

Abstract
Amelogenins make up over 90 percent of the protein present during enamel formation and have been demonstrated to be critical in proper enamel development, but the mechanism governing this control is not well understood. Leucine-rich amelogenin peptide (LRAP) is a 59-residue splice variant of amelogenin and contains the charged regions from the full protein thought to control crystal regulation. In this work, we utilized neutron reflectivity (NR) to investigate the structure and orientation of LRAP adsorbed from solutions onto molecularly smooth COOH-terminated self-assembled monolayers (SAMs) surfaces. Sedimentation velocity (SV) experiments revealed that LRAP is primarily a monomer in saturated calcium phosphate (SCP) solutions (0.15 M NaCl) at pH 7.4. LRAP adsorbed as ~32 Å thick layers at ~70% coverage as determined by NR. Rosetta simulations of the dimensions of LRAP in solution (37 Å diameter) indicate that the NR determined z dimension is consistent with an LRAP monomer. SV experiments and Rosetta simulation show that the LRAP monomer has an extended, asymmetric shape in solution. The NR data suggests that the protein is not completely extended on the surface, having some degree of structure away from the surface. A protein orientation with the C-terminal and inner N-terminal region (residues ~8–24) located near the surface is consistent with the higher scattering length density (SLD) found near the surface by NR. This work presents new information on the tertiary and quaternary structure of LRAP in solution and adsorbed onto surfaces. It also presents further evidence that the monomeric species may be an important functional form of amelogenin proteins.
Keywords: Neutron Reflectivity (NR), biomineralization, amelogenin, LRAP, surface interface, protein orientation
INTRODUCTION
Biomineralization proteins are responsible for the unusual strength and morphological properties seen in biominerals.1 A great deal of experimental effort has been undertaken to demonstrate that these proteins are essential to controlling both crystal nucleation and growth processes.1–6 Specific protein-crystal interactions and electrostatic interactions by charged residues, both of which depend on the orientation of protein on the crystal, are thought to be important in this process.5,7–9 Unfortunately, details of the interaction mechanism have remained unknown due to a limited number of experimental techniques capable of providing quantitative insights for the structure and orientation of proteins bound to surfaces. Understanding these interactions, however, is critical toward the development of a basic knowledge of how the elegant biomineralized structures found in nature are formed.
Enamel is an example of a biomineral exquisitely controlled by the organic matrix during development. Its exceptional hardness is attributed to the high aspect ratio of crystallites (25 nm × 70 nm by × 100 Em)10 and the cross-weaved crystallite pattern, both of which are controlled by the organic matrix during formation.6 Amelogenins are a family of proteins that consist of over 90% of the protein present during enamel formation6 and have been found to be critical to proper enamel development.4 The parent protein is a hydrophobic 180 residue protein (~21 kD), rich in proline, glutamine and histidine.11 Genetic splice variation or proteolytic cleavage during enamel development results in amelogenins as small as 5 kD,11 including the 59 residue leucine-rich amelogenin peptide (LRAP).12 Amelogenins are observed to form into self-assemblies 20 – 60 nm in diameter consisting of 20 to 100 monomers.13 These self-assemblies or “nanospheres” are thought to be an important functional form of the protein.
Unlike other biomineralization proteins, there are only 14 charged residues in amelogenin, nine of which are found in the C-terminus and five in the N-terminus. Both of these regions are highly conserved across many species and are thought to have a specific function. Because of the increased content of charged residues in the C-terminus, this region of the protein is thought to be responsible for the interaction of this very hydrophobic protein with the charged surface of hydroxyapatite (HAP).14 Indeed, early studies reported reduced binding affinities in the absence of the C-terminus, leading to the suggestion that this region promoted protein-HAP interactions.14 Computational studies also suggested that the carboxyl groups in the C-terminus were critical for binding LRAP to HAP.15,16
Direct experimental evidence for the interaction of the C-terminus with HAP has only recently been reported based on solid-state NMR (SSNMR) studies of LRAP bound to HAP under physiological conditions.16–20 LRAP was used as a model protein for full-length amelogenin since it contains the highly conserved N- and C-termini from the parent protein21 thought to control protein-protein and protein-crystal interactions. LRAP’s small size allows for the incorporation of 13C or 15N isotopic labels into specific residues of the protein using solid phase peptide synthesis techniques. Studies of heteronuclear coupling between 13C or 15N nuclei and 31P nuclei on the HAP surface resulted in the determination of distances between the labeled protein residues and the HAP surface. These distances were in the range of 4–6.5 Å for residues 49 to 58, suggesting that the end of the C-terminus interacts directly with the HAP surface and lies flat on the surface. This orientation would allow the C-terminus of LRAP to control the growth of HAP. In addition, heteronuclear coupling between 13C and 15N residues within the protein (L42-A46, A49-T53, K54-V58) resulted in distances that suggest the C-terminal domain is in a random coil conformation on the surface.
