitudinally monitoring treatment efficacy is available. Traditional imaging methods may not be adequate for rapid monitoring of molecular drug action and response, since in many cases molecular drug action results in tumor stasis rather than shrinkage [9], [10]. Magnetic resonance spectroscopy (MRS) presents a non-invasive and non-destructive method that can provide biochemical information of normal and tumor tissues, presenting pharmacodynamic biomarkers of oncogenesis and response to anticancer therapies [11]–[14]. As emerging targeted therapies continue to be used more prevalently, the development of accompanying biomarkers is equally important. For this reason, identifying novel biomarkers of target inhibition that are detectable by non-invasive methods is essential for determining the efficacy of treatment and correlation with antitumor effects. Furthermore, the validation of a biomarker through the characterization of the underlying biochemical mechanism(s) provides context necessary for effective interpretation, and may suggest the strengths and limitations of the biomarker.
We have previously used MRS to develop and mechanistically validate biomarkers of response to emerging targeted therapies [15]–[21]. In particular, we have shown that treatment with the HDAC inhibitor SAHA is associated with increased PC levels [22]. Consistent with this finding, administration of LAQ-824 or Belinostat, two other HDAC inhibitory compounds, led to increased PC levels not only in cells but also in tumors in vivo [23], [24]. These observations are particularly interesting, as they run counter to conventional wisdom. Typically, an increase in the endogenous choline-containing metabolites is associated with cell transformation, while a decrease is observed in response to cytotoxic treatments [11]–[14], [25]–[27]. Importantly though, treatment with 17AAG, an inhibitor of the heat shock protein 90 (HSP90), also leads to increased PC levels [16], [28], and HSP90 is one of the proteins affected by HDAC inhibition [22]. We have recently shown that in the case of MCF7 cells, the increase in PC following 17AAG treatment is due to an increase in expression of the choline transporter SLC44A1 [29]. Accordingly, HSP90 inhibition and the resulting choline transport upregulation offers a possible mechanism by which PC levels increase with SAHA. The goal of this study was therefore to assess the mechanisms by which SAHA elevates PC levels in MCF7 breast cancer cells. To this end, 1H, 31P and 13C MRS of MCF7 cells labeled with [1,2-13C]choline was performed to characterize choline metabolism and its modulation following treatment with the HDAC inhibitor SAHA. In parallel, the expression and activity of enzymes involved in choline metabolism were also probed. We found that, similar to HSP90 inhibition, SAHA treatment led to an increase in the expression of the choline transporter SLC44A1, but that in addition the expression and activity of choline kinase α`s also increased in treated cells.
Materials and Methods
Cell Culture
MCF7 cells were obtained from ATCC. Unique DNA “fingerprint” identities (i.e., variable number tandem repeat PCR products) have been established for the cell line used in this study, and the identity of the cell line was confirmed in association with its use in the experiments described here.
Cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 units/mL penicillin and 100 µg/mL streptomycin. For MRS labeling studies, custom DMEM without choline (UCSF Cell Culture Facility) was supplemented with [1,2-13C]choline chloride at a final concentration of 28 µM (the concentration normally present in DMEM). For all experiments, cells were harvested in their logarithmic phase of proliferation.
For HDAC inhibition, cell cultures were incubated with 10 µM SAHA (courtesy of Drs. W. Bornmann and A. Pal, University of Texas M.D. Anderson Cancer Center for initial studies, and Cyman Chemical Company for subsequent studies; findings were within experimental error), replenishing drug and medium every 24 h. The final concentration of DMSO was 1∶1000 in culture medium.
Cell Proliferation
The effect of drug treatment on cell proliferation was determined using the WST-1 reagent assay (Roche). Cells were seeded in 96-well plates and treated for 4 to 48 h with 0, 5, 10, and 20 µM SAHA. After treatment, WST-1 reagent was incubated in wells for 1 h and cell viability was determined by quantification of absorbance at 440 nm using a spectrophotometer (Tecan).
HDAC Inhibition
The effect of SAHA on HDAC activity was determined using the Fluor de Lys fluorometric assay (Biomol), following the manufacturer’s instructions. Cells were seeded in 96-well plates and incubated with SAHA for 48 h. The Fluor de Lys substrate was added for 1.5 h, medium was transferred to adjacent wells, and cells were rinsed with PBS. Fluor de Lys developer was added to both the wells containing cells and medium. After 20 min, fluorescence was measured at 460 nm using a spectrofluorometer (Tecan).
Cell Cycle and Cell Size Analysis
Control and treated cells were harvested by trypsinization, washed in PBS and fixed in 95% ethanol. After fixation cells were washed and resuspended in PBS supplemented with 10 µg/ml RNase A (Sigma) and 20 µg/mL propidium iodide (Biotium). After 30 minutes of staining, cells were analyzed using a fluorescence-activated cell sorting (FACS) Calibur flow cytometer and CellQuest Pro software (BD Biosciences). Single cells were gated away from clumped cells by using forward light scattering on an FL2-width versus FL2-area dot plot. Percentages of cells in the G1, S, and G2/M phases were determined by plotting a histogram of FL2-A. Cell size distribution was determined by Coulter counter. Approximately 5×106 cells per sample were diluted into 10 mL of Isoton solution in a clean cuvette. Coulter counter readings provided cell number and cell size.
MRS Acquisition and Analysis
For initial MRS studies, MCF7 cells were treated for 48 h with 0, 5, 10 and 20 µM SAHA (n = 2). For mechanistic studies, MCF7 cells were treated for 48 h with 10 µM SAHA. Culture medium was replaced with [1,2-13C]choline-labeled medium 6 h prior to extraction. Cells (∼3×107) were extracted using the dual-phase extraction method, as previously described [22], [30]. Briefly, cells were harvested and washed with saline to remove residual medium. Cells were fixed in 10 mL ice-cold methanol, 10 mL ice-cold chloroform was added, followed by 10 mL ice-cold deionized water. Following phase separation and solvent removal by lyophilization, the aqueous fraction was reconstituted in 400 µL deuterium oxide (D2O) for 1H and 13C measurements. Before performing 31P measurements, EDTA was added to a final concentration of 10 µM. MR studies were performed on a 600-MHz Varian spectrometer at 25°C. 13C and 31P spectra were obtained using a 30° acquisition pulse with proton decoupling and a 3-s relaxation delay. 1H spectra were acquired with a 90° pulse and 3-second relaxation with suppression of the water signal. Metabolite concentrations were determined by integration, normalized to the area of the external reference (TMS, 1H and 13C; MDPA, 31P) and cell number, and corrected for saturation effects.
