

Abstract
Background
Endothelial progenitor cells (EPCs) augment neovascularization and repair of damaged tissues, but may undergo functional changes during exposure to cardiovascular risk factors. This study tested the hypothesis that early renovascular hypertension (RVH) modulates the temporal pattern of EPC function that deteriorates with disease duration.
Methods
RVH was induced in domestic pigs by unilateral renal artery stenosis. EPCs were cultured after 3, 6, and 12 weeks of RVH or normal control to evaluate EPC function, growth factor, and homing receptor expression. Plasma renin activity (PRA), vascular endothelial growth factor (VEGF), and its soluble receptor-1 (sFlt-1) were measured in plasma. EPCs (10 × 106) isolated from 3-week RVH or from normal pigs were also injected into control kidneys (n = 6–7, each group), and 4 weeks later single-kidney renal blood flow (RBF) and glomerular filtration rate (GFR) were evaluated. Microvascular density was studied ex vivo using microcomputed tomography.
Results
Blood pressure peaked at 3 weeks and remained higher than normal throughout the study. Systemic PRA also peaked after 3 weeks of RVH and declined thereafter, whereas sFlt-1 showed a reciprocal pattern. In vivo, only RVH but not normal EPCs increased RBF, GFR, and microvascular density. RVH–EPCs showed in vitro enhanced proliferation, tube formation, VEGF, and homing receptor expression that peaked at 3 weeks, which were abolished by valsartan and returned to baseline levels after 12 weeks of RVH. EPC number remained unchanged throughout the study.
Conclusion
A transient enhancement of EPC function, mediated by angiotensin II, may contribute to compensatory vascular adaptation in early RVH, but is lost as hypertension persists.
Keywords: angiogenesis, angiotensin II, endothelial progenitor cell, growth factor, renal hypertension
Introduction
Endothelial progenitor cells (EPCs) have been implicated in adult neovascularization and play an important role in vascular repair during exposure to cardiovascular risk factors. Functional utility of EPCs relies on the availability of an adequate number of EPCs with robust ability to home, engraft, migrate, and proliferate in target tissues. An important element of EPC function derives from their ability to secrete and respond to vascular endothelial growth factor (VEGF). Alas, systemic disease processes that impose vascular injury and generate a need for EPCs may also interfere with their function.
Hypertension is an important risk factor that impairs vascular endothelial function and structural integrity, leading to a cascade of events that progress to cardiovascular diseases. However, the effect of hypertension on EPC function has not been fully elucidated. In patients with hypertension and coronary artery disease, blunted migration and reduced number of EPCs have been reported [1], and EPC senescence is accelerated in both hypertensive rats and patients with essential hypertension [2]. On the contrary, Salguero et al.[3] found enhanced EPC mobilization in renovascular hypertension (RVH), possibly as a compensatory vascular adaptive mechanism. This differential effect of hypertension on EPC function could be due to different causes, timing, or durations of exposure. However, the temporal pattern of EPC function in hypertension remains unclear.
At the early stage of RVH, increased production of angiotensin II (AngII) contributes greatly to the elevation of blood pressure. Exposure to AngII or hypertension may affect the bioavailability of EPCs in several opposing ways. Although AngII potentiates VEGF-induced human EPC proliferation and vascular network formation through the upregulation of VEGF receptor 2 (KDR) [4], soluble Flt-1 (sFlt-1), or VEGF receptor-1, may sequester and inhibit VEGF and has been reported to be increased in hypertensive patients [5]. AngII may affect EPC function through AngII type 1 (AT1) receptor [6], and enhanced mechanical stretch-induced vascular reactive oxygen species production may lead to EPC mobilization during the early phase of AngII-dependent hypertension [3]. Conversely, AngII may decrease EPC levels and accelerates the onset of EPC senescence [7]. It is not unlikely that the divergent effects of AngII and hypertension on EPC function are related to the duration and level of exposure.
We have shown that EPCs isolated after early (3 weeks) RVH show enhanced angiogenic activity in vitro[8]. However, whether enhanced EPC function is detectable in vivo, and persists with longer duration of RVH, remains unknown. We hypothesize that transient activation of the renin–angiotensin–aldosterone system (RAAS) in early hypertension is accompanied by temporary enhancement of EPC activity, both ex vivo and in vivo, which is lost in later stage of hypertension.