Although SSNMR is a powerful tool that has revealed very important molecular-level details about the secondary structure and orientation of LRAP on a HAP surface, we are also interested in understanding how LRAP adsorbs onto surfaces at the tertiary and quaternary level. For example, do nanospheres or other structures adsorb on the surface? What is the thickness and coverage of the adsorbed protein? Neutron reflectivity (NR) is well suited to answer these types of questions since scattering of neutrons off the surface can provide a depth profile of multilayer structures, with a resolution that can be on the order of angstroms.22 In a reflectivity experiment, a sample is exposed to an incident neutron beam that is then deflected by the surface of interest. This deflection corresponds to the change in momentum, Q, of the incident beam. In specular reflection, the angle of the incident beam (θ) with respect to the surface of interest equals the angle of the reflected beam, and the change in momentum is related to θ by Q = (4π/λ) sinθ, where λ is the wavelength of the neutron. The pattern of reflected intensity as a function of Q will then depend on the scattering length density (SLD) profile of the sample, which is directly related to the chemical/isotopic composition profile of the sample along the surface normal. The SLD of an element or molecule relates to the degree to which the material scatters neutrons. NR has promise in providing insights into the structure of biomineralization proteins at interfaces as demonstrated by previous investigations of non-biomineralization proteins.23–26
In this work, we investigated the tertiary and quaternary structure of LRAP adsorbed onto COOH-terminated self-assembled monolayers (SAMs) on gold on iron (Fe) on silicon, a surface that provides the flatness (≈5 Å root mean square (rms) roughness) required for NR. Several residues of the protein in the C-terminal region were deuterated to increase the SLD scattering length density of the protein so it could be differentiated from the underlying SAM layer and to probe for protein orientation. LRAP was adsorbed under physiological conditions at pH 7.4. The quaternary structures of LRAP in physiological solutions were studied using sedimentation velocity experiments. Neutron reflectivity studies were performed using a neutron beam in two polarization states since this results in two distinct reflectivity curves in the magnetic Fe region. Also, we exposed the adsorbed protein to three humid atmospheres with varying amounts of D2O and H2O. This resulted in the collection of six reflectivity curves for each sample that could be simultaneously fit to determine a SLD profile that was used to obtain protein thicknesses. Further modeling allowed the determination of protein coverage and degree of hydration. Since the size and dimensions of the LRAP monomer are not known, Rosetta computer simulations of LRAP were performed and compared to the surface structures determined by neutron reflectivity. This work demonstrates that LRAP adsorbs as a monomer and that NR can be a useful technique for the study of biomineralization proteins.
EXPERIMENTAL METHODS
Materials
Deuterium oxide (99.9%), proline-d7 (98%), tryptophan-indole-d5 (98%), alanine-d3 (99%) and leucine-d3 (98%) were purchased from Cambridge Isotopes Laboratories and used as received. 16-Mercaptohexadecanoic acid was purchased from Asemblon, Inc. Absolute ethanol was obtained from United Chemicals. All other chemicals were reagent grade and obtained from Sigma-Aldrich.
Protein synthesis and purification
All labeled amino acids were FMOC-protected using standard protocols.27 LRAP-D (bovine) was synthesized using solid phase peptide synthesis by United Biochemical Research (Seattle, WA) and is shown in Table 1. The serine residue, S16, was phosphorylated and was designated pS. LRAP-D had six amino acids that were deuterium labeled (L42, P43, L44, A46, W47, and A48) for a total of 24 deuterium atoms. Proteins were purified using prep scale reverse phase HPLC, buffer A (0.1% trifluoroacetic acid in water), and buffer B (0.1% trifluoroacetic acid in acetonitrile). LRAP eluted at 54% B and was analyzed for purity and molecular weight using electrospray mass spectroscopy (MS).
Table 1.
LRAP-D (bovine) used for NR studies. Bold residues were deuterium labeled.
| LRAP-D (bovine) | MPLPPHPGHPGYINFpSYEVLTPLKWYQSMIRHPPLPPMLPDLPLEAWPATDKTKREEVD |
Self-assembled monolayer (SAM) substrate formation
For the NR experiments, 20 nm of gold was deposited on 6 nm of magnetic iron (Fe) on 2″ diameter single crystal silicon substrates using a magnetron sputtering chamber. The substrates were placed into 1 mM thiol in absolute ethanol for at least 24 hours. Samples were removed and cleaned in acetic acid/ethanol solutions and then ethanol solutions. By convention, the end group name was given as the non-ionized group even though the groups may be protonated or deprotonated in solution. Advancing contact angles of water were typically 20° for the COOH SAMs. A schematic of the multilayer substrate used for NR experiments is shown in Figure 1.
Figure 1.
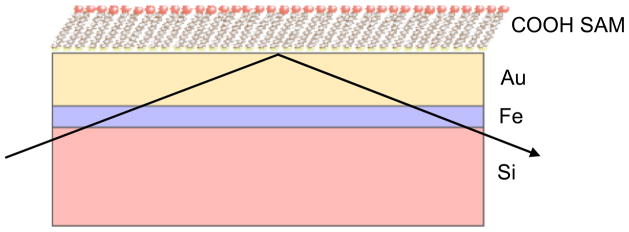
Schematic of the molecularly smooth, multilayer substrate used for protein adsorption studies showing COOH SAMs on gold on silicon with an Fe interlayer. The NR beam geometry is shown.
LRAP adsorption
LRAP solutions were prepared using conditions that we have previously found to give significant protein adsorption (equivalent ellipsometric protein thicknesses of ~19–23 Å) onto COOH SAMs.28 LRAP was prepared in “SCP” solution, 0.15 M NaCl saturated with respect to calcium phosphate. The SCP buffer was prepared by adding hydroxyapatite powder to 0.15 M NaCl, stirring for several days, and filtering out the particles as described previously.17 LRAP was dissolved in 0.01 M HCl, diluted into the SCP buffer to 0.28 mg/ml and adjusted to pH 7.4 using KOH. COOH-terminated SAM substrates were placed into the solutions for at least two hours, enough time to reach maximum adsorption.28 The substrates were removed, rinsed with a stream of deionized water, briefly dried in a stream of nitrogen, and placed in the NR sample cell.
Sedimentation velocity (SV)
SV is an analytical ultracentrifugation technique that can be used to obtain information on the distribution of protein species in solution as reviewed elsewhere.29 LRAP samples were prepared at 0.21 mg/ml in pH 7.4 SCP solutions. Samples were loaded into cells with 2-channel charcoal-epon centerpieces with 12 mm optical pathlength and the corresponding buffer was loaded into the reference channel. The cells were placed into an AN-60Ti analytical rotor and loaded into a Beckman-Coulter ProteomeLab XL-I analytical ultracentrifuge at 20 °C. The rotor was then brought to 3,000 rpm and the samples were scanned at 280 nm to confirm proper cell loading. After accelerating to 60,000 rpm the cells were scanned every 4 minutes for 3.2 hours (48 scans of each sample) and then the scan rate was dropped to every 16 minutes for another 14.4 hours (54 additional scans of each sample). A second velocity run (37 scans collected at 3.1 minute intervals) was made at 25,000 rpm to test for any large, rapidly sedimenting species.
