

Abstract
The cleavage of covalent C-H bonds is one of the most energetically demanding, yet biologically essential, chemical transformations. Two C-H bond cleavages are involved in the reaction catalyzed by thymidylate synthase (TSase), which provides the sole de novo source of thymidylate (i.e. the DNA base T) for most organisms. Our QM/MM free energy calculations show that the C-H→O proton transfer has three transition states that are energetically similar but structurally diverse. These characteristics are different from our previous calculation results on the C-H→C hydride transfer, providing an explanation for differences in temperature dependences of KIEs on these two C-H bond activation steps. The calculations also suggest that the traditionally-proposed covalent bond between the protein and substrate (the C6-S bond) is very labile during the multi-step catalytic reaction. Collective protein motions not only assist cleavage of the C6-S bond to stabilize the transition state of the proton transfer step, but also rearrange the H-bond network at the end of this step to prepare the active site for subsequent chemical steps. These computational results illustrate functionalities of specific protein residues that reconcile many previous experimental observations, and provide guidance for future experiments to examine the proposed mechanisms. The synchronized conformational changes in the protein and ligands observed in our simulations demonstrate participation of protein motions in the reaction coordinate of enzymatic reactions. Our computational findings suggest the existence of new reaction intermediates not covalently bound to TSase, which may lead to a new class of drugs targeting DNA biosynthesis.
The covalent bond between carbon and hydrogen atoms (C-H bond) is one of the strongest and most ubiquitous chemical bonds in organic compounds. Most metabolic pathways, including those involved in DNA biosynthesis, require enzyme catalyzed activation of C-H bonds.1–3 Among the four DNA nucleotides, 2'-deoxythymidine-5'-monophosphate (dTMP, deoxythymidylate) is the only precursor unique to DNA (the three others have the same bases as in RNA). The only intracellular de novo source of dTMP is the reductive methylation of deoxyuridine-5'-monophosphate (dUMP) catalyzed by thymidylate synthase (TSase), which requires a C-H bond activation in the substrate (Scheme 1). Since TSase is present in most living organisms, and it is overexpressed in tumor cells,4, 5 this enzyme has been an attractive target for both antibiotic and anticancer drugs. Classical drugs that target TSase are analogues of precursors of either the substrate (e.g. 5-fluorouracil) or the folate cofactor (e.g. raltitrexed, also known as ZD1694). These drugs often interact with other proteins involved in the metabolic pathways of either nucleosides/nucleotides or folates, leading to toxicity and development of resistance in cells.6,7 Therefore, current drug designs focus on selectively targeting TSase activity in malignant tumor cells or in specific pathogenic species, which can be aided by careful inspection of the mechanistic features of TSase.6,8
Scheme 1.
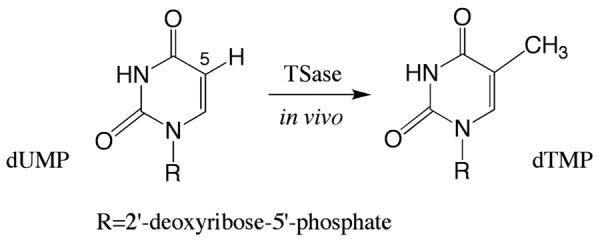
Thymidylate synthase catalyzes the reductive methylation of dUMP to produce dTMP in vivo, where the C-H bond at the C5 position of the pyrimidine is replaced by a C-C bond to a methyl group.
In the TSase-catalyzed reaction, the folate cofactor, 5,10-methylene-5,6,7,8-tetrahydrofolate (CH2H4folate), donates the methylene and hydride to the substrate dUMP, producing 7,8-dihydrofolate (H2folate) and dTMP.9 2 The mechanism of TSase proposed herein involves two C-H bond activation steps (Scheme 2): a C-H→B (B: general base) proton abstraction from C5 of dUMP (step 4), and a C-H→C hydride transfer from C6F of CH2H4folate to the exocyclic-methylene intermediate (step 5). We have previously studied those two C-H bond activation steps in the wild-type (WT) Escherichia coli TSase (ecTSase) by measuring the temperature dependence of their intrinsic kinetic isotope effects (KIEs). The KIE on the hydride transfer is temperature independent (EaT - EaH = −0.02 ± 0.25 kcal/mol)10 while that on the proton transfer is temperature dependent (EaT – EaH = 8.0 ± 0.1 kcal/mol)11, implying a better-organized transition state (TS) for the former.12–14 In order to investigate the molecular details underlying those experimental observations, we have recently simulated the catalytic mechanism of WT ecTSase using hybrid quantum mechanics/molecular mechanics (QM/MM) potentials.12, 15–17 The calculations suggested that the hydride transfer is concerted with cleavage of the covalent bond between C6 of dUMP and S of the C146 residue (noted “C6-S bond” hereafter), which regenerates the free form of the protein. Progressively, important protein motions accommodate the changes in geometric and electrostatic characteristics of the ligands during the hydride transfer, thereby reducing the free energy barrier. Particularly important in this choreography is the approach of a highly conserved arginine residue, R166, to C146 as the reaction proceeds, which polarizes the electron density around the S atom to stabilize the TS.16, 17 It appears that, owing to the adaptability of the protein environment, the average TS structure is invariant within the examined temperature range (5–40°C), which leads to a temperature-independent KIE that is consistent with our experimental findings.12
Scheme 2.
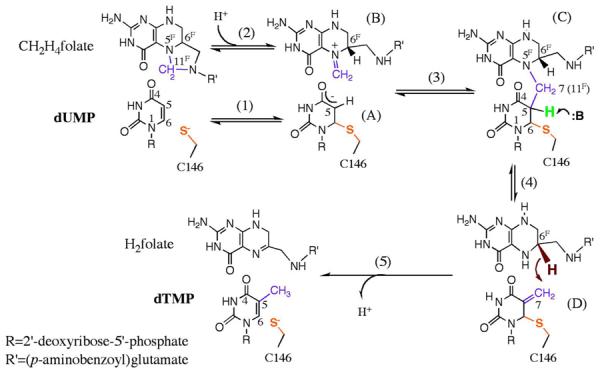
The traditionally proposed reaction mechanism of TSase9 (adapted from Ref11 with permission from ACS). The numbering of atoms on CH2H4folate is marked with an F superscript (F) to discriminate from those on the nucleotides. The proton (green) abstraction and C7-N5F (-CH2- in purple) cleavage steps are tightly coupled in WT TSase and thus is represented by one step (step 4). The following hydride (dark red) transfer is rate limiting for the catalytic turnover10, 47 and has been carefully investigated in our previous calculations12 (step 5).
In contrast to the well-resolved mechanism for the hydride transfer, the general base that abstracts the proton (H5) from C5 remains unidentified, due to the complex network of H-bonds at the active site (indicated by previous structural and kinetic studies18, 19). Furthermore, the deprotonation of C5 is tightly coupled with the subsequent β-elimination of the cofactor in WT TSase (step 4 in Scheme 2),9, 20 making it difficult to expose the intrinsic nature of this C-H bond activation. Earlier work exploited model reactions in solution,21–23 as well as the tritium release and dehalogenation from C5 of dUMP and analogues, respectively, in the binary enzyme complexes,24, 25 and proposed that an enol/enolate forms at C5-C4=O4 during the proton transfer step. In contrast, our previous QM/MM calculations with the semiempirical AM1 method suggested that the deprotonation of C5 leads to cleavage of the C6-S bond, with minimal polarization of the C4=O4 carbonyl.15 This computational result implicates a new reaction intermediate where the ligands are not covalently attached to the protein, providing new directions for drug designs that target TSase activity.