Choline Kinase Activity
Experiments were performed to assay the total cellular activity of choline kinase as previously described [31], [32]. In brief, cell pellets (2×107) were resuspended in 540 µL lysis buffer containing 100 mM Tris-HCl (pH 8.0), 10 mM DTT and 1 mM EDTA in D2O. Lysate was homogenized on ice by repeated passage through fine-tipped needle (27G 1/2) and further disrupted by sonication 10×1s at 20 kHz. The homogenate was centrifuged at 16,000 rpm for 30 minutes at 4°C. The choline kinase activity of the supernatant fraction was measured immediately after addition of 60 µL reaction mixture (final concentrations: 50 mM Tris-HCl (pH 8.0), 5 mM choline chloride, 10 mM ATP and 10 mM MgCl2). MR experiments were performed at 25°C on a 600-MHz Varian spectrometer equipped with a 5-mm broadband observe probe. 1H MR spectra were obtained over the course of 1 hour using 90° acquisition pulses and 3-second relaxation delay with water presaturation. Choline and PC concentrations were simultaneously measured from peak areas. Choline kinase activity was determined by regression analysis of the linear portion of the time course of PC formation.
Cytidylyltransferase Activity
Experiments were performed to measure the activity of phosphatidylcholine-specific CTP:phosphocholine cytidylyltransferase, as previously described [29]. In brief, cell pellets (2×107) were resuspended in 540 µL lysis buffer containing 50 mM HEPES (pH 7.0), 5 mM EDTA, 5 mM EGTA, 1 µL/mL protease inhibitor cocktail (Calbiochem) and 5.5 mM sodium bisulfite. Lysate was repeatedly passed through a fine-tipped needle (27G 1/2) for homogenization and was sonicated 10×1s at 20 kHz for membrane solubilization. The homogenate was centrifuged at 16,000 rpm for 30 minutes at 4°C. The cytidylyltransferase activity of the supernatant fraction was measured immediately after addition of 60 µL reaction mixture (final concentrations: 50 mM Tris-HCl (pH 8.0), 10 mM cytidine triphosphate (CTP), 5 mM PC, 5 mM DTT and 25 mM MgCl2). MR experiments were performed at 33°C on a 600-MHz Varian spectrometer equipped with a 5-mm broadband observe probe. 31P MR spectra were obtained using 30° acquisition pulse with proton decoupling, 2.6-s relaxation delay and 128 transients per FID. PC and CDP-choline concentrations were simultaneously measured from peak areas normalized to the area of the external reference (MDPA) and cell number, and corrected for saturation effects. Cytidylyltransferase activity was determined by regression analysis of the linear portion of the time course of CDP-choline formation.
Phospholipase A Activity
The activities of phospholipase A1 and A2 were determined using the EnzChek phospholipase A assay kits (Invitrogen) according to manufacturer’s instructions. The fluorescent cleavage product was measured (495 nm excitation, 515 nm emission) by an Infinite M200 fluorescence spectrophotometer (Tecan).
Immunoblotting
Control and treated cells were lysed using Cell Lysis Buffer (Cell Signaling). Lysates, were run on 4%–20% Tris-HCl Gels (Bio-Rad) by SDS-PAGE and electrotransferred onto PVDF membranes (Millipore). Blots were blocked and incubated with primary antibodies, anti-PLA2G4C (Abcam), anti-PLA2G10 (Abnova), anti-ChoK α (Sigma), anti-SLC44A1 (Abcam) or anti-β-Actin (Cell Signaling), then incubated with secondary antibody anti-IgG HRP-linked antibody (Cell Signaling). Immunocomplexes were visualized using ECL Western Blotting Substrate (Pierce). Quantification was performed using ImageJ (NIH) by measuring band intensities in scanned blots, and normalizing to loading control. Results are expressed as percentage of controls.
Statistical Analysis of Immunoblotting, Extract and Activity Assay Results
All results are expressed as mean ± SD and represent an average of three repeats unless otherwise stated. Two-tailed unpaired Student’s t test was used to verify statistical significance of the results, with P≤0.05 considered to be significant.
Gene Expression Analysis
Total cellular RNA was isolated from approximately 1×107 cells after 48 h of treatment with SAHA or vehicle (DMSO), using the RNeasy Mini Kit (Qiagen) according to manufacturer’s instructions. RNA quality was determined by Bioanalyzer (Agilent), considering RNA integrity number (RIN) values of ≥8.0 acceptable (most values were 9.5 or higher). Microarray hybridization was performed at the UCSF Genomics Core Laboratories using the Human Gene 1.0 ST assay (Affymetrix). Arrays were analyzed by fluorescence detection using the Agilent GeneArray Scanner (Agilent). Data acquisition was performed using the MicroArray Suite 5.0 software (Affymetrix). Microarray experiments were performed with 4 repeats of each condition. Microarray data is available through the ArrayExpress public repository [EMBL:E-MTAB-339] in compliance with standards of the Microarray Data Gene Expression Society.
Results
SAHA Inhibits Cell Proliferation and Elevates Levels of PC and GPC
Figure 1A illustrates the effects of 5, 10 and 20 µM SAHA on MCF7 cells, and demonstrates that all treatment doses resulted in significant inhibition of cell proliferation by 48 h of treatment. Next, we carried out a small scale MRS study to verify previous observations by our lab, which had found increased PC and glycerophosphocholine (GPC) levels in response to SAHA treatment [22]. Our findings are illustrated in Figure 1B, with typical MR spectra displayed in Figure 2. In line with previous observations, cellular PC and GPC levels increased following 48 h of treatment at all SAHA concentrations investigated.
Figure 1. Efficacy of SAHA treatment on cellular proliferation and PC levels.

(A) WST-1 assay showing anti-proliferative effects of 0, 5, 10 and 20 µM over a 48-hour treatment period; (B) Cellular PC and GPC levels following treatment with 0, 5, 10 and 20 µM SAHA at 48-hour time point.