The data were analyzed using the c(s) method developed by Peter Schuck and implemented in his analysis program SEDFIT (version 11.3).30 The raw data scans (~37,000 data points) are directly fit to derive the distribution of sedimentation coefficients, while modeling the influence of diffusion on the data in order to enhance the resolution. This method assigns a diffusion coefficient to each value of sedimentation coefficient based on an assumption that all species have the same overall hydrodynamic shape. That hydrodynamic shape is defined by f/f0 ratio, where f is the frictional coefficient of the macromolecule and f0 is the frictional coefficient of an anhydrous sphere with the same volume as the macromolecule. The f/f0 values are varied to find the best overall fit of the data to the monomer peak. Maximum entropy regularization probabilities of 0.683 (1 σ) were used and time-independent noise was removed.
The velocity data were also analyzed using the time derivative (∂c/∂t) method, as implemented in the program DCDT+ (version 2.4.0).31,32 The ∂c/∂t curve derived from 16 scans taken when the boundary was near the middle of the cell was directly fit to determine the molar mass of the major species. The partial specific volume (v̄) for LRAP at 20 °C was calculated as 0.7476 ml/g, using the program SEDNTERP.33 Solvent densities and viscosities at 20 °C for the SCP buffer were calculated using SEDNTERP as 1.00442 g/ml and 1.0165 cp respectively. SEDNTERP was also used to convert raw sedimentation coefficients to standardized s20,w values and to calculate f/f0 ratios from the measured sedimentation coefficients.
RosettaSurface simulations
RosettaSurface is a multiscale Metropolis Monte Carlo-plus minimization algorithm developed to predict the structure of biomineralization proteins both in the surface-adsorbed and solution state.34 Here, RosettaSurface was used to predict the solution-state structure of LRAP. Each execution of the RosettaSurface algorithm began by folding a low-resolution (united-atom) representation of LRAP from a fully extended structure in implicit solvent. Next, all atoms (including hydrogen) were added to the model and high-resolution energy minimization was implemented to generate candidate solution-state structures. We generated large ensembles of 105 candidate solution-state structures from which the 100 lowest-energy structures were chosen for further analysis. A complete description of these methods can be found in the publication by Masica and Gray.34
We built an extended molecular structure of LRAP using PyMol (Delan Scientific). The atomic parameter set was the same as the set used previously.34 The structural designations “Helix” and “Turn” were assigned using the dictionary of secondary structure of proteins (DSSP) definitions.35 Classification relied on Rosetta’s hydrogen-bond function36 rather than the generalized hydrogen-bond function implemented by the DSSP package. The structural designation “Other” indicates that hydrogen bonding was either long range or absent at that residue.
NR measurements
Measurements were performed at the NG1 vertical stage reflectometer at the National Institute of Standards and Technology (NIST) Center for Neutron Research (NCNR). A schematic of the reflectometer neutron focusing optics and detector is shown in Figure S1. The wavelength of the neutron beam, λ, was fixed at 5 Å with ΔQ/Q ~ 0.025 over the Q range for which the reflectivity was measured. The “footprint” of the beam on the sample was kept approximately constant by varying the series of apertures that define the incident beam size and angular divergence. The reflectivity data from a sample was obtained by subtracting the off-specular background coming from the incoherently scattered neutrons from the sample and substrate as well as stray neutrons from other sources from the specularly reflected intensity and then dividing by the incident intensity.
A polarized neutron beam was used to take advantage of the fact that a magnetic Fe layer was used as an adhesion layer between gold and silicon as shown in Figure 1. The Fe layer has different SLDs depending on the polarization of the neutron beam. Additionally, the sample was exposed to three humid atmospheres consisting of argon gas saturated with water (100% H2O, 50% D2O: 50% H2O, and 100% D2O) at 92% humidity.37 The 92% controlled humidity apparatus is described in detail elsewhere.38 For the protein/substrate sample, six data sets were collected – two polarization states for each of the three atmospheres.
Simultaneous fitting of the NR data
SLD depth profiles (SLD as a function of layer depth, z) were obtained from the reflectivity data by modeling each layer of the multilayer system shown in Figure 1 as a box with constant SLD. Error functions were used to describe the interfacial layers between the boxes. All data sets (3 humid atmospheres, 2 polarizations each) were fit simultaneously and self-consistently. The SLDs and thicknesses of the Fe, Au, SAM, and protein boxes were allowed to vary within a range of expected values. For each iteration of SLD and thickness, reflectivities as a function of Q were calculated and evaluated for fit to the experimental reflectivity data. Iterations continued until the fit converged to a minimum chi-squared (χ2) value. The protein layer was initially modeled as one box with a uniform SLD (one layer protein) as shown in the schematic in Figure 2a. This model was used to determine if the protein was adsorbing as a monomer, dimer, or other quaternary structure based on the resulting thickness. The protein layer consists of protein and water adsorbed from vapor so that the SLD that was fit, a total SLD, was a function of the protein coverage, protein SLD, degree of hydration, and water SLD.
Figure 2.