Methods
All procedures were approved by Mayo Clinic Institutional Animal Care and Use Committee. Domestic pigs (n = 6, 35–40 kg) were induced with RVH by implanting a local irritant coil in the main renal artery to induce unilateral renal artery stenosis, as we previously described [9,10]. A PhysioTel telemetry system (Data Sciences, St. Paul, Minnesota, USA) was also implanted in the left femoral artery to measure mean arterial pressure (MAP) [10,11]. Peripheral blood (50 ml) was collected at baseline and after 3, 6, and 12 weeks of RVH to isolate and characterize EPCs, as we previously described [8,12]. In addition, plasma renin activity (PRA), creatinine, VEGF, and sFlt-1 levels in peripheral blood were measured at each time point using ELISA or western blotting [12].
The characteristics, number, and function of EPCs collected after different durations of RVH was evaluated by cell surface marker expression, the number of colony forming units (CFUs), migration, proliferation, and tube formation analysis [8,13], as well as by growth factor and homing signaling protein expression, as we previously described [12].
To evaluate the contribution of exposure to endogenous AngII, EPC function ex vivo was also assessed after co-incubation of the AngII AT1 receptor inhibitor valsartan.
Furthermore, to evaluate whether enhanced EPC function in early RVH was detectable in vivo, EPCs collected from either normal pigs or after 3 weeks of RVH were transplanted into control pig kidneys. Four weeks later, renal function was estimated using multidetector computed tomography (MDCT) scanning, and microvascular density using micro-CT.
Endothelial progenitor cell function ex vivo
Late outgrowth EPCs were cultured as described previously [8,12] from mononuclear cells obtained from peripheral blood of normotensive pigs or pigs after 3, 6, and 12 weeks of RVH. CFUs were counted after 7 days of culture to assess EPC number, as previously shown [14,15]. EPCs were characterized by the expression of CD34 and KDR, no expression of CD45 and CD14, as well as by uptake of acetylated low-density lipoprotein (Ac-LDL) [8]. EPC function was studied with or without co-incubation with valsartan (200 nmol/l; Novartis, New Jersey, USA) dissolved in culture media [4] for 16–24 h.
Migration assay
EPC migration was tested using a QCM Haptotaxis cell migration kit (Millipore, Billerica, Massachusetts, USA), following manufacturer’s instructions. Briefly, EPCs (1 × 106 cells/ml) were placed in fibronectin-coated or BSA-coated (negative control) wells and incubated for 24 h; nonmigrated cells were then removed, and migrated cells stained [13]. After solubilizing stained EPCs with extraction buffer, 100 μl buffers were transferred to a 96-well plate, and read at absorbance 562 nm.
Proliferation assay
EPC proliferative activity was determined by MTS assay (CellTiter 96 Non-Radioactive Cell Proliferation Assay, Promega, Madison, Wisconsin, USA), as we previously described [13].
Matrigel tube formation assay
The ability of EPCs to incorporate into endothelial cells and form vascular structures was evaluated in matrigel (BD Biosciences, Bedford, Massachusetts, USA) and quantified as we previously described [13,16].
Endothelial progenitor cell protein expression
Protein expression of stromal cell-derived factor-1 (SDF-1) and C-X-C chemokine receptor type 4 (CXCR-4) (both 1 : 500; Abcam, Cambridge, Massachusetts, USA), important homing signals for EPC migration to injury sites [12], was measured by western blotting in homogenized EPCs. In addition, expression of VEGF-A, the AT1 receptor, and endothelial nitric oxide synthase (eNOS) (all 1 : 200, Santa Cruz Biotechnology, Santa Cruz, California, USA) were determined using standard western blotting. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1 : 5000; Covance, Emeryville, California, USA) was used as loading control.
Endothelial progenitor cell angiogenic activity in vivo
To compare the angiogenic activity in vivo of EPCs harvested from RVH or normal (N) pigs, additional normal pigs were anesthetized, a 7F balloon catheter advanced to one renal artery, and allogeneic RVH–EPCs or N–EPCs (late EPCs, 10×106) labeled with CM-DiI were then injected into the kidneys (n = 7, each group) [8,12]. Four weeks later, single-kidney renal blood flow (RBF) and glomerular filtration rate (GFR) were evaluated using MDCT, as we previously described [8,12,17]. Pigs were euthanized a few days later with a lethal intravenous dose of sodium pentobarbital (100 mg/kg Sleepaway; Fort Dodge Laboratories Inc, Fort Dodge, Iowa, USA). Kidneys were removed and kidney lobes were either shock-frozen in liquid nitrogen and stored at −80°C, or preserved in formalin, and a segmental artery perfusing the intact end of the kidney cannulated and prepared for micro-CT. Microvascular density in the renal cortex was studied ex vivo using micro-CT, as previously described [10,18–20]. EPC retention rate was estimated as we reported before [8,12]. Immuostaining of CD3 in frozen slides was used to evaluate immune responses to allogeneic EPCs [21]. To assess kidney protein expression, standard western blotting protocols were followed [22], using specific polyclonal antibodies against VEGF, KDR (all 1 : 200; Santa Cruz Biotechnology), and SDF-1 (1 : 1000, Abcam). GAPDH (1 : 5000; Covance) was used as loading control.