Figure 2. Detection of endogenous metabolites by magnetic resonance spectroscopy.
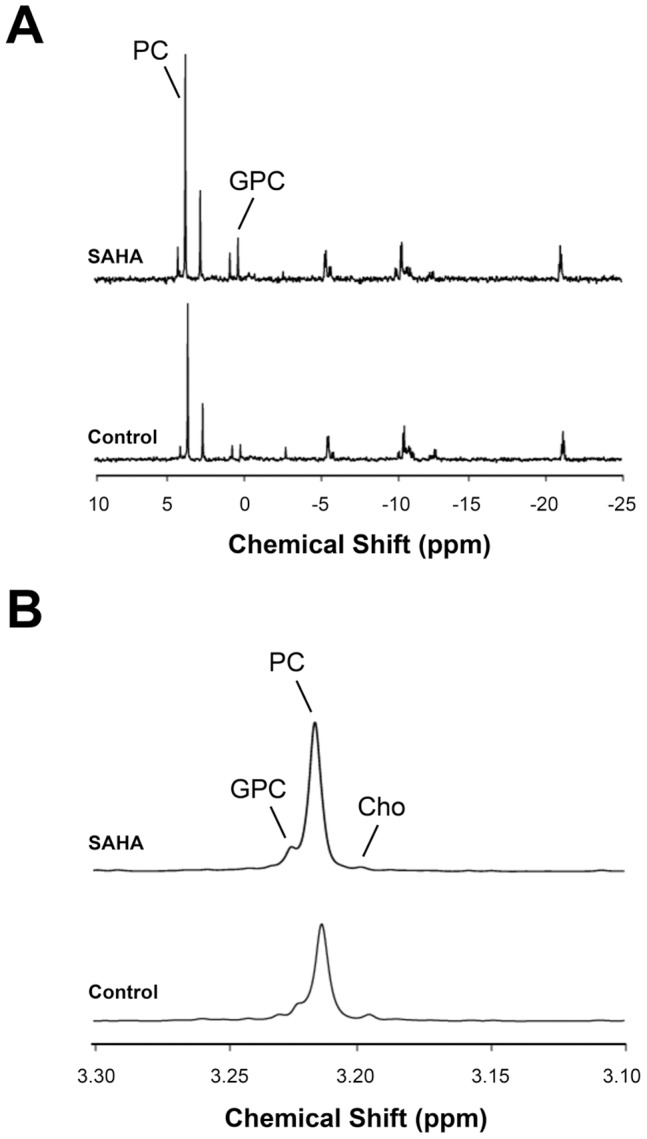
(A) Representative 31P spectra of control (bottom) and SAHA-treated (top) MCF7 cell extracts, showing increases in PC and GPC after 48-hour treatment; (B) Representative 1H spectra (3.10–3.30 ppm) of choline-containing metabolites, highlighting increased tCho levels after 48-hour treatment.
In order to determined the underlying mechanism of this effect we chose to focus further studies on treatment with 10 µM SAHA for 48 h, a dose that led to a reduction in cell proliferation to 50±5% (P<0.001, n = 3) of control (Fig. 1A). The direct deacetylase assay indicated that this treatment reduced HDAC activity from 74±2 pmol deacetylated substrate/hr to 39±4 pmol/hr, or 53±4% of control (P = 0.0002, n = 3). Cell cycle analysis indicated that the fraction of cells in the G1 phase remained unchanged following SAHA treatment (73±2% in control cells and 73±1% in SAHA-treated cells). The fraction of cells in G2 increased following treatment from 8±1% to 17±1% (P<0.002, n = 3), while the fraction of cells in the S phase dropped from 18±2% to 7±1% (P = 0.001). This slight change in cell cycle distribution did not lead to a significant change in average cell size following SAHA treatment (control: 19.2±3.8 µm, SAHA-treated: 18.9±3.2 µm).
31P MRS demonstrated that PC increased to 129±7% of control (P = 0.006, n = 3; Fig. 2A). Control cells had 17.8±1.2 fmol PC/cell, while SAHA-treated cells had 22.5±1.9 fmol PC/cell. Additionally, the concentration of GPC increased significantly with SAHA treatment to 210±45% of control, from 1.6±0.3 fmol GPC/cell to 3.7±0.4 fmol GPC/cell (P = 0.01, n = 3). The increased levels of choline-containing metabolites were also evident in 1H MR spectra (Fig. 2B). Total choline (tCho), a peak that consists of choline (Cho), PC and GPC significantly increased from 19.7±1.5 fmol/cell to 27.2±1.1 fmol/cell with treatment, or 138±9% of control (P = 0.002, n = 3), consistent with the 31P MRS findings.
SAHA Increases Choline Uptake
To evaluate the contribution of de novo synthesis to increased endogenous PC levels, the uptake of [1,2-13C]choline was measured after short-term incubation (Fig. 3). 13C MR spectra show PC peaks from the incorporation and phosphorylation of labeled choline. The level of 13C-labeled PC was significantly higher in treated cells representing 154±19% of control. The labeled PC pool in control cells was 8.5±1.5 fmol/cell, and 13.1±0.9 fmol/cell in treated cells (P = 0.01, n = 3). It is worth noting that this increase in de novo PC was within standard deviation of the increase in total endogenous PC levels. Relative to the total PC levels determined from the 31P spectra, 47±6% of the PC pool was 13C-labeled in control cells and 59±8% of the pool was labeled in SAHA-treated after 6 hours of labeling, with no significant difference in the percentage of labeling between control and treated cells (P = 0.13, n = 3). Over this time period, no label was distributed beyond PC into PtdCho, GPC or other phospholipids, which is consistent with previous studies [29].
Figure 3. Measurement of choline uptake by magnetic resonance spectroscopy.