To further investigate the findings from previous calculations, the current study thoroughly explores different proposed mechanisms for the proton transfer step with higher-level QM/MM calculations (Figure 1). We first calculated the potential energy profiles of different proposed mechanisms, and then studied the free energy profile in order to delineate features of the TS for the proton transfer. Our calculations not only provide the molecular details underlying previous experimental observations, but also suggest future experiments that can examine the proposed mechanisms.
Figure 1.
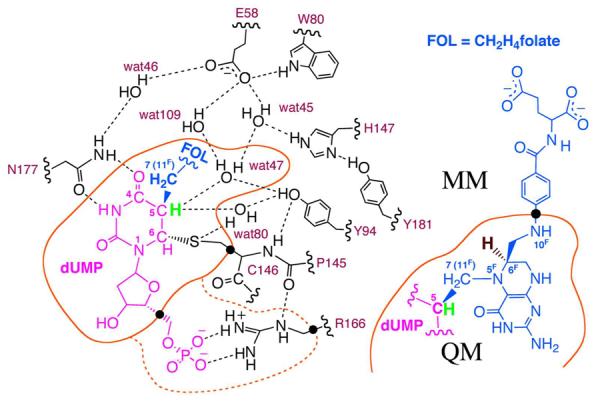
The H-bond network at the active site of the ternary ecTSase-dUMP-CH2H4folate complex (Intermediate C in Scheme 2) is shown based on the final equilibrated structure after 600 ps MD simulation (see Computation Details). This study focuses on the proton (green) abstraction from C5 of dUMP, and compares the computational results with the rate-limiting hydride (dark red) transfer studied in our previous calculations.12 The QM region for the PES calculations included 57 atoms and 3 hydrogen link atoms (black dots) on the boundary between the QM and MM regions. The QM region for the PMF calculations included 77 atoms (additional atoms in the dashed orange curve) and 3 hydrogen link atoms. The color code in this figure matches that for the structures in Figures 4B and 4C.
COMPUTATION DETAILS
Computation Model
The starting point for the calculations was a X-ray crystal structure solved for ecTSase (PDB code 1TLS, resolution 2.60 Å),26 which had both 5F-dUMP and CH2H4folate bound at the active site of each protomer. We used PyMOL27 to replace the fluorine with a hydrogen atom at C5 of dUMP, and to add hydrogen atoms to both ligands; and we used fDYNAMO28, 29 to add hydrogen atoms to the protein with a defined pH of 7.5. To account for the influence of the local protein environment, we used the empirical PROPKA3 program to recalculate the pKa values of the titratable protein residues to verify their protonation states.30 To neutralize the overall charge of the system, we added 22 counter ions (Na+) to the system at optimal electrostatic positions, at least 10.5 Å away from the periphery of the protein and 5 Å away from each other, using a regular grid of 0.5 Å. Finally, we used a pre-relaxed water box (100 × 80 × 80 Å3) to solvate the system by removing the water molecules that had their oxygen atoms within 2.8 Å of any non-hydrogen atoms.
The hybrid QM/MM calculations require division of the whole system into a QM part and a MM part.31 This work examines the proton abstraction from the substrate, which can be affected substantially by the network of H-bonds in the protein. Therefore, initial calculations on the PESs used a QM region of 157 atoms (Figure S1), which contained all the residues involved in the H-bond network at the active site. Further studies examined the effects of various QM regions, and the results suggested that the smaller QM region (57 atoms) in Figure 1 was sufficient for calculations on the proposed mechanisms (Scheme 3). This QM region was used for the final PES calculations presented in Figure 2 and Scheme 3. The PMF calculations included R166 in the QM region (77 atoms in total), and the PES calculations of the H147-involved mechanism included H147 in the QM region (68 atoms in total). The remaining atoms (including the rest of the protein and ligands, the crystallized and solvent water molecules, and the sodium counter ions) were simulated by MM. Link (hydrogen) atoms were added at the boundary between QM and MM regions to satisfy the valence of the QM/MM frontier atoms.31, 32 Based on the “half-of-the-sites-activity” proposed for ecTSase,33, 34 the calculations only modeled one active site in the QM region, leaving the second active site (ligands removed) in the MM region.
Scheme 3.

Our calculations examined three proposed mechanisms for conversion of Intermediate C to Intermediate D (step 4 in Scheme 2) in ecTSase-catalyzed reaction. This conversion involves the proton abstraction from C5 (PA), N5F protonation (NP), and C7-N5F cleavage (CN). The combinations of Roman and Arabic numbers (I.1, I.2, …) indicate the proposed steps of each mechanism as described in the text. The numbers near the reaction arrows for each step denote the activation energy barriers (kcal/mol) calculated with the AM1/MM or B3LYP/MM (in parenthesis) method. Mechanisms I and II are traditionally proposed mechanisms, where a water molecule (wat47) serves as a proton transfer conduit during the deprotonation of C5 and protonation of O4 or N5F (Step I.1 or II.1, where Intermediates F and G have different chirality of N5F). The current study suggests Mechanism III where the deprotonation of C5 leads to the cleavage of C6-S bond is the lowest energy path. The PMF calculations suggest that residue R166 stabilizes the S anion upon cleavage of the C6- S bond in Mechanism III.
Figure 2.
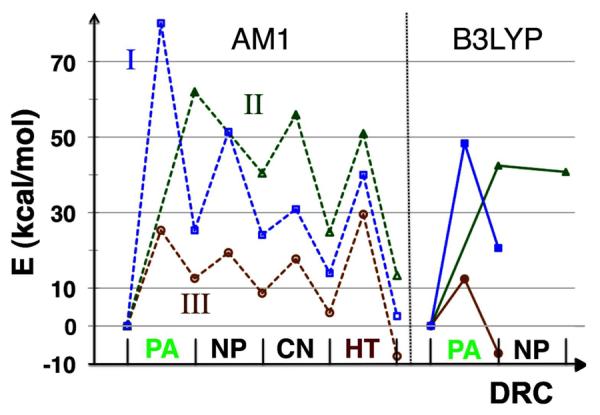
PES calculations of the proposed mechanisms (Scheme 3) suggest that cleavage of the C6-S bond lowers the activation energy barrier for the proton transfer step (Mechanism III). The energy profile for the hydride transfer (HT) is adopted from Ref 15. The horizontal axis is the DRC, which differs for each chemical step as described in the Computation Details. Dashed lines (left panel) present the energy profiles calculated with AM1/MM, and solid lines (right panel) present the energy profiles of the first step of each mechanism calculated with B3LYP/MM.
The QM region was used to investigate various proposed mechanisms (Scheme 3) with either the Austin Model 1 (AM1) method35 or the density functional theory with the Becke, three-parameter, Lee-Yang-Parr (B3LYP) exchange-correlation functional and the 6–31G(d,p) basis set36, 37. The MM region used the OPLS-AA force field38 for the protein and ligand atoms, and the TIP3P force field39 for water molecules. The nonbonding interactions were simulated using periodic boundary conditions and a switching function with a cutoff radius in the range of 14.5–16 Å. The MD simulations were performed using the canonical ensemble (NVT, with a reference temperature of 298 K) and the Langevin-Verlet integrator with a time step of 1 fs.40
Molecular Dynamics
After setting up the model, we optimized the complete system using the Adopted Basis Newton Raphson (ABNR)41 method with the backbone of the protein frozen, and then equilibrated it by 600 ps MD simulation.40 Analysis of the final structures and the time evolution of the root mean square deviation (RMSD) suggested that the system was equilibrated (Figure S2). The last structure from this 600 ps MD simulation was fully optimized again to serve as the reaction intermediate (Intermediate C in Scheme 2) for the PES calculations. The main interactions between the active site residues and water molecules in the final optimized structure resemble those proposed by Stroud and Finer-Moore.20 Considering the large size of the system, we froze all the residues farther than 25 Å from the QM center (a total of 53425 out of 60826 atoms) for the following PES and PMF calculations.
