

Abstract
Cathelicidins form a family of small host defense peptides distinct from another class of cationic antimicrobial peptides, the defensins. They are expressed as large precursor molecules with a highly conserved pro-domain known as the cathelin-like domain (CLD). CLDs have high degrees of sequence homology to cathelin, a protein isolated from pig leukocytes and belonging to the cystatin family of cysteine protease inhibitors. In this report, we describe for the first time the X-ray crystal structure of the human CLD (hCLD) of the sole human cathelicidin, LL-37. The structure of hCLD, determined at 1.93 Å resolution, shows the cystatin-like fold and is highly similar to the structure of the CLD of the pig cathelicidin, protegrin-3. We assayed the in vitro antibacterial activities of hCLD, LL-37 and the precursor form, pro-cathelicidin (also known as hCAP18), and we found that the unprocessed protein inhibited the growth of Gram-negative bacteria with efficiencies comparable to the mature peptide, LL-37. In addition, the antibacterial activity of LL-37 was not inhibited by hCLD intermolecularly, since exogenously added hCLD had no effect on the bactericidal activity of the mature peptide. hCLD itself lacked antimicrobial function and did not inhibit the cysteine protease, cathepsin L. Our results contrast with previous reports of hCLD activity. A comparative structural analysis between hCLD and the cysteine protease inhibitor stefin A showed why hCLD is unable to function as an inhibitor of cysteine proteases. In this respect, the cystatin scaffold represents an ancestral structural platform from which proteins evolved divergently, with some losing inhibitory functions.
Keywords: cathelin-like domain, cathelicidin, LL-37, antimicrobial peptide, precursor form, x-ray crystallography, cystatin scaffold
Cathelicidin antimicrobial peptides (CAMPs) are major components of innate immunity, acting directly against microbial infections (1–3). As the most abundant host defense peptides, CAMPs are found in many mammals including primates, rodents, ungulates and rabbits (1). Recently, the cathelicidin-like genes have also been identified in non-mammals including chickens, Atlantic salmon and hagfish (4–6). The family of CAMPs is highly heterogeneous and covers a wide range of peptides that vary in length, share low sequence similarity and display marked structural diversity (3, 7, 8).
CAMPs are synthesized as precursors with a significantly larger (94–114 aa) and highly conserved N-terminal pro-domain known as the cathelin-like domain or CLD (9–15). The CLD shows high sequence homology to cathelin, a protein of 96 aa residues isolated from porcine neutrophils and classified initially into the cystatin family of cysteine protease inhibitors based on its inhibitory activity against cathepsin L (13, 14). Due to low sequence similarities among C-terminal antimicrobial domains, the evolutionarily conserved CLD defines these peptides as belonging to the cathelicidin family (7, 16). The pro-forms of CAMPs, by definition, are considered to be storage forms and, as inactive precursors, require proteolytic processing to become biologically functional (11, 17–20). During the activation process, a specific enzyme removes the CLD and frees the C-terminal antimicrobial peptide to fulfill its cathelicidin function (3).
Despite the abundance of mammalian CAMPs, only one CAMP is found in humans (17, 21–23). Human cathelicidin is stored as a 16-kDa pro-form, pro-cathelicidin, in the secondary granules of neutrophils. Pro-cathelicidin is also known as human CAP18 (hCAP18) based on its close relationship to CAP18, a cationic antimicrobial protein of 18 kDa isolated from the rabbit (22, 23). Pro-cathelicidin consists of an N-terminal human CLD (hCLD) of 103 amino acid residues and the C-terminal cathelicidin peptide, LL-37, of 37 residues. Upon stimulation, pro-cathelicidin is processed into hCLD and LL-37 by proteinase 3, a serine protease originating from azurophilic granules (17, 19). In addition to neutrophils, which are the major source of LL- 37, the cathelicidin gene is also expressed in various blood cell populations including monocytes and certain lymphocytes (24), in the squamous epithelial cells of the airways, intestine and vagina (25), in the epididymis and testis (26), and in keratinocytes in inflammatory skin diseases. The prostate-specific protease, gastricsin, has been identified to process pro-cathelicidin in seminal plasma into ALL-38, which contains an additional Ala residue at the N-terminus as compared to LL-37 (27, 28).
The multiple roles of LL-37 in host immune defense have been studied extensively during the two decades since its discovery (7, 29, 30). LL-37 was initially recognized for its broad-spectrum antimicrobial activity against bacteria, fungi, and viral pathogens. Other biologically important functions of LL-37 were subsequently reported, including immunomodulatory and chemotactic properties, stimulation of angiogenesis, and LPS-neutralizing activities (22, 29–31). Recently, the high-quality solution structure of LL-37 in dodecylphosphocholine (DPC) and sodium dodecyl sulfate (SDS) micelles has been determined using multi-dimensional NMR spectroscopy, showing an α-helical and amphipathic structure of LL-37 upon target membrane binding (32, 33).
In contrast to LL-37, little is known about the biological functions, if any, of pro-cathelicidin and hCLD despite the presence of high concentrations of pro-cathelicidin in many biological settings including human blood plasma (25, 34) and seminal plasma (26). In fact, pro-cathelicidin is considered to be a storage form of LL-37 endowed with no biological activity (19, 35). Recently, hCLD has been shown to inhibit cathepsin L activity in vitro and possess antibacterial activity comparable to that of LL-37 (35). It has been proposed that hCLD may possess dual functions strictly related to the LL-37 maturation process: (1) intramolecularly inhibiting the antimicrobial function of LL-37 prior to proteolytic processing (7, 17, 22), and (2) contributing to host defense through direct antimicrobial and protease inhibitory activities upon proteolytic processing (35).
In this report, we describe for the first time the X-ray crystal structure of the CLD of human cathelicidin determined at 1.93 Å resolution. The structure of hCLD shows the expected cystatin family fold and is highly similar to the structure of ProS – the CLD of the pig cathelicidin protegrin-3 and the only mammalian CLD studied so far by X-ray crystallography and NMR spectroscopy. However, we could not confirm the inhibitory activity of hCLD against cathepsin L or three strains of bacteria. Instead, we found that pro-cathelicidin is capable of killing Gram-negative bacteria in vitro with efficiency comparable to that of the mature cathelicidin peptide LL-37.
MATERIALS AND METHODS
Expression and purification of pro-cathelicidin
The cDNA fragment coding for the human cathelicidin precursor protein was amplified using a forward primer, 5′-CATATGCAGGTCCTCAGCTACAAGGAAGCT, paired with a reverse primer, 5′-GGATCCCTAGGACTCTGTACGAGGTACAAGATT. The PCR product was ligated into the pCR2.1-TOPO vector (Invitrogen) and sequenced to verify the sequence. The correct insert was cloned in the NdeI/BamHI sites of the pET15b Vector (Novagen). The translation product consists of an N-terminal His6-Tag followed by a thrombin digestion site and the full-length pro-cathelicidin sequence. A single colony of E. coli BL21(DE3) carrying the recombinant plasmid was used to initiate growth of a 50-ml overnight culture at 37°C in Luria-Bertani broth (LB) supplemented with ampicillin (100 μg/ml). Each culture was then diluted 1:100 into fresh LB medium and grown to A600 = 0.8 at 37°C when the expression was induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. The cells were harvested 4 h after induction by centrifugation at 6,000g for 20 min and subjected to lysis with BugBuster® Protein Extraction Reagent (Novagen). His6-pro-cathelicidin protein was produced exclusively as inclusion bodies. The pellet (insoluble fraction) was separated by centrifugation at 20,000 g at 4°C for 15 min and washed several times with 2% (v/v) Triton X-100, 50 mM Tris (pH 7.5), and then with 50 mM Tris, pH 7.5 alone. Finally, the insoluble aggregates were dissolved under denaturing conditions in the binding buffer (6 M GuHCl, 20 mM sodium phosphate buffer, pH 7.4) and loaded onto a 5 ml HiTrap Chelating HP column (GE Amersham) charged with Ni and equilibrated with the binding buffer. Weakly-bound proteins were removed with binding buffer supplemented with 50 mM imidazole and His6-pro-cathelicidin was eluted with binding buffer supplemented with 500 mM imidazole. The denatured His6- pro-cathelicidin solution was supplemented with dithiothreitol (DTT) to a final concentration of 20 mM and stirred for 20 min. Fully reduced His6-pro-cathelicidin was next precipitated under reducing conditions by overnight dialysis against 50 mM Tris-HCl, pH 8.4, 1 mM DTT. Insoluble His6-pro-cathelicidin protein was separated by centrifugation at 20,000 g for 15 min, dissolved in 6 M GuHCl and subjected to overnight oxidative folding through thiol-disulfide shuffling in the presence of reduced (3 mM) and oxidized (0.3 mM) glutathione, in 1.5 M GuHCl, 50 mM Tris-HCl, pH 8.4. The oxidized sample was purified by preparative C18 reversed phase liquid chromatography (RP-HPLC) on a Waters Delta Prep 600 system and freeze-dried. The His-tag was cleaved from the His6-pro-cathelicidin protein by thrombin, which specifically recognizes and cleaves a sequence located upstream of the pro-cathelicidin sequence, i.e., LVPR↓GSHM. The digestion was carried out in thrombin digestion buffer (150 mM of NaCl, 2.5 mM CaCl2 and 20 mM Tris-HCl, pH 8.4) with 2.5 U of thrombin (Invitrogen)/mg of His6-pro-cathelicidin. The cleavage proceeded to completion in 30 minutes and the full-length pro-cathelicidin was purified to homogeneity by RP-HPLC and subsequently lyophilized. Of note, the recombinant pro-cathelicidin protein contained four extra amino acid residues (GSHM) at its N-terminus – part of the thrombin cleavage sequence
Generation and purification of the hCLD and LL-37
hCLD and LL-37 were generated by enzymatic cleavage of pro-cathelicidin with proteinase 3. Briefly, lyophilized pro-cathelicidin (1mg) was dissolved in 0.1M HEPES, pH 7.5 and treated with 0.5 U Proteinase 3 (Elastin Products Company, Inc, USA) for 3–4 hours, at room temperature. The two desired products were separated by preparative RP-HPLC and purified to homogeneity. The purity and identity of pro-cathelicidin, hCLD and LL-37 preparations were confirmed by analytical RP-HPLC and electrospray ionization mass spectrometry (ESI-MS, Micromass ZQ-4000) (Figure 1).