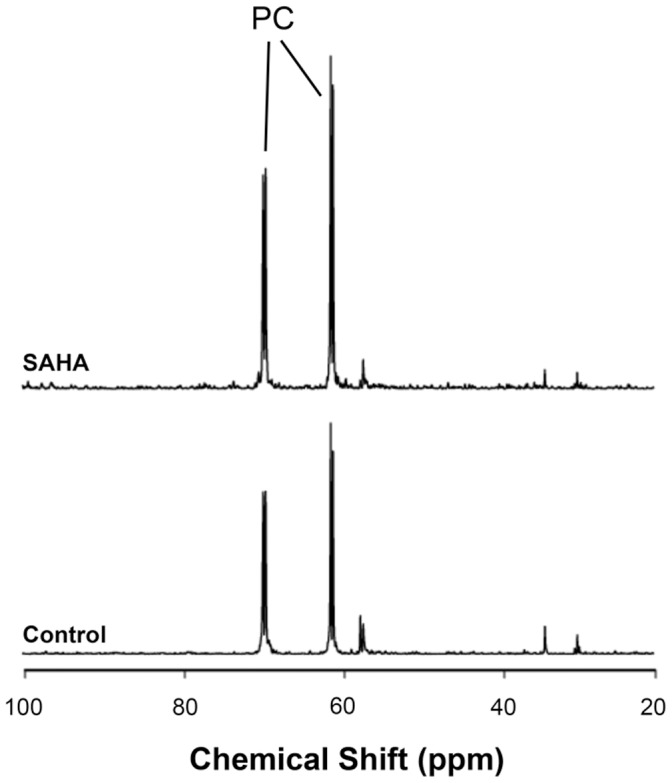
Representative 13C spectra of control (bottom) and SAHA-treated (top) MCF7 cell extracts labeled with [1,2-13C]choline, showing increased uptake and incorporation of labeled choline as [1,2-13C]PC.
Interestingly, there is no [1,2-13C]choline peak visible in the 13C spectrum. The concentration of labeled choline does not reach the detection limit, which suggests that choline is phosphorylated shortly after being transported into the cell. The relative rates of the two steps involved in PC synthesis can be estimated using the signal-to-noise ratio of the PC peaks (SNR values ≥20) [33]. Thus, choline phosphorylation is at least 20-fold faster than choline transport, indicating that transport is the rate-limiting step in PC synthesis in MCF7 cells.
SAHA Modulates Activities of Enzymes Involved in Choline Metabolism
In order to explain the observed changes in choline-containing metabolite concentrations, we analyzed the activities of choline kinase and CTP: phosphocholine cytidylyltransferase, the enzymes directly involved in PC production and consumption respectively. The cellular activity of choline kinase was measured by 1H NMR assay in cell lysates. Figure 4 shows a representative time course of the choline kinase-mediated PC production and choline consumption. These assays revealed a choline kinase rate of 3.6±0.8 fmol PC/cell per hr in control cells and an increase to 6.3±1.4 fmol PC/cell per hr in treated cells (177±31% of control; P = 0.04).
Figure 4. Quantification of choline kinase activities.

(A) Simultaneous detection of PC and choline peaks in MCF7 cell cytosolic preparations over 1 hour; (B) Time courses of PC production in control and SAHA-treated cells in representative experiments, showing increased choline kinase activity with treatment.
The activity of CTP:phosphocholine cytidylyltransferase was measured in total cell lysates by 31P NMR assay. These assays revealed a rate of 10.0±2.2 fmol CDP-choline/cell per hr and no significant change in treated cells (P = 0.15). Figure 5 shows a representative time course of the cytidylyltransferase-mediated CDP-choline production.
Figure 5. Quantification of CTP:phosphocholine cytidylyltransferase activities.

(A) Buildup of CDP-choline peaks in MCF7 cell lysates over 1.5 hours; (B) Time courses of CDP-choline production in control and SAHA-treated cells, showing no significant change in cytidylyltransferase activity with treatment.
Fluorimetric assessment of PLA activities measured in total cell lysates revealed a 162±18% (P = 0.03) increase in PLA2, while no significant change was seen in PLA1 (115±12%; P = 0.13).
SAHA Alters Expression of Genes Associated with Metabolic Modulation
To verify that the increases in enzyme activities are due to elevated levels of enzymes, we determined the relative expressions of annotated genes associated with choline metabolism [34] by mRNA microarray analysis (Table 1). SAHA treatment resulted in changes in gene expression of several proteins directly involved in choline metabolic pathway. Expression of intermediate-affinity choline transporters SLC44A1 and SLC44A3 increased (144% and 126%, respectively), while SLC44A2 decreased (69%). No significant changes were observed in high-affinity choline transporter SLC5A7 or low-affinity polyspecific organic-cation transporters SLC22A1-3. Expression of choline kinase α (CHKA) increased (161%), while choline kinase β (CHKB) decreased (77%). No significant changes were seen in the expression of CTP:phosphocholine cytidylyltransferase 1α and β (PCYT1A and PCYT1B). Expression of several PLA2 isoforms (PLA2) increased significantly, most notably PLA2G4C (271%) and PLA2G10 (366%). There were conflicting changes in expression of GDPDs, with an increase in GDPD1 and a decrease in GDPD3. It is worth noting that the gene sequences of phosphatidylcholine-specific phosholipase C has not yet been determined, and therefore was not included in the microarray analysis.
Table 1. Summary of microarray data for relative changes of expression for enzymes involved in choline metabolism.