Statistical analysis
Continuous data are expressed as mean ± SEM. Multiple group comparisons utilized analysis-of-variance, followed by post-hoc Tukey test. Statistical significance was accepted if P value was 0.05 or less.
Results
Compared with normal, MAP started increasing in RVH pigs 3–7 days after coil implantation, peaked at 3 weeks, and remained elevated throughout the study (Table 1). Systemic PRA also peaked at 3 weeks of RVH and declined thereafter, whereas sFlt-1 reciprocally decreased after 3 weeks of RVH and returned to basal levels by 12 weeks. Plasma creatinine and VEGF were not significantly changed during the study (Table 1).
Table 1.
Systemic characteristics (mean ± SEM) in pigs during the evolution of renovascular hypertension
| Baseline | 3 weeks | 6 weeks | 12 weeks | |
|---|---|---|---|---|
| Mean arterial pressure (mmHg) | 104.4 ± 1.4 | 192.3 ± 1.9* | 151.5 ± 3.7*,† | 130.1 ± 1.1*,† |
| Plasma renin activity (ng/ml) | 0.17 ± 0.04 | 8.48 ± 8.03* | 0.36 ± 0.16* | 0.24 ± 0.05 |
| Plasma creatinine (mg/dl) | 1.4 ± 0.2 | 1.3 ± 0.2 | 1.5 ± 0.1 | 1.5 ± 0.2 |
| Plasma VEGF (relative protein expression) | 1.3 ± 0.2 | 1.5 ± 0.3 | 1.6 ± 0.2 | 1.2 ± 0.2 |
| sFlt-1 (ng/ml) | 46.8 ± 13.5 | 10.5 ± 2.9* | 28.8 ± 8.8 | 42.0 ± 2.8 |
| Colony forming units (per 10 cm2) | 56.2 ± 4.9 | 61.3 ± 6.2 | 55.1 ± 5.8 | 53.6 ± 8.4 |
VEGF, vascular endothelial growth factor.
P < 0.05 vs. baseline.
P < 0.05 vs. 3 weeks.
Endothelial progenitor cell function ex vivo
EPCs were characterized by cobblestone morphology, uptake of Ac-LDL, positive expression of CD34 and KDR, and negative expression of CD14 and CD45 (Fig. 1).
Figure 1.

Representative images of endothelial progenitor cell morphology, CD34-positive and KDR-positive expression, Ac-LDL uptake, and negative CD14 and CD45 expression.
The numbers of CFUs have not changed during the 12-week observation period and were similar to normal (Table 1). In contrast, RVH-EPC showed enhanced migration, proliferation, and tube formation at 3 weeks (Fig. 2), but all returned to normal levels after 12 weeks of RVH.
Figure 2.

Images of endothelial progenitor cells and their quantification. (a) Representative images of endothelial progenitor cells (EPCs) incorporated into tube structures in matrigel. (b) Quantifications of EPC migration, proliferation, and tube formation. All of these functional attributes peaked at 3 weeks of renovascular hypertension (RVH) and returned to baseline by 12 weeks of RVH, and all were abolished by co-incubation with valsartan, implicating the angiotensin II AT1 receptor. *P<0.05 vs. normal. †P<0.05 vs. valsartan. ‡P<0.05 vs. 3 weeks.
In addition, at 3 weeks EPCs showed increased expression of SDF-1 and CXCR-4 compared with baseline and with 12 weeks of RVH (Fig. 3), suggesting enhanced homing capability to injury sites at this stage. Furthermore, VEGF, AT1, and eNOS expressions were all transiently elevated at 3 weeks, but decreased by 6 weeks of RVH, and CXCR-4 and eNOS expression declined below normal levels. All of the enhanced EPC function and protein expression at 3 weeks of RVH were significantly attenuated after treatment with valsartan (Figs. 2 and 3), indicating that AngII partly mediated EPC functional enhancement in early RVH through the AT1 receptor. Valsartan had no effect on EPC function from normotensive pigs or after 6 and 12 weeks of RVH.
Figure 3.