We also conducted MD simulations of several active site mutants to explore their potential effects on the reaction intermediate prior to the proton transfer (Intermediate C in Scheme 2). In particular, our calculations with the WT ecTSase revealed important roles played by C146, R166, E58, and Y94 during the proton transfer (see Results and Discussions); consequently, we conducted MD simulations of C146S, R166K, E58Q, and Y94F mutants. We also simulated the mutant H147V, because this His residue seems to stabilize the intermediate in the traditionally proposed mechanism for the proton transfer (Mechanism I in Scheme 3), while B. subtilis TSase has a Val residue as the natural variance at this position. We used PyMOL to introduce the mutation to the final optimized structure of the WT reaction complex. Afterwards, the resulting mutant complexes were optimized, equilibrated for 600 ps, and optimized again, following the same procedure as for the WT reaction intermediate.
Potential Energy Surface
We generated the QM/MM PESs for the proton transfer in the WT ecTSase using the final optimized WT reaction intermediate. Previous experiments suggested that the whole network of H-bonds serves as the general base for the deprotonation of C5.19 After a careful inspection of the protein structure around the active site, we found that two water molecules can serve as the direct acceptor for H5 transfer, both of them H-bonded with Y94 (wat47 and wat80 in Figure 1). However, due to the proximity of wat80 to C146, wat80 is inclined to protonate the S anion of C146 once it accepts H5. Therefore, all the calculations presented in this paper used wat47 as the proton acceptor. The DRC used for simulation of each chemical step is the corresponding interatomic distance (when describing a bond cleavage/formation) or antisymmetric combination of two distances (when describing an atom transfer). The PES scanning for each proposed mechanism provided saddle point structures that were used in the calculations of stationary-point location and characterization of TS structure. The obtained TS structure and corresponding intrinsic reaction coordinate (IRC, i.e. the vibrational mode with an imaginary frequency) were then used to optimize to the reactant and product structures. To authenticate the AM1/MM results, we also used the B3LYP/6–31G(d,p) method to calculate the TS structures for the first step of each proposed mechanism, followed by calculations with the IRC and full optimization of the corresponding reactants and products. Those PES calculations followed the micro-macro iterations scheme42 that is implemented in the fDynamo library28, 29, as described in our previous publications.15 In our calculations, the core space contained all the QM atoms, and the environment space included all the MM atoms, and the Hessian matrix was explicitly calculated only for the core atoms.
Potential of Mean Force (PMF)
To obtain the free energy profile for the proton transfer, we employed the umbrella sampling approach and the weighted histogram analysis method (WHAM)43, 44 to calculate the PMFs with AM1/MM. We followed the same procedure as for our previous calculations of the hydride transfer15, 16, and used an umbrella force constant of 2500 kJ·mol−1·Å−2 (597 kcal·mol−1·Å−2) for each window. Each window carried out a 5 ps equilibration and 10 ps production with a time step of 0.5 fs. In all PMF windows, the resulting structure had a total energy fluctuation lower than 0.6%, a kinetic energy fluctuation lower than 1%, and a change in temperature lower than 3 K over the production period of time (the last 10 ps). We plotted all the sampled values on the DRC and also checked all the resulting structures to make sure trajectories in adjacent windows have similar structures, i.e. good overlaps between adjacent PMF windows.
We first calculated the PMF using the antisymmetric combination of distances describing the proton transfer (dC5-H5-dH5-Ow) as the DRC. This DRC was varied from −1.50 to 1.50 Å with a window width of 0.05 Å, generating 60 windows. The starting structure for all the windows in this 1D-PMF calculation was a TS structure located in the PES calculations. In order to obtain a more precise TS for the proton transfer, we calculated a 2D-PMF using the interatomic C6-S distance as the additional dimension of the DRC. There were 45 simulation windows along the dC5-H5-dH5-Ow coordinate (from −1.48 to 1.82 Å with a window width of 0.075 Å) and 31 simulation windows along the dC6-S coordinate (from 1.8 to 4.8 Å with a window width of 0.1 Å), generating 1395 windows in total. The contour plot of the 2D-PMF results was generated with Surfer (R) Version 8.04 from Golden Software, Inc.
RESULTS AND DISCUSSION
Proton Transfer Mechanism
In order to resolve the proton transfer mechanism, we thoroughly explored the potential energy surface (PES) of this reaction using various combinations of distinguished reaction coordinates (DRCs). Although experiments were unable to isolate the deprotonation of C5 from the subsequent β-elimination of the cofactor in WT TSase,20 our 2D-PES mapping suggests that cleavage of the C7-N5F bond only occurs after deprotonation of C5 and protonation of N5F. Scheme 3 summarizes three major proposed mechanisms that our calculations have investigated.
In the traditional mechanism proposed by Carreras and Santi (Mechanism I),9 the C4=O4 carbonyl forms an enol with C5 during the deprotonation step (Intermediate E), which then transfers the proton to N5F through a water molecule (wat47) and becomes an enolate (Intermediate F). The subsequent cleavage of C7-N5F generates H4folate, which donates a hydride to the exocyclic methylene intermediate (Intermediate D). In addition, Hardy et al. found that 5-deazatetrahydrofolate cannot stimulate TSase-catalyzed tritium release, and thus proposed that N5F is the general base that deprotonates C5 through wat47 (Mechanism II).45 Both Mechanisms I and II involve formation of an enol/enolate at C5-C4=O4, in accordance with the classical catalytic mechanism proposed for abstraction of an α–proton adjacent to a carbonyl group.46 However, our previous AM1/MM PES calculations suggested that an active site water molecule (wat47) abstracts H5 from C5, leading to cleavage of the C6-S bond (Intermediate H in Mechanism III).15 This protonated water molecule (associated with a broader network of hydrogen bonds) then transfers a proton to N5F (Intermediate K), followed by S re-attacking C6 to eliminate H4folate.
Figure 2 plots the potential energy profiles of these three proposed mechanisms, which shows that Mechanism III is the lowest energy path and thus is the most probable mechanism for the proton transfer. Among these proposed mechanisms, only Mechanism III presents the hydride transfer step as the highest potential energy barrier in the AM1 PES calculations, which agrees with previous experimental observations that the hydride transfer is rate limiting for the catalytic turnover.10, 47
Since calculations using the semiempirical AM1 Hamiltonian usually overestimate the energy barriers for chemical bond cleavages/formations, we employed the B3LYP/6-31G(d,p) method to calculate the structures of a TS and the corresponding reactants and products for the first step of each proposed mechanism (Figure S4). The potential energy profiles calculated with the B3LYP/MM method are also presented in Figure 2. The B3LYP/MM calculations significantly reduced the heights of the activation barriers, and confirmed Mechanism III as the lowest energy path, which supported the use of the much cheaper AM1/MM method in this case.