Figure 1.

C18 RP-HPLC and ESI-MS analysis of purified recombinant pro-cathelicidin, LL-37 and hCLD. Proteins were analyzed on a Waters XBridge™ BEH130 C18 column (4.6 × 150 mm, 3.5μm) using a linear gradient of 5 – 65% acetonitrile at a flow rate of 1 mL/min over 30 min. The experimental values of the molecular masses of the desired products are shown together with expected values calculated based on the average isotope compositions (in parentheses).
Crystallization and Data Collection
The crystallization experiment was performed at room temperature using the hanging-drop vapor diffusion method. Drops were prepared by mixing 1 μl stock solution of hCLD at 10 mg/ml in water and 1 μl reservoir solution (0.1 M sodium HEPES pH 7.5, 0.2 M sodium citrate, 20% isopropanol). The best quality crystals were cryoprotected in a solution containing the reservoir solution and 25% glycerol, and flash-cooled with liquid nitrogen at 100K. The data were collected at Stanford Synchrotron Radiation Laboratory (Menlo Park, CA) (beamline 9-1) using an ADSC Quantum-315R CCD detector (Area Detector Systems Corp). The data were integrated with DENZO and scaled with SCALPACK using the HKL2000 package (36). Crystals of hCLD belonged to space group p21212 with unit cell dimensions of a=91.15 Å, b=57.99 Å, c=39.55 Å and two molecules of hCLD per asymmetric unit. The data statistics are summarized in Table 1.
Table 1.
Crystallographic data collection and refinement statistics
| Data collection | |
|---|---|
|
| |
| Wavelength, Ǻ | 0.98 |
| Space group | P21212 |
| Cell parameters | a=91.15 Å, b=57.99 Å, c=39.55 Å 2 |
| Molecules/a.u. | 91.15-1.93 (2.0-1.93) |
| Resolution, Åa | |
| Number of reflections | 30982 |
| Total | 16740 |
| Unique | 9.8 (37.2) |
| Rsymb, % | 14.4 (6.1) |
| I/σ | 99.4 (98.7) |
| Completeness, % | 6.2 (6.8) |
| Redundancy | |
|
| |
| Refinement statistics | |
|
| |
| Resolution, Å | 20-1.93 |
| Rcrystc, % | 19.1 |
| Rfreed, % | 24.2 |
| Number of atoms | |
| Protein | 1629 |
| Water | 133 |
| Root mean square deviation | |
| Bond lengths, Å | 0.02 |
| Bond angles, ° | 2.0 |
| Ramachandran plot | |
| Most favored region, % | 92.3 |
| Additional allowed region, % | 7.7 |
| Generously allowed region, % | 0.0 |
| Disallowed region, % | 0.0 |
| PDB code | 4EYC |
All data (outer shell).
Rsym = Σ|I − <I>|/ΣI, where I is the observed intensity and <I> is the average intensity obtained from multiple observations of symmetry-related reflections after rejections
Rcryst = Σ||Fo| − k| Fc||/Σ|Fo |, where Fo and Fc are the observed and calculated structure factors, respectively
Rfree = defined by by Brünger (91)
Structure Determination and Refinement
The structure of hCLD was solved by molecular replacement with Phaser (37) from the CCP4 suite (38) using the CLD of protegrin-3 (PDB code 1PFP, (39)) as a search model. The structure was refined with the program Refmac5 (40) and rebuilt using the program COOT (41). Final model statistics are shown in Table 1. The coordinates and structure factors have been deposited in the Protein Data Bank (PDB code). Ramachandran statistics were calculated with MolProbity (42) and all illustrations were prepared with the the PyMol Molecular Graphic suite (DeLano Scientific, San Carlos, CA, USA)
Antimicrobial assays
Pro-cathelicidin, hCLD and LL-37 were tested for antibacterial activity against Escherichia coli ATCC 25922, Enterobacter aerogenes ATCC 13048 and Staphylococcus aureus ATCC 29213. Assays for antimicrobial activity were conducted using the 96-well turbidimetric method known as virtual colony count (vCC), as previously described for α-defensins (43). Briefly, the bacteria (1×106 CFU/ml) were incubated at 37°C for 2h with various concentrations of protein. After addition of twice-concentrated Mueller-Hinton broth, the kinetics of bacterial growth were measured at 650 nm for 12 hours to determine the time necessary to reach a threshold change in optical density of 0.02. Calibration experiments were conducted per Table 1 and Figure 2 of (43) using a Tecan infinite M1000 plate reader and 10 mM phosphate buffer without supplementation by tryptic soy broth or other nutrients, resulting in a slope of −70.124 and y-intercept of 520.4 for E. coli, a slope of −65.315 and a y-intercept of 484.3 for E. aerogenes, and a slope of −74.365 and y-intercept of 558.54 for S. aureus. Because pro-cathelicidin and LL-37 in phosphate buffer were found to react with Mueller-Hinton broth to produce turbidity in the absence of cells, all four peptides were tested in the absence of cells. These background optical density experiments indicated that only pro-cathelicidin and LL-37 turbidity fluctuated during the assay. Except for one isolated point of the virtual survival plot at 256 μg/mL (57 μM) caused by an anomalous S. aureus growth curve with a slightly slower growth rate than controls that was not corrected, LL-37 background turbidity only fluctuated significantly at 256 μg/mL, which completely inhibited growth in all eight other vCC experiments with the three strains. This anomalous S. aureus growth curve could have been an experimental artifact resulting from cross-contamination leading to one or more cells adhering to the side of the well above the liquid until after the abrogation of LL-37 activity due to the addition of twice-concentrated Mueller Hinton broth to the well. The 256-1 twofold dilution series of pro-cathelicidin was included on each microplate with buffer added in place of cells diluted in buffer, and optical density readings were subtracted from the experimental pro-cathelicidin data exposed to cells at each 5-minute time point. In addition, a data processing procedure called translocation was developed to correct for S. aureus growth curves that had reduced growth rates compared to the controls after the addition of Mueller-Hinton broth. The threshold times at a change in optical density of 0.01 were calculated, and then the doubling times of each curve between 0.01 and 0.02 and standard deviation (SD) were calculated. Curves with doubling times more than 3×SD greater than the mean of the controls were corrected by the translocation procedure, which replaced absorbance readings after a change in optical density of 0.001 from a representative average control curve from the calibration experiment. All assays were carried out in triplicate, except for hCLD and the lower concentration range of pro-cathelicidin (diluted twofold from 128 to 0.5 μg/mL), which were assayed in duplicate. Some points on the virtual survival plot represent less than triplicate or duplicate results, as explained in the Table ST1. Human α-defensin 1 (HNP1) was used as a positive control. The virtual LD50 (vLD50), vLD90, vLD99 and vLD99.9 were reported as the protein concentrations that resulted in virtual survival rates of 0.5, 0.1, 0.01 and 0.001, respectively.