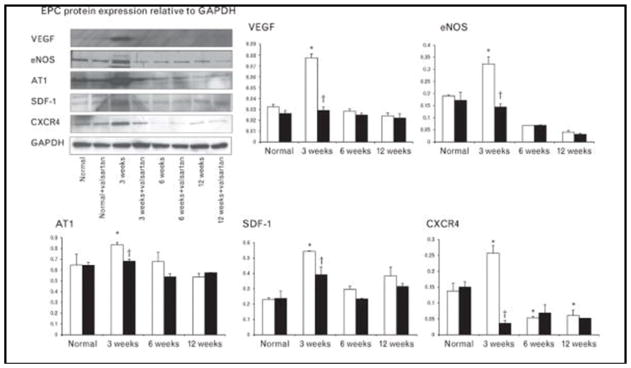
Representative western blotting bands and quantifications of vascular endothelial growth factor, eNOS, angiotensin II type 1, stromal cell-derived factor-1, and CXCR4 showing that endothelial progenitor cell expression of growth factors and homing signals peaked at 3 weeks of renovascular hypertension (RVH), and decreased thereafter. All of the enhanced protein expressions were normalized by valsartan (*P<0.05 vs. normal, †P<0.05 vs. valsartan). AT1, angiotensin II type 1; EPC, endothelial progenitor cell; SDF-1, stromal cell-derived factor-1; VEGF, vascular endothelial growth factor;
Renovascular hypertension–endothelial progenitor cells promote angiogenesis in the pig kidney
The retention rates of normal EPCs and RVH–EPCs in the kidney were similar (8.3 ± 3.1 vs. 8.9 ± 2.7%, P > 0.05). In kidneys implanted with EPCs isolated after 3 weeks of RVH, RBF and GFR were significantly increased compared with kidneys implanted with EPCs isolated from normotensive pigs (Fig. 4), as was renal microvascular density (Fig. 4). Moreover, only RVH–EPC-treated kidneys showed increased SDF-1 and VEGF expression compared with normal, whereas KDR remained similar among the three groups (Fig. 5). CD3 staining did not show clustering around EPCs, arguing against rejection of allogeneic EPCs (Fig. 5).
Figure 4.

Microcomputed tomography images and cortical microvascular density of kidneys. (a) Representative microcomputed tomography images of kidneys that received normal or renovascular hypertension (RVH)–endothelial progenitor cell (EPCs), and control kidneys. (b) Cortical microvascular density in these kidneys, showing that RVH–EPCs had better angiogenic ability in vivo. (c) Renal blood flow (RBF) and glomerular filtration rate (GFR) were higher in normal kidneys that received RVH–EPCs than those received normal EPCs (*P<0.05 vs. normal).
Figure 5.
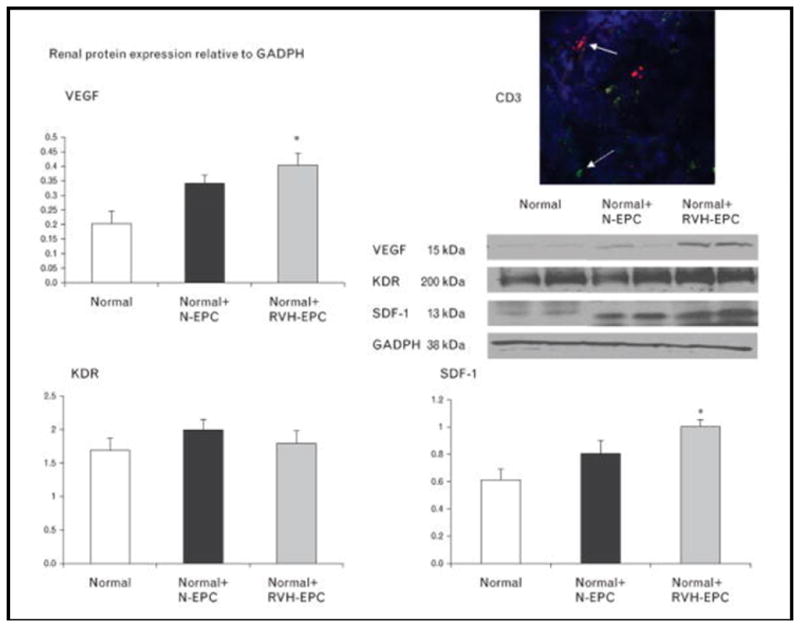
Western blotting showing that kidneys that received renovascular hypertension–endothelial progenitor cells had higher vascular endothelial growth factor and stromal cell-derived factor-1 expression than normal, whereas VEGF receptor KDR expression was similar among the groups. Representative immunohistochemical staining of CD3-positive lymphocytes (green) that do not spatially correspond to transplanted EPCs (red), arguing against rejection of allogenic EPCs (right top). EPCs, endothelial progenitor cells; RVH, renovascular hypertension; SDF-1, stromal cell derived factor-1; VEGF, vascular endothelial growth factor. *P<0.05 vs. normal.