| Function | Gene Symbol | Gene Title | MA | % Control | FDR |
| Choline Transporters | SLC5A7 | solute carrier family 5, member 7 | −0.13 | 91 | 0.3 |
| SLC44A1 | solute carrier family 44, member 1 | 0.53 | 144 | 1.10E–05 | |
| SLC44A2 | solute carrier family 44, member 2 | −0.54 | 69 | 9.70E–06 | |
| SLC44A3 | solute carrier family 44, member 3 | 0.33 | 126 | 0.014 | |
| SLC44A4 | solute carrier family 44, member 4 | 0.04 | 103 | 0.75 | |
| SLC44A5 | solute carrier family 44, member 5 | 0.08 | 106 | 0.54 | |
| SLC22A1 | solute carrier family 22, member 1 | 0.07 | 105 | 0.66 | |
| SLC22A2 | solute carrier family 22, member 2 | 0.00 | 100 | 0.99 | |
| SLC22A3 | solute carrier family 22, member 3 | −0.01 | 99 | 0.96 | |
| Choline Kinases | CHKA | choline kinase alpha | 0.69 | 161 | 3.00E–06 |
| CHKB | choline kinase beta | −0.38 | 77 | 0.0075 | |
| Cytidylyltransferases | PCYT1A | phosphocholine | −0.01 | 99 | 0.96 |
| cytidylyltransferase 1 alpha | |||||
| PCYT1B | phosphcholine | 0.08 | 106 | 0.53 | |
| cytidylyltransferase 1 beta | |||||
| Choline Phosphotransferase | CHPT1 | choline phosphotransferase 1 | −0.52 | 70 | 0.00036 |
| Phospholipase A | PLA1A | phospholipase A1 member A | 0.11 | 108 | 0.48 |
| PLA2G1B | phospholipase A2, group IB | 0.24 | 118 | 0.074 | |
| PLA2G2A | phospholipase A2, group IIA | −0.16 | 90 | 0.13 | |
| PLA2G2C | phospholipase A2, group IIC | 0.05 | 104 | 0.8 | |
| PLA2G2D | phospholipase A2, group IID | 0.03 | 102 | 0.83 | |
| PLA2G2E | phospholipase A2, group IIE | −0.22 | 86 | 0.22 | |
| PLA2G3 | phospholipase A2, group III | −0.06 | 96 | 0.74 | |
| PLA2G4C | phospholipase A2, group IVC | 1.44 | 271 | 4.50E–08 | |
| PLA2G4E | phospholipase A2, group IVE | −0.04 | 97 | 0.83 | |
| PLA2G4F | phospholipase A2, group IVF | −0.22 | 86 | 0.058 | |
| PLA2G5 | phospholipase A2, group V | −0.13 | 91 | 0.53 | |
| PLA2G6 | phospholipase A2, group VI | 0.27 | 121 | 0.042 | |
| PLA2G7 | phospholipase A2, group VII | −0.04 | 97 | 0.83 | |
| PLA2G10 | phospholipase A2, group X | 1.87 | 366 | 1.20E–06 | |
| PLA2G12A | phospholipase A2, group XIIA | 0.05 | 104 | 0.61 | |
| PLA2G12B | phospholipase A2, group XIIB | 0.12 | 109 | 0.3 | |
| PLA2G15 | phospholipase A2, group XV | 0.74 | 167 | 7.50E–06 | |
| PLA2G16 | phospholipase A2, group XVI | 0.57 | 148 | 8.90E–05 | |
| Phospholipase D | PLD1 | phospholipase D family, member 1 | 1.95 | 386 | 4.50E–09 |
| PLD2 | phospholipase D family, member 2 | 0.11 | 108 | 0.52 | |
| PLD3 | phospholipase D family, member 3 | 1.20 | 230 | 2.20E–08 | |
| PLD4 | phospholipase D family, member 4 | 0.11 | 108 | 0.59 | |
| PLD5 | phospholipase D family, member 5 | 0.26 | 120 | 0.054 | |
| PLD6 | phospholipase D family, member 6 | −0.45 | 73 | 1.00E–04 | |
| Lysophospholipase | LYPLA1 | lysophospholipase I | −0.62 | 65 | 1.50E–05 |
| LYPLAL1 | lysophospholipase-like 1 | 1.13 | 219 | 4.80E–07 | |
| LYPLA2 | lysophospholipase II | 0.20 | 115 | 0.061 | |
| Glycerophosphocholine | GDE1 | glycerophosphodiester | 0.32 | 125 | 0.017 |
| Phosphodiesterase | phosphodiesterase 1 | ||||
| GDPD1 | glycerophosphodiester | 2.07 | 420 | 7.00E–09 | |
| phosphodiesterase domain 1 | |||||
| GDPD2 | glycerophosphodiester | −0.21 | 86 | 0.18 | |
| phosphodiesterase domain 2 | |||||
| GDPD3 | glycerophosphodiester | −1.47 | 36 | 1.70E–07 | |
| phosphodiesterase domain 3 | |||||
| GDPD5 | glycerophosphodiester | 0.03 | 102 | 0.89 | |
| phosphodiesterase domain 5 |
Genes for which log2<3 were considered as not expressed.
To further confirm the most significant microarray findings, Western blot analysis was used to monitor the protein levels of SLC44A1, ChoK α, PLA2G4C and PLA2G10. Our findings are illustrated in Figure 6. Quantification of the immunoblots indicated that the expression of SLC44A1, CHKA, and PLA2G4C all increased significantly, in line with mRNA microarray analysis. SLC44A1 increased significantly from below detection level in control cells, to clearly detectable levels in treated cells. ChoK α increased to 270±27% of control (P = 0.01) and PLA2G4C increased to 138±19% of control (P = 0.03). PLA2G10 also increased to 143±23%, but this increase did not reach statistical significance (data not shown).
Figure 6. Immunoblots probing the protein expression of PLA2G4C, ChoKα SLC44A1.
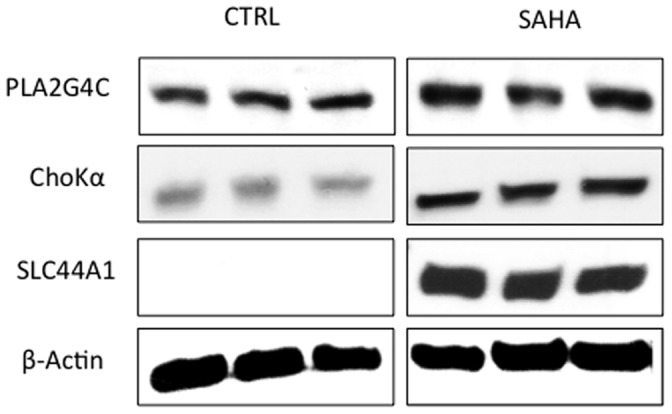
β-actin served a loading control.
Discussion
Novel therapeutic approaches are increasingly developed to target specific molecular genetic events associated with cancer. These advances will lead to more personalized cancer treatment and are expected to result in improved response and reduced toxicity. However, several challenges remain. Most significantly, many targeted therapies result in tumor stasis rather than shrinkage. Consequently, there is a critical need for non-invasive functional imaging biomarkers that confirm drug delivery and molecular drug activity at the tumor site.