Free Energy Profile of the Proton Transfer
Our previous experiments10, 11 found different temperature dependences of KIEs for the proton and hydride transfer steps. Our calculations12, 16, 17 have revealed important mechanistic features for the latter. In order to afford a good starting point to compare the nature of those two C-H bond activations, we calculated the potential of mean force (PMF) of the deprotonation of C5 at 25 °C. In this calculation, the DRC was the antisymmetric combination of the distances describing the proton transfer (DRC = dC5-H5 - dH5-Ow, where Ow refers to the oxygen of wat47). The obtained free energy profile describes an endothermic chemical reaction (ΔG = 13.5 kcal·mol−1), with an activation free energy (ΔG‡) of 25.5 kcal·mol−1 (Figure 3A).
Figure 3.
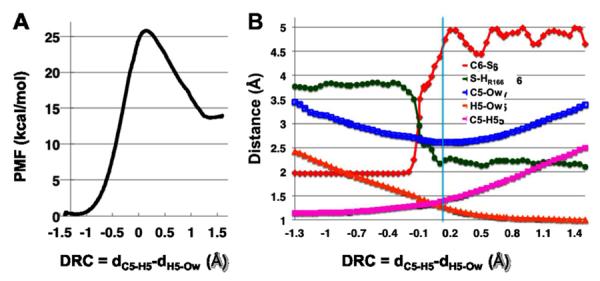
The AM1/MM PMF calculation at 25 °C provided (A) the free energy profile for the deprotonation of C5 (step III.1 in Scheme 3), and (B) changes in interatomic distances along the DRC. Atom labels: C5, H5, and C6 refer to the atoms on dUMP; S--the S atom of C146; HR166--the H atom of R166 that is closest to S (see the structures in Figure 4); Ow--the O atom of wat47. The cyan line indicates the average value of the DRC at the TS.
We analyzed the important interatomic distances that changed along the DRC in this PMF calculation (Figure 3B). As the proton is transferred from the donor to the acceptor (dC5-H5 and dH5-Ow), the distance between the donor and acceptor (dC5-Ow) reaches the minimum value at the TS. Figure S3 in SI provides additional analysis of key geometric parameters around C5 of dUMP, which confirms formation of a double bond (C5=C6) after the deprotonation of C5 and elimination of the S anion from C6. An interesting finding of this PMF calculation is that R166 approaches C146 (dS-HR166 in Figure 3B) during the proton transfer, stabilizing the thiolate anion upon the C6-S cleavage (dC6-S). This is similar to the findings of calculations on the hydride transfer.12, 16, 17 However, in contrast to the hydride transfer step, the C6-S bond is fully broken (4.5 Å) before the proton transfer reaches the TS in this 1D-PMF calculation. This observation suggests that the C6-S distance is an important component of the reaction coordinate; thus, the 1D-PMF calculation that excludes the C6-S distance from the DRC may not give a complete picture of the reaction. Consequently, we calculated the 2D-PMF of this proton transfer step at 25 °C, using the C6-S bond distance as an additional dimension of the DRC (Figure 4A).
Figure 4.
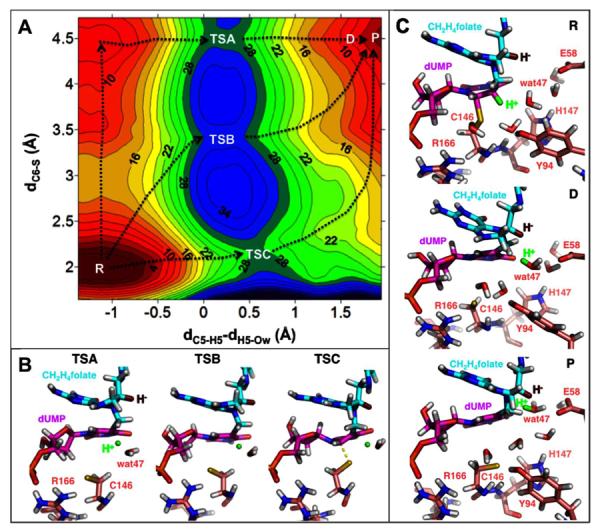
(A) The 2D-PMF calculation used the C6-S bond distance (dC6-S, vertical axis) and the antisymmetric combination describing the C-→O proton transfer (dC5-H5 - dH5-Ow, horizontal axis) as the DRC. There are various reaction paths (exemplified by the black dotted lines) with three TSs for the proton transfer (labeled as TSA, TSB, and TSC on the plot). TSA, TSB, and TSC have similar free energies (28, 30, and 28 kcal/mol, respectively) relative to the reactant state (R). The C6-S bond is fully broken (4.5 Å) in the product state (P), suggesting a reaction intermediate where the ligands are not covalently attached to the protein. (B) Representative structures for the three TSs revealed by this 2D-PMF. Only residues R166 and C146 are shown for the protein for clarity. R166 forms a strong H-bond (2.1 Å, Table S1) with C146 to stabilize the S anion in TSA and TSB. The transferred proton is in green and the hydride to be transferred (in step 5 of Scheme 2) is in dark red. (C) Representative structures for the R and P states, and a representative structure (D) along the reaction path. H5 has been transferred to wat47 in D (see the H5-Ow distance in Table S1), however, the system does not reach energy minimum (P) until the protonated wat47 moves to an optimal position that is ready to protonate N5F (the next step). This process is accompanied by rearrangement of the H-bond network that not only relocates wat47 but also destabilizes the S anion, making the active site favorable for the subsequent chemical steps (see text). Only the protein residues discussed in the text are shown for clarity. Table S1 in SI provides structural analyses of the six states labeled in this figure.
Our previous PMF calculations on the hydride transfer found one TS that was invariant at temperatures from 5 to 40 °C.12, 16 By contrast, the 2D-PMF surface for the proton transfer is more complex, with multiple saddle points for different reaction paths (Figure 4A), including the E1 (TSA), E2 (TSB) and E1cB (TSC) mechanisms. All these saddle points comprise the ensemble-averaged TS that is probed by experiments for the proton transfer step. Since TSA, TSB, and TSC have similar free energies, the reaction can go through these three TSs with comparable probabilities at 25 °C. Given this complex free energy surface, a change in temperature could alter the distribution among the possible reaction paths, leading to different geometric properties of the ensemble-averaged TS. Therefore, this 2D-PMF provides a possible explanation for the experimentally-measured temperature dependence of the KIE on the proton transfer11; these results contrast with the simpler 2D-PMF surface16 and the temperature-independent KIE10, 12 found for the hydride transfer. The complexity of the free energy surface for the proton transfer most likely arises from the mobility of the proton acceptor – a water molecule – in the versatile network of H-bonds at the active site of TSase. In contrast, the hydride is transferred between two C atoms of the ligands, and is very sensitive to the orientations of dUMP and CH2H4folate10, 18, 20, 48. This dissimilarity is probably the major determinant for the difference in temperature dependences of KIEs on those two C-H bond activations. The multiple TSs found for the proton transfer step corroborates a recent numerical analysis based on the Marcus-like model, which suggested that steeply temperature-dependent KIEs can be explained only by multiple TSs with different structures.14
In addition, the 2D-PMF calculations revealed conformational changes in both dUMP and CH2H4folate after the deprotonation of C5 (from D to P in Figure 4C), which are accompanied by rotations of protein residues and rearrangement of the H-bond network at the active site. These changes not only bring the protonated wat47 to a favorable location and orientation for the subsequent protonation of N5F, but also destabilize the S anion to prepare C146 for the next nucleophilic attack onto dUMP during the later cleavage of C7-N5F (discussed in more detail below). These synchronized motions of the protein, ligands, and solvent molecules provide an explanation for the previous experimental observations that the deprotonation of C5 is tightly coupled with ß-elimination of the cofactor in WT TSase.9, 20
Our 1D- and 2D-PMF results demonstrate the importance of 2D-PMF calculations in studying enzymatic reactions. The 1D-PMF calculations shown in Figure 3 provided a TS equivalent to TSA (Figure 4) and revealed important features for the proton transfer, i.e. cleavage of the C6-S bond and the interaction between R166 and C146. In comparison, the 2D-PMF calculations also exposed the multi-path nature of the proton transfer step, as well as other interesting information. The disparity is rooted in the fact that 2D-PMF calculations allow explicit control of more than one geometric parameter in the DRC; this level of control aids the study of a complex system whose real reaction coordinate involves many degrees of freedom. It is particularly useful in studying an enzyme-catalyzed reaction, in which the protein's motions (e.g. movements of C146 and R166 residues in this case) can occur on the same timescale as the chemical transformation and are part of the reaction coordinate. Consequently, in order to accurately access the mechanism of the chemical reaction, the simulations should include those important protein motions in the DRC.