Discussion
Hypertension is a major risk factor for cardiovascular diseases and induces injury in critical target organs such as the brain, heart, and kidneys. Vascular adaption in response to hypertension may relate to and determine the extent of end-organ damage. This study demonstrates that the angiogenic function of EPCs in early hypertension shows a temporal pattern that is related to the duration of the disease. Hence, the angiogenic potency of EPCs, an important determinant of their capacity for vascular repair, is transiently augmented in early RVH both in vivo and ex vivo in association with transient activation of the systemic RAAS and with a pro-angiogenic profile of circulating growth factors. However, this compensatory mechanism is subsequently lost with longer duration of hypertension, which might conceivably permit vascular injury and target-organ damage.
EPCs may play an important role in replacing damaged endothelial cells, and thereby maintaining vascular tone and integrity. At the early stage of RVH, the increase in blood pressure is mainly dependent on increased circulating levels of AngII, which subsequently decline as alternative pressor mechanisms develop [9]. A recent study has shown that a 3-week murine RVH and AngII mobilize EPCs from the bone marrow in a mechanism dependent on mechanical stretch and reactive oxygen species [3]. Remarkably, although we did not observe an increase in EPC numbers in the swine model, our study shows that EPC migration, proliferation, and tube formation capability, as well as VEGF and eNOS expression, also peaked after 3 weeks of RVH. Moreover, we observed that this phenomenon is transient, as most indices of enhanced angiogenic potential of EPCs gradually returned to baseline as RVH continued, whereas in fact eNOS expression declined to levels below normal. Importantly, these were accompanied by a similar pattern of systemic (PRA) and tissue (AT1 receptor) RAAS activation, implicating AngII in the transient potentiation of angiogenic function in our study. Furthermore, enhanced EPC function in early RVH was attenuated ex vivo by valsartan, underscored by a previous study showing that valsartan attenuated AngII-mediated EPC angiogenesis [6]. Enhanced EPC migration was not completely abolished by valsartan, suggesting that factors other than AngII may be also involved, such as mechanical stretch or oxidative stress. These observations also imply that in addition to sustaining GFR in the stenotic kidney, AngII also activates mechanisms to repair it, at least at the early stage of renovascular disease.
The mechanisms by which AngII enhances EPC angiogenic potential may be multifactorial. AngII infusion in mice increases local and systemic expression of VEGF and its receptors, an effect possibly mediated by hypoxia-inducible factor-1alpha [23]. Furthermore, AngII potentiates VEGF-induced human EPC proliferation and network formation through upregulation of the VEGF receptor KDR [4]. We did not observe significant changes in circulating VEGF levels, suggesting that AngII upregulated VEGF mainly in EPCs, and VEGF release from EPCs may not suffice to affect systemic level. In addition to VEGF release, circulating VEGF might have also been affected by sFlt-1. Elevated plasma levels of both VEGF and sFlt-1 were found in untreated patients with essential hypertension [5]. A decrease in sFlt-1 at early RVH might be induced by AngII, which can downregulate Flt-1 [24], as we observed that the RAAS was significantly activated at that stage. Furthermore, AngII induces renal protective factor hemeoxygenase-1 (HO-1) [25], a recent study also demonstrated that HO-1 negatively regulates sFlt-1 [26], and our previous study indicated that HO-1 expression was upregulated in RVH [27]. Interestingly, recombinant sFlt-1 has antiangiogenic activity, induces endothelial cell apoptosis, and decreases nitric oxide generation in endothelial cells [28]. Therefore, gradually increased sFlt-1 levels in our RVH pigs may not only sequester VEGF but also blunt EPC angiogenic potential in the established stage of hypertension.
AngII also directly stimulates the angiogenic function of EPCs through AT1 receptors in a process involving VEGF-induced eNOS activation [29]. After 12 weeks of RVH, EPC angiogenic function returned to baseline levels accompanied with downregulation of eNOS to below-normal levels. Therefore, in early RVH, RAAS activation may augment EPC function partly through the physiological AT1 receptor/eNOS pathway, a process that is possibly initiated by the release of growth factors. However, chronic RVH may result a gradual decline in relative EPC function and expression of eNOS, possibly mediated by oxidative stress.