Enzyme inhibition assays
A kinetic assay with fluorogenic substrates was used to determine the inhibitory activity of pro-cathelicidin, hCLD and LL-37 against human liver cathepsin L (Calbiochem, CA), as described earlier for the inhibition of the cysteine proteinases by cystatins (44). In brief, the enzyme (0.1 mU) was incubated for 2 min at 30°C with increasing concentrations (1 – 1000 nM) of pro-cathelicidin, hCLD and LL-37 in 0.1 M acetate buffer, pH 5.5, 1 mM EDTA, 1 mM DTT, 0.1% Brij 35, followed by the addition of Z-Phe-Arg-AMC (where Z stands for benzyloxycarbonyl and AMC for 7-amino-4-methylcoumarin) to a final concentration of 20 μM. The fluorescence increase due to substrate cleavage was measured continuously for 20 min at excitation and emission wavelengths of 360 and 460 nm, respectively, using a Varian (Cary) Eclipse fluorescence spectrophotometer accessorized with a Cary Eclipse microplate reader. Human urine cystatin C was used as a positive control.
RESULTS
Protein preparation
Pro-cathelicidin (140 aa) with an N-terminal His6Tag (in the pET15b expression vector) was expressed in E. coli as an insoluble protein and purified under denaturing conditions by immobilized nickel affinity chromatography. His6-tagged pro-cathelicidin was fully reduced with DTT and reoxidized through thiol-disulfide shuffling in the presence of reduced and oxidized glutathione. Oxidative folding resulted in one major peak on analytical C18 RP-HPLC, which corresponds to the oxidized product with an observed molecular mass of 18175.9 Da (the expected value was 18175.6 Da, calculated on the basis of the average isotope compositions). The His6Tag was removed by thrombin cleavage and recombinant pro-cathelicidin was purified to homogeneity by RP-HPLC. Quantitative cleavage was achieved within 30 min of incubation at room temperature, and nonspecific cleavage or further degradation of the desired product was not observed. The molecular mass of the cleaved product was determined to be 16424.5 Da, in agreement with the calculated value of 16424.7 Da (Figure 1). Biosynthesis on a one-liter culture scale typically yielded 3 mg of highly pure and correctly folded protein.
Proteinase 3 is the processing enzyme of pro-cathelicidin in vivo. It recognizes with high specificity a cleavage site within the pro-cathelicidin sequence (between Ala133 and Leu134) to generate hCLD (residues 31–133) and LL-37 (residues 134–170) (19). We treated recombinant pro-cathelicidin with proteinase 3 and, as expected, two chromatographically distinct species corresponding to hCLD and LL-37 were recovered. The two desired products were purified by RP-HPLC to homogeneity, and their molecular masses were ascertained by ESI-MS (Figure 1). For hCLD, its determined molecular mass of 11949.2 Da was in agreement with the expected value of 11949.4 Da calculated from the average isotopic compositions of hCLD; for LL-37, its determined molecular mass of 4492.9 Da also agreed with its theoretical value of 4493.3 Da.
hCLD adopts the cystatin-like structural fold
The crystal structure of hCLD was determined in space group P21212 and refined to 1.93 Å resolution (Table 1, Figure 2 and S1). The final model consists of two hCLD molecules in one asymmetric unit, both missing Pro65 located in the L1 loop region. In molecule A the four extra amino acid residues as part of the His6Tag were assigned as −3 (Gly), −2 (Ser), −1 (His) and 0 (Met). The two hCLD molecules present in the asymmetric unit could be aligned with a root-mean-square (rms) deviation value of 0.61 Å for 95 Cα atoms (Figure S1). The structure was refined to a final Rcrys of 19.6% and an Rfree of 24.9% with all non-glycine and non-proline residues falling in the most favored or additionally allowed regions of the Ramachandran plot (Table 1).
Figure 2.

The overall structure of the human cathelin-like domain. (A) The stereoscopic representation of the fold of hCLD drawn as a cartoon diagram. Disulfide bonds are depicted as yellow balls and sticks. (B) Alignment of hCLD and ProS, the cathelicidin-like domain of the pig. A stereo view of the superposition of crystal structures (hCLD in green, ProS in orange) is shown at the top of the Figure, with the amino acid sequence alignment at the bottom. Amino acids identical in both sequences are indicated by stars. Topology diagrams depicting a distribution of secondary structure elements as calculated with DSSP (87) are shown above the aligned sequences and assisted by a plot of the average RMS deviations (in Å) of the corresponding Cα positions.
Figure 2A shows the molecule of hCLD. It adopts a cystatin-like fold (45, 46) and contains a compact core formed by a twisted four-stranded antiparallel β-sheet and a long N-terminal α-helix packing against each other. From the top, the core is flanked by flexible loops L1 (connecting β1 to β2) and L2 (connecting β3 to β4), and from the bottom, by an appending domain (connecting β2 to β3) consisting of a short 310-helix (Pro94-Glu95-Asp96) and two β turns (Arg88-Thr89 and Lys101-Asp102). This well-defined hCLD structure is stabilized by the Cys86–Cys97 disulfide bond, which additionally connects the appending domain to the β2-strand and the core. The second disulfide bond Cys108–Cys125 is formed within the core and connects the β3 and β 4 strands.
The hCLD structure shows remarkable similarity to ProS, the only other CLD protein studied so far by X-ray crystallography and NMR spectroscopy (47–49). hCLD and ProS share 66% sequence identity and, as shown in Figure 2B, their overall crystal structures are highly similar, with a root-mean-square deviation (rmsd) of 0.88Å for 81 Cα atom pairs (as compared with 1KWI) (47). Comparison of the distribution of secondary structure elements in both proteins confirms almost identical global folds and reveals only minor differences in the length of the main α-helix and the β4 strand. In fact, both ends of the β4 strand show the largest rmsd for corresponding Ca atoms (Figure 2B).
Of note, the crystal structure of ProS lacks the L1 and L2 loops due to poorly defined electron density (47). We were able to resolve the conformation of the L2 loop in two hCLD molecules present in the asymmetric unit, which adopts a hydrogen-bonded tight turn spanning residues Gln116-Ala117-Arg118 (Figure 3). Comparison of the hCLD crystal structure and a solution structure of ProS ((49), rmsd of 1.1 Ǻ) shows a lack of such a turn in the highly mobile and unstructured L2 loop region of ProS. Analysis of the crystal contacts reveals that the Gln116-Arg118 region of hCLD is partially constrained by intermolecular contacts. Thus, the L2 loop structure seen in hCLD may only partially represent a native conformation.
Figure 3.

Comparison of the crystal structure of hCLD and the solution structure of ProS. Left panel: Superposition of Ca ribbon diagrams of hCLD (green) and ProS (pink) Right panel: 90° rotated close-up view of the L1 and L2 loop regions. Disulfide bonds and residues of hCLD involved in a hydrogen-bonded turn are shown as balls and sticks.
hCLD does not inhibit cathepsin L
Recently, Zaiou et al. (35) and Zhu at al.(50) have reported intriguing results of cysteine proteinase inhibitory activities of human and porcine CLDs. Recombinant hCLD and ProS were reported to inhibit and activate cathepsin L, respectively. In light of these paradoxical reports, we tested the inhibition of human liver cathepsin L by our recombinant hCLD protein using the same protocol as described earlier for kinetic measurements of inhibitory activity of cystatins against cysteine proteases (44), and we failed to replicate these findings.
We incubated cathepsin L (0.1 mU) for 2 min at 30°C with increasing concentrations of hCLD (from 0 to 1,000 nM) followed by addition of the fluorescent substrate Z-Phe-Arg-AMC, and the enzymatic reaction was kinetically monitored over a period of 20 min. As shown in Figure 4A, hCLD had little effect on cathepsin L activity at concentrations of up to 1,000 nM, while 10 nM of human urine cystatin C nearly quantitatively inhibited this protease under identical conditions. Since weak inhibitory activity against cathepsin L has been reported earlier for bovine cathelicidins ProBac5 (51, 52), ProBac7 and proBMP-28 (52), we also tested cathepsin L inhibition by human pro-cathelicidin and the LL-37 peptide. Indeed, pro-cathelicidin and LL-37 showed similarly weak but dose-dependent inhibition of cathepsin L at concentrations above 10 nM (Figure 4B). At the highest concentration used (1,000 nM), over 50% inhibition of protease activity was observed for pro-cathelicidin and LL-37. Since hCLD was inactive against cathepsin L, inhibition of cathepsin L by pro-cathelicidin and LL-37 likely resulted from the C-terminal cathelicidin domain.
Figure 4.