Previous studies have reported increased PC levels in response to HDAC inhibition in cell lines and in tumors of several cancer types. We showed that PC levels were inversely correlated with HDAC activity in SAHA-treated PC3 cells [22], [23], and show here that a similar trend is detectable in MCF7 cells. A study in colorectal and prostate cells using belinostat – an HDAC inhibitor with an alternative chemical scaffold – also reported an increase in PC, both in cells and in in vivo [24]. Although promising, the use of PC as a validated biomarker requires an understanding of the mechanistic underpinning of metabolic modulation. This is particularly important because the change in PC is counter to that observed in response to chemotherapy or treatment with specific inhibitors of oncogenic signaling pathways. As recently reviewed, PC levels are controlled by multiple factors and are typically higher in proliferating cells when compared to cells in which proliferation is inhibited following treatment [34]. Consequently, an elevation in PC, while providing a potential biomarker of drug action, also requires robust mechanistic validation.
Our studies focused on treatment with 10 µM SAHA, a dose that led to a 50% inhibition in cell proliferation, and thus could be considered as mimicking response to treatment. This dose was consistent with a previous study on 8 breast cancer cell lines, in which the IC50 ranged from 0.81 to 18.6 µM [35]. Early clinical trials, in which SAHA was administered intravenously, resulted in similar plasma concentrations, although more recent trials, in which SAHA is used in combination with other therapies, report plasma concentrations around 1µM [36].
Drug-induced metabolic changes are the result of altered enzyme activities. Elevated PC levels in response to HDACi reflect the modulation of enzymes involved in the Kennedy pathway of phospholipid biosynthesis [34]. An increase in the endogenous PC pool could be caused by several changes: elevation of choline uptake and phosphorylation, via choline transporters and choline kinases; decrease in PC-to-PtdCho conversion, via cytidylyltransferase and choline phosphotransferase; and/or increase in PtdCho breakdown, via phospholipases C and D.
Our results suggest that PC increases as the result of increased choline transport and phosphorylation. The use of 13C-labeled choline provided a means to characterize the incorporation of choline. After short-term choline incubation in SAHA-treated cells, an increase in [1,2-13C]PC was observed, indicating that the elevated levels of endogenous PC are the result of elevated PC synthesis via choline transport and choline phosphorylation. Choline can be transported into the cell by several classes of proteins: high-affinity choline transporters (SLC5A7), intermediate-affinity choline transporter-like proteins (SLC44A1, etc.), and low-affinity polyspecific organic-cation transporters (SLC22A1, etc.) [37]. The microarray-based gene expression analysis found significant increases in the expression of the intermediate-affinity choline transporters SLC44A1 and SLC44A3 to 144% and 126% of control and a decrease in the expression of SLC44A2 to 69%, while no significant changes in expression were seen in other choline transporters. The increase in expression of SLC44A1, the most significantly altered transporter, was also confirmed at the protein level.
Choline phosphorylation is catalyzed by choline kinase. The activity of the enzyme depends on which subunits (ChoK α and ChoK β) form the dimer, as a recent study demonstrated that the activity of ChoK α is much greater than that of the β isoform [38]. Our activity assay measuring total cellular ChoK activity indicated an increase in cellular ChoK activity, consistent with significant increases in both ChoK α protein and mRNA levels.
However, interpretation of the 13C MR results and previous studies suggest that elevated PC levels following treatment are more likely a result of increased choline uptake, at least in our system. Previous studies in MCF7 cells have reported that choline transport is the rate-limiting step in PC synthesis [33], [39]. In our studies, choline was not visible in the 13C spectra in either control or treated cells, indicating that [1,2-13C]choline was at an intracellular concentration below detection. The proportion of labeled [PC] to [Cho] is greater than 20 (the lowest measured SNR of PC peaks), suggesting that upon transport of choline into the cell it is immediately phosphorylated by ChoK at a rate that is at least 20 times faster than transport and therefore choline transport is the rate-limiting step in PC synthesis. Taken together, our findings indicate that the increase in PC is therefore primarily due to an overall increase in transporter expression and activity following HDAC inhibition.
Our findings with regard to ChoK α are in line with studies of belinostat that demonstrated that the increase in PC was associated with an induction of ChoK α expression, not only in cells but also in the in vivo setting. However, the authors of that study did not report on choline transport, and thus it is unclear whether our observations with regard to transport are specific to our system or are more generally applicable [24].
Increased expression of the choline transporter SLC44A1 was observed in MCF7 cells following treatment with the HSP90 inhibitor 17AAG, which led to an increase in PC both in cells and in vivo [16], [29]. The increase in SLC44A1 observed in our study following HDAC inhibition could therefore be mediated by an HSP90-associated mechanism. However, 17AAG treatment had no reported effect on choline transporters SLC44A2 and SLC44A3 or choline kinase, suggesting that HDAC inhibition causes additional HSP90-independent changes to transcriptional regulation. Furthermore, reported results of the effect of HDAC inhibition in the in vivo setting have been variable. An increase in PC was observed both in cells and in vivo in response to LAQ-824 and belinostat [23], [24]. In contrast, the increase in PC did not translate to the in vivo setting in the case of PC3 cells treated with SAHA [40]. In this context, it is worth noting that the rate of choline transport is dependent on extracellular choline concentration. The culture conditions used in these studies used 28 µM of choline, the concentration typically used in culture medium. This is higher than physiological concentration, which ranges from 5 to 10 µM. Since choline transport is undertaken by several transporter classes with different affinities, the relative contributions of specific transporters to the overall transport rate are likely to vary over a range of concentrations. Further studies are therefore needed to fully understand the factors contributing to modulation of choline metabolism in the in vivo setting.
An additional metabolic change was observed in the MR results – increased GPC. The increase in GPC following treatment could have been caused by the increased catabolic breakdown of PtdCho by PLA2 (and lysophospholipase) and/or the inhibition of GPC phosphodiesterase. Indeed, measurement of total PLA2 activity showed a significant increase, and expression of several PLA2 isoforms increased. Only a limited number of studies have looked at the modulation of PLA2 isoforms with oncogenesis or response to treatment. We have previously found that PLA2G10 was significantly elevated in MCF7 cells following HSP90 inhibition [29]. A study comparing MDA-MD-231 breast cancer cells observed a significant decrease in PLA2G4A - a PLA2 isoform that was not significantly detectable in our cells [41]. In the case of ovarian cells, PLA2G4A as well as PLA2G4C were lower in cancer cell lines compared to normal [31]. The role of each of the PLA2 isoforms in breast and other cancer types thus remains to be fully elucidated.
