

Abstract
GnRH regulation of pituitary gonadotropin gene transcription is critical for fertility, and metabolic dysregulation is associated with reproductive disorders and altered hypothalamic-pituitary responses. Here, we examined signaling pathways in gonadotropes through which GnRH modulates gonadotropin levels, and potential common signaling pathways with insulin. Using LβT2 cells, we show that GnRH rapidly (5 minutes) triggers activating phosphorylation of AMP-activated protein kinase (AMPK) up to 5-fold; this stimulation is enhanced by insulin through increased total AMPKα levels and activity. GnRH also stimulated c-Jun N-terminal kinase (JNK) and ERK activation, whereas insulin alone stimulated Akt. Inhibition of AMPK activity by compound C, or diminishing AMPK levels by small interfering RNA against AMPKα, prevented GnRH-stimulated transcription of the endogenous LHβ gene and transfected LHβ promoter. Egr-1 (early growth response-1), a transcription factor required for LHβ expression, is synthesized in response to GnRH, and compound C prevents this induction. However, overexpression of Egr-1 in the presence of compound C did not restore GnRH stimulation of LHβ, suggesting that AMPK stimulation of transcription also occurs through additional mechanisms or signaling pathways. One such pathway may be JNK activation, because GnRH stimulation of JNK activity and LHβ transcription occurs more slowly than stimulation of AMPK activity, and AMPK inhibition by compound C or small interfering RNA also prevented GnRH-stimulated JNK phosphorylation. Finally, in primary mouse pituitary cells, GnRH also stimulates AMPK, and AMPK inhibition suppresses GnRH-stimulated LHβ transcription. These studies indicate a novel role for AMPK in GnRH-stimulated transcription in pituitary gonadotropes and a potential common mechanism for GnRH and metabolic modulation of fertility.
Temporally regulated secretion and synthesis of the pituitary gonadotropins LH and FSH is critical for ovarian steroidogenesis and gamete production (1–4). Gonadotropin secretion and gene transcription are driven by hypothalamic GnRH pulses of defined frequency and amplitude and by physiological feedback from ovarian sex steroids and other hormones (1–4). There is also increasing evidence that metabolic signals influence fertility and that interaction between steroids and metabolism plays a role in regulating reproduction or in contributing to infertility (5, 6). For example, polycystic ovarian syndrome (PCOS) affects between 5% and 10% of women of reproductive age and is characterized by infertility and hyperandrogenism; in addition, many PCOS women are also obese and insulin resistant (6). The etiology of PCOS is unknown, but a neuroendocrine characteristic of many PCOS women is persistently rapid LH and GnRH pulses, contributing to elevated LH and increased LH to FSH serum ratios (6–9). The direct role of insulin or insulin resistance on LH serum levels in this population is not fully defined, but treatment of PCOS women with the insulin-sensitizing drug metformin has some success in reducing LH and insulin levels and in restoring menstrual cycles and fertility (8–12).
In rodent models, metabolic status and insulin action clearly impact reproduction, and a brain-specific knockout of the insulin receptor in mice results in both metabolic defects and infertility due to decreased LH synthesis and secretion (13). Similarly, mice in which the gene for the insulin signaling substrate, insulin receptor substrate (IRS)-2, was disrupted developed type 2 diabetes and infertility with reduced LH (14). Anterior pituitary cells contain insulin receptors (15), and insulin treatment can increase LH secretion and synthesis from cultured normal rodent or clonal pituitary cells (12, 16, 17) or augment GnRH-stimulated LH secretion and synthesis (17–19). The mechanism for this modulation has not been fully explained, but GnRH binding to its receptor on gonadotropes activates many intracellular signaling pathways, some of which are shared with insulin and other growth factors (18–28). For example, GnRH stimulation of gonadotropin secretion and gene transcription involves activation of protein kinase C, protein kinase A, Ca2+/calmodulin-dependent protein kinase, and MAPK and stress kinase pathways (21–29). The MAPK and stress kinases include ERK, p38, and c-Jun N-terminal kinase (JNK), with JNK stimulation by GnRH specifically favoring LHβ transcription (23–26). Insulin, acting via its cell surface receptor, stimulates intracellular phosphorylation of IRSs and other proteins (14, 20). These phosphorylation cascades result in activation of signaling pathways, including the phosphatidylinositol 3-kinase–Akt/protein kinase B pathway, which is responsible for many metabolic actions of insulin, the Ras-MAPK pathway, and stress kinases (20). Thus, MAPK and stress kinases may be at least one potential point of intersection in signaling between insulin and GnRH.
Basal and GnRH-stimulated LHβ gene transcription requires participation of several transcription factors, including the nuclear receptor steroidogenic factor 1 (SF1), the homeoprotein pituitary homeobox 1, and the early growth response protein (Egr-1), whose expression is dramatically regulated by GnRH (27–33). In one study, insulin exerted effects on LHβ promoter activity via Egr-1 synthesis, although the effects were much less robust than with GnRH (17). In another investigation (33), insulin treatment stimulated the LHβ promoter with no effect on Egr-1 or SF1 levels and was suggested to work via either protein-protein interactions or intracellular signaling pathways. In these studies, we demonstrate for the first time that GnRH acts via the AMP-activated protein kinase (AMPK) pathway. Inhibition of AMPK activity suppresses GnRH-stimulated LHβ transcription, activity of the stress kinase JNK, and enhancement of Egr-1 protein levels. These studies indicate a novel role for AMPK in GnRH-stimulated transcription in pituitary gonadotropes and a potential mechanism for metabolic signals to influence GnRH modulation of LH levels and fertility.
Materials and Methods
Reagents and plasmid constructs
Insulin, GnRH, and enzyme inhibitors including the AMPK inhibitor (compound C, 40μM), and MAPK kinase inhibitor (U0126, 10μM), were obtained from Sigma Chemical Company (St. Louis, Missouri). The JNK-specific inhibitor SP600125 (50nM; Calbiochem, San Diego, California) or the inactive isoform (termed vehicle in figures; 50nM JNK inhibitor II negative control/0.25% dimethylsulfoxide) were obtained from Calbiochem (San Diego, California). A rat LHβ promoter (from −617 to +44 bp relative to the transcriptional start site; −617LHβLuciferase) luciferase reporter construct has been described previously (25) and used to evaluate promoter activity. A cytomegalovirus (CMV) promoter-driven expression vector for Egr-1 (34) or empty vector pcDNA 3.1 was used in cotransfection studies to alter Egr-1 expression as indicated. For normalization studies, some cells were cotransfected with 0.2 μg pRL-TK Renilla luciferase (a kind gift of Dr Hui Li, University of Virginia) as a cotransfection control (35). Cell lysates were harvested 48 hours after transfection for analyses of firefly and Renilla luciferases with the Promega (Madison, Wisconsin) dual-luciferase reporter assay system. Similar normalization results were obtained by cotransfection and normalization of firefly luciferase activity with pRL-TK Renilla luciferase, 1.0 μg CMV–β-galactosidase, or cellular protein. Transfected empty vector resulted in no detectable luciferase activity.
Transfection and small interfering RNA studies
Clonal LβT2 gonadotropes were originally obtained from Dr Pamela Mellon (University of California, San Diego, California) and were maintained in DMEM supplemented with 10% fetal bovine serum and 1% antibiotic/antimycotic (GIBCO, Grand Island, New York). For transfection, cells were plated using phenol red-free DMEM with 5% charcoal-stripped newborn calf serum and 2% l-glutamine at a concentration of 500 000 cells per 20-mm well. Transfections with Lipofectamine 2000 (Invitrogen, Carlsbad, California) and analysis were as previously described (34). The LHβ promoter luciferase reporter construct (−617LHβLuciferase) was used at a concentration of 1.0 μg/well. After transfection, cells were treated for 6 hours with 100nM GnRH or with 20nM insulin as noted, alone or in combination, with GnRH added for the last 6 hours. For inhibitor studies, compounds were added 20 minutes before GnRH or insulin, unless otherwise described. For contransfection studies, the luciferase construct and expression vectors, with or without empty vector pcDNA 3.1 to normalize total DNA levels or with 0.2 μg pRL-TK Renilla luciferase, were added simultaneously. Luciferase assays and normalization and assessment of Egr-1 protein levels, were performed as described (34, 36) To knock down AMPK protein expression, siGENOME SMARTpool small interfering RNA (siRNA) reagents (0.1nM and 0.2nM; Dharmacon RNA Technologies, Lafayette, Colorado) targeting AMPKα were introduced into cells by nucleofection, simultaneously with −617LHβLuciferase, as previously described (34). A pool of siControl nontargeting siRNA #1 (0.2nM; Dharmacon) was used as a negative control. After incubation of cells for 24 or 48 hours, some wells were treated with GnRH for 6 hours. After treatment, some lysates were collected to assess expression of total AMPKα (antibody from Cell Signaling Technology, Beverly, Massachusetts) on 10% SDS-PAGE gels, and parallel wells used to measure LHβ promoter activity.