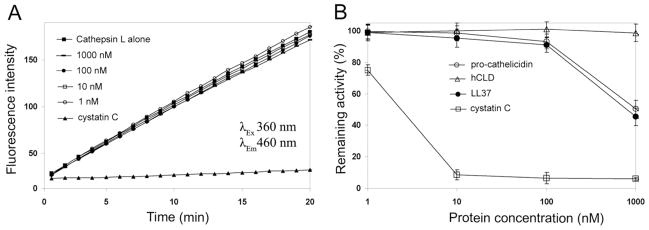
Effect of pro-cathelicidin, hCLD and LL-37 on the activity of human cathepsin L. (A) Hydrolysis of Z-Phe-Arg-AMC substrate by human liver cathepsin L (0.1 mU) in the presence of hCLD (concentration ranging from 0–1,000 nM). Human urine cystatin C at 10 nM concentration was used as a positive control. (B) Dose-dependent inhibition of cathepsin L (0.1 mU) by pro-cathelicidin, hCLD and LL-37 and human urine cystatin C. The remaining activity of cathepsin L was measured 20 minutes after the reaction and hydrolysis was initiated by the addition of the Z-Phe-Arg-AMC substrate. Each curve is the mean of three independent experiments.
hCLD shows no antibacterial activity
Zaiou et al reported that recombinant hCLD protein was bactericidal when tested in a radial diffusion assay and a standard liquid phase AMP test (35). At concentrations ranging from 16 to 32 μM, hCLD was shown to kill E. coli and S. aureus strains efficiently whereas pro-cathelicidin and mature LL-37 were inactive against S. aureus (35). These findings were surprising in light of the fact that LL-37 has been widely reported as a potent bactericidal peptide that acts effectively against both Gram-positive and Gram-negative strains of bacteria (30, 53, 54). We were therefore motivated to test the antibacterial activity of pro-cathelicidin, hCLD and LL-37 against E. coli and E. aerogenes (Gram-negative) and S. aureus (Gram-positive) using the previously established vCC turbidimetric method (43). To verify the vCC results, we also conducted an antibacterial assay using traditional colony count methods in the same 10 mM phosphate buffer used as the vCC experiment, and with the same 2-hour incubation time. The a-defensin human neutrophil peptide 1 (HNP1) was used as a positive control, and the data are shown in Figure 5A–C and S2 and Table ST1 and ST2.
Figure 5.

Antibacterial activity of pro-cathelicidin, hCLD and LL-37 determined by the virtual colony count method. Virtual survival curves are shown of (A) E. coli ATCC 25922, (B) E. aerogenes ATCC 13048 and (C) S. aureus ATCC 29213 exposed to protein concentrations varying twofold from 1–256 μg/mL in triplicate, except hCLD was analyzed in duplicate. In addition, because the E. aerogenes results included a paradoxical point at 256 μg/mL, and because the background turbidity in the absence of cells was much greater when pro-cathelicidin in phosphate buffer was mixed with 2× Mueller-Hinton broth at 256 μg/mL than at 128 μg/mL or below, pro-cathelicidin was also analyzed in a separate series from 0.5–128 μg/mL. Some points are reported in duplicate (3.6 μM LL-37 vs. both E. coli and E. aerogenes, 18.6 μM HNP1 vs. S. aureus) or singly (7.1, 14.2 and 57.0 μM LL-37 vs. S. aureus) because the other replicates were scored with threshold times of >720 minutes (virtual survival = 0) in the VCC assay. Virtual survival was also zero for the four highest concentrations tested (7.1–57.0 μM) of LL-37 vs. both E. coli and E. aerogenes, and the two highest concentrations of HNP1 (37.2 and 74.4 μM) vs. S. aureus and these points could not be plotted on a logarithmic scale. Protein concentrations were converted from μg/mL to μM for plotting. HNP1 was used as positive control. Each curve is the mean of three separate experiments (±S.D.). Points scored as zero virtual survival could not be plotted. (D) Antibacterial activity of LL-37 in the presence of the hCLD protein. The virtual survival curves of E. coli ATCC 25922 are shown in the presence of 1.4 μM LL-37 alone (open circle) or titrated with hCLD at concentrations varying twofold from 0.23 to 60 μM (open squares). The data are an average of two independent experiments.
As expected, and consistent with previously reported data (30, 53, 54), LL-37 displayed potent antibacterial activity, and LL-37 was more potent than HNP1 against the tested bacteria in the region of the virtual lethal doses. Importantly, the precursor protein pro-cathelicidin also efficiently inhibited the growth of two Gram-negative strains tested, E. coli and E. aerogenes, with similar potency (Figure 5A–C and Table ST1). Apparently, hCLD intramolecularly does not abrogate the antibacterial action of LL-37 except in the case of the Gram-positive S. aureus. Compared to other cathelicidins, our results with human pro-cathelicidin are distinct in that other members of the family are inactive before proteolytic processing. In this respect, human pro-cathelicidin may be an exception to the rule. In sharp contrast, no antibacterial activity was detected for the hCLD protein even at the highest concentration used (60 μM). We confirmed our findings on pro-cathelicidin, hCLD and LL-37 with respect to their anti-E. coli activity or lack thereof using the standard radial diffusion method (55) and a lawn-spotting assay in a second laboratory. As shown in Figure S3 and S4, while LL-37 and pro-cathelicidin showed dose-dependent inhibition of the growth of E. coli colonies, hCLD was completely inactive at the highest concentration tested (50 μM). In addition, the traditional colony count results supported vCC qualitatively (Figure S2 and Table ST2). HNP1 activity at high concentrations was greater in the vCC assay than the colony count assay, indicating that vCC measures lag times in addition to bacteriostatic growth inhibition and/or bactericidal killing, as initially reported (43).
hCLD does not inhibit LL-37
Human α-defensins are synthesized in vivo as inactive precursors, and defensin activation requires proteolytic removal of the N-terminal pro-domains (56). The pro-domains functionally neutralize defensin activity through both intramolecular and intermolecular interactions with defensins (55, 57). To better understand the functional aspects of the hCLD in relation to LL-37 in pro-cathelicidin, we tested the ability of different concentrations of hCLD to inhibit the anti-E. coli activity of LL-37 at the LD90 concentration of 1.4 μM. As shown in (Figure 5D), no decrease of antibacterial activity was observed across the entire concentration range (up to 60 μM), demonstrating that exogenously added hCLD does not inhibit the antibacterial activity of the mature LL-37 peptide.
DISCUSSION
Cathelicidin antimicrobial peptides form a distinct family of host defense peptides known to share a characteristic gene architecture (1). At the protein level, they are organized in such a way that the highly diverse C-terminal antimicrobial domain is attached to the large and highly conserved pro-domain known as the cathelin-like domain, which is constrained by two invariant disulfide bonds and shows high homology to the cathelin of pig leukocytes (1, 9, 69). The structures of C-terminal antimicrobial peptides are highly variable, including short tryptophan-, proline-rich or α-helical peptides and β-hairpin peptides stabilized by one or two disulfide bonds. Although the exact mechanism of the antimicrobial action of individual CAMPs may vary, they all appear to act on microbial membranes. Despite their diverse sizes and structures, nearly all of them display a net positive charge and maintain an overall amphipathic character – the common features which allow them to attach to and disrupt the negatively charged microbial membranes (1, 8). It is broadly accepted that precursor forms of CAMPs are functionally inactive and require proteolytic cleavage to free the C-terminal active peptide (12, 14, 16, 20). Recently, the recombinant pro-cathelicidin hCAP18 has been shown to be inactive against a panel of Gram-positive and Gram-negative bacteria tested in standard radial diffusion and liquid phase AMP testing assays (35). In contrast, the mature LL-37 peptide and hCLD demonstrated antibacterial activity against distinct spectrums of bacteria (35). We produced full-length pro-cathelicidin precursor protein by a recombinant method in E. coli and generated the cathelin-like domain protein (hCLD) and the mature LL-37 peptide by cleavage of the precursor holoprotein with the in vivo processing enzyme, proteinase 3 (19). The homogeneity and correct folding of recombinant proteins were confirmed by detailed ESI-MS and structural studies of hCLD. We used recombinant pro-cathelicidin, hCLD and LL-37 proteins in a functional analysis, the results of which were distinct from those of Zaiou et al (35) and were in contrast to an earlier prevailing view of the relationship between the processing of CAMPs and their function. The precursor holoprotein pro-cathelicidin displayed antibacterial activity similar to mature LL-37 against E. coli and E. aerogenes tested by the vCC method and E. coli tested by the traditional colony count method. The lack of activity against S. aureus suggests that different mechanisms of action exist between Gram-positive and Gram-negative bacteria, perhaps in a way similar to defensins (70– 72). The cathelin-like domain protein, hCLD, lacked any antibacterial function. Taken together, our studies demonstrated for the first time that the antibacterial activity against gram negative bacteria of the LL-37 cathelicidin is not inhibited by the hCLD pro-region, both intramolecularly within the precursor holoprotein, and intermolecularly, since an exogenously added pro-region protein had no effect on the bactericidal activity of the mature peptide. In this respect, human cathelicidin differs significantly from defensins, for which the processing of inactive precursors was shown to be definitively required for antibacterial function (56, 73, 74). The pro-peptides of defensins are anionic, especially in the case of the cryptdins, the family of α-defensins from the Paneth cells of the mouse (56, 75). It has been shown that nine N-terminal acidic amino acids of pro-cryptdin 4 are primarily responsible for inhibition of the defensin domain (76, 77). Unlike the pro-peptides of defensins, the hCLD pro-region has no net charge. Although it has been shown for human a-defensin, HNP1 that hydrophobic forces rather than electrostatic interactions have a dominant role in mediating the interaction between the anionic pro-peptide and the cationic defensins (57), the absence of charge complementarity between the neutral hCLD pro-region and its cationic LL-37 counterpart should not be overlooked. The lack of electrostatic potential may directly determine the low affinity intermolecular binding of the hCLD protein for the LL-37 peptide and the lack of intramolecular inhibition of LL-37 activity within the precursor protein. Further, whereas pro-defensins have a net neutral charge, the pro-cathelicidin precursor preserves the cationicity of the mature LL-37 peptide (+6 net charge). Therefore, if cationicity is the most important factor determining the antimicrobial activity of the mature LL-37 peptide, the unprocessed pro-cathelicidin protein should retain effectiveness.