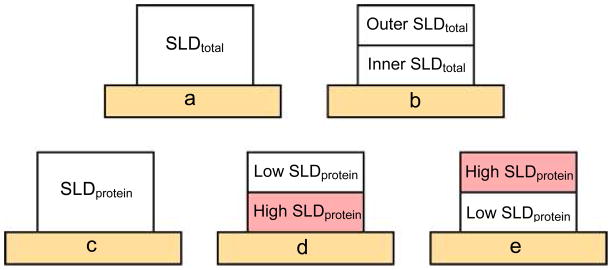
Schematics of models tested for simultaneous fitting showing the protein layer (protein plus hydration) on the substrate (light orange). Fits to a total SLD are shown in (a) and (b): a) one layer protein - protein layer was fit to one total SLD, b) two layer monomer – adsorbed monomer was modeled as two layers each fit to a unique total SLD. Fits using a model where total SLD is a function of protein SLD, protein coverage, water SLD, and hydration are shown in (c), (d), and (e): c) one layer protein with calculated protein SLDs, d) two layer monomer with calculated protein SLDs. The inner protein layer was assumed to be 8–24, 42–59 and had a higher protein SLD than the outer layer, e) two layer monomer with calculated protein SLDs. The outer protein layer was 8–24, 42–59 and had a higher protein SLD than the inner layer protein SLD. See the text for further details.
Once it was found that LRAP was adsorbing as a monomer (see SV, NR, and Rosetta results), a refinement of the model was performed. The monomer was modeled as two layers (two layer monomer) in order to determine if more highly deuterated regions of the monomer could be located relative to the substrate (Figure 2b). For example, the C-terminal and inner N-terminal regions were more highly deuterated due to site-specific labeling and H to D exchange with the deuterated solvents. A more highly deuterated protein region would result in a higher SLD for the layer. The total SLD and thickness of each layer in the two layer monomer were allowed to vary freely. A two layer model was used to give a general idea of the location of more highly deuterated residues of the protein monomer. We chose to use as simple a model as possible to test for protein orientation in this study.
In addition to fitting to the total SLD and thickness, we also did fits assuming an underlying model based on the following equation for the total SLD, SLDt :
| (1) |
where V is the volume fraction for the protein (p), protein hydration (w), and air (a). The data was fit to obtain the volume fraction of LRAP (Vp) and volume fraction protein hydration (Vw) using calculated protein SLDs. The outputs were required to be self-consistent between the six data sets. Hydration of the COOH SAM was also considered but NR measurements found that SAM hydration did not contribute significantly to the total hydration. Schematics of the modeling with calculated protein SLDs are shown in Figures 2c, d, and e. We also did simultaneous fitting to an underlying model where we allowed the protein SLDs to vary freely over a range. This resulted in much higher standard errors for the resulting protein SLDs, protein coverages, and hydrations because of the many possible combinations of protein SLD and hydration. Fixing the protein SLD constrained the system and gave much lower standard errors, good fits, and outcomes that were consistent with the physicochemical model.
Simultaneous fitting was performed using the DREAM program, which employs Bayesian analysis using the Markov Chain Monte Carlo method with Differential Evolution update steps.39 The resulting Markov chains converged to an underlying probability distribution. The expected value, the maximum likelihood value, and uncertainty of the value were estimated from this distribution. A chi-squared value for the fitting was also obtained. This method was useful in cases where the optimized parameters were correlated and could not be determined independently. DREAM fitting has significant advantages over least-squares techniques. For example, from the obtained distribution it is possible to determine statistically the degree of distinguishability between two models that differ in the number of fit parameters, such as between the one layer protein and two layer monomer models. Also, the DREAM analysis gives correlations between parameters in a model and gives accurate error analysis of all fit parameters. In addition to the DREAM fitting data analysis, we also used the Hamilton test to compare various models as previously described.26,40
Briefly, R was compared to R*b,ν,α where
| (2) |
and χ12 and χ22 are chi-squared values obtained from the DREAM fitting for model 1 and model 2, respectively. The factor R*b,ν,α is described as
| (3) |
where b is the difference in degrees of freedom between models, ν is the number of data points minus the greatest number of degrees of freedom, and α is the confidence level, which was chosen to be 0.005 (0.5%). The term, Rb,120,α is obtained from the confidence tables in the Hamilton paper.40 If R > R*, model 1 is not as good as model 2 with 99.5% confidence.
RESULTS
Sedimentation velocity
Raw data scans of absorbance versus radius for LRAP in SCP solutions at 60,000 rpm are shown in Figure S2. The high-resolution sedimentation coefficient distributions for LRAP in SCP buffer derived from runs at 60,000 rpm are shown in Figure 3. These distributions are normalized so the area under each peak represents its species fraction. Figure 3 indicates that the SCP solution contains one major species at 98.0% with a sedimentation coefficient of 0.743 S. That sedimentation coefficient, together with the best-fit frictional coefficient ratio relative to an anhydrous sphere ((f/f0 ratio), imply this peak has a mass of approximately 6.3 kD, consistent with an assignment as an LRAP monomer. The assignment of the major component as a monomer was also confirmed by time derivative analysis. The monomer’s f/f0 ratio, as determined from the measured sedimentation coefficient, is 1.59 and the hydrodynamic radius is 20.2 Å. The rest of material is distributed among 10 peaks or shoulders between 5.6 and 44 S, each at levels of 0.4% or less. The presence of the half-peak at 50 S, the upper limit of the data analysis, indicates that there is some material sedimenting faster than 50 S. A second run at lower rotor speed (25,000 rpm) confirms there are species sedimenting between 50 S and 200 S at 0.2% concentration (not shown). Experiments were also done with LRAP in 0.15 M NaCl, pH 7.4 SCP solution at higher concentrations, 0.5 to 2.0 mg/ml LRAP. All solutions contained monomers with a very low concentration (less than 2%) of oligomers.
Figure 3.

Normalized sedimentation coefficient distribution for LRAP in SCP solution with expanded inset. Peaks are labeled with % amount and sedimentation coefficient, s, value in svedbergs (S). The sedimentation coefficient of the main monomer peak is the average over the peak.