Enzyme immunoblotting and assays
Enzyme assays were performed on LβT2 cells treated with 100nM GnRH for 0 to 60 minutes (25, 37). LβT2 cell lysates were then collected in 2× gel loading buffer (100mM Tris-HCl [pH 6.8], 4% sodium dodecyl sulfate [SDS], 20% glycerol, 25mM NaF, 1mM Na3VO4). Proteins (20 μg) were resolved on 10% SDS-acrylamide gels at 140 V for 1.5 to 2 hours and then transferred to nitrocellulose membranes. Membranes were blocked in 5% nonfat dry milk in 20mM Tris base, 137mM NaCl, 3.8mM HCl, 0.1% Tween 20 and then incubated with primary antibodies specific for phosphorylated ERK1 and -2 (1:2000) and total (phosphorylated and unphosphorylated) ERK (1:2000), phosphorylated and total AKT (both at 1:1000), phosphorylated and total AMPKα (both at 1:1000), and phosphorylated and total JNK (all from Cell Signaling Technology). Antibody for mouse β-actin (1:50 000) was obtained from Sigma (St Louis, Missouri). Secondary antibodies were either horseradish peroxidase-conjugated donkey antirabbit Fab fragment IgG (1:5000; GE HealthCare, Piscataway, New Jersey) for ERK, Akt, and AMPK or sheep antimouse IgG (1:5000; Jackson Immunoresearch, West Grove, Pennsylvania) for β-actin and JNK. Protein was detected with SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, Illinois) followed by autoradiography. Films were analyzed by densitometry, and immunoreactive protein levels in each sample were normalized for either total enzyme or β-actin on the same blot. All samples for each assay were run at the same time on the same gel.
Primary transcript assay
Primary transcript assays for mouse LHβ and Egr-1 were performed as previously described (27, 34, 38). LβT2 cells were plated at a density of 2.5 × 106 cells per 40-mm well. Cells were treated for 60 minutes with 20nM insulin, 100nM GnRH, or both and then washed with PBS and collected using the QIAGEN (Valencia, California) RNeasy Mini Kit. In some studies, the AMPK inhibitor compound C was added 30 minutes before GnRH. Samples were then treated with 2 μL deoxyribonuclease for 1 hour at 37°C (Roche Applied Sciences, Indianapolis, Indiana). After phenol-chloroform extraction, samples were subjected to reverse transcription using Bio-Rad (Hercules, California) iScript cDNA synthesis kit and real-time PCR. Negative controls, in which reverse transcriptase was not added, were performed for each RNA sample; no DNA contamination was observed. LHβ primary transcript was measured by a forward primer crossing the first intron/exon border and a reverse primer located entirely within intron 1. The primers for LHβ were forward AGAGGCTCCAGGTAAGATGGTA and reverse CCACTCAGTATAATACAGAAAC. Primers for Egr-1 were as previously described (17, 34). Each sample was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA; normalized LHβ/GAPDH mRNA levels were expressed relative to values for vehicle-only–treated cells, which were set at 1.0.
LH secretory studies
LH secreted into the media from LβT2 cells was measured essentially as previously described (25, 39). Briefly, cells were plated (1 × 106 cells per well) in 12-well dishes and then treated overnight in DMEM containing 0.2% BSA. After washing, cells were incubated in the same media for secretory studies, including pretreatment with or without 20nM insulin for 1 hour, followed by 30 minutes with or without 100nM GnRH or 40μM compound C. After treatment, medium was collected and centrifuged to remove debris, and cells were lysed for total protein measurement. Mouse LH was measured by sandwich ELISA by the Ligand Core of the Center for Research in Reproduction at the University of Virginia. Results are from 6 experiments. All secreted LH values are normalized to total cellular protein (39) expressed as picograms LH per microgram cellular protein.
Mouse pituitary cell culture
Pituitaries from adult male mice were pooled and dissociated in medium containing 0.35% collagenase, 0.1% hyaluronidase, and 0.01% deoxyribonuclease as previously described (25, 27, 32). After dissociation, cells were suspended phenol red-free DMEM with 10% charcoal-stripped newborn calf serum and 2% l-glutamine and distributed into culture wells (1 × 106 to 2 × 106 cells per 35-mm well) as previously described (25). Cells were cultured for 24 hours before beginning each experiment, and all treatments were contained in the same experiment. Similar studies for AMPK activation were also conducted in pituitary fragments as previously described (27). Cells were treated with 2nM GnRH for indicated times and collected for AMPK activity or mRNA as for LβT2 cells.
Statistical analysis
The data for primary transcript mRNA, gonadotropin subunit primary transcripts, and rat LH promoter activity were analyzed by ANOVA, with differences between treatment groups determined by Duncan's multiple-range test, as previously described (27, 38).
Results
Insulin enhances GnRH-stimulated transcription of LHβ
To understand how insulin and GnRH may functionally interact on the gonadotrope, we examined the transcription of LHβ, because it is the gonadotropin subunit with the most robust transcriptional response to GnRH (3, 4), in LβT2 cells. Treatment with insulin alone had modest effects on the activity of the transfected LHβ promoter (Figure 1A, left panel) but significantly enhanced the stimulatory effect of GnRH on promoter activity, from 24- to 46-fold. To verify that this response occurred with the endogenous LHβ gene, we measured primary mRNA transcript levels under identical hormonal treatment conditions and found similar results. Insulin had no significant effect on LHβ transcription alone. However, insulin plus GnRH stimulated LHβ primary transcript levels 7.2-fold over vehicle control versus 3.2-fold for GnRH alone.
Figure 1.

A, Insulin enhances GnRH-stimulated LHβ promoter activity and endogenous gene transcription. Left panel, LβT2 cells were transfected with −617LHβLuciferase and treated with 20nM Insulin overnight (18–20 hours) and then with 100nM GnRH for 6 hours. Normalized luciferase activity was calculated and expressed as the mean ± SEM for 3 experiments. Right panel, GnRH-stimulated endogenous LHβ transcription is enhanced by insulin. LβT2 cells were treated overnight with 20nM insulin and then with 100nM GnRH for 90 minutes. LHβ primary mRNA transcripts (PT) were then measured using intron/exon-spanning primers and quantitative real-time PCR. Data are normalized to GAPDH mRNA levels and expressed relative to untreated controls. Error bars reflect SEM for triplicate samples in 3 experiments. Calculated GnRH stimulation over control is shown in parentheses. For both panels: *P ≤ .05 vs control; #P ≤ .05, insulin + GnRH vs GnRH. B, ERK, JNK, and AKT activities in LβT2 cells stimulated with GnRH, insulin, or both hormones. LβT2 cells were pretreated with 20nM insulin for either 1 or 23.5 hours and then with 100nM GnRH for an additional 30 minutes, with or without insulin. Time of total insulin treatment, with GnRH, is shown. Cell lysates (20 μg protein) were prepared for SDS-PAGE and immunoblots, and active, phosphorylated enzyme (P-enzyme), total enzyme (T-enzyme), or β-actin was detected with specific antibodies. Representative blots of 3 experiments are shown.
Because GnRH and insulin both act via cell surface receptors, we next evaluated whether they exerted their actions in gonadotrope cells through distinct or common intracellular signaling pathways. We first analyzed activation of ERK1/2 and JNK because both pathways are involved in GnRH signaling and gonadotropin transcription, and JNK specifically in GnRH-stimulated LHβ transcription (23–25). GnRH treatment stimulated ERK1/2-activating phosphorylations at Thr202/Tyr 204 (Figure 1B, upper left panel). Pretreatment with insulin before GnRH stimulation did not alter levels of phospho-ERK1/2 in basal or GnRH-stimulated conditions. Total levels of ERK1/2 remained stable through the time course tested. Similarly, GnRH increased the JNK-activating phosphorylation at Tyr185 (Figure 1B, upper right panel), whereas insulin pretreatment had no further effect. We next explored whether GnRH can stimulate pathways classically associated with insulin. Insulin stimulated phosphorylation of AKT at Ser473 within 1.5 hours of treatment (Figure 1B, lower left panel). Overnight insulin treatment had no sustained effect on phospho-AKT levels, and GnRH had no effect at either time of insulin treatment.