Further studies are needed to assess our findings in other cancer types in both cell and tumor models. Nonetheless, this study was the first to systematically assess a range of enzymes that, collectively, lead to the increased PC and GPC levels associated with HDAC inhibition. Additional studies are also needed to fully understand the complex interplay between HDAC and choline metabolism. Comparison of results to other similar studies suggest that HSP90 may possibly be responsible for mediating some of the effects observed in our study, including the elevation in SLC44A1 and PLA2 [7], [29]. Alternatively, a previous study into the transcriptional regulation of SLC44A1 identified the binding sites of transcription factors E2F and Sp1 within the promoter sequence [42]. It has also been shown that E2F and Sp1 interact directly with HDAC, causing alterations in their activities [43]. E2F and Sp1 are also affected by cell cycle and MAPK pathway, which have been found to be modulated in response to HDAC inhibition [3], [44], [45]. However, besides these reports studies on the regulation of choline metabolism are surprisingly sparse. Accordingly, a mechanism connecting SAHA molecular action with expression of other enzymes involved in choline metabolism remains unaddressed.
In summary, this study is a first step in developing the necessary mechanistic understanding that can lead to establishing clinically applicable non-invasive imaging methods, which can help inform on target modulation and potential response to HDAC inhibitor treatment.
Acknowledgments
We thank Drs. Christopher Barker and Linda Ta of UCSF Genomic Core for expert technical assistance in performing gene arrays, and Dr. Alisha Holloway of the Biostatistics Core for performing data analysis of microarray results.
Funding Statement
Funding by National Institutes of Health (NIH) (www.NIH.gov) grants R01 CA130819 and R21 120010-01A1. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, et al. (1999) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401: 188–193. [DOI] [PubMed] [Google Scholar]
- 2. Xu WS, Parmigiani RB, Marks PA (2007) Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26: 5541–5552. [DOI] [PubMed] [Google Scholar]
- 3. Lane AA, Chabner BA (2009) Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 27: 5459–5468. [DOI] [PubMed] [Google Scholar]
- 4. Mai A, Massa S, Rotili D, Cerbara I, Valente S, et al. (2005) Histone deacetylation in epigenetics: an attractive target for anticancer therapy. Med Res Rev 25: 261–309. [DOI] [PubMed] [Google Scholar]
- 5. Hess-Stumpp H (2005) Histone deacetylase inhibitors and cancer: from cell biology to the clinic. Eur J Cell Biol 84: 109–121. [DOI] [PubMed] [Google Scholar]
- 6. Epping MT, Bernards R (2009) Molecular basis of the anti-cancer effects of histone deacetylase inhibitors. Int J Biochem Cell Biol 41: 16–20. [DOI] [PubMed] [Google Scholar]
- 7. Glaser KB (2007) HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol 74: 659–671. [DOI] [PubMed] [Google Scholar]
- 8. Prince HM, Bishton MJ, Harrison SJ (2009) Clinical studies of histone deacetylase inhibitors. Clin Cancer Res 15: 3958–3969. [DOI] [PubMed] [Google Scholar]
- 9. Marks PA, Richon VM, Rifkind RA (2000) Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst 92: 1210–1216. [DOI] [PubMed] [Google Scholar]
- 10. Stimson L, La Thangue NB (2009) Biomarkers for predicting clinical responses to HDAC inhibitors. Cancer Lett 280: 177–183. [DOI] [PubMed] [Google Scholar]
- 11. de Certaines JD, Larsen VA, Podo F, Carpinelli G, Briot O, et al. (1993) In vivo 31P MRS of experimental tumours. NMR Biomed 6: 345–365. [DOI] [PubMed] [Google Scholar]
- 12. Leach MO, Verrill M, Glaholm J, Smith TA, Collins DJ, et al. (1998) Measurements of human breast cancer using magnetic resonance spectroscopy: a review of clinical measurements and a report of localized 31P measurements of response to treatment. NMR Biomed 11: 314–340. [DOI] [PubMed] [Google Scholar]
- 13. Kvistad KA, Bakken IJ, Gribbestad IS, Ehrnholm B, Lundgren S, et al. (1999) Characterization of neoplastic and normal human breast tissues with in vivo (1)H MR spectroscopy. J Magn Reson Imaging 10: 159–164. [DOI] [PubMed] [Google Scholar]
- 14. Evelhoch JL, Gillies RJ, Karczmar GS, Koutcher JA, Maxwell RJ, et al. (2000) Applications of magnetic resonance in model systems: cancer therapeutics. Neoplasia 2: 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ronen SM, Jackson LE, Beloueche M, Leach MO (2001) Magnetic resonance detects changes in phosphocholine associated with Ras activation and inhibition in NIH 3T3 cells. Br J Cancer 84: 691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chung YL, Troy H, Banerji U, Jackson LE, Walton MI, et al. (2003) Magnetic resonance spectroscopic pharmacodynamic markers of the heat shock protein 90 inhibitor 17-allylamino,17-demethoxygeldanamycin (17AAG) in human colon cancer models. J Natl Cancer Inst 95: 1624–1633. [DOI] [PubMed] [Google Scholar]
- 17. Beloueche-Babari M, Jackson LE, Al-Saffar NM, Eccles SA, Raynaud FI, et al. (2006) Identification of magnetic resonance detectable metabolic changes associated with inhibition of phosphoinositide 3-kinase signaling in human breast cancer cells. Mol Cancer Ther 5: 187–196. [DOI] [PubMed] [Google Scholar]
- 18. Beloueche-Babari M, Jackson LE, Al-Saffar NM, Workman P, Leach MO, et al. (2005) Magnetic resonance spectroscopy monitoring of mitogen-activated protein kinase signaling inhibition. Cancer Res 65: 3356–3363. [DOI] [PubMed] [Google Scholar]