RosettaSurface simulations of LRAP solution-state structure
Figure 4 shows the predicted structure of LRAP in solution. The predicted ensemble structure in the N-terminal 20 residues of LRAP is mostly extended, with increasing populations of locally hydrogen boned turn-like secondary structure and some helical structure from residues 10–20 (Figure 4a). The ensemble structure in the middle segment is almost entirely helical from residues 23 to 31, with a mix of locally hydrogen bonded and extended structure from residues ~32 to 40. RosettaSurface predicts the C-terminal 20 residues adopt a mix of turn-like and extended structures, with a higher propensity for helix formation from residues ~48 to 55. Coordinates for the 10 lowest-energy structures show the dominant helical structures adopted from residues 23 to 31 and residues 48 to 55, connected by flexible and loosely structured segments (Figure 4b). The radius of gyration, calculated from the 100 lowest energy structures, is 18.5 ± 2.9 Å (37 Å equivalent diameter).
Figure 4.

Rosetta simulation of the LRAP monomer showing a) the distribution of three basic secondary structure motifs for the low energy ensemble of 100 solution state LRAP structures going from residue 1 (N-terminus) to residue 59 (C-terminus) and b) coordinates for the 10 lowest energy, solution state structures superimposed about the helical middle segment (residues 23–31). Structural cartoons go from left N-terminus (blue) to right C-terminus (Red) and one structure is emboldened for clarity. SSNMR experimental studies of LRAP are showing a stable helix at K24-S28, consistent with the Rosetta simulations. The experimental studies and simulations also both show lower degrees of helical character in the C-terminus and N-terminus.
Neutron Reflectivity
Neutron reflectivity experiments were performed on LRAP adsorbed onto COOH SAMs from solutions similar to physiological solutions at pH 7.4. Figure 5a shows the reflectivity versus Q for adsorbed LRAP in three 92% humidity atmospheres and two polarizations each using the one layer protein model. Figure 5b shows the SLD profile (SLD versus depth, z) for each layer of the multilayer film on silicon using the one layer protein model. The solid lines in Figure 5a show the calculated reflectivities obtained from the SLD profile shown in Figure 5b. The SLD profiles give thicknesses of 50 Å for the Fe layer and 230 Å for the gold layer. The COOH SAM layer is indicated by a reduction in SLD and a thickness of 20 Å. The adsorbed LRAP is revealed by an increase in SLD over the SAM region resulting in an adsorbed protein thickness of 29.76 Å (Table 2). The increase in SLD of the protein layer in H2O atmosphere is due to the incorporation of deuterium labels into the protein. A further increase in SLD with increasing amount of D2O in the humid atmosphere indicates that the protein is hydrated by D2O and/or that some deuterium exchange for labile protons has occurred.
Figure 5.

a) reflectivity data (symbols) and corresponding fits (lines) for LRAP adsorbed from SCP solutions under three humid atmosphere conditions (92% RH 100% D20, 50% H2O:50% D2O, 100% H2O). The fits are the calculated reflectivities corresponding to the SLD profiles in Figure 5b. b) SLD profiles obtained from simultaneous fitting of reflectivity data using the one layer protein model fit to the total SLD.
Table 2.
LRAP layer z dimensions determined by NR for the one layer protein and two layer monomer models and outcome of the Hamilton analysis comparison between models.a
| Model | Layer | Z dimension (Å) |
|---|---|---|
| one layer protein | 29.76±0.14 | |
| two layer monomer | Inner1 | 21.1±1.4 |
| Outer | 10.6±1.0 |
Layer next to the SAM surface
Hamilton analysis: R = 1.047, R* = 1.005. Since R>R*, the two layer monomer model is considered to be a better model with 99.5% confidence.
We concluded that LRAP was adsorbing as a monomer since the solutions contained monomers (SV) and the NR thickness from the one layer protein model was ~30 Å, consistent with the Rosetta simulation monomer size (18.5 Å radius, 37 Å diameter) and SV hydrodynamic size (20.2 Å radius, 40.4 Å diameter). Once we knew the protein was adsorbing as a monomer, the monomer was modeled as two layers (two layer monomer) in order to probe for the tertiary structure of the protein. This was enabled by determining the location of more highly deuterated regions of the protein. Parts of the protein are deuterated by site-specific deuterium labeling at residues 42–48 while other regions are deuterated due to exchange with D2O solvent. It is well known that proteins exposed to water vapor at high humidity will be hydrated and that D2O hydration will lead to exchange of deuterium for labile protons in the protein.41 The labile protons are found in the side chains as well as in the amide backbone.42 Amide protons that are not in a canonical secondary structure are more easily exchanged than amide protons in α-helical or β-sheet structure.43 Although the degree of deuterium exchange in our system is unknown, we expect that some deuteration has occurred over the 24 hour time period of each atmosphere exposure.
The reflectivity fits and SLD profile in the protein region for the two layer monomer model are shown in Figure 6. The SLD profile shows that the protein monomer consists of two layers with different total SLDs. The “inner” layer, the layer closest to the surface, has a higher SLD than the “outer” layer. The inner part of the monomer was 21.1 Å thick while the outer part of the monomer was 10.6 Å thick resulting in a total thickness of 31.7 Å (Table 2). The two layer monomer model was compared to the one layer protein model by the DREAM analysis, by comparing chi-squared values, and by using the Hamilton test and was found to be a better model with a 99.5% confidence level.
Figure 6.


a) reflectivity data (symbols) and corresponding fits (lines) for LRAP adsorbed from SCP solutions under three humid atmosphere conditions (92% RH 100% D20, 50% H2O:50% D2O, 100% H2O). The fits are the calculated reflectivities corresponding to the SLD profiles in Figure 6b. b) SLD profiles obtained from simultaneous fitting of reflectivity data using the two layer protein model to the total SLD. c) Expanded view of the SLD profile of the LRAP layer in 100% D2O humid atmosphere.
For both the one layer protein and two layer monomer models, the data was fit to the total SLD of the protein layer (see schematics in Figures 2a and 2b). The protein layer, however, consists of the protein and any hydration of the protein by H2O or D2O vapor. The total SLD, therefore, is a function of the coverage of the protein, the protein SLD, the degree of hydration, and the water vapor SLD. Further fitting using models described by Equation 1 gave estimates of the protein coverage and percent hydration using calculated protein SLDs.