Identification of AMPK as a new GnRH signaling pathway and common pathway for insulin and GnRH signaling
We then analyzed the effect of insulin and GnRH on the phosphorylation and activation of AMPK, a fuel-sensing enzyme that is activated by increases in the intracellular AMP to ATP ratio (40, 41). Insulin has been reported to modulate AMPK activity, either via influence on other signaling pathways or indirectly via influencing the energy charge of the cell, and drugs that target AMPK are used to treat insulin-resistant patients (9–12, 40, 41). Treatment of LβT2 cells with GnRH alone for 5 to 60 minutes showed that phosphorylation of the AMPKα subunit at Thr172 was rapidly stimulated by GnRH, reaching maximal levels (2- to 5-fold in several experiments) at 5 minutes; activity then subsided although remained elevated over basal levels through 60 minutes of treatment (Figure 2). The time course of AMPK activation is similar to that of GnRH stimulation of ERK1/2 and much faster than activation of Ca2+/calmodulin-dependent protein kinase II and JNK (22–27) and suggests that AMPK is activated early in the GnRH signaling pathway. GnRH treatment has no significant effect on total AMPK protein during this time period as shown in Figures 2 and 3, and in 5 separate experiments, total AMPK was unchanged relative to β-actin (vehicle = 1.02 ± 0.22; 30 minutes GnRH = 0.98 ± 0.15; 60 minutes GnRH = 1.02 ± 0.18 vs β-actin control).
Figure 2.
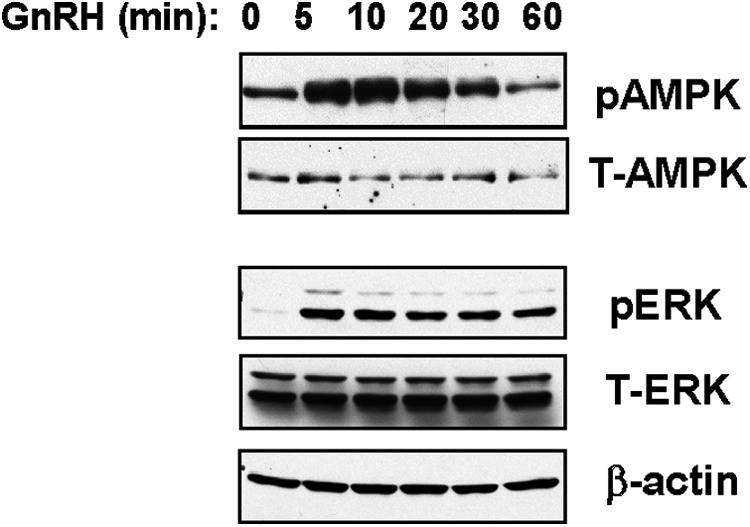
GnRH rapidly stimulates AMPK phosphorylation in LβT2 cells. LβT2 cells were treated with 100nM GnRH for 5 to 30 minutes and then analyzed for activated phosphorylated AMPK (P-AMPK) and total AMPK (T-AMPK) or on a separate blot for ERK (P-ERK), total ERK (T-ERK), and β-actin by immunoblots of cell lysates (20 μg protein).
Figure 3.
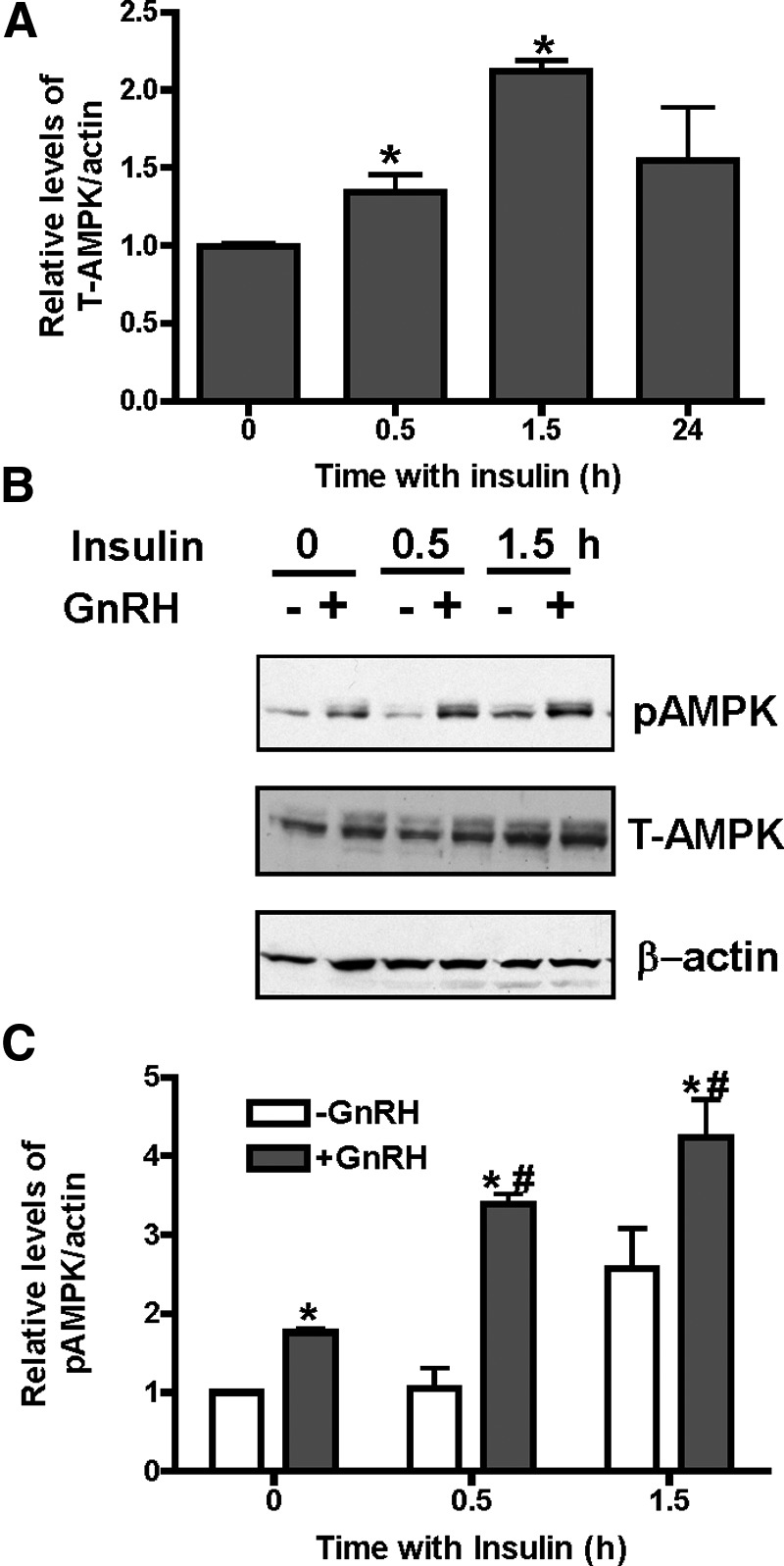
GnRH and insulin stimulate AMPK activity, whereas insulin increases total AMPKα levels. A, LβT2 cells were treated with 20nM insulin for 0, 30, or 90 minutes or 24 hours and then analyzed for total (T-AMPK) AMPKα and β-actin by immunoblots of cell lysates (20 μg protein). Densitometric analysis was performed, and T-AMPK was normalized for β-actin. Bars represent values averaged for 6 samples each. *P ≤ .05, vehicle vs insulin. B, LβT2 cells were treated with 20nM insulin simultaneously with GnRH (0 time) or for 20 or 80 minutes before adding 100nM GnRH for 10 minutes and then analyzed for activated phosphorylated (P-AMPK) or total (T-AMPK) αAMPK and β-actin by immunoblots of cell lysates (20 μg protein). Time of insulin treatment includes pretreatment plus cotreatment with GnRH. C, Densitometric analysis was performed on 3 experiments as in B, and P-AMPK was normalized for β-actin. Bars represent values averaged from 3 experiments with 3 to 6 individual samples each. *P ≤ .05, vehicle vs GnRH; #, P ≤ .05, insulin + GnRH vs GnRH.
Figure 5.