- 19. Ross J, Najjar AM, Sankaranarayanapillai M, Tong WP, Kaluarachchi K, et al. (2008) Fatty acid synthase inhibition results in a magnetic resonance-detectable drop in phosphocholine. Mol Cancer Ther 7: 2556–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ward CS, Venkatesh HS, Chaumeil MM, Brandes AH, Vancriekinge M, et al. (2010) Noninvasive detection of target modulation following phosphatidylinositol 3-kinase inhibition using hyperpolarized 13C magnetic resonance spectroscopy. Cancer Res 70: 1296–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koul D, Shen R, Kim YW, Kondo Y, Lu Y, et al. (2010) Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro Oncol 12: 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sankaranarayanapillai M, Tong WP, Maxwell DS, Pal A, Pang J, et al. (2006) Detection of histone deacetylase inhibition by noninvasive magnetic resonance spectroscopy. Mol Cancer Ther 5: 1325–1334. [DOI] [PubMed] [Google Scholar]
- 23. Chung YL, Troy H, Kristeleit R, Aherne W, Jackson LE, et al. (2008) Noninvasive magnetic resonance spectroscopic pharmacodynamic markers of a novel histone deacetylase inhibitor, LAQ824, in human colon carcinoma cells and xenografts. Neoplasia 10: 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beloueche-Babari M, Arunan V, Troy H, te Poele RH, te Fong AC, et al. (2012) Histone deacetylase inhibition increases levels of choline kinase alpha and phosphocholine facilitating noninvasive imaging in human cancers. Cancer Res 72: 990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beloueche-Babari M, Chung YL, Al-Saffar NM, Falck-Miniotis M, Leach MO (2010) Metabolic assessment of the action of targeted cancer therapeutics using magnetic resonance spectroscopy. Br J Cancer 102: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Negendank W (1992) Studies of human tumors by MRS: a review. NMR Biomed 5: 303–324. [DOI] [PubMed] [Google Scholar]
- 27. Podo F (1999) Tumour phospholipid metabolism. NMR Biomed 12: 413–439. [DOI] [PubMed] [Google Scholar]
- 28. Milkevitch M, Shim H, Pilatus U, Pickup S, Wehrle JP, et al. (2005) Increases in NMR-visible lipid and glycerophosphocholine during phenylbutyrate-induced apoptosis in human prostate cancer cells. Biochim Biophys Acta 1734: 1–12. [DOI] [PubMed] [Google Scholar]
- 29. Brandes AH, Ward CS, Ronen SM (2010) 17-allyamino-17-demethoxygeldanamycin treatment results in a magnetic resonance spectroscopy-detectable elevation in choline-containing metabolites associated with increased expression of choline transporter SLC44A1 and phospholipase A2. Breast Cancer Res 12: R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tyagi RK, Azrad A, Degani H, Salomon Y (1996) Simultaneous extraction of cellular lipids and water-soluble metabolites: evaluation by NMR spectroscopy. Magn Reson Med 35: 194–200. [DOI] [PubMed] [Google Scholar]
- 31. Iorio E, Mezzanzanica D, Alberti P, Spadaro F, Ramoni C, et al. (2005) Alterations of choline phospholipid metabolism in ovarian tumor progression. Cancer Res 65: 9369–9376. [DOI] [PubMed] [Google Scholar]
- 32. Gabellieri C, Beloueche-Babari M, Jamin Y, Payne GS, Leach MO, et al. (2009) Modulation of choline kinase activity in human cancer cells observed by dynamic 31P NMR. NMR Biomed 22: 456–461. [DOI] [PubMed] [Google Scholar]
- 33. Katz-Brull R, Degani H (1996) Kinetics of choline transport and phosphorylation in human breast cancer cells; NMR application of the zero trans method. Anticancer Res 16: 1375–1380. [PubMed] [Google Scholar]
- 34. Glunde K, Bhujwalla ZM, Ronen SM (2011) Choline metabolism in malignant transformation. Nat Rev Cancer 11: 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chang H, Jeung HC, Jung JJ, Kim TS, Rha SY, et al. (2011) Identification of genes associated with chemosensitivity to SAHA/taxane combination treatment in taxane-resistant breast cancer cells. Breast Cancer Res Treat 125: 55–63. [DOI] [PubMed] [Google Scholar]
- 36. Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, et al. (2011) A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer 104: 1828–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Michel V, Yuan Z, Ramsubir S, Bakovic M (2006) Choline transport for phospholipid synthesis. Exp Biol Med (Maywood) 231: 490–504. [DOI] [PubMed] [Google Scholar]
- 38. Gallego-Ortega D, Gomez del Pulgar T, Valdes-Mora F, Cebrian A, Lacal JC (2011) Involvement of human choline kinase alpha and beta in carcinogenesis: a different role in lipid metabolism and biological functions. Adv Enzyme Regul 51: 183–194. [DOI] [PubMed] [Google Scholar]
- 39. Eliyahu G, Kreizman T, Degani H (2007) Phosphocholine as a biomarker of breast cancer: molecular and biochemical studies. Int J Cancer 120: 1721–1730. [DOI] [PubMed] [Google Scholar]
- 40. Sankaranarayanapillai M, Tong WP, Yuan Q, Bankson JA, Dafni H, et al. (2008) Monitoring histone deacetylase inhibition in vivo: noninvasive magnetic resonance spectroscopy method. Mol Imaging 7: 92–100. [PubMed] [Google Scholar]
- 41. Glunde K, Jie C, Bhujwalla ZM (2004) Molecular causes of the aberrant choline phospholipid metabolism in breast cancer. Cancer Res 64: 4270–4276. [DOI] [PubMed] [Google Scholar]
- 42. Yuan Z, Tie A, Tarnopolsky M, Bakovic M (2006) Genomic organization, promoter activity, and expression of the human choline transporter-like protein 1. Physiol Genomics 26: 76–90. [DOI] [PubMed] [Google Scholar]
- 43. Banchio C, Lingrell S, Vance DE (2006) Role of histone deacetylase in the expression of CTP:phosphocholine cytidylyltransferase alpha. J Biol Chem 281: 10010–10015. [DOI] [PubMed] [Google Scholar]
- 44. Sugimoto H, Banchio C, Vance DE (2008) Transcriptional regulation of phosphatidylcholine biosynthesis. Prog Lipid Res 47: 204–220. [DOI] [PubMed] [Google Scholar]
- 45. Geilen CC, Wieder T, Boremski S, Wieprecht M, Orfanos CE (1996) c-Ha-ras oncogene expression increases choline uptake, CTP: phosphocholine cytidylyltransferase activity and phosphatidylcholine biosynthesis in the immortalized human keratinocyte cell line HaCaT. Biochim Biophys Acta 1299: 299–305. [DOI] [PubMed] [Google Scholar]