Protein SLDs were calculated using the NIST calculator assuming two levels of deuterium exchange - no deuterium exchange by solvent and 100% exchange of labile protons (Table S2). The labile protons involved in exchange were determined by the method of Epimova44,45 and are shown in the supplementary section. We assumed all backbone amide protons were exchanged since LRAP does not have much secondary structure. The amount of exchange was assumed to be 100% for the 100% D2O atmosphere and was adjusted for the 50% D2O:50% H2O atmosphere.
Table 3 shows that the coverage of the protein on the surface in the one layer model assuming no protein deuterium exchange is ~70 v%. There is a significant amount of hydration (adsorbed water) at ~30 v%. The protein coverage is ~74 v% and the hydration is ~21 v% assuming 100% deuterium exchange with the protein. The balance of volume percent coverage is air. The hydration amounts in v% convert to 0.32 and 0.20 g H2O/g protein, respectively. These values are within the range of hydration found for a number of different proteins in 92% relative humidity vapor (0.24 to 0.42 g H2O/g protein).46
Table 3.
LRAP layer protein coverage and hydration obtained by assuming protein SLD values. The protein SLDs were calculated assuming 0% or 100% deuteration of LRAP by exchange of labile protons with D2O solvent (Table S2).
| Model | %D exchange | Layer | Protein SLD × 106 (Å−2) | LRAP coverage (v%) | Hydration (v%) | gH2O/g protein1 |
|---|---|---|---|---|---|---|
| One layer protein | 0 | 2.16 | 70.2±0.04 | 29.8±0.4 | 0.32 | |
| 100 | 3.13 | 74.1±1.2 | 20.7±0.5 | 0.21 | ||
| Two layer monomer-inner high protein SLD2 | 0 | Inner | 2.31 | 65.5±1.2 | 32.0±0.6 | 0.37 |
| Outer | 1.81 | 80.5±5.0 | 18.7±1.4 | 0.17 | ||
| 100 | Inner | 3.82 | 63.5±1.7 | 25.0±1.1 | 0.29 | |
| Outer | 2.55 | 73.6±1.6 | 13.1±0.9 | 0.13 | ||
| Two layer monomer-inner low protein SLD3 | 0 | Inner | 1.81 | 30.24±22.5 | 60±18 | 1.49 |
| Outer | 2.31 | 66.3±3.4 | 29.5±3.1 | 0.33 | ||
| 100 | Inner | 2.55 | 74.65±1.31 | 25.2±0.94 | 0.26 | |
| Outer | 3.82 | 89.22±1.63 | 10.6±1.2 | 0.09 |
Degree of hydration calculated in terms of grams of water (H2O) per grams of protein.
Protein SLDs were calculated assuming the inner layer was the C-terminus (42–59) and inner N-terminus (8–24) and the outer layer was the outer N-terminus (1–7) and middle hydrophobic region (25–41) as described in the text and supplementary section.
Protein SLDs were calculated assuming the protein was flipped - inner layer was the N-terminus (1–7) and middle hydrophobic region (25–41) and the outer layer was the C-terminus (42–59) and inner N-terminus (8–24).
For the two layer monomer model, the protein SLDs were first calculated assuming that the inner layer was composed of the C-terminal region (residues 42–59) and inner N-terminal region (residues 8–24) since these regions were found close to HAP surfaces by SSNMR20 and the outer layer was composed of the N-terminus (1–7) and middle hydrophobic region (25–41). The inner layer had a higher protein SLD than the outer layer. Conditions of no deuteration by solvent exchange and 100% deuteration were considered. For both levels of deuteration that we assumed, the inner layer has higher degrees of hydration than the outer layer as shown in Table 3. For example, assuming 0% deuteration the inner layer hydration is 32.0 v% compared to 18.7 v% for the outer layer. Assuming 100% deuteration, the inner layer hydration is 25 v% compared to 13.1 v% hydration for the outer layer. Therefore, the total hydration over both layers is lower assuming 100% deuteration of the protein compared to no deuteration of the protein by solvent exchange. A model that flipped the protein so that the lower SLD regions were close to the surface (Figure 2e) was rejected by a chi-squared value comparison and the Hamilton test in comparison to the model with higher SLD regions close to the surface. Also, the outputs from this model were not consistent with what would be expected physicochemically. For example, unusually high values of hydration were obtained assuming 0% deuteration (60% in the inner layer) and unusually high protein coverages were obtained assuming 100% deuteration (89% in the outer layer). Also, the inner protein layer had higher amounts of hydration than the outer layer (assuming 0% deuteration and 100% deuteration) even though the inner layer was more hydrophobic.
DISCUSSION
LRAP structure in solution
The SV experiments show that the SCP solutions at pH 7.4 consist primarily of LRAP monomers with a very small percentage of oligomers and larger structures. The monomeric quaternary structure for LRAP at pH 7.4 is in contrast to nanospheres typically found in full length amelogenin solutions at pH 7.4 over a range of buffer conditions and protein concentrations.47,48 The monomer size was found to be 18.5 Å radius of gyration by Rosetta simulations and 20.2 Å hydrodynamic radius by SV. We note that the radius of gyration is not the same physical parameter as the hydrodynamic radius although the two are related to each other. To our knowledge, this is the first work to identify monomers as the dominant species in LRAP solutions at pH 7.4 and to determine the monomer size. Previously, it has been suggested that the nanosphere is the most important physiological quaternary structure for amelogenin proteins.13,48,49 Our work, however, has found that the monomer is an important functional form of LRAP.