Sanchez et al (47) first elucidated the structural basis of interactions between the cathelin-like domain pro-region and the cathelicidin antimicrobial peptide based on the modeled structure of the porcine ProS-PG1 precursor protein. Precursor forms of protregrins were shown previously to be inactive against bacteria, requiring cleavage by elastase to perform their antimicrobial functions, (78) and the proposed model of the precursor holoprotein fully explains the mechanism by which the N-terminal antibacterial peptide is inhibited functionally by its C-terminal pro-region (47). In contrast to LL-37, which is unstructured in solution and adopts a-helical structure only in structure-inducing environments such as the presence of SDS (54, 79), PG1 consists of a rigid β-hairpin structure constrained by two disulfide bonds (80). In the proposed minimized model of the holoprotein, the PG1 peptide docks as an extension of the central β-sheet of ProS and establishes multiple hydrophobic and electrostatic interactions with the ProS core. In addition, the high content of anionic residues (17 acidic residues) and the negative net charge (−4) of ProS suggest that electrostatic interactions with the positively charged antimicrobial peptide are likely. A predicted tight packing of the holoprotein and multiple intramolecular interactions between ProS and PG1 has been suggested to be responsible for inactivation of the antibacterial precursor (47). A similar model of a compact and well-stabilized structure of the holoprotein cannot be easily proposed for the hCLD-LL-37 precursor based on the available hCLD structure. In fact, the lack of charge complementarity between hCLD and LL-37 and the low intermolecular affinity of the cathelin-like pro-region for the LL-37 peptide preclude that a compact conformation would be formed when assembled into a single polypeptide chain. A similar susceptibility for degradation in vitro and in vivo in human wound fluid by elastase-producing P. aeruginosa has been recently reported for both the mature LL-37 peptide and the precursor pro-cathelicidin, suggesting that the antimicrobial peptide is not protected in the holoprotein from proteolytic degradation in protease-rich environments by the cathelin-like platform (3, 81). Nevertheless, LL-37 is present primarily as the uncleaved holoprotein (3) in a variety of locations including wound and blister fluids (82), phagocytic vacuoles (19), psoriatic skin (83), and seminal plasma (27). In this sense, perhaps it is not surprising that the pro-cathelicidin is active, since it is so widespread.
A high degree of genetic similarity of the cathelin-like motif of cathelicidins to the cystatin family of protease inhibitors suggested that CLDs may act as protease inhibitors as well (12, 15, 21). Cathelin, a 96-residue peptide isolated from porcine neutrophils and sharing about 70% identity with CLDs, initially was shown to inhibit cathepsin L. The inhibition was later ascribed to contamination by cystatins, calling into question the classification of porcine cathelin as a cysteine protease inhibitor (58). On the other hand, very recent studies suggested that human and porcine CLD may function as inhibitors or activators of cathepsin L, respectively (35, 50). Based on the modeled structures of hCLD and ProS in complex with cysteine protease, the functional switch from inhibition to activation was attributed to the sequence variations in the L2 loop region of human and porcine CLDs (35, 50, 59). Indeed, hCLDs and cystatins adopt similar three-dimentional structures, known as the cystatin-like fold, but our structural analysis does not confirm that hCLD is a functional inhibitor of cysteine proteases (60). First of all, hCLD and other CLDs differ substantially from cystatins at the protein sequence level. hCLD lacks the functional residues that are involved in the specific binding of cysteine protease inhibitors to their targets, which are highly conserved among cystatins. It was shown that effective inhibitor binding requires presence of the GAP11, QVVAG57 and PW104 motifs (stefin B numbering) within the extended N-terminus and the L1 and L2 loops of cystatins, respectively (61–63). These motifs are not present in the hCLD sequence. The crystallographic studies of complexes formed by cysteine proteases and their cystatin inhibitors indicate that the cystatin-like fold of cystatins allows them to form a characteristic wedge-shaped structure with the N-terminal trunk and two hairpin loops at its narrow edge that fits into the substrate binding groove of the target protease (64–67). The extended N-terminus of cystatin occupies the S1 and S2 sites of the substrate binding area and occludes the catalytic Cys of the protease, whereas the L1 and L2 loops bind into the S1′ and S2′ sites, respectively. We aligned structurally the hCLD to the monomeric human Cystatin C in the unliganded state (PDB code: 3GAX, (68), Figure 6). In addition, since there is no available complex structure of Cystatin C with any of its targeted proteases, we superimposed the hCLD model to that of stefin A in the stefin A-cathepsin H complex (Figure 7, (67)). The structural similarities between hCLD and Cystatin C/stefin A are limited only to a common global fold, which clearly varies at the N-terminus as well as the L1 and L2 loop regions. As shown in Figure 2, the hCLD has a relatively short N-terminus with only four residues (the 30QVLS segment) proceeding the N-terminal a-helix. Although the hCLD protein used in our studies has a four amino acid extension at the N-terminus as a remnant of the cleavage by thrombin (shown in grey in Figure 6), its presence should not have much effect on its conformation due to rigidifying interactions of the neighboring helix. The short N-terminus of hCLD faces away from the S1 and S2 sites which are occupied in the complex by the elongated N-terminal trunk of cystatin C/stefin A. These substantial differences are observed also for the conformation of the L1 and L2 loops of hCLD as compared to the equivalent hairpin loops of cystatin c/stefin A. hCLD, with its longer and ‘relaxed’ L1 loop and L2 loop constrained by the hydrogen-bonded turn at the top, is unable to form a characteristic wedge that could fit into the substrate-binding groove of cathepsin H or other cysteine proteases. (47, 52).
Figure 6.

Structural alignment of hCLD and the cysteine protease inhibitor Cystatin C. Backbone atoms of hCLD were superimposed on monomeric human Cystatin C in the unliganded state (PDB code: 3GAX). hCLD is shown in green, Cystatin C in yellow and, in both, the N-terminus is colored in red. Four additional residues at the N-terminus of hCLD as remnants of the cleavage by thrombin are colored in grey. The crystal structure of monomeric human cystatin C stabilized against aggregation was chosen for comparison since in all crystal structures of Cystatin C studied to date, the protein has been found to form 3D domain-swapped dimers, created through a conformational change of a beta-hairpin loop, L1 (68, 88–90). 3D domain swapping free structure defines the conformation of loop L1, which is essential for the inhibition of papain-like cysteine proteases.
Figure 7.

Structural comparison of hCLD and the cysteine protease inhibitor stefin A. (A) Backbone atoms of hCLD were superimposed on stefin A in the stefin A-cathepsin H complex (PDB code: 1NB5, (67)). hCLD is shown in green, stefin A in blue and cathepsin H in grey. Catalytic Cys-25 and His-159 of cathepsin H are shown in yellow and residues of the N-termini and hairpin L1 and L2 loops of stefin A with motifs highly conserved among cysteine protease inhibitors are shown in light blue. The disordered Pro65 region of the L1 loop is indicated by a green dotted line (B) The same as (A) with the cathepsin H surface shown in grey and colored yellow around active site residues.
Interestingly, we detected a moderate inhibition of cathepsin L activity in the presence of high doses of the precursor pro-cathelicidin holoprotein and the mature LL-37 peptide. The fact that the same inhibitory effect is observed for both pro-cathelicidin and LL-37 suggests that the LL-37 peptide part of the precursor, rather than the cathelin-like motif, interacts with the cathepsin L active site. Similar results were reported in the early 1990s when the weak inhibition of cathepsin L by the precursor forms of bovine cathelicidins ProBac5, ProBac7 and proBMP-28 was detected (51, 52). These authors suggested that a weak inhibitory activity observed for some cathelicidin precursors is functionally irrelevant and may be due to nonspecific interactions that block access to the enzyme active site (52). In fact, we confirmed that the LL-37 peptide may serve as a cathepsin L substrate (data not shown).