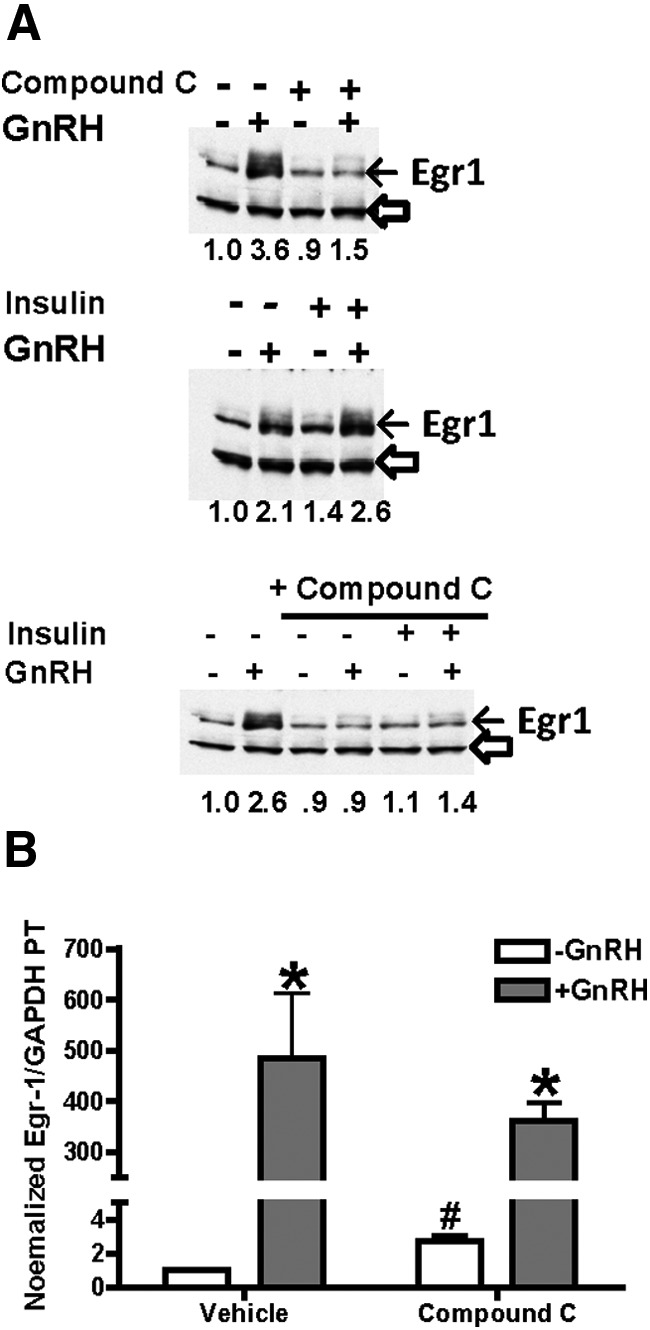
GnRH-stimulated Egr-1 protein levels are inhibited by compound C. A, LβT2 cells were treated with 20nM insulin for 1 hour and then with 100nM GnRH for 30 minutes. Some cells were also pretreated with 40μM compound C for 30 minutes before GnRH. After treatment, cells were collected for protein and RNA measurement. Protein lysates (20 μg) were analyzed on SDS-PAGE gels and immunoblots with antibodies against Egr-1 and β-actin on the same blot. The migration of Egr-1 is shown by the closed arrow and the loading control by the open arrowhead on each blot. Calculated normalized Egr-1 levels are shown under each lane relative to untreated control expressed as 1.0. B, In some cells, Egr-1 mRNA primary transcript was measured by RT-PCR and normalized for GAPDH mRNA in the same samples. Values are expressed as the mean ± SEM for 3 experiments. *P < .05, treatment vs GnRH; #P < .05, vehicle vs compound C.
Treatment of LβT2 cells with insulin alone results in significantly increased levels of AMPKα protein (total AMPK), normalized for β-actin, after 30 and 60 minutes (Figure 3A), from 2.0- to 2.5-fold over several experiments. Insulin also slightly stimulated AMPK phosphorylation (Figure 3B), but never to the extent of GnRH. Pretreatment of cells with insulin before GnRH treatment thus resulted in increased total and activated AMPK compared with untreated controls and increased overall phosphorylated AMPK when normalized to cellular protein and actin (Figure 3C). Thus, the AMPK signaling pathway is stimulated by both GnRH and insulin, with only insulin pretreatment significantly increasing total AMPKα protein.
Transcription of LHβ requires AMPK activity
To determine whether AMPK activation is necessary for GnRH-stimulated transcription of LHβ, we performed both AMPK inhibitor (compound C) and AMPK siRNA knockdown studies. Treatment of LβT2 cells transfected with −617LHβLuciferase (Figure 4A) with GnRH, insulin, and inhibitor showed that compound C inhibited simulated transcription from the LHβ promoter by over 80%. Basal transcription from the transfected promoter was very low and unaffected by compound C. To corroborate these data, we also examined expression of endogenous LHβ primary transcript in LβT2 cells treated with or without GnRH or compound C (Figure 4B). In agreement with the luciferase data, compound C inhibited GnRH-stimulated LHβ primary transcript mRNA from 4.7-fold to 2.1-fold.
Figure 4.

Inhibition of AMPK with compound C or siRNA suppresses GnRH-stimulated LHβ promoter activity and endogenous gene transcription. A, LβT2 cells were transfected with −617LHβLuciferase and treated with 20nM insulin with or without 100nM GnRH for 6 hours. The AMPK inhibitor compound C (40μM) or vehicle (dimethylsulfoxide) was added 30 minutes before GnRH. After treatment, cells were collected and luciferase activity measured. Data are averaged from 3 experiments. *P ≤ .05, vehicle vs GnRH; #P ≤ .05, insulin + GnRH vs GnRH; a, P ≤ .05, treatment vs treatment + compound C. B, LβT2 cells were treated with 100nM GnRH, with 40μM compound C added for 10 minutes before 90 minutes GnRH. After treatment, cells were collected and levels of LHβ primary transcript (PT) were quantified by RT-PCR, using primers to exon 1 and intron 1. Samples were from 3 independent experiments and were normalized against GAPDH mRNA in the same samples. *P ≤ .05 GnRH vs vehicle; #P ≤ .05, treatment vs treatment + compound C. C, AMPK expression was decreased by introduction of siGENOME SMARTpool siRNA reagents (0.1nM and 0.2nM; Dharmacon) targeting AMPKα1/2 by nucleofection, simultaneously with −617LHβLuciferase; parallel wells were treated with siControl nucleotides. After 42 hours, cells were treated with 100nM GnRH and collected for either luciferase activity or immunoblots for AMPKα1/2 or β-actin (20 μg total protein). A representative blot for AMPK knockdown is shown in Figure 7. Data are averaged from 3 experiments. *P ≤ .05 GnRH vs vehicle; #, P ≤ .05, GnRH vs GnRH + siAMPK.
As another test for the role of AMPK activity, we decreased cellular levels of AMPKα, the catalytic subunit, by siRNA treatment. Treatment of cells with 0.2nM siRNA for AMPKα reduced cellular AMPKα levels to approximately 50% of control (blot shown in Figure 7) and resulted in inhibition of GnRH-stimulated LHβ transcription by approximately 40% (Figure 4C). Overall, these data support a role for AMPK in the GnRH-stimulated transcription of LHβ.
Figure 7.
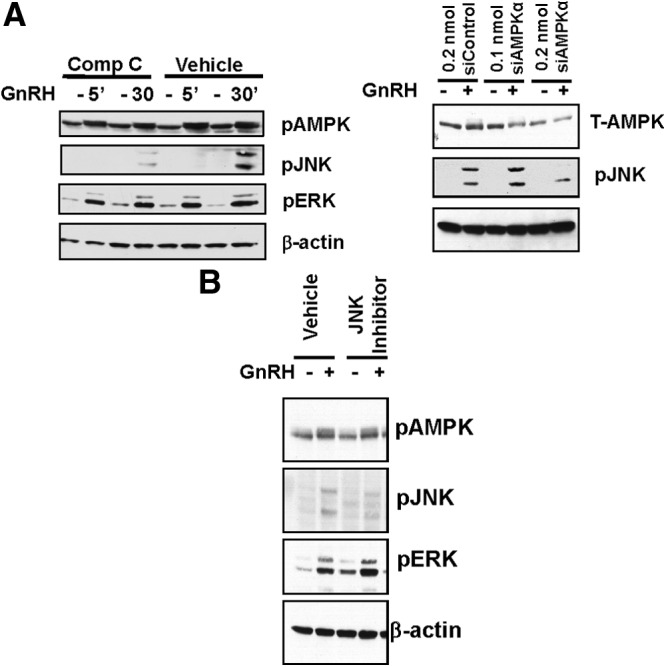
Inhibition of AMPK suppresses GnRH-stimulated JNK phosphorylation. LβT2 cells were preincubated for 30 minutes with vehicle or specific inhibitors for AMPK (40μM compound [Comp] C) (A, left panel) or JNK (50nM SP600125) (B); cells were then treated with vehicle or 100nM GnRH for 15 minutes or indicated times. Cell lysates (20 μg protein) were prepared for SDS-PAGE and immunoblots, and active, phosphorylated enzyme (P-enzyme), total enzyme (T-enzyme), or β-actin was detected with specific antibodies. Representative blots of 3 experiments are shown. Some cells were also treated with siRNA for AMPKα or control siRNA (A, right panel) at indicated times and concentrations and then treated with GnRH and analyzed for total AMPK or JNK phosphorylation on SDS gels (20 μg protein per well). A representative blot (of 3 independent experiments) is depicted.