Interestingly, our study showed that early RVH not only improved EPC function but also significantly increased homing signals to attract EPCs to injury sites. SDF-1 and its receptor CXCR-4 expression in EPCs both peaked at 3 weeks of RVH and subsequently declined, with CXCR-4 reaching levels, to lower than normal. SDF-1[alpha] is a chemokine for EPCs that enhances ischemia-induced vasculogenesis and angiogenesis in vivo through a VEGF/eNOS-related pathway [30] and by augmenting EPC recruitment to ischemic tissues [31]. Our study further demonstrated that SDF-1/CXCR-4-rich EPCs may also augment angiogenesis in normal tissues, as normal kidneys infused with EPCs cultured after 3 weeks of RVH showed a striking elevation in function and microvascular density. Therefore, the augmentation of EPC angiogenic potency observed ex vivo translated into consequential functional benefits in vivo. In addition to the physical contribution of EPCs to newly formed vessels, paracrine secretion of growth factors may be a supportive mechanism to improve blood vessel formation and tissue regeneration after cell therapy [32]. These potential mechanisms are consistent with the high expression of VEGF in early-RVH–EPCs and in kidneys that had received them, compared with those that received and retained similar numbers of normal EPCs. Furthermore, a previous study found that the kidney subjected to chronic ischemia may become a source rather than a target of EPCs [33], thereby possibly contributing to altered EPC function in RVH.
Our studies may be limited by using young animals and relatively short duration of hypertension. No changes in renal function were observed in this model of early unilateral RVH, devoid of comorbidities. In addition, some decrease in blood pressure following the 3-week peak, possibly secondary to renal artery recanalization and remodeling, might have affected EPC function as well, yet blood pressure remained significantly elevated. Temporal variation in EPC functional capacity also needs to be tested in additional disease models because alternative mechanisms likely contribute to deterioration of EPC function under different disease conditions.
In conclusion, the current study demonstrated a marked yet transient enhancement of EPC function at early experimental RVH, both ex vivo and in vivo, which may contribute to compensatory vascular adaptation at the early phase of RVH, but is lost as hypertension persists. EPC function augmentation might be secondary to a transient activation of the systemic RAAS and a pro-angiogenic profile of circulating growth factors. These findings may be critical in future application of cell-based therapy in hypertensive patients and support strategies to boost EPC function. Additional studies are also needed to explore the mechanisms for decrease in EPC number and function observed in patients after longer duration of hypertension [34].
Acknowledgments
This study was partly supported by NIH grants DK73608, DK77013, HL77131, and HL085307. The authors are grateful to Novartis for kindly providing valsartan.
Abbreviations
- AngII
angiotensin II
- CXCR-4
C-X-C chemokine receptor type 4
- EPC
endothelial progenitor cell
- GFR
glomerular filtration rate
- KDR
VEGF receptor 2
- MAP
mean arterial pressure
- MDCT
multidetector computed tomography
- PRA
plasma renin activity
- RAAS
renin–angiotensin–aldosterone system
- RBF
renal blood flow
- RVH
renovascular hypertension
- SDF-1
stromal cell-derived factor-1
- sFlt-1
soluble VEGF receptor-1
- VEGF
vascular endothelial growth factor
Footnotes
Conflicts of interest
There are no conflicts of interest.
References
- 1.Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:E1–E7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 2.Imanishi T, Moriwaki C, Hano T, Nishio I. Endothelial progenitor cell senescence is accelerated in both experimental hypertensive rats and patients with essential hypertension. J Hypertens. 2005;23:1831–1837. doi: 10.1097/01.hjh.0000183524.73746.1b. [DOI] [PubMed] [Google Scholar]
- 3.Salguero G, Akin E, Templin C, Kotlarz D, Doerries C, Landmesser U, et al. Renovascular hypertension by two-kidney one-clip enhances endothelial progenitor cell mobilization in a p47phox-dependent manner. J Hypertens. 2008;26:257–268. doi: 10.1097/HJH.0b013e3282f09f79. [DOI] [PubMed] [Google Scholar]