Our previous dynamic light scattering (DLS) studies only detected larger, 100 nm structures in the SCP LRAP solutions.28 This indicates that larger structures may dominate DLS measurements because of the large effect of size on scattering intensity and the low scattering coefficient from monomeric protein molecules. Recent studies of LRAP in solutions that were 2 mg/ml protein and 20 mM buffer also found relatively small structures in solution at pH values around 7: ~20 Å radius structures as determined by DLS.50 The 20 Å radius structures may represent monomers because the hydrodynamic radius is consistent with sizes we obtained by Rosetta simulations and SV. Further studies will be necessary to examine the quaternary structures of LRAP in a range of possible protein concentrations, ionic strengths, and pH values found for LRAP in-vivo to determine if the monomer is the dominant species or if other species such as nanospheres become more important.
Both the Rosetta simulations and sedimentation velocity experiments suggest that the LRAP monomer is asymmetric and extended in structure under pH 7.4 physiological solution conditions. The SV f/fo ratio is 1.59, a value similar to asymmetric proteins such as antibodies, and higher than 1.2–1.3 values typical of globular proteins.51 The Rosetta simulations of LRAP in implicit solvent shown in Figure 3 support this less compact structure.
The Rosetta simulations suggest that LRAP is globally unstructured with little tertiary structure, consistent with what would be expected from a low molecular weight, proline-rich protein. The simulations predict some secondary structure with a stable helix in the central residue 23–31 region. SSNMR experimental studies of hydrated LRAP also showed a highly helical region at 24–28 with an average 13C-15N distance of 4.5 Å, close to the 4.2 Å expected for a perfect helix.20 Interestingly, when LRAP was adsorbed onto HAP the experimental 13C-15N distance of residues 24–28 increased to 5.7 Å, suggesting some degree of unfolding of the helix to a more random structure. The Rosetta simulations predict two additional partially helical regions – one in the N-terminus at residues 12–17 and one in the C-terminus at residues 48–55. These regions are predicted to be dynamic, sampling both helical and turn-like conformations. The SSNMR results also showed less structure in these regions with 13C-15N distances of 5.9 Å for residues 15–19 and 5.9 Å for residues 49–53.
An extended, relatively unstructured conformation for amelogenins may have important advantages in promoting their suggested functions as nucleation promoters and crystal growth modulators in biomineralization processes. A more flexible structure may be better at promoting nucleation compared to a rigid protein lattice that may have structural mismatches with the lattice of the calcium phosphate critical nucleus. An extended structure may also promote better control over the growth of HAP crystals, allowing coverage onto more surface sites of the (100) and (010) faces, blocking growth of these faces.
LRAP quaternary structure at the surface
The NR results show that LRAP adsorbs from physiological solutions as a monomer. The adsorbed protein layer z dimension of 31.7 Å determined by NR is consistent with the diameter of the LRAP monomer obtained from the Rosetta simulations (diameter ~36 Å) and the SV data (diameter ~40 Å). The protein thickness was similar over the different models used for the simultaneous fitting.
Monomer adsorption is consistent with our previous study on the adsorption of LRAP onto SAM surfaces where we used tapping mode AFM to detect adsorbates onto SAMs on atomically smooth gold on mica.28 Small adsorbates were observed by AFM, much smaller than the size of a typical nanosphere. In addition, single wavelength ellipsometry (SWE) measurements resulted in equivalent protein thicknesses of ~21 Å at saturation adsorption suggestive of a monomer or dimer-sized adsorbate. Both AFM and ellipsometry have limitations in obtaining the true size of a structure, however. AFM diameters are overestimated because of tip broadening effects.52 Also, AFM height measurements of soft, viscoelastic structures can be underestimated by up to six times because of nonlinear effects due to the oscillating tip – soft structure interaction.53 This resulted in AFM measurements of 10–15 nm diameter × 0.5–.75 nm height for the adsorbed monomers. SWE measurements are also limited in assuming a uniform thickness of a given adsorbate resulting in the measurement of an equivalent thickness - a true protein thickness times coverage. NR, on the other hand, gives accurate measurements of the z-dimension of the protein to be ~32 Å, giving direct evidence for the adsorption of monomer-sized structures.
LRAP orientation at the surface
The two layer monomer model shows that the adsorbed monomer consists of an inner layer next to the SAM surface with higher total SLD layer and an outer layer with lower total SLD. This result is consistent with the monomer having a preferred orientation with the more highly deuterated C-terminal (42–58) and inner N-terminal regions (8–24) located closest to the surface. Figure 7 shows a hand-docked example of LRAP on COOH SAMs with this orientation. We note that this is a schematic and not a structure derived from simulations of LRAP on COOH SAMs. The domains closest to the surface are more highly deuterated because of the site specific deuterium labeling in residues 42–48 and because of the higher numbers of side chain labile protons in these regions available for solvent deuterium exchange. These regions are also more hydrophilic because they contain charged polar (R, H, K, D, E) and uncharged polar (S, T, N, Q) amino acids. For example, the 8–24 and the 41–58 residues have 17 polar residues compared to 6 polar residues in the remaining parts of the protein. It is well known that polar residues adsorb higher amounts of water from humid atmospheres compared to hydrophobic residues and that hydration correlates strongly with the fraction of polar residues in a protein.46,54,55 Polar residues also have higher amounts of hydrogen bonded water when the protein is in solution.56,57 For example, the number of adsorbed water molecules per amino acid was found to be 6–7.5 for ionized acidic amino acid groups, 2–3 for unionized acidic groups, 3–4.5 for basic groups, 2–4 for polar uncharged groups, and 0–1 for hydrophobic groups.58 Higher amounts of protein hydration near the surface, therefore, would be consistent with hydrophilic domains (C-terminal domain and inner part of the N-terminal domain) being close to the surface and hydrophobic domains being away from the surface. NR studies have also found variation in hydration within polymer lipid bilayers used as cell membrane mimics under 92% RH atmospheres.38 Hydration was 40 v% in the inner polyelectrolyte layer, 10 v% in the middle terpolymer, and 10 v% for the outer phospholipid layer.