GnRH stimulation of Egr-1 protein requires AMPK activity
Expression of LHβ requires several transcription factors such as SF1 and Egr-1, and a critical component of GnRH stimulation of LHβ transcription is increased expression of the early response protein Egr-1 (30–33). Insulin has been reported both to independently stimulate Egr-1 (17) and to have no effect on transcription factor levels (33). We observed only a modest (approximately 1.3- to 1.8-fold stimulation over 3 experiments) stimulation of Egr-1 protein by insulin alone vs control (Figure 5A), and this effect was never as robust as that of GnRH. Treatment with the AMPK inhibitor compound C eliminated the increase in Egr-1 protein by GnRH. To evaluate whether AMPK is also required for the transcription of Egr-1, we concentrated on the robust GnRH response and analyzed the levels of Egr-1 primary transcript mRNA. As shown in Figure 5B, GnRH greatly enhanced the transcription of Egr-1. Compound C treatment alone increased basal transcription of Egr-1 by 3.4-fold (white bars). However, in the presence of compound C, Figure-stimulated Egr-1 transcription was approximately 130-fold, compared with over 500-fold stimulation by GnRH alone (gray bars). Thus, these data together suggest AMPK is critical for the stimulation of Egr-1 levels in response to GnRH, possibly through both transcription and posttranscriptional mechanisms.
Because Egr-1 is essential to support GnRH-induced transcription of LHβ, we performed experiments to determine whether restoration of Egr-1 levels can overcome the effects of AMPK inhibition. Increasing amounts of an Egr-1 expression vector were introduced into LβT2 cells along with the LHβ promoter-luciferase reporter. In the absence of exogenous Egr-1, compound C inhibited GnRH-induced transcription from the LHβ promoter (Figure 6, top panel). As observed previously, increased expression of Egr-1 decreased GnRH stimulation (due to increased basal expression from the promoter) (34) but failed to restore GnRH stimulation in the presence of compound C (Figure 6, middle and bottom panels). Thus, although Egr-1 synthesis in response to GnRH is crucial for the transcription of LHβ, increased Egr-1 levels alone cannot bypass compound C inhibition.
Figure 6.
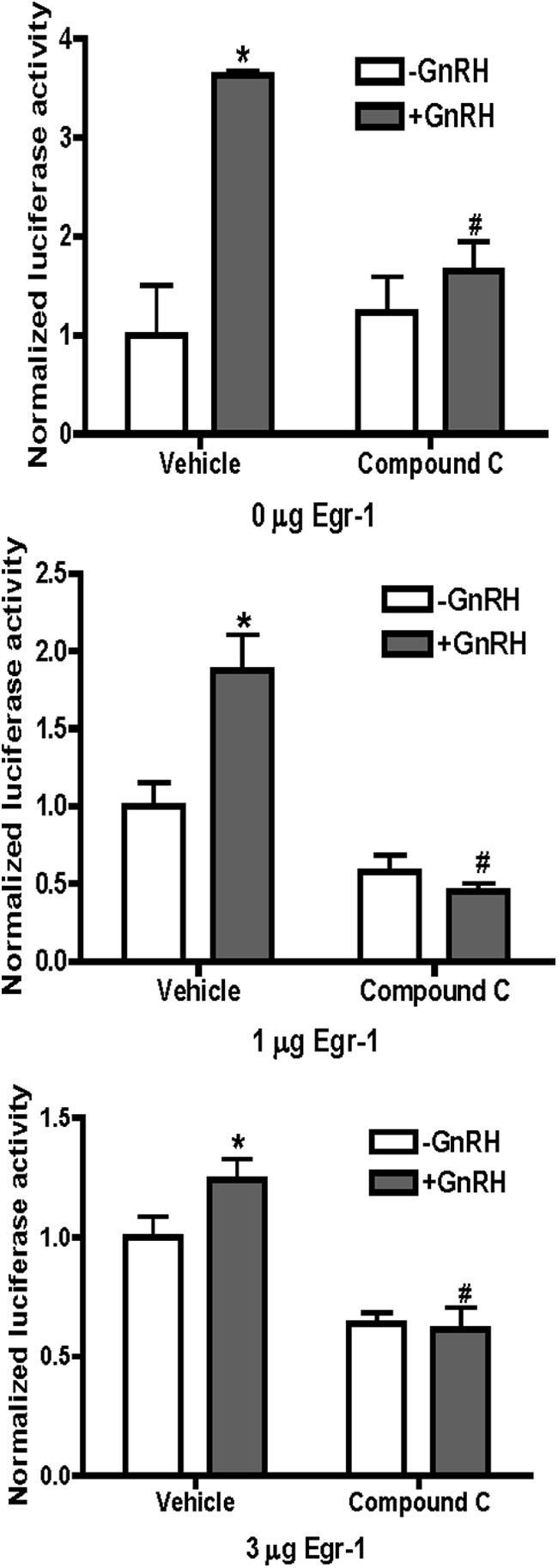
Restoration of Egr-1 protein levels does not overcome AMPK suppression of GnRH-stimulated LHβ. LβT2 cells were transfected with −617LHβLuciferase and constant amounts (3 μg) of total DNA, including the empty CMV expression vector (pcDNA 3.1) or CMV-Egr-1. Cells were treated with GnRH and/or compound C as in Figure 5. Normalized luciferase activity was calculated and expressed as the mean ± SEM for 3 experiments in triplicate. *P < .05, treatment vs GnRH; #P < .05, GnRH vs GnRH plus compound C.
Inhibition of AMPK suppresses JNK phosphorylation in response to GnRH
Because GnRH stimulation of AMPK is so rapid, we tested whether there might be potential cross talk between the AMPK and other downstream signaling pathways. We treated LβT2 cells with GnRH in the absence or presence of the AMPK inhibitor compound C or JNK inhibitor SP600125 and performed immunoblots for phosphorylated enzymes ERK1/2 and JNK as a surrogate for enzyme activation. Compound C can bind to unphosphorylated and phosphorylated mimics of AMPK; it dramatically alters the conformation of the activation loop, which partially overlaps with the putative ATP-binding pocket and prevents enzyme activity (42). Thus, compound C does not need to completely abolish AMPK phosphorylation to suppress AMPK activity. Because JNK stimulation by GnRH is not maximal until approximately 15 to 30 minutes of GnRH treatment and ERK1/2 stimulation is much faster (23–25), JNK was a potential candidate for influence by AMPK. As shown in Figure 7A, left panel, GnRH stimulates AMPK and ERK1/2 phosphorylation at 5 minutes, but GnRH stimulation of JNK requires longer time frames and was maximal only at 30 minutes of GnRH, as previously observed (25). Compound C treatment (note left side of panel) nearly abolished GnRH stimulation of JNK phosphorylation but had no effect on GnRH-stimulated ERK1/2 phosphorylation. Basal levels of ERK phosphorylation, however, were slightly increased. When AMPKα levels were decreased by siRNA by approximately 40% (Figure 7A, right panel), GnRH stimulation of JNK phosphorylation was also significantly suppressed. In contrast, treatment of cells with the JNK inhibitor SP600125 did not alter the increase in phospho-AMPK levels induced by GnRH, although it did significantly suppress JNK phosphorylation (Figure 7B). The ERK1/2 inhibitor U0126 similarly had no effect on AMPK phosphorylation (not shown). Together, these data support the hypothesis of cross talk between the AMPK and JNK pathways, with AMPK influencing JNK activity.
Inhibition of AMPK activity influences LH secretion
Given the significant effects of AMPK inhibition on LHβ gene transcription, we tested the LH secretory response with and without compound C. As shown in Figure 8, GnRH significantly stimulated LH secretion from LβT2 cells. The addition of compound C significantly increased basal secretion of LH. There was no significant effect of compound C on the amount of LH secreted in response to GnRH among the various conditions, but the effect of increasing basal secretion resulted in a lower calculated fold stimulation for GnRH in the presence of compound C (from 5.0- to 2.2-fold or 3.8- to 1.7-fold, in the absence or presence of GnRH, regardless of the presence of insulin).
Figure 8.