In conclusion, our results contrast with the commonly held view that cathelicidin precursors are unable to display the biological functions of the mature cathelicidin peptide due to intramolecular inhibition by the cathelin-like domain pro-region. We limited our functional analysis to antibacterial activity testing, so we cannot exclude that the pro-cathelicidin precursor holoprotein may also exhibit other biological activities of LL-37, which were initially assigned only to the mature peptide. Further studies are required to determine the biological activities of pro-cathelicidin and fully analyze the spectrum of functional overlap between LL-37 and its precursor protein. Since the CAMP family is highly heterogeneous and covers a wide range of structurally distinct peptides, we cannot conclude that observations we made of the precursor protein of human cathelicidin could be generalized over all CAMP family members. To the contrary, we strongly believe that other family members may undergo functional inactivation through interactions with the cathelin-like pro-region in the precursor holoprotein. As an example, a cysteine-bridged β-hairpin of porcine protregrin was proposed to dock effectively on the cathelin-like motif platform. In this respect, human cathelicidin may be the only exception. It would not be the only antimicrobial peptide to be active in the human version in ways that differ from other mammals. For example, defensins bind and inactivate anthrax lethal factor (71, 84, 85), and the defensin HD6 self-assembles to form nanonets that trap bacteria (86).
To this end, the significance of the strong evolutionary pressure for the conservation of the cathelin-like pro-region among the CAMP family is still poorly explored and a matter of debate. By combined structural and functional studies, we proved that hCLD does not exhibit a protease inhibitory function regardless of its overall structural similarity to cystatins. CLDs share a common cystatin-like fold with a wide range of distinct proteins which play a variety of roles other than protease inhibitory biological functions (47). Although the evolutionary record contains only one human CLD among the great diversity of CLDs found in mammals, it appears that the cystatin scaffold may represent an ancestral structural platform which has diverged in the various species to form proteins both with and without inhibitory functions.
Supplementary Material
Acknowledgments
Funding Sources:
This work was supported by American Cancer Society Research Scholar Grant CDD112858 and National Institutes of Health Grants AI056264 and AI061482 (to W.L.)
We would like thank Dr. Sergey G. Tarasov and Ms. Marzena Dyba of Structural Biophysics Laboratory, NCI-Frederick for participation in the discussion of structural models and Dr. Erik de Leeuw from the Institute of Human Virology, University of Maryland School of Medicine, who assisted with vCC data recording and calculation. We thank the X-ray Crystallography Core Facility of the University of Maryland at Baltimore for providing crystallographic equipment and resources. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209), and the National Institute of General Medical Sciences.
ABBREVIATIONS
- CAMP
Cathelicidin antimicrobial peptide
- hCLD
human cathelin-like domain
- hCAP18
human cationic antimicrobial protein
- SPR
surface plasmon resonance
- RP-HPLC
reversed-phase high-performance liquid chromatography
- ESI-MS
electrospray ionization mass spectrometry
- vCC
virtual colony count
Footnotes
Author Contributions:
All authors have given approval to the final version of the manuscript.
Supporting Information. Table ST1 and ST2, Figures S1, S2 and S3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lehrer RI, Ganz T. Cathelicidins: a family of endogenous antimicrobial peptides. Curr Opin Hematol. 2002;9:18–22. doi: 10.1097/00062752-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Zanetti M. The role of cathelicidins in the innate host defenses of mammals. Curr Issues Mol Biol. 2005;7:179–196. [PubMed] [Google Scholar]
- 3.Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol. 2004;75:39–48. doi: 10.1189/jlb.0403147. [DOI] [PubMed] [Google Scholar]
- 4.Lynn DJ, Higgs R, Gaines S, Tierney J, James T, Lloyd AT, Fares MA, Mulcahy G, O’Farrelly C. Bioinformatic discovery and initial characterisation of nine novel antimicrobial peptide genes in the chicken. Immunogenetics. 2004;56:170–177. doi: 10.1007/s00251-004-0675-0. [DOI] [PubMed] [Google Scholar]
- 5.Uzzell T, Stolzenberg ED, Shinnar AE, Zasloff M. Hagfish intestinal antimicrobial peptides are ancient cathelicidins. Peptides. 2003;24:1655–1667. doi: 10.1016/j.peptides.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 6.Chang CI, Zhang YA, Zou J, Nie P, Secombes CJ. Two cathelicidin genes are present in both rainbow trout (Oncorhynchus mykiss) and atlantic salmon (Salmo salar) Antimicrob Agents Chemother. 2006;50:185–195. doi: 10.1128/AAC.50.1.185-195.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zanetti M, Gennaro R, Scocchi M, Skerlavaj B. Structure and biology of cathelicidins. Adv Exp Med Biol. 2000;479:203–218. doi: 10.1007/0-306-46831-X_17. [DOI] [PubMed] [Google Scholar]
- 8.Tomasinsig L, Zanetti M. The cathelicidins--structure, function and evolution. Curr Protein Pept Sci. 2005;6:23–34. doi: 10.2174/1389203053027520. [DOI] [PubMed] [Google Scholar]
- 9.Scocchi M, Wang S, Zanetti M. Structural organization of the bovine cathelicidin gene family and identification of a novel member. FEBS Lett. 1997;417:311–315. doi: 10.1016/s0014-5793(97)01310-0. [DOI] [PubMed] [Google Scholar]
- 10.Zhao C, Ganz T, Lehrer RI. Structures of genes for two cathelin-associated antimicrobial peptides: prophenin-2 and PR-39. FEBS Lett. 1995;376:130–134. doi: 10.1016/0014-5793(95)01237-3. [DOI] [PubMed] [Google Scholar]
- 11.Zhao C, Ganz T, Lehrer RI. The structure of porcine protegrin genes. FEBS Lett. 1995;368:197–202. doi: 10.1016/0014-5793(95)00633-k. [DOI] [PubMed] [Google Scholar]
- 12.Zanetti M, Gennaro R, Romeo D. Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 1995;374:1–5. doi: 10.1016/0014-5793(95)01050-o. [DOI] [PubMed] [Google Scholar]
- 13.Storici P, Zanetti M. A novel cDNA sequence encoding a pig leukocyte antimicrobial peptide with a cathelin-like pro-sequence. Biochem Biophys Res Commun. 1993;196:1363–1368. doi: 10.1006/bbrc.1993.2403. [DOI] [PubMed] [Google Scholar]
- 14.Ritonja A, Kopitar M, Jerala R, Turk V. Primary structure of a new cysteine proteinase inhibitor from pig leucocytes. FEBS Lett. 1989;255:211–214. doi: 10.1016/0014-5793(89)81093-2. [DOI] [PubMed] [Google Scholar]
- 15.Kopitar M, Ritonja A, Popovic T, Gabrijelcic D, Krizaj I, Turk V. A new type of low-molecular mass cysteine proteinase inhibitor from pig leukocytes. Biol Chem Hoppe Seyler. 1989;370:1145–1151. doi: 10.1515/bchm3.1989.370.2.1145. [DOI] [PubMed] [Google Scholar]
- 16.Gennaro R, Zanetti M. Structural features and biological activities of the cathelicidin-derived antimicrobial peptides. Biopolymers. 2000;55:31–49. doi: 10.1002/1097-0282(2000)55:1<31::AID-BIP40>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 17.Sorensen O, Arnljots K, Cowland JB, Bainton DF, Borregaard N. The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood. 1997;90:2796–2803. [PubMed] [Google Scholar]
- 18.Zanetti M, Litteri L, Griffiths G, Gennaro R, Romeo D. Stimulus-induced maturation of probactenecins, precursors of neutrophil antimicrobial polypeptides. J Immunol. 1991;146:4295–4300. [PubMed] [Google Scholar]
- 19.Sorensen OE, Follin P, Johnsen AH, Calafat J, Tjabringa GS, Hiemstra PS, Borregaard N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood. 2001;97:3951–3959. doi: 10.1182/blood.v97.12.3951. [DOI] [PubMed] [Google Scholar]
- 20.Zarember KA, Katz SS, Tack BF, Doukhan L, Weiss J, Elsbach P. Host defense functions of proteolytically processed and parent (unprocessed) cathelicidins of rabbit granulocytes. Infect Immun. 2002;70:569–576. doi: 10.1128/iai.70.2.569-576.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cowland JB, Johnsen AH, Borregaard N. hCAP-18, a cathelin/pro-bactenecin-like protein of human neutrophil specific granules. FEBS Lett. 1995;368:173–176. doi: 10.1016/0014-5793(95)00634-l. [DOI] [PubMed] [Google Scholar]