The Labile C6-S Bond
C146 is the nucleophile that attacks C6 of dUMP to initiate the TSase-catalyzed reaction (step 1 in Scheme 2). It has been proposed that the C6-S bond remains intact until the last chemical transformation in the catalytic turnover: the hydride transfer step (step 5 in Scheme 2).20 The deprotonation of C5 of dUMP can occur in solution (e.g. with high concentrations of L-cysteine or other non-enzymatic catalysts)21–23 as well as in the binary TSase-dUMP complex (Intermediate A in Scheme 2)25. Evidence suggested that those reactions occur via formation of an enol/enolate at C5-C4=O4.9 In the ternary enzyme complex, the binding of CH2H4folate induces conformational changes in the protein as well as formation of a new C-C bond at C5 (Intermediate C in Scheme 2). These changes may impose geometric strain on the pyrimidine ring of dUMP and weaken the C6-S bond, increasing the acidity of the C5-H5 bond and facilitating the deprotonation.26 Therefore, formation of the methylene bridge with CH2H4folate alters the reaction coordinate for the proton abstraction from C5, which then occurs via a mechanism different from that of the model reactions in solution and the off-pathway deprotonation of C5 in the binary complex.
Once the C6-S bond is broken after the proton-transfer step, the question remains whether and when this labile bond forms again in the catalytic turnover. We examined the possibility of the subsequent C7-N5F cleavage (step III.3 in Scheme 3) without restoration of the C6-S bond and found a much higher energy barrier (33 kcal/mol with the AM1/MM method), indicating that the S anion attacks C6 to assist ß-elimination of the cofactor. Furthermore, our previous calculations suggest that cleavage of the C6-S bond also lowers the energy barrier for the hydride transfer step. Therefore, the lability of the C6-S bond is important to TSase's catalysis of multiple chemical steps in the reaction.
We scrutinized all the available electron density maps for the crystal structures of ecTSase on http://www.rcsb.org/ and found a very diffuse electron density between C6 and S in almost all the ternary complexes with dUMP and folate analogues (e.g. PDB IDs 2G8O, 1KZI, and 3BHL). These observations comport with our calculations and suggest that conformations of the ternary enzyme complex allow the C6-S bond to be labile while catalyzing the chemical transformations of the ligands. Since the C6-S bond is the only covalent bond formed between TSase and the ligands during the catalyzed reaction, our calculations suggest new reaction intermediates not covalently attached to ecTSase (Intermediates H and K in Scheme 2), providing a potential structural lead for new antibiotic drugs targeting DNA synthesis.
Functionalities of Several Key Residues during the Proton Transfer
Many other enzymes use specific protein residues as the general acid/base, but both experiments19, 49–51 and computations15 suggested that TSase exploits active site water molecules as the proton donors/acceptors in the catalytic turnover. To examine the functionalities of active site residues, we conducted molecular dynamics (MD) simulations of the reaction intermediates (corresponding to Intermediate C in Scheme 2) in several single site mutants of ecTSase (see Computational Details). The results suggest that the network of H-bonds in TSase rearranges in response to the mutation, which could partially recover the functionalities of the mutated residue. This suggestion agrees with saturation mutagenesis studies that concluded only five protein residues are essential to TSase activity.52 Our calculations revealed that four of those five essential residues are involved in the proton transfer step, including C146, R166, E58, and Y94, where the particular roles of C146 and R166 have been discussed above.
Although a water molecule (wat47 in Figure 1) is the direct acceptor of the proton abstracted from C5, several protein residues can modulate the basicity of this water to influence the reaction. Particularly, the negatively charged E58 forms the largest number of H-bonds with other active site residues and water molecules, and thus is critical to the entire H-bond network at the active site. In the reactant state of the proton transfer step, E58 is connected to wat47 via H-bonds through other water molecules (Figure 1 and Figure 4C, R). Conformational fluctuations of the protein environment enable formation of a direct H-bond between E58 and wat47, which not only increases the basicity of wat47 for the proton abstraction, but also brings the protonated wat47 to a favorable position for the subsequent protonation of N5F (Figure 4C). These results agree with previous experimental observations that mutations of E58 caused moderately slower deprotonation of C5 and dramatically slower ß-elimination of the cofactor, leading to accumulation of the ternary enzyme complex.45
In addition, the Y94 residue was postulated to act as the general base to deprotonate C5,53, 54 based on its proximity to the fluorine of 5-fluoro-dUMP (5F-dUMP) in the crystal structure of the ternary TSase-(5F-dUMP)-CH2H4folate complex.26 Our calculations suggest that Y94 is connected to the S of C146 via H-bonds through a water molecule (wat80 in Figure 1) before and during the proton transfer step (Figure 4, R, D, and TSs). However, once the proton is transferred to wat47, Y94 replaces R166 to form a direct H-bond with C146 (from D to P in Figure 4), which is involved in the rearrangement of active site H-bonds that moves the protonated wat47 to a favorable position for the next step. Since Y94 stabilizes the S anion to a lesser extent than does the positively charged R166, this rearrangement also prepares C146 for the next nucleophilic attack on C6 of dUMP, which facilitates elimination of the cofactor. These results corroborate our recent experiments which suggested Y94 is not the general base deprotonating C5, but rather plays an important role in the steps between the proton transfer and hydride transfer.19
Although H147 is not one of the five essential residues, it is highly conserved and was proposed to assist the enol formation at C5-C4=O4 during the proton transfer (step I.1 in Scheme 3).19 During our 600 ps MD simulation of the WT reaction intermediate (Intermediate C in Scheme 2), H147 was connected to O4 via H-bonds through a water molecule rather than directly. We calculated a TS and the corresponding reactant and product structures for the mechanism in which H147 donates a proton to O4, facilitating the enol formation during the deprotonation of C5. The results suggest that this mechanism has a lower activation energy barrier (42 kcal/mol with the AM1/MM method) than do Mechanisms I and II, but its activation energy is still higher than that of Mechanism III. In addition, the in silico H147V mutation of ecTSase caused large displacements of atoms in C146 as well as in the 2'-deoxyribose-5'-phosphate moiety of dUMP (by comparison with the WT structure, in the final optimized structure after the 600 ps MD simulation). Those atomic displacements are accompanied by alterations in the H-bonds around the C6-S bond, while the residues around O4 of dUMP are marginally affected. These results suggest that, in contrast to the traditional proposal, H147 (in concert with E58 and other residues) is most likely involved in coordinating active site water molecules. This concerted coordination maintains the proper conformations of C146 and dUMP for the deprotonation of C5 and cleavage of C6-S.