Compound C influences LH secretion. LβT2 cells were incubated overnight in DMEM containing 0.2% BSA. After washing, cells were incubated in the same media for secretory studies, including pretreatment with or without 20nM insulin for 1 hour, followed by 30 minutes with or without 100nM GnRH or 40μM compound C. All secreted LH values are normalized to total cellular protein and expressed as picograms LH per microgram cellular protein. Data are expressed as mean ± SEM for 4 experiments. GnRH-stimulated secretion is shown as fold change in parentheses over the GnRH bars. *P < .05, GnRH vs vehicle control; a, P < .05, compound C vs vehicle control.
AMPK is stimulated by GnRH and is required for LHβ transcription in primary mouse pituitary cells
To assess the involvement of AMPK on GnRH-induced transcription of LHβ in a more physiological system, normal mouse pituitary cells were used. We first tested whether AMPK was activated in response to GnRH. After 10 or 20 minutes with GnRH, AMPK phosphorylation was clearly stimulated in primary cells (5.1- and 1.8-fold, respectively) in Figure 9A. Over several experiments, similar results were noted in acute treatment of freshly isolated pituitaries or dispersed cells. A potential role of AMPK on gonadotropin transcription in mouse pituitaries was investigated by measuring LHβ mRNA primary transcript in dispersed mouse pituitary cells (27, 34). Pituitary cells were treated in culture with or without GnRH and either vehicle or compound C (Figure 9B). As in LβT2 cells, GnRH robustly stimulated LHβ primary transcript mRNA, and this stimulation was significantly suppressed by AMPK inhibition. Thus, normal mouse gonadotropes also respond to GnRH stimulation with increased activating AMPK phosphorylation, and AMPK activity appears to be required for GnRH stimulation of LHβ gene transcription.
Figure 9.

AMPK is activated by GnRH in mouse pituitary cells and is required for GnRH stimulation of LHβ transcription. A, Mouse primary pituitary cells were treated with 2nM GnRH for 10 or 20 minutes for AMPK activation and then collected for SDS-PAGE and immunoblots. B, To measure transcription of LHβ, pituitary cells from male mice were dissociated by enzymatic digestion and allowed to attach to culture wells overnight. Cells were treated with 2nM GnRH for 20 minutes with or without preincubation for 10 minutes with 40μM compound C. After treatment, total RNA was prepared and primary transcript mRNA (PT-mRNA) for LHβ was measured by quantitative real-time RT-PCR, normalized to mRNA for GAPDH. *P < .05, treatment vs GnRH; #P < .05, GnRH vs GnRH plus compound C.
Discussion
Appropriate regulation of GnRH pulse patterns and pulsatile GnRH regulation of pituitary gonadotropin subunit gene transcription are required for fertility (1–4). Pituitary responses to GnRH may be influenced in conditions such as polycystic ovarian disease by altered levels of or responses to androgens and insulin or by metabolic phenotypes such as insulin resistance (3–8). Many PCOS women in fact exhibit increased LH secretion and increased LH to FSH ratios in serum, which could occur from altered biological responses at the level of the hypothalamus, pituitary, or both; similar results are observed in some animal models that mimic PCOS (7–9, 43). However, the mechanisms by which androgens, insulin, and GnRH interact to control gonadotropin under these conditions are unclear. In these studies, we have identified AMPK as an important signaling molecule for GnRH and found that its activity is important for GnRH stimulation of LHβ transcription. We suggest that the AMPK pathway may be a common site for signaling between GnRH and metabolic hormones and a potential mechanism for metabolic signals to influence GnRH modulation of LH levels and fertility.
In some studies, insulin directly influenced secretion of LH from isolated pituitary cells (16–19) and stimulated LHβ promoter activity (17, 33), at least in part via stimulation of Egr-1 levels. Other investigators found that GnRH and insulin may modulate each other's signaling in gonadotrope cells, showing that insulin alone had no effect on ERK1/2 phosphorylation but enhanced GnRH stimulation of ERK1/2 and cap-dependent protein translation (44). Under similar conditions in our studies, insulin enhanced GnRH-stimulated LHβ gene transcription (Figure 1A), but we did not observe cross talk via previously identified GnRH signaling pathways, such as ERK1/2 (21–23). Insulin treatment of LβT2 gonadotrope cells had no effect on the time course or extent of GnRH-stimulated JNK or ERK1/2 activity (Figure 1B), and GnRH itself did not alter activation of AKT, a major stimulatory pathway of insulin.
In contrast, GnRH alone robustly stimulated AMPK activity, and AMPK may be a major common pathway by which GnRH may intersect with insulin and metabolic signaling in the gonadotrope (Figures 2 and 3). Insulin treatment alone modestly stimulated phosphorylated and thus activated AMPKα (at Thr172) but also increased levels of total AMPKα protein. This correlates with 2.4- ± 0.2-fold increased levels of AMPKα1 and -2 mRNA in microarray studies of LβT2 cells treated for 1 hour with 20nM insulin (not shown) but does not rule out posttranscriptional pathways of regulation, such as the translational control with insulin and GnRH noted by Navratil et al (44). GnRH rapidly (5 minutes) stimulated AMPK activity by 5-fold and maintained 3-fold stimulated AMPK levels at 1 hour, but no changes in total AMPKα protein were observed in any study. Thus, treatment with both hormones increases total and active AMPK and thus overall AMPK activity.
Treatment with the AMPK inhibitor compound C or knockdown of AMPKα suppressed GnRH-stimulated LHβ promoter activity and transcription of the endogenous LHβ gene in LβT2 cells (Figure 4), suggesting that AMPK activity plays a significant role in GnRH regulation of gonadotropes. In support of this hypothesis, compound C also prevented GnRH stimulation of Egr-1 protein (Figure 5), which is required for increased LHβ expression (17, 29–34). AMPK has also been implicated in the stimulation of Egr-1 levels in liver cells, where it contributes to the inhibition of gluconeogenesis through transcription of several genes (45). Alteration of Egr-1 levels alone was not enough to explain effects of AMPK in gonadotropes, however, because overexpression of Egr-1 failed to rescue LHβ transcription in the presence of compound C (Figure 6), suggesting that either additional proteins or additional posttranscriptional modifications of Egr-1 or other proteins were required. This agrees with previous studies suggesting that GnRH (21, 46) can act via posttranscriptional modifications of, or interactions between, Egr-1 and SF1.
AMPK is a fuel-sensing enzyme activated by decreases in a cell's energy state, reflected by an increased AMP to ATP ratio (40, 41). When activated, it initiates metabolic and genetic events to restore ATP levels. AMPK achieves its downstream effects by direct phosphorylation of enzyme substrates such as acetyl-coenzyme A carboxylase, glycogen synthase, IRS-1, and hydroxyl-methylglutaryl-coenzyme A reductase but can also have long-term effects on gene expression and on specific transcription factors (40, 45–48). For example, AMPK directly phosphorylates hepatocyte nuclear factor 4α and represses its transcriptional activity (48). APMK can also phosphorylate the transcriptional cofactor p300 at serine 89 and dramatically reduce its interaction with nuclear receptors such as peroxisome proliferator-activated receptorγ and thyroid hormone receptor, without affecting p300 interaction with the non-nuclear receptor transcription factors adenovirus early region 1a protein, p53, or GATA4 (49). Such a phosphorylation might favor Egr-1 interactions with p300 in gonadotrope cells, as suggested by Berasi et al. (45). In the pituitary, AMPK stimulates transcription of the POMC gene, potentially via AP1 proteins (50). In contrast, Tosca et al (51) found that AMPK stimulation by metformin decreased gonadotropin secretion and MAPK (ERK1/2) phosphorylation induced by GnRH and suppressed FSHβ subunit expression and SMA and MAD related protein-2 (SMAD-2) phosphorylation induced by activin.
Based on the suppression of activin-stimulated FSHβ gene transcription by AMPK noted by Tosca et al (51) and these studies showing AMPK stimulation is required to increase LHβ transcription, it appears that AMPK could act to influence stimulated LHβ and FSHβ transcription in opposite directions. Thus, AMPK stimulation might amplify other differences in physiological regulation that might occur through steroids such as androgens or growth factors such as insulin. AMPK stimulation also precedes GnRH stimulation of JNK activation and AMPK inhibition suppresses JNK activation (Figure 7). JNK activation specifically favors GnRH-stimulated LHβ transcription in normal pituitary cells, with only FSHβ basal expression requiring JNK activity (24). Other investigators have demonstrated that AMPK stimulation increases activity of stress kinases, including ERK p42/44, p38, and JNK in liver (52); however, direct links between AMPK, these enzymes, and Egr-1 induction or regulated transcription was not tested.