- 22.Larrick JW, Hirata M, Balint RF, Lee J, Zhong J, Wright SC. Human CAP18: a novel antimicrobial lipopolysaccharide-binding protein. Infect Immun. 1995;63:1291–1297. doi: 10.1128/iai.63.4.1291-1297.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agerberth B, Gunne H, Odeberg J, Kogner P, Boman HG, Gudmundsson GH. FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc Natl Acad Sci U S A. 1995;92:195–199. doi: 10.1073/pnas.92.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agerberth B, Charo J, Werr J, Olsson B, Idali F, Lindbom L, Kiessling R, Jornvall H, Wigzell H, Gudmundsson GH. The human antimicrobial and chemotactic peptides LL-37 and alpha-defensins are expressed by specific lymphocyte and monocyte populations. Blood. 2000;96:3086–3093. [PubMed] [Google Scholar]
- 25.Frohm Nilsson M, Sandstedt B, Sorensen O, Weber G, Borregaard N, Stahle-Backdahl M. The human cationic antimicrobial protein (hCAP18), a peptide antibiotic, is widely expressed in human squamous epithelia and colocalizes with interleukin-6. Infect Immun. 1999;67:2561–2566. doi: 10.1128/iai.67.5.2561-2566.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malm J, Sorensen O, Persson T, Frohm-Nilsson M, Johansson B, Bjartell A, Lilja H, Stahle-Backdahl M, Borregaard N, Egesten A. The human cationic antimicrobial protein (hCAP-18) is expressed in the epithelium of human epididymis, is present in seminal plasma at high concentrations, and is attached to spermatozoa. Infect Immun. 2000;68:4297–4302. doi: 10.1128/iai.68.7.4297-4302.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andersson E, Sorensen OE, Frohm B, Borregaard N, Egesten A, Malm J. Isolation of human cationic antimicrobial protein-18 from seminal plasma and its association with prostasomes. Hum Reprod. 2002;17:2529–2534. doi: 10.1093/humrep/17.10.2529. [DOI] [PubMed] [Google Scholar]
- 28.Sorensen OE, Gram L, Johnsen AH, Andersson E, Bangsboll S, Tjabringa GS, Hiemstra PS, Malm J, Egesten A, Borregaard N. Processing of seminal plasma hCAP-18 to ALL-38 by gastricsin: a novel mechanism of generating antimicrobial peptides in vagina. J Biol Chem. 2003;278:28540–28546. doi: 10.1074/jbc.M301608200. [DOI] [PubMed] [Google Scholar]
- 29.Durr UH, Sudheendra US, Ramamoorthy A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta. 2006;1758:1408–1425. doi: 10.1016/j.bbamem.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 30.Turner J, Cho Y, Dinh NN, Waring AJ, Lehrer RI. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob Agents Chemother. 1998;42:2206–2214. doi: 10.1128/aac.42.9.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Y, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. 2000;192:1069–1074. doi: 10.1084/jem.192.7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Porcelli F, Verardi R, Shi L, Henzler-Wildman KA, Ramamoorthy A, Veglia G. NMR structure of the cathelicidin-derived human antimicrobial peptide LL-37 in dodecylphosphocholine micelles. Biochemistry. 2008;47:5565–5572. doi: 10.1021/bi702036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J Biol Chem. 2008;283:32637–32643. doi: 10.1074/jbc.M805533200. [DOI] [PubMed] [Google Scholar]
- 34.Sorensen O, Bratt T, Johnsen AH, Madsen MT, Borregaard N. The human antibacterial cathelicidin, hCAP-18, is bound to lipoproteins in plasma. J Biol Chem. 1999;274:22445–22451. doi: 10.1074/jbc.274.32.22445. [DOI] [PubMed] [Google Scholar]
- 35.Zaiou M, Nizet V, Gallo RL. Antimicrobial and protease inhibitory functions of the human cathelicidin (hCAP18/LL-37) prosequence. J Invest Dermatol. 2003;120:810–816. doi: 10.1046/j.1523-1747.2003.12132.x. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W, Carter Charles W., Jr . Methods in Enzymology. Academic Press; 1997. Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 37.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 39.Strub MP, Hoh F, Sanchez JF, Strub JM, Bock A, Aumelas A, Dumas C. Selenomethionine and selenocysteine double labeling strategy for crystallographic phasing. Structure. 2003;11:1359–1367. doi: 10.1016/j.str.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 40.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica Section D, Biological crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ericksen B, Wu Z, Lu W, Lehrer RI. Antibacterial activity and specificity of the six human {alpha}-defensins. Antimicrob Agents Chemother. 2005;49:269–275. doi: 10.1128/AAC.49.1.269-275.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abrahamson M. Cystatins. Methods Enzymol. 1994;244:685–700. doi: 10.1016/0076-6879(94)44051-4. [DOI] [PubMed] [Google Scholar]
- 45.Staniforth RA, Giannini S, Higgins LD, Conroy MJ, Hounslow AM, Jerala R, Craven CJ, Waltho JP. Three-dimensional domain swapping in the folded and molten-globule states of cystatins, an amyloid-forming structural superfamily. EMBO J. 2001;20:4774–4781. doi: 10.1093/emboj/20.17.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murzin AG. Sweet-tasting protein monellin is related to the cystatin family of thiol proteinase inhibitors. J Mol Biol. 1993;230:689–694. doi: 10.1006/jmbi.1993.1186. [DOI] [PubMed] [Google Scholar]
- 47.Sanchez JF, Hoh F, Strub MP, Aumelas A, Dumas C. Structure of the cathelicidin motif of protegrin-3 precursor: structural insights into the activation mechanism of an antimicrobial protein. Structure. 2002;10:1363–1370. doi: 10.1016/s0969-2126(02)00859-6. [DOI] [PubMed] [Google Scholar]
- 48.Sanchez JF, Wojcik F, Yang YS, Strub MP, Strub JM, Van Dorsselaer A, Martin M, Lehrer R, Ganz T, Chavanieu A, Calas B, Aumelas A. Overexpression and structural study of the cathelicidin motif of the protegrin-3 precursor. Biochemistry. 2002;41:21–30. doi: 10.1021/bi010930a. [DOI] [PubMed] [Google Scholar]
- 49.Yang Y, Sanchez JF, Strub MP, Brutscher B, Aumelas A. NMR structure of the cathelin-like domain of the protegrin-3 precursor. Biochemistry. 2003;42:4669–4680. doi: 10.1021/bi027133c. [DOI] [PubMed] [Google Scholar]
- 50.Zhu S, Wei L, Yamasaki K, Gallo RL. Activation of cathepsin L by the cathelin-like domain of protegrin-3. Mol Immunol. 2008;45:2531–2536. doi: 10.1016/j.molimm.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verbanac D, Zanetti M, Romeo D. Chemotactic and protease-inhibiting activities of antibiotic peptide precursors. FEBS Lett. 1993;317:255–258. doi: 10.1016/0014-5793(93)81287-a. [DOI] [PubMed] [Google Scholar]
- 52.Storici P, Tossi A, Lenarcic B, Romeo D. Purification and structural characterization of bovine cathelicidins, precursors of antimicrobial peptides. Eur J Biochem. 1996;238:769–776. doi: 10.1111/j.1432-1033.1996.0769w.x. [DOI] [PubMed] [Google Scholar]
- 53.Larrick JW, Hirata M, Zhong J, Wright SC. Anti-microbial activity of human CAP18 peptides. Immunotechnology. 1995;1:65–72. doi: 10.1016/1380-2933(95)00006-2. [DOI] [PubMed] [Google Scholar]
- 54.Travis SM, Anderson NN, Forsyth WR, Espiritu C, Conway BD, Greenberg EP, McCray PB, Jr, Lehrer RI, Welsh MJ, Tack BF. Bactericidal activity of mammalian cathelicidin-derived peptides. Infect Immun. 2000;68:2748–2755. doi: 10.1128/iai.68.5.2748-2755.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lehrer RI, Rosenman M, Harwig SS, Jackson R, Eisenhauer P. Ultrasensitive assays for endogenous antimicrobial polypeptides. J Immunol Methods. 1991;137:167–173. doi: 10.1016/0022-1759(91)90021-7. [DOI] [PubMed] [Google Scholar]
- 56.Valore EV, Martin E, Harwig SS, Ganz T. Intramolecular inhibition of human defensin HNP-1 by its propiece. J Clin Invest. 1996;97:1624–1629. doi: 10.1172/JCI118588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zou G, de Leeuw E, Lubkowski J, Lu W. Molecular determinants for the interaction of human neutrophil alpha defensin 1 with its propeptide. J Mol Biol. 2008;381:1281–1291. doi: 10.1016/j.jmb.2008.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lenarcic B, Ritonja A, Dolenc I, Stoka V, Berbic S, Pungercar J, Strukelj B, Turk V. Pig leukocyte cysteine proteinase inhibitor (PLCPI), a new member of the stefin family. FEBS Lett. 1993;336:289–292. doi: 10.1016/0014-5793(93)80822-c. [DOI] [PubMed] [Google Scholar]