Future Directions to Test the Proposed Mechanisms
The calculations presented above do not only delineate the molecular mechanisms underlying previous experimental observations, they also provide new insights for further consideration and investigation. Although our previous calculation suggested that the C6-S bond could break during the deprotonation of C5,15 it did not provide enough details of the new mechanism to allow the prediction of feasible experiments examining the computational outcomes. It was, for example, suggested that due to the role of H147 in stabilizing enol intermediate at carbonyl C4, the examination of mutations at H147 could assist in distinguishing between proposed mechanisms.18, 20 However, the current calculations indicate that H147 is involved in mediating the H-bond network at the active site, which can affect the lability of C6-S bond. Therefore, the experimentally measured H147 mutational effects would require more careful interpretation. Accordingly, we suggest that future experiments may take multiple approaches to verify the cleavage of the C6-S bond during the proton transfer. A most convincing experiment would be to chemically trap any of the proposed intermediates (H or K in Scheme 3) using a quench-flow experimental assay similar to the one recently developed for the flavin-dependent thymidylate synthase.55 Alternatively, the unquenched intermediates might be detected using time-resolved mass spectrometry.56 Another relevant experiment would be the examination of the C146S mutant of ecTSase. This mutant can form a stiffer C6-O bond upon the initial nucleophilic attack, which, according to the proposed mechanism, would increase the energy barrier for the proton abstraction from C5. In light of this prediction, previous kinetic studies of this mutant can be reanalyzed. The C146S mutation caused a 1000-fold decrease in the kcat57 and a 5000-fold decrease in the rate of tritium release from 5-3H-dUMP58. Those results reflect the mutation's more substantial effect on proton abstraction from C5 than on the overall turnover. Our current calculations also indicate that R166 is critical in stabilizing the thiol anion of C146 during the proton transfer, while E58 and Y94 are important for the H-bond rearrangements toward the end of this step. Future experimental studies of R166, E58, and Y94 mutations will examine their effects on proton abstraction in attempt to differentiate the proposed mechanisms.
CONCLUSIONS
Understanding the mechanisms of enzyme-catalyzed C-H bond activation has been a focus of biochemistry research for the past few decades. The current study used QM/MM calculations to investigate a proton abstraction catalyzed by ecTSase, which is one of the two C-H bond activations in this enzymatic reaction. Experimental evidence suggested that an analogous proton transfer occurs in solution and in the binary enzyme complex TSase-dUMP via formation of an enol/enolate in the substrate. However, our calculations suggest that ecTSase uses a different strategy, the labile C6-S bond between the protein and substrate, to catalyze the proton transfer step. The cleavage of the C6-S bond during the proton transfer leads to new reaction intermediates that are not covalently bound to the enzyme, revealing a new potential target for mechanism-based inhibitor design. Such inhibitors may serve as leads for a new class of antibiotic or chemotherapeutic drugs. In addition, our calculations suggest that the proton transfer has a complex free energy surface with multiple TSs, which provides an explanation for the temperature-dependent KIE measured experimentally.11 In contrast to the well defined TS for the hydride transfer step,12 the more complex PMF for the proton transfer step leads to steep temperature dependence of intrinsic KIEs.
The comparison between the 1D- and 2D-PMFs of the proton transfer suggests that some protein motions are part of the reaction coordinate, and should be included in the DRC in simulations to obtain complete information for the reaction mechanism. Collective protein motions rearrange the network of H-bonds at the active site to accommodate structural changes in the ligands during and after the proton transfer. In particular, the positively charged R166 approaches C146, rendering it a better leaving group, and thereby stabilizes the TS of the proton transfer step. After that step, the conformational changes in the active site prepare the C146 residue and the protonated water for the subsequent chemical steps in the catalyzed reaction. These synchronized conformational changes in the ligands and protein residues provide another example of the indispensable roles of protein motions in enzyme-catalyzed chemical reactions. Further analysis of the global network of H-bonds that accommodates those local changes is an interesting future direction, one that may reveal important long-range interactions in TSase activity.
Our calculations not only illustrate the functionalities of specific protein residues that reconcile previous experimental observations, but also suggest future directions for experiments to further test and sort the proposed mechanisms. The catalytic mechanism of thymidylate synthase is representative of electrophilic substitutions at C5 of the pyrimidine heterocycle; this substitution is catalyzed by many other enzymes, including deoxyuridylate,59 deoxycytidylate hydroxymethylase,60 DNA methyltransferase,61 and RNA methyltransferase62. All these enzymes also catalyze proton abstraction from the C5 of the pyrimidine ring upon nucleophillic attack of the catalytic cysteine residue at C6. Thus, the current findings for thymidylate synthase may provide insights into the catalytic mechanisms of other important enzymes involved in nucleic acid biochemistry.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Adrian Elcock and Dr. Sergio Martí for technical assistance, and Dr. Janet Finer-Moore for insightful discussions.
Funding Source Statement
This work was supported by NIH GM065368 to AK, Spanish Ministerio de Economía y Competitividad project CTQ2012-36253-C03-01, Generalitat Valenciana projects ACOMP/2012/119 and Prometeo/2009/053, and Universitat Jaume I-Bancaixa project P1·1B2011-23. ZW was also supported by the T. Anne Cleary International Dissertation Research Fellowship, a Graduate College Strategic Investment Fund Fellowship, a fellowship from the University of Iowa Center for Biocatalysis and Bioprocessing, and by activities of the Predoctoral Training program in Biotechnology, NIH grant 2T32GM008365.