This is to our knowledge the first demonstration of GnRH signaling via AMPK and, because GnRH also stimulates AMPK activity in normal mouse gonadotropes (Figure 9), suggests that AMPK may be an important intermediary of GnRH-regulated transcription in gonadotropes and a key link between energy pathways and reproduction. Adiponectin has also been reported to stimulate AMPK in LβT2 cells, although with a slower time course than with GnRH noted here, and to selectively decrease LH versus FSH secretion (39). This is in contrast to the work of Tosca et al (51) in which treatment with metformin increased AMPK activity in gonadotropes but decreased secretion of both LH and FSH while suppressing FSHβ transcription. Our results (Figure 8), showing that AMPK inhibition increased basal LH secretion agrees with the results of Lu et al (39) in which introduction of a dominant-negative AMPK increased basal LH secretion and overcame effects of AMPK stimulation by adiponectin. AMPK inhibition did not suppress the amount of hormone secreted in response to GnRH, but because basal LH secretion was higher, the fold stimulated LH secretion can be less (Figure 8). The less robust effect of adiponectin on AMPK activity and slower time course of activation may play a role in overall cellular response compared with GnRH; alternatively, additional signaling pathways stimulated by GnRH may also modify responses. Interestingly, under some conditions, AMPK activation was reported to suppress hormone secretion (ACTH) even if transcription was stimulated, as for pro-opiomelanocortin/ACTH (49), and indicates that both cell context and other hormonal signals and signaling pathways will play a role in overall responses of both gene expression and hormone secretion.
Recent studies suggest that coordinated responses to metabolic and reproductive hormones will be complicated, with tissue-specific effects. Hypothalamic GnRH neurons sense low glucose via AMPK activation, an effect that is attenuated in the presence of dihydrotestosterone (53), suggesting interplay of steroids, metabolic cues, and GnRH secretion. Diet-induced obesity in mice results in infertility; mice with a pituitary-specific knockout of the insulin receptor are immune to diet-induced obesity (54), suggesting that insulin signaling in the pituitary is required for the reproductive impairment and that the gonadotrope maintains insulin sensitivity in a setting of peripheral insulin resistance. Both the ovary and the pituitary were shown to maintain normal insulin signaling in obese mice, where metabolic tissues exhibited insulin resistance (55). In this setting, retained sensitivity of the pituitary and ovary to insulin could contribute to the pathophysiology of reproductive disorders, such as might occur in PCOS. Insulin can modulate total AMPK levels and shares the AMPK signaling pathway with GnRH. Clearly, additional studies are needed to fully understand the interplay of hormones and metabolic signals. Our studies show that the reproductive hormone GnRH can rapidly activate AMPK in the gonadotrope and that AMPK activation is required for the transcription of LHβ. AMPK effects are mediated, at least in part, by the control of Egr-1 protein levels and by the activation of JNK, both required for LHβ transcription. This places AMPK activity in a critical regulatory role for gonadotrope activity and a potentially important intermediary in the control of fertility.
Acknowledgments
We gratefully acknowledge support from the National Institutes of Health through a project (to M.A.S) and the Ligand Core laboratory of the Center for the Study of Reproduction at the University of Virginia supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development/National Institutes of Health cooperative agreement (U54 HD28934) as part of the Specialized Cooperative Centers Program in Infertility and Reproductive Research.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AMPK
- AMP-activated protein kinase
- CMV
- cytomegalovirus
- Egr-1
- early growth response protein
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- IRS
- insulin receptor substrate
- JNK
- c-Jun N-terminal kinase
- PCOS
- polycystic ovarian syndrome
- SF1
- steroidogenic factor 1
- SDS
- sodium dodecyl sulfate
- siRNA
- small interfering RNA.
References
- 1. Belchetz PE, Plant TM, Nakai Y, Keough EJ, Knobil E. Hypophysial responses to continuous and intermittent delivery of hypothalamic gonadotropin-releasing hormone. Science. 1978;202:631–633 [DOI] [PubMed] [Google Scholar]
- 2. Crowley WF, Jr, Filicori M, Spratt DI, Santoro NF. The physiology of gonadotropin- releasing hormone (GnRH) secretion in men and women. Recent Prog Horm Res. 1985;41:473–531 [DOI] [PubMed] [Google Scholar]
- 3. Shupnik MA. Gonadotropin gene modulation by steroids and gonadotropin-releasing hormone. Biol Reprod. 1996;54:279–286 [DOI] [PubMed] [Google Scholar]
- 4. Ferris HA, Shupnik MA. Mechanisms for pulsatile regulation of the gonadotropin subunit genes by GNRH1. Biol Reprod. 2006;74:993–998 [DOI] [PubMed] [Google Scholar]
- 5. Wade GN, Schneider JE, Li HY. Control of fertility by metabolic cues. Am J Physiol. 1996;270:E1–E19 [DOI] [PubMed] [Google Scholar]
- 6. Dunaif A, Thomas A. Current concepts in the polycystic ovary syndrome. Annu Rev Med. 2001;52:401–419 [DOI] [PubMed] [Google Scholar]
- 7. McCartney CR, Eagleson CA, Marshall JC. Regulation of gonadotropin secretion: implications for polycystic ovary syndrome. Semin Reprod Med. 2002;20(4):317–326 [DOI] [PubMed] [Google Scholar]
- 8. Baptiste CG, Battista MC, Trottier A, Baillargeon JP. Insulin and hyperandrogenism in women with polycystic ovary syndrome. J Steroid Biochem Mol Biol. 2010;122(1–3):42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Legro RS, Barnhart HX, Schlaff WD, et al. ; Cooperative Multicenter Reproductive Medicine Network Clomiphene, metformin, or both for infertility in the polycystic ovary syndrome. N Engl J Med. 2007;356:622–624 [DOI] [PubMed] [Google Scholar]
- 10. Nestler JE, Jakubowicz DJ, Evans WS, Pasquali R. Effects of metformin on spontaneous and clomiphene-induced ovulation in the polycystic ovary syndrome. N Engl J Med. 1998;338:1876–1880 [DOI] [PubMed] [Google Scholar]
- 11. Genazzani AD, Battaglia C, Malavasi B, Strucchi C, Tortolani F, Gamba O. Metformin administration modulates and restores luteinizing hormone spontaneous episodic secretion and ovarian function in nonobese patients with polycystic ovary syndrome. Fertil Steril. 2004;81:114–119 [DOI] [PubMed] [Google Scholar]
- 12. Oride A, Kanasaki H, Purwana IN, Miyazaki K. Effects of metformin administration on plasma gonadotropin levels in women with infertility, with an in vitro study of the direct effects on the pituitary gonadotrophs. Pituitary. 2010;13:236–241 [DOI] [PubMed] [Google Scholar]
- 13. Bruning JC, Gautam D, Burks DJ, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125 [DOI] [PubMed] [Google Scholar]
- 14. Burks DJ, Font de Mora J, Schubert M, et al. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature. 2000;407:377–382 [DOI] [PubMed] [Google Scholar]
- 15. Werther GA, Hogg A, Oldfield BJ, et al. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology. 1987;121:1562–1570 [DOI] [PubMed] [Google Scholar]
- 16. Weiss JM, Polack S, Diedrich K, Ortmann O. Effects of insulin on luteinizing hormone and prolactin secretion and calcium signaling in female rat pituitary cells. Arch Gynecol Obstet. 2003;269:45–50 [DOI] [PubMed] [Google Scholar]
- 17. Buggs C, Weinberg F, Kim E, Wolfe A, Radovick S, Wondisford F. Insulin augments GnRH-stimulated LHβ gene expression by Egr-1. Mol Cell Endocrinol. 2006;249:99–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Soldani R, Cagnacci A, Yen SS. Insulin, insulin-like growth factor I (IGF-I) and IGF-II enhance basal and gonadotrophin-releasing hormone-stimulated luteinizing hormone release from rat anterior pituitary cells in vitro. Eur J Endocrinol. 1994;131:641–645 [DOI] [PubMed] [Google Scholar]
- 19. Xia YX, Weiss JM, Polack S, Diedrich K, Ortmann O. Interactions of insulin-like growth factor-I, insulin and estradiol with GnRH-stimulated luteinizing hormone release from female rat gonadotrophs. Eur J Endocrinol. 2001;144:73–79 [DOI] [PubMed] [Google Scholar]