- 59.Zhu S. Did cathelicidins, a family of multifunctional host-defense peptides, arise from a cysteine protease inhibitor? Trends Microbiol. 2008;16:353–360. doi: 10.1016/j.tim.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 60.Zerovnik E, Staniforth RA, Turk D. Amyloid fibril formation by human stefins: Structure, mechanism & putative functions. Biochimie. 2010;92:1597–1607. doi: 10.1016/j.biochi.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 61.Ochieng J, Chaudhuri G. Cystatin superfamily. J Health Care Poor Underserved. 2010;21:51–70. doi: 10.1353/hpu.0.0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Turk V, Bode W. The cystatins: protein inhibitors of cysteine proteinases. FEBS letters. 1991;285:213–219. doi: 10.1016/0014-5793(91)80804-c. [DOI] [PubMed] [Google Scholar]
- 63.Machleidt W, Thiele U, Assfalg-Machleidt I, Forger D, Auerswald EA. Molecular mechanism of inhibition of cysteine proteinases by their protein inhibitors: kinetic studies with natural and recombinant variants of cystatins and stefins. Biomed Biochim Acta. 1991;50:613–620. [PubMed] [Google Scholar]
- 64.Stubbs MT, Laber B, Bode W, Huber R, Jerala R, Lenarcic B, Turk V. The refined 2.4 A X-ray crystal structure of recombinant human stefin B in complex with the cysteine proteinase papain: a novel type of proteinase inhibitor interaction. The EMBO journal. 1990;9:1939–1947. doi: 10.1002/j.1460-2075.1990.tb08321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bode W, Engh R, Musil D, Thiele U, Huber R, Karshikov A, Brzin J, Kos J, Turk V. The 2.0 A X-ray crystal structure of chicken egg white cystatin and its possible mode of interaction with cysteine proteinases. The EMBO journal. 1988;7:2593–2599. doi: 10.1002/j.1460-2075.1988.tb03109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alvarez-Fernandez M, Liang YH, Abrahamson M, Su XD. Crystal structure of human cystatin D, a cysteine peptidase inhibitor with restricted inhibition profile. The Journal of biological chemistry. 2005;280:18221–18228. doi: 10.1074/jbc.M411914200. [DOI] [PubMed] [Google Scholar]
- 67.Jenko S, Dolenc I, Guncar G, Dobersek A, Podobnik M, Turk D. Crystal structure of Stefin A in complex with cathepsin H: N-terminal residues of inhibitors can adapt to the active sites of endo- and exopeptidases. J Mol Biol. 2003;326:875–885. doi: 10.1016/s0022-2836(02)01432-8. [DOI] [PubMed] [Google Scholar]
- 68.Kolodziejczyk R, Michalska K, Hernandez-Santoyo A, Wahlbom M, Grubb A, Jaskolski M. Crystal structure of human cystatin C stabilized against amyloid formation. Febs J. 2010;277:1726–1737. doi: 10.1111/j.1742-4658.2010.07596.x. [DOI] [PubMed] [Google Scholar]
- 69.Shinnar AE, Butler KL, Park HJ. Cathelicidin family of antimicrobial peptides: proteolytic processing and protease resistance. Bioorg Chem. 2003;31:425–436. doi: 10.1016/s0045-2068(03)00080-4. [DOI] [PubMed] [Google Scholar]
- 70.Pazgier M, Wei G, Ericksen B, Jung G, Wu Z, de Leeuw E, Yuan W, Szmacinski H, Lu WY, Lubkowski J, Lehrer RI, Lu W. Sometimes it takes two to tango: contributions of dimerization to functions of human alpha-defensin HNP1 peptide. The Journal of biological chemistry. 2012;287:8944–8953. doi: 10.1074/jbc.M111.332205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wei G, de Leeuw E, Pazgier M, Yuan W, Zou G, Wang J, Ericksen B, Lu WY, Lehrer RI, Lu W. Through the looking glass, mechanistic insights from enantiomeric human defensins. The Journal of biological chemistry. 2009;284:29180–29192. doi: 10.1074/jbc.M109.018085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rajabi M, Ericksen B, Wu X, de Leeuw E, Zhao L, Pazgier M, Lu W. Functional determinants of human enteric alpha-defensin HD5: crucial role for hydrophobicity at dimer interface. The Journal of biological chemistry. 2012;287:21615–21627. doi: 10.1074/jbc.M112.367995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, Hultgren SJ, Matrisian LM, Parks WC. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286:113–117. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
- 74.Shirafuji Y, Tanabe H, Satchell DP, Henschen-Edman A, Wilson CL, Ouellette AJ. Structural determinants of procryptdin recognition and cleavage by matrix metalloproteinase-7. The Journal of biological chemistry. 2003;278:7910–7919. doi: 10.1074/jbc.M210600200. [DOI] [PubMed] [Google Scholar]
- 75.Ouellette AJ. Paneth cell alpha-defensins in enteric innate immunity. Cellular and molecular life sciences : CMLS. 2011;68:2215–2229. doi: 10.1007/s00018-011-0714-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Figueredo SM, Weeks CS, Young SK, Ouellette AJ. Anionic amino acids near the pro-alpha-defensin N terminus mediate inhibition of bactericidal activity in mouse pro-cryptdin-4. The Journal of biological chemistry. 2009;284:6826–6831. doi: 10.1074/jbc.M807024200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Figueredo SM, Ouellette AJ. Inhibition of bactericidal activity is maintained in a mouse alpha-defensin precursor with proregion truncations. Peptides. 2010;31:9–15. doi: 10.1016/j.peptides.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cole AM, Shi J, Ceccarelli A, Kim YH, Park A, Ganz T. Inhibition of neutrophil elastase prevents cathelicidin activation and impairs clearance of bacteria from wounds. Blood. 2001;97:297–304. doi: 10.1182/blood.v97.1.297. [DOI] [PubMed] [Google Scholar]
- 79.Johansson J, Gudmundsson GH, Rottenberg ME, Berndt KD, Agerberth B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J Biol Chem. 1998;273:3718–3724. doi: 10.1074/jbc.273.6.3718. [DOI] [PubMed] [Google Scholar]
- 80.Aumelas A, Mangoni M, Roumestand C, Chiche L, Despaux E, Grassy G, Calas B, Chavanieu A. Synthesis and solution structure of the antimicrobial peptide protegrin-1. Eur J Biochem. 1996;237:575–583. doi: 10.1111/j.1432-1033.1996.0575p.x. [DOI] [PubMed] [Google Scholar]
- 81.Schmidtchen A, Frick IM, Andersson E, Tapper H, Bjorck L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol Microbiol. 2002;46:157–168. doi: 10.1046/j.1365-2958.2002.03146.x. [DOI] [PubMed] [Google Scholar]
- 82.Frohm M, Gunne H, Bergman AC, Agerberth B, Bergman T, Boman A, Liden S, Jornvall H, Boman HG. Biochemical and antibacterial analysis of human wound and blister fluid. European journal of biochemistry / FEBS. 1996;237:86–92. doi: 10.1111/j.1432-1033.1996.0086n.x. [DOI] [PubMed] [Google Scholar]
- 83.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347:1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 84.Kim C, Gajendran N, Mittrucker HW, Weiwad M, Song YH, Hurwitz R, Wilmanns M, Fischer G, Kaufmann SH. Human alpha-defensins neutralize anthrax lethal toxin and protect against its fatal consequences. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4830–4835. doi: 10.1073/pnas.0500508102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang W, Mulakala C, Ward SC, Jung G, Luong H, Pham D, Waring AJ, Kaznessis Y, Lu W, Bradley KA, Lehrer RI. Retrocyclins kill bacilli and germinating spores of Bacillus anthracis and inactivate anthrax lethal toxin. The Journal of biological chemistry. 2006;281:32755–32764. doi: 10.1074/jbc.M603614200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chu H, Pazgier M, Jung G, Nuccio SP, Castillo PA, de Jong MF, Winter MG, Winter SE, Wehkamp J, Shen B, Salzman NH, Underwood MA, Tsolis RM, Young GM, Lu W, Lehrer RI, Baumler AJ, Bevins CL. Human alpha-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets. Science. 2012;337:477–481. doi: 10.1126/science.1218831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 88.Janowski R, Abrahamson M, Grubb A, Jaskolski M. Domain swapping in N-truncated human cystatin C. Journal of molecular biology. 2004;341:151–160. doi: 10.1016/j.jmb.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 89.Janowski R, Kozak M, Abrahamson M, Grubb A, Jaskolski M. 3D domain-swapped human cystatin C with amyloidlike intermolecular beta-sheets. Proteins. 2005;61:570–578. doi: 10.1002/prot.20633. [DOI] [PubMed] [Google Scholar]
- 90.Orlikowska M, Jankowska E, Kolodziejczyk R, Jaskolski M, Szymanska A. Hinge-loop mutation can be used to control 3D domain swapping and amyloidogenesis of human cystatin C. J Struct Biol. 2011;173:406–413. doi: 10.1016/j.jsb.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 91.Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.