Abbreviations
- TSase
thymidylate synthase
- ecTSase
Escherichia coli thymidylate synthase
- dUMP
deoxyuridine-5'-monophosphate
- dTMP
2'-deoxythymidine-5'-monophosphate
- CH2H4folate
5,10-methylene-5,6,7,8-tetrahydrofolate
- WT
wild type
- PES
potential energy surface
- PMF
potential of mean force
- DRC
distinguished reaction coordinate
- MD
molecular dynamics
- QM/MM
quantum mechanics/molecular mechanics
- AM1
Austin Model 1 method
- B3LYP
the Becke, three-parameter, Lee-Yang-Parr exchange-correlation functional
- RMSD
root mean square deviation
Footnotes
SUPPORTING INFORMATION AVAILABLE
Initial QM region that contains 157 atoms; time evolution of the RMSD during 600 ps MD simulation of the WT ecTSase complex; additional geometric analysis of dUMP during the proton transfer; structures calculated with the B3LYP/MM method; tables comparing the geometric parameters of the reactant, TS, and product structures. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Bollinger JM, Jr., Broderick JB. Frontiers in enzymatic C-H-bond activation. Curr. Opin. Chem. Biol. 2009;13:51–57. doi: 10.1016/j.cbpa.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 2.Rittle J, Green MT. Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 3.Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Pujar A, Shearer AG, Travers M, Weerasinghe D, Zhang P, Karp PD. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012;40:D742–753. doi: 10.1093/nar/gkr1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen C-Y, Chang Y-L, Shih J-Y, Lin J-W, Chen K-Y, Yang C-H, Yu C-J, Yang P-C. Thymidylate synthase and dihydrofolate reductase expression in non-small cell lung carcinoma: The association with treatment efficacy of pemetrexed. Lung Cancer. 2011;74:132–138. doi: 10.1016/j.lungcan.2011.01.024. [DOI] [PubMed] [Google Scholar]
- 5.Johnston PG, Lenz HJ, Leichman CG, Danenberg KD, Allegra CJ, Danenberg PV, Leichman L. Thymidylate synthase gene and protein expression correlate and are associated with response to 5-fluorouracil in human colorectal and gastric tumors. Cancer Res. 1995;55:1407–1412. [PubMed] [Google Scholar]
- 6.Costi MP. Thymidylate synthase inhibition: a structure-based rationale for drug design. Medicinal research reviews. 1998;18:21–42. doi: 10.1002/(sici)1098-1128(199801)18:1<21::aid-med2>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 7.Longley DB, Johnston PG. 5-Fluorouracil: Molecular Mechanisms of Cell Death. In: Srivastava R, editor. Apoptosis, Cell Signaling, and Human Diseases: Molecular Mechanisms. Humana Press; 2007. pp. 263–278. [Google Scholar]
- 8.Costi MP, Tondi D, Rinaldi M, Barlocco D, Pecorari P, Soragni F, Venturelli A, Stroud RM. Structure-based studies on species-specific inhibition of thymidylate synthase. Biochimica et biophysica acta. 2002;1587:206–214. doi: 10.1016/s0925-4439(02)00083-2. [DOI] [PubMed] [Google Scholar]
- 9.Carreras CW, Santi DV. The catalytic mechanism and structure of thymidylate synthase. Annu. Rev. Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 10.Agrawal N, Hong BY, Mihai C, Kohen A. Vibrationally enhanced hydrogen tunneling in the Escherichia coli thymidylate synthase catalyzed reaction. Biochemistry. 2004;43:1998–2006. doi: 10.1021/bi036124g. [DOI] [PubMed] [Google Scholar]
- 11.Wang Z, Kohen A. Thymidylate Synthase Catalyzed H-Transfers: Two Chapters in One Tale. J. Am. Chem. Soc. 2010;132:9820–9825. doi: 10.1021/ja103010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanaan N, Ferrer S, Martí S, Garcia-Viloca M, Kohen A, Moliner V. Temperature Dependence of the Kinetic Isotope Effects in Thymidylate Synthase. A Theoretical Study. J. Am. Chem. Soc. 2011;133:6692–6702. doi: 10.1021/ja1114369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu H, Warshel A. Origin of the temperature dependence of isotope effects in enzymatic reactions: the case of dihydrofolate reductase. J. Phys. Chem. B. 2007;111:7852–7861. doi: 10.1021/jp070938f. [DOI] [PubMed] [Google Scholar]
- 14.Roston D, Cheatum CM, Kohen A. Hydrogen donor-acceptor fluctuations from kinetic isotope effects: a phenomenological model. Biochemistry. 2012;51:6860–6870. doi: 10.1021/bi300613e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanaan N, Martí S, Moliner V, Kohen A. A quantum mechanics/molecular mechanics study of the catalytic mechanism of the thymidylate synthase. Biochemistry. 2007;46:3704–3713. doi: 10.1021/bi061953y. [DOI] [PubMed] [Google Scholar]
- 16.Kanaan N, Martí S, Moliner V, Kohen A. QM/MM study of thymidylate synthase: enzymatic motions and the temperature dependence of the rate limiting step. J. Phys. Chem. A. 2009;113:2176–2182. doi: 10.1021/jp810548d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanaan N, Roca M, Tuñón I, Martí S, Moliner V. Theoretical study of the temperature dependence of dynamic effects in thymidylate synthase. Phys. Chem. Chem. Phys. 2010;12:11657–11664. doi: 10.1039/c003799k. [DOI] [PubMed] [Google Scholar]
- 18.Stroud RM, Finer-Moore JS. Conformational Dynamics along an Enzymatic Reaction Pathway: Thymidylate Synthase, "the Movie". Biochemistry. 2003;42:239–247. doi: 10.1021/bi020598i. [DOI] [PubMed] [Google Scholar]
- 19.Hong B, Maley F, Kohen A. Role of Y94 in proton and hydride transfers catalyzed by thymidylate synthase. Biochemistry. 2007;46:14188–14197. doi: 10.1021/bi701363s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finer-Moore JS, Santi DV, Stroud RM. Lessons and Conclusions from Dissecting the Mechanism of a Bisubstrate Enzyme: Thymidylate Synthase Mutagenesis, Function, and Structure. Biochemistry. 2003;42:248–256. doi: 10.1021/bi020599a. [DOI] [PubMed] [Google Scholar]
- 21.Wataya Y, Hayatsu H, Kawazoe Y. Cysteine-catalyzed hydrogen isotope exchange at the 5 position of uridylic acid. J. Am. Chem. Soc. 1972;94:8927–8928. doi: 10.1021/ja00780a059. [DOI] [PubMed] [Google Scholar]
- 22.Santi DV, Brewer CF. Model studies of thymidylate synthetase. Intramolecular catalysis of 5-hydrogen exchange and 5-hydroxymethylation of 1-substituted uracils. Biochemistry. 1973;12:2416–2424. doi: 10.1021/bi00737a008. [DOI] [PubMed] [Google Scholar]
- 23.Pogolotti AL, Jr., Santi DV. Model studies of the thymidylate synthetase reaction. Nucleophilic displacement of 5-p-nitrophenoxymethyluracils. Biochemistry. 1974;13:456–466. doi: 10.1021/bi00700a010. [DOI] [PubMed] [Google Scholar]
- 24.Garrett C, Wataya Y, Santi DV. Thymidylate synthetase. Catalysis of dehalogenation of 5-bromo- and 5-iodo-2'-deoxyuridylate. Biochemistry. 1979;18:2798–2804. doi: 10.1021/bi00580a017. [DOI] [PubMed] [Google Scholar]
- 25.Pogolotti AL, Jr., Weill C, Santi DV. Thymidylate synthetase catalyzed exchange of tritiumfrom [5–3H]-2'-deoxyuridylate for protons of water. Biochemistry. 1979;18:2794–2798. doi: 10.1021/bi00580a016. [DOI] [PubMed] [Google Scholar]
- 26.Hyatt DC, Maley F, Monfort WR. Use of strain in a stereospecific catalytic mechanism: Crystal structures of Escherichia coli thymidylate synthase bound to FdUMP and methylenetetrahydrofolate. Biochemistry. 1997;36:4585–4594. doi: 10.1021/bi962936j. [DOI] [PubMed] [Google Scholar]
- 27.Schrodinger LLC. The PyMOL Molecular Graphics System. Version 1.5.0.4 2010. [Google Scholar]
- 28.Field MJ. A practical introduction to the simulation of molecular systems. Cambridge University Press; Cambridge: 1999. [Google Scholar]
- 29.Field MJ, Albe M, Bret C, Proust-De Martin F, Thomas A. The dynamo library for molecular simulations using hybrid quantum mechanical and molecular mechanical potentials. J. Comput. Chem. 2000;21:1088–1100. [Google Scholar]