- 4.Imanishi T, Hano T, Nishio I. Angiotensin ii potentiates vascular endothelial growth factor-induced proliferation and network formation of endothelial progenitor cells. Hypertens Res. 2004;27:101–108. doi: 10.1291/hypres.27.101. [DOI] [PubMed] [Google Scholar]
- 5.Belgore FM, Blann AD, Li-Saw-Hee FL, Beevers DG, Lip GY. Plasma levels of vascular endothelial growth factor and its soluble receptor (sflt-1) in essential hypertension. Am J Cardiol. 2001;87:805–807.A809. doi: 10.1016/s0002-9149(00)01512-5. [DOI] [PubMed] [Google Scholar]
- 6.Yin T, Ma X, Zhao L, Cheng K, Wang H. Angiotensin ii promotes no production, inhibits apoptosis and enhances adhesion potential of bone marrow-derived endothelial progenitor cells. Cell Res. 2008;18:792–799. doi: 10.1038/cr.2008.69. [DOI] [PubMed] [Google Scholar]
- 7.Imanishi T, Kobayashi K, Kuroi A, Ikejima H, Akasaka T. Pioglitazone inhibits angiotensin ii-induced senescence of endothelial progenitor cell. Hypertens Res. 2008;31:757–765. doi: 10.1291/hypres.31.757. [DOI] [PubMed] [Google Scholar]
- 8.Chade AR, Zhu X, Lavi R, Krier JD, Pislaru S, Simari RD, et al. Endothelial progenitor cells restore renal function in chronic experimental renovascular disease. Circulation. 2009;119:547–557. doi: 10.1161/CIRCULATIONAHA.108.788653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lerman LO, Nath KA, Rodriguez-Porcel M, Krier JD, Schwartz RS, Napoli C, Romero JC. Increased oxidative stress in experimental renovascular hypertension. Hypertension. 2001;37:541–546. doi: 10.1161/01.hyp.37.2.541. [DOI] [PubMed] [Google Scholar]
- 10.Zhu XY, Chade AR, Rodriguez-Porcel M, Bentley MD, Ritman EL, Lerman A, Lerman LO. Cortical microvascular remodeling in the stenotic kidney: Role of increased oxidative stress. Arterioscler Thromb Vasc Biol. 2004;24:1854–1859. doi: 10.1161/01.ATV.0000142443.52606.81. [DOI] [PubMed] [Google Scholar]
- 11.Zhu XY, Daghini E, Chade AR, Napoli C, Ritman EL, Lerman A, Lerman LO. Simvastatin prevents coronary microvascular remodeling in renovascular hypertensive pigs. J Am Soc Nephrol. 2007;18:1209–1217. doi: 10.1681/ASN.2006090976. [DOI] [PubMed] [Google Scholar]
- 12.Chade AR, Zhu XY, Krier JD, Jordan KL, Textor SC, Grande JP, et al. Endothelial progenitor cells homing and renal repair in experimental renovascular disease. Stem Cells. 2010;28:1039–1047. doi: 10.1002/stem.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu XY, Daghini E, Chade AR, Lavi R, Napoli C, Lerman A, Lerman LO. Disparate effects of simvastatin on angiogenesis during hypoxia and inflammation. Life Sci. 2008;83:801–809. doi: 10.1016/j.lfs.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 15.Choi JH, Kim KL, Huh W, Kim B, Byun J, Suh W, et al. Decreased number and impaired angiogenic function of endothelial progenitor cells in patients with chronic renal failure. Arterioscler Thromb Vasc Biol. 2004;24:1246–1252. doi: 10.1161/01.ATV.0000133488.56221.4a. [DOI] [PubMed] [Google Scholar]
- 16.Daghini E, Zhu XY, Versari D, Bentley MD, Napoli C, Lerman A, Lerman LO. Antioxidant vitamins induce angiogenesis in the normal pig kidney. Am J Physiol. 2007;293:F371–F381. doi: 10.1152/ajprenal.00475.2006. [DOI] [PubMed] [Google Scholar]
- 17.Zhu XY, Chade AR, Krier JD, Daghini E, Lavi R, Guglielmotti A, et al. The chemokine monocyte chemoattractant protein-1 contributes to renal dysfunction in swine renovascular hypertension. J Hypertens. 2009;27:2063–2073. doi: 10.1097/HJH.0b013e3283300192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu XY, Rodriguez-Porcel M, Bentley MD, Chade AR, Sica V, Napoli C, et al. Antioxidant intervention attenuates myocardial neovascularization in hypercholesterolemia. Circulation. 2004;109:2109–2115. doi: 10.1161/01.CIR.0000125742.65841.8B. [DOI] [PubMed] [Google Scholar]
- 19.Bentley MD, Rodriguez-Porcel M, Lerman A, Sarafov MH, Romero JC, Pelaez LI, et al. Enhanced renal cortical vascularization in experimental hypercholesterolemia. Kidney Int. 2002;61:1056–1063. doi: 10.1046/j.1523-1755.2002.00211.x. [DOI] [PubMed] [Google Scholar]
- 20.Chade AR, Bentley MD, Zhu X, Rodriguez-Porcel M, Niemeyer S, Amores-Arriaga B, et al. Antioxidant intervention prevents renal neovascularization in hypercholesterolemic pigs. J Am Soc Nephrol. 2004;15:1816–1825. doi: 10.1097/01.asn.0000130428.85603.6b. [DOI] [PubMed] [Google Scholar]