- 20. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96 [DOI] [PubMed] [Google Scholar]
- 21. Bliss SP, Navratil AM, Xie J, Roberson MS. GnRH signaling, the gonadotrope and endocrine control of fertility. Front Neuroendocrinol. 2010;31:322–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haisenleder DJ, Ferris HA, Shupnik MA. The calcium component of GnRH-stimulated LH subunit gene transcription is mediated by calcium/calmodulin-dependent protein kinase type II. Endocrinology. 2003;144:2409–2416 [DOI] [PubMed] [Google Scholar]
- 23. Harris D, Bonfil D, Chuderland D, Kraus S, Seger R, Naor Z. Activation of MAPK cascades by GnRH: ERK and Jun N-terminal kinase are involved in basal and GnRH-stimulated activity of the glycoprotein hormone LHβ-subunit promoter. Endocrinology. 2002;143:1018–1025 [DOI] [PubMed] [Google Scholar]
- 24. Yokoi T, Ohmichi M, Tasaka K, et al. Activation of the luteinizing hormone β promoter by gonadotropin-releasing hormone requires c-Jun NH2-terminal protein kinase. J Biol Chem. 2000;275:21639–21647 [DOI] [PubMed] [Google Scholar]
- 25. Haisenleder DJ, Burger LL, Walsh HE, et al. Pulsatile gonadotropin-releasing hormone stimulation of gonadotropin subunit transcription in rat pituitaries: evidence for the involvement of Jun N-terminal kinase but not p38. Endocrinology. 2008;149:139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kanasaki H, Bedecarrats GY, Kam KY, Xu S, Kaiser UB. Gonadotropin-releasing hormone pulse frequency-dependent activation of extracellular signal-regulated kinase pathways in perifused LβT2 cells. Endocrinology. 2005;146:5503–5513 [DOI] [PubMed] [Google Scholar]
- 27. Ferris HA, Walsh HE, Stevens J, Fallest PC, Shupnik MA. Luteinizing hormone β promoter stimulation by adenylyl cyclase and cooperation with gonadotropin-releasing hormone 1 in transgenic mice and LβT2 cells. Biol Reprod. 2007;77:1073–1080 [DOI] [PubMed] [Google Scholar]
- 28. Moore JP, Jr, Yang RQ, Winters SJ. Targeted pituitary overexpression of pituitary adenylate-cyclase activating polypeptide alters postnatal sexual maturation in male mice. Endocrinology. 2012;153:1421–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Halvorson LM, Kaiser UB, Chin WW. The protein kinase C system acts through the early growth response protein 1 to increase LHβ gene expression in synergy with steroidogenic factor-1. Mol Endocrinol. 1999;13:106–116 [DOI] [PubMed] [Google Scholar]
- 30. Tremblay JJ, Drouin J. Egr-1 is a downstream effector of GnRH and synergizes by direct interaction with Ptx1 and SF-1 to enhance luteinizing hormone β gene transcription. Mol Cell Biol. 1999;19:2567–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weck J, Anderson AC, Jenkins S, Fallest PC, Shupnik MA. Divergent and composite gonadotropin-releasing hormone-responsive elements in the rat luteinizing hormone subunit genes. Mol Endocrinol. 2000;14:472–485 [DOI] [PubMed] [Google Scholar]
- 32. Burger LL, Haisenleder DJ, Aylor KW, Marshall JC. Regulation of Lhb and Egr1 gene expression by GNRH pulses in rat pituitaries is both c-Jun N-terminal kinase (JNK)- and extracellular signal-regulated kinase (ERK)-dependent. Biol Reprod. 2009;81:1206–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dorn C, Mouillet JF, Yan X, Ou Q, Sadovsky Y. Insulin enhances the transcription of luteinizing hormone-beta gene. Am J Obstet Gynecol. 2004;191:132–137 [DOI] [PubMed] [Google Scholar]
- 34. Walsh HE, Shupnik MA. Proteasome regulation of dynamic transcription factor occupancy on the GnRH-stimulated luteinizing hormone β-subunit promoter. Mol Endocrinol. 2009;23:237–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu B, Yang WH, Gerin I, Hu CD, Hammer GD, Koenig RJ. Dax-1 and steroid receptor RNA activator (SRA) function as transcriptional coactivators for steroidogenic factor 1 in steroidogenesis. Mol Cell Biol. 2009;29:1719–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Curtin D, Ferris HA, Häkli M, et al. Small nuclear RING finger protein stimulates the rat luteinizing hormone-β promoter by interacting with Sp1 and steroidogenic factor-1 and protects from androgen suppression. Mol Endocrinol. 2004;18:1263–1276 [DOI] [PubMed] [Google Scholar]
- 37. Haisenleder DJ, Burger LL, Aylor KW, et al. Testosterone stimulates follicle-stimulating hormone β transcription via activation of extracellular signal-regulated kinase: evidence in rat pituitary cells. Biol Reprod. 2005;72:523–529 [DOI] [PubMed] [Google Scholar]
- 38. Kowase T, Walsh HE, Darling DS, Shupnik MA. Estrogen enhances gonadotropin-releasing hormone stimulated transcription of the luteinizing hormone subunit promoters via altered expression of stimulatory and suppressive transcription factors. Endocrinology. 2007;148:6083–6091 [DOI] [PubMed] [Google Scholar]
- 39. Lu M, Tang Q, Olefsky JM, Mellon PL, Webster NJ. Adiponectin activates adenosine monophosphate-activated protein kinase and decreases luteinizing hormone secretion in LβT2 gonadotropes. Mol Endocrinol. 2008;22:760–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol. 1999;277:E1–E10 [DOI] [PubMed] [Google Scholar]
- 42. Handa N, Takagi T, Saijo S, et al. Structural basis for compound C inhibition of the human AMP-activated protein kinase α2 subunit kinase domain. Acta Crystallogr D Biol Crystallogr. 2011;67:480–487 [DOI] [PubMed] [Google Scholar]
- 43. Sullivan SD, Moenter SM. Prenatal androgens alter GABAergic drive to gonadotropin-releasing hormone neurons: implications for a common fertility disorder. Proc Natl Acad Sci U S A. 2004;101:7129–7134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Navratil AM, Song H, Hernandez JB, et al. Insulin augments gonadotropin-releasing hormone induction of translation in LβT2 cells. Mol Cell Endocrinol. 2009;311:47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Berasi SP, Huard C, Li D, et al. Inhibition of gluconeogenesis through transcriptional activation of EGR1 and DUSP4 by AMP-activated kinase. J Biol Chem. 2006;281:27167–27177 [DOI] [PubMed] [Google Scholar]
- 46. Dorn C, Ou Q, Svaren J, Crawford PA, Sadovsky Y. Activation of luteinizing hormone β gene by gonadotropin-releasing hormone requires the synergy of early growth response-1 and steroidogenic factor-1. J Biol Chem. 1999;274:13870–13876 [DOI] [PubMed] [Google Scholar]
- 47. Clarke PR, Hardie DG. Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J. 1990;9:2439–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hong YH, Varanasi US, Yang W, Leff T. AMP-activated protein kinase regulates HNF4α transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem. 2003;278:27495–27501 [DOI] [PubMed] [Google Scholar]
- 49. Yang W, Hong YH, Shen XQ, Frankowski C, Camp HS, Leff T. Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J Biol Chem. 2001;276:38341–38334 [DOI] [PubMed] [Google Scholar]
- 50. Iwasaki Y, Nishiyama M, Taguchi T, et al. Activation of AMP-activated protein kinase stimulates proopiomelanocortin gene transcription in AtT20 corticotroph cells. Am J Physiol Endocrinol Metab. 2007;292:E1899–E1905 [DOI] [PubMed] [Google Scholar]
- 51. Tosca L, Froment P, Rame C, et al. Metformin decreases GnRH- and activin-induced gonadotropin secretion in rat pituitary cells: potential involvement of adenosine 5′ monophosphate-activated protein kinase (PRKA). Biol Reprod. 2011;84:351–362 [DOI] [PubMed] [Google Scholar]
- 52. Meisse D, Van de Casteele M, Beauloye C, et al. Sustained activation of AMP-activated protein kinase induces c-Jun N-terminal kinase activation and apoptosis in liver cells. FEBS Lett. 2002;526:38–42 [DOI] [PubMed] [Google Scholar]
- 53. Roland AV, Moenter SM. Glucosensing by GnRH neurons: inhibition by androgens and involvement of AMP-activated protein kinase. Mol Endocrinol. 2011;25:847–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brothers KJ, Wu S, DiVall SA, et al. Rescue of obesity-induced infertility in female mice due to a pituitary-specific knockout of the insulin receptor. Cell Metab. 2010;12:295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu S, Divall S, Wondisford F, Wolfe A. Reproductive tissues maintain insulin sensitivity in diet-induced obesity. Diabetes. 2012;61:114–123 [DOI] [PMC free article] [PubMed] [Google Scholar]