- 30.Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 31.Warshel A, Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 32.Singh UC, Kollman PA. A combined ab initio quantum mechanical and molecular mechanical method for carrying out simulations on complex molecular systems: Applications to the CH3Cl + Cl− exchange reaction and gas phase protonation of polyethers. J. Comput. Chem. 1986;7:718–730. [Google Scholar]
- 33.Maley F, Pedersenlane J, Changchien LM. COMPLETE RESTORATION OF ACTIVITY TO INACTIVE MUTANTS OF ESCHERICHIA-COLI THYMIDYLATE SYNTHASE - EVIDENCE THAT ESCHERICHIA-COLI THYMIDYLATE SYNTHASE IS A HALF-THE-SITES ACTIVITY ENZYME. Biochemistry. 1995;34:1469–1474. doi: 10.1021/bi00005a001. [DOI] [PubMed] [Google Scholar]
- 34.Saxl RL, Changchien LM, Hardy LW, Maley F. Parameters affecting the restoration of activity to inactive mutants of thymidylate synthase via subunit exchange: Further evidence that thymidylate synthase is a half-of-the-sites activity enzyme. Biochemistry. 2001;40:5275–5282. doi: 10.1021/bi002925x. [DOI] [PubMed] [Google Scholar]
- 35.Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP. Development and use of quantum mechanical molecular models. 76. AM1: a new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985;107:3902–3909. [Google Scholar]
- 36.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 37.Miehlich B, Savin A, Stoll H, Preuss H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989;157:200–206. [Google Scholar]
- 38.Kaminiski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA Force Field for proteins via comparison with accurate Quantum Chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474–6487. [Google Scholar]
- 39.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. COMPARISON OF SIMPLE POTENTIAL FUNCTIONS FOR SIMULATING LIQUID WATER. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 40.Verlet L. COMPUTER EXPERIMENTS ON CLASSICAL FLUIDS. I. THERMODYNAMICAL PROPERTIES OF LENNARD-JONES MOLECULES. Phys. Rev. 1967;159:98–103. [Google Scholar]
- 41.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM - A PROGRAM FOR MACROMOLECULAR ENERGY, MINIMIZATION, AND DYNAMICS CALCULATIONS. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 42.Martí S, Moliner V, Tuñón I. Improving the QM/MM description of chemical processes: a dual level strategy to explore the potential energy surface in very large systems. J. Chem. Theory Comput. 2005;1:1008–1016. doi: 10.1021/ct0501396. [DOI] [PubMed] [Google Scholar]
- 43.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. THE WEIGHTED HISTOGRAM ANALYSIS METHOD FOR FREE-ENERGY CALCULATIONS ON BIOMOLECULES.1. THE METHOD. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 44.Torrie GM, Valleau JP. Non-physical sampling distributions in monte-carlo free-energy estimation - umbrella sampling. J. Comput. Phys. 1977;23:187–199. [Google Scholar]
- 45.Hardy LW, Graves KL, Nalivaika E. Electrostatic guidance of catalysis by a conserved glutamic acid in Escherichia coli dTMP synthase and bacteriophage T4 dCMP hydroxymethylase. Biochemistry. 1995;34:8422–8432. doi: 10.1021/bi00026a025. [DOI] [PubMed] [Google Scholar]
- 46.Gerlt JA, Gassman PG. Understanding the rates of certain enzyme-catalyzed reactions: proton abstraction from carbon acids, acyl-transfer reactions, and displacement reactions of phosphodiesters. Biochemistry. 1993;32:11943–11952. doi: 10.1021/bi00096a001. [DOI] [PubMed] [Google Scholar]
- 47.Spencer HT, Villafranca JE, Appleman JR. Kinetic scheme for thymidylate synthase from Escherichia coli: determination from measurements of ligand binding, primary and secondary isotope effects and pre-steady-state catalysis. Biochemistry. 1997;36:4212–4222. doi: 10.1021/bi961794q. [DOI] [PubMed] [Google Scholar]
- 48.Wang Z, Abeysinghe T, Finer-Moore JS, Stroud RM, Kohen A. A remote mutation affects the hydride transfer by disrupting concerted protein motions in thymidylate synthase. J. Am. Chem. Soc. 2012;134:17722–17730. doi: 10.1021/ja307859m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fauman EB, Rutenber EE, Maley GF, Maley F, Stroud RM. Water-mediated substrate/product discrimination: the product complex of thymidylate synthase at 1.83 Å. Biochemistry. 1994;33:1502–1511. doi: 10.1021/bi00172a029. [DOI] [PubMed] [Google Scholar]
- 50.Sage CR, Rutenber EE, Stout TJ, Stroud RM. An essential role for water in an enzyme reaction mechanism: the crystal structure of the thymidylate synthase mutant E58Q. Biochemistry. 1996;35:16270–16281. doi: 10.1021/bi961269r. [DOI] [PubMed] [Google Scholar]
- 51.Huang W, Santi DV. Active Site General Catalysts Are Not Necessary for Some Proton Transfer Reactions of Thymidylate Synthase‚Ć. Biochemistry. 1997;36:1869–1873. doi: 10.1021/bi961982g. [DOI] [PubMed] [Google Scholar]
- 52.While the sequence of TSase has several hundreds of amino acids, there are only five residues essential for the catalytic activity: E58, Y94, C146, R166, and D169 (the numbering refers to ecTSase) [Google Scholar]
- 53.Liu Y, Barrett JE, Schultz PG, Santi DV. Tyrosine 146 of thymidylate synthase assists proton abstraction from the 5-position of 2'-deoxyuridine 5'-monophosphate. Biochemistry. 1999;38:848–852. doi: 10.1021/bi9822877. [DOI] [PubMed] [Google Scholar]
- 54.Saxl RL, Reston J, Nie Z, Kalman TI, Maley F. Modification of Escherichia coli thymidylate synthase at tyrosine-94 by 5-Imidazolylpropynyl-2'-deoxyuridine 5'-monophosphate. Biochemistry. 2003;42:4544–4551. doi: 10.1021/bi0268089. [DOI] [PubMed] [Google Scholar]
- 55.Mishanina TV, Koehn EM, Conrad JA, Palfey BA, Lesley SA, Kohen A. Trapping of an intermediate in the reaction catalyzed by flavin-dependent thymidylate synthase. J. Am. Chem. Soc. 2012;134:4442–4448. doi: 10.1021/ja2120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roberts A, Furdui C, Anderson KS. Observation of a chemically labile, noncovalent enzyme intermediate in the reaction of metal-dependent Aquifex pyrophilus KDO8PS by time-resolved mass spectrometry. Rapid Commun. Mass Spectrom. 2010;24:1919–1924. doi: 10.1002/rcm.4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fauman EB, Rutenber EE, Maley GF, Maley F, Stroud RM. Water-mediated substrate/product discrimination: the product complex of thymidylate synthase at 1.83 A. Biochemistry. 1994;33:1502–1511. doi: 10.1021/bi00172a029. [DOI] [PubMed] [Google Scholar]
- 58.Dev IK, Yates BB, Leong J, Dallas WS. Functional role of cysteine-146 in Escherichia coli thymidylate synthase. P. Natl. Acad. Sci. USA. 1988;85:1472–1476. doi: 10.1073/pnas.85.5.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kunitani MG, Santi DV. On the mechanism of 2'-deoxyuridylate hydroxymethylase. Biochemistry. 1980;19:1271–1275. doi: 10.1021/bi00548a001. [DOI] [PubMed] [Google Scholar]
- 60.Graves KL, Butler MM, Hardy LW. Roles of Cys148 and Asp179 in catalysis by deoxycytidylate hydroxymethylase from bacteriophage T4 examined by site-directed mutagenesis. Biochemistry. 1992;31:10315–10321. doi: 10.1021/bi00157a020. [DOI] [PubMed] [Google Scholar]
- 61.Bestor TH, Verdine GL. DNA methyltransferases. Curr. Opin. Chem. Biol. 1994;6:380–389. doi: 10.1016/0955-0674(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 62.Santi DV, Hardy LW. Catalytic mechanism and inhibition of tRNA (uracil-5-)methyltransferase: evidence for covalent catalysis. Biochemistry. 1987;26:8599–8606. doi: 10.1021/bi00400a016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.