- 21.Price CM, Colman SM, Kanfer EJ. Persistence of multilineage host haemopoiesis following allogeneic bone marrow transplantation. Br J Haematol. 1995;90:465–468. doi: 10.1111/j.1365-2141.1995.tb05177.x. [DOI] [PubMed] [Google Scholar]
- 22.Chade AR, Rodriguez-Porcel M, Grande JP, Zhu X, Sica V, Napoli C, et al. Mechanisms of renal structural alterations in combined hypercholesterolemia and renal artery stenosis. Arterioscler Thromb Vasc Biol. 2003;23:1295–1301. doi: 10.1161/01.ATV.0000077477.40824.52. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Q, Ishibashi M, Hiasa K, Tan C, Takeshita A, Egashira K. Essential role of vascular endothelial growth factor in angiotensin ii-induced vascular inflammation and remodeling. Hypertension. 2004;44:264–270. doi: 10.1161/01.HYP.0000138688.78906.6b. [DOI] [PubMed] [Google Scholar]
- 24.Anton L, Merrill DC, Neves LA, Gruver C, Moorefield C, Brosnihan KB. Angiotensin ii and angiotensin-(1–7) decrease sflt1 release in normal but not preeclamptic chorionic villi: an in vitro study. Reprod Biol Endocrinol. 2010;8:135–144. doi: 10.1186/1477-7827-8-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haugen EN, Croatt AJ, Nath KA. Angiotensin ii induces renal oxidant stress in vivo and heme oxygenase-1 in vivo and in vitro. Kidney Int. 2000;58:144–152. doi: 10.1046/j.1523-1755.2000.00150.x. [DOI] [PubMed] [Google Scholar]
- 26.Cudmore M, Ahmad S, Al-Ani B, Fujisawa T, Coxall H, Chudasama K, et al. Negative regulation of soluble flt-1 and soluble endoglin release by heme oxygenase-1. Circulation. 2007;115:1789–1797. doi: 10.1161/CIRCULATIONAHA.106.660134. [DOI] [PubMed] [Google Scholar]
- 27.Zhu XY, Daghini E, Chade AR, Versari D, Krier JD, Textor KB, et al. Myocardial microvascular function during acute coronary artery stenosis: effect of hypertension and hypercholesterolaemia. Cardiovasc Res. 2009;83:371–380. doi: 10.1093/cvr/cvp140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Marco GS, Reuter S, Hillebrand U, Amler S, Konig M, Larger E, et al. The soluble vegf receptor sflt1 contributes to endothelial dysfunction in ckd. J Am Soc Nephrol. 2009;20:2235–2245. doi: 10.1681/ASN.2009010061. External Resolver Basic Bibliographic Links [Context Link] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamarat R, Silvestre JS, Durie M, Levy BI. Angiotensin ii angiogenic effect in vivo involves vascular endothelial growth factor- and inflammation-related pathways. Lab Invest. 2002;82:747–756. doi: 10.1097/01.lab.0000017372.76297.eb. [DOI] [PubMed] [Google Scholar]
- 30.Hiasa K, Ishibashi M, Ohtani K, Inoue S, Zhao Q, Kitamoto S, et al. Gene transfer of stromal cell-derived factor-1alpha enhances ischemic vasculogenesis and angiogenesis via vascular endothelial growth factor/endothelial nitric oxide synthase-related pathway: next-generation chemokine therapy for therapeutic neovascularization. Circulation. 2004;109:2454–2461. doi: 10.1161/01.CIR.0000128213.96779.61. [DOI] [PubMed] [Google Scholar]
- 31.Yamaguchi J, Kusano KF, Masuo O, Kawamoto A, Silver M, Murasawa S, et al. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation. 2003;107:1322–1328. doi: 10.1161/01.cir.0000055313.77510.22. [DOI] [PubMed] [Google Scholar]
- 32.Urbich C, Aicher A, Heeschen C, Dernbach E, Hofmann WK, Zeiher AM, Dimmeler S. Soluble factors released by endothelial progenitor cells promote migration of endothelial cells and cardiac resident progenitor cells. J Mol Cell Cardiol. 2005;39:733–742. doi: 10.1016/j.yjmcc.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 33.Fadini GP, Miotto D, Baesso I, Facco M, Miorin M, Tiengo A, et al. Arterio-venous gradient of endothelial progenitor cells across renal artery stenosis. Atherosclerosis. 2005;182:189–191. doi: 10.1016/j.atherosclerosis.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 34.Giannotti G, Doerries C, Mocharla PS, Mueller MF, Bahlmann FH, Horvath T, et al. Impaired endothelial repair capacity of early endothelial progenitor cells in prehypertension: relation to endothelial dysfunction. Hypertension. 2010;55:1389–1397. doi: 10.1161/HYPERTENSIONAHA.109.141614. [DOI] [PubMed] [Google Scholar]