Figure 7.

Hand-docked picture of LRAP placed on top of a COOH SAM with blue N-terminus on the left and red C-terminus on the right. The protein is placed onto the surface with the C-terminal domain and inner part of the N-terminus near the surface and the N-terminus and middle hydrophobic region away from the surface. The protein secondary structure and the range of structure over the100 lowest energy conformations represented by the white surface are derived from Rosetta simulations. The orientation of the protein, with the C-terminal and inner N-terminal region located near the surface, is suggested by the NR experimental data. Note that this is a schematic of a possible orientation and not a computer simulation of LRAP adsorbed onto COOH SAMs.
Good fits and reasonable values for the protein coverages and hydration amounts were obtained when we modeled the protein as two layers with a higher protein SLD near the surface compared to away from the surface (Figure 2d schematic). We also modeled the protein by flipping the layers so that the lower protein SLD layer (N-terminus and middle hydrophobic region) was close to the surface and the higher protein SLD layer (C-terminus and inner N-terminus) was away from the surface as shown in the schematic in Figure 2e. This model was rejected by chi-squared and Hamilton comparisons and because it resulted in values for hydration and protein coverage that were out of range of what would be reasonable from a physicochemical perspective. We tested for random orientation of the adsorbed protein in two ways. If the protein was randomly oriented on the surface, the “inner” and “outer” layers of the two layer monomer model would have the same total SLD which was not seen in Figure 6. Random orientation of the protein was also tested by the one layer protein model that assumes the protein layer has a uniform SLD (Figure 4). This model, however, is rejected by the Hamilton test in comparison to the two layer monomer model. It was necessary to have a higher protein SLD in the inner layer in order to obtain the higher total SLD in the inner layer. The protein regions with higher SLD were also more polar and expected to be more hydrated, further contributing to the higher total SLDs in the inner layer.
The adsorbed LRAP thickness is ~ 32 Å by NR, lower than the diameters predicted by Rosetta simulations (36 Å) and SV (40.4 Å). This result is consistent with the orientation of the protein shown in the Figure 7 schematic where the long dimension of LRAP (~50 Å in the Figure 4 simulation) is parallel to the surface and the short dimension (~26 Å) is perpendicular to the surface. The NR thickness reflects the asymmetric shape of LRAP while the SV diameter is an equivalent spherical diameter, a diameter assuming the protein shape is spherical.
The degree of protein deuteration due to solvent exchange is typically not known in neutron reflectivity studies of adsorbed protein layers.26,59 Layer thicknesses can be obtained from the total SLD profiles, however, because there is a difference in SLD between the deuterated protein and solvent. To get further information such as protein coverage, however, it is necessary to experimentally determine or calculate an SLD for the protein.26,44,59 In our studies, models that assumed different degrees of protein deuteration (different calculated protein SLDs) primarily resulted in different degrees of protein hydration. For example, increasing the assumed protein SLD resulted in a lower degree of protein hydration. Therefore, uncertainties in the degree of deuteration of the protein did not affect the overall conclusions of our work: a monomer adsorbs at ~70 v% coverage with preferred orientation.
Our NR studies gave the general location of more deuterated regions of the protein relative to the surface using a two layer model. There was too much D2O hydration and/or deuteration by solvent exchange to be more specific. We believe NR has the potential to give more specific information on the location of deuterated protein residues but only if we decrease the amount of deuteration that occurs by solvent exchange and increase the amount of deuteration by site-specific protein labeling. This can be accomplished by using a series of solvents that have much lower amounts of D2O and by site-specific deuterium labeling larger sections of the protein.
Previous SSNMR studies showed that the C-terminal region (residues 42–58) and the inner N-terminus (residues 8–24) are located near hydroxyapatite crystal surfaces by measuring distances between 13C in the protein backbone and 31P in the hydroxyapatite surface.20 Rosetta simulations predicted that the hydrophobic N-terminal region (~residues 1–7) and central hydrophobic region (~25–41) would be located away from the surface. We propose, therefore, that LRAP has a similar orientation on COOH SAMs as on hydroxyapatite.
It would not be surprising for LRAP to have a similar orientation on the HAP surfaces studied previously and the COOH surfaces used in this study. Both surfaces are hydrophilic and expected to have a negative charge at pH 7.4. Both surfaces have charged groups that may promote specific adsorption interactions between the LRAP protein and surface. For example, positive amino acid residues in the C-terminus (two lysine, one arginine) may bind to the negative COOH groups in SAMs or phosphate groups in HAP. Calcium bridging may also be involved in the adsorption of negative amino acid residues in the C-terminus (three glutamic acid, three aspartic acid) with negative surface sites.28 Ideally we would like to do NR studies of protein structure on hydroxyapatite. Unfortunately, large molecularly smooth thin films of hydroxyapatite overlying Fe layers are not available for these studies. The SAM layers, however, are very good model systems with some surface properties similar to hydroxyapatite. A range of experimental techniques such as NR, ellipsometry, and AFM can be used to probe surface structures of proteins using SAMs on atomically smooth gold on mica.
Relevance to other studies
Our previous SSNMR studies gave detailed information on the location of LRAP residues in the C-terminus and near N-terminus relative to the surface and the secondary structure of domains near the surface.20 There were gaps in our knowledge, however, of the quaternary form of adsorbed LRAP as well as the structure of LRAP away from the surface, not accessible by the SSNMR techniques. This study fills the gaps, giving a more complete picture of the structure of adsorbed LRAP. We now have direct evidence that the adsorbed LRAP studied by NMR is not part of a nanosphere, but adsorbs as a monomer. In addition, LRAP is not completely flat on the surface but has folding away from the surface as evidenced by the ~32 Å z-dimension determined by NR. This folding away from the surface is shown in the schematic in Figure 7. If LRAP was completely flat and extended along the surface, the z dimension would be the average diameter of the amino acid residues, around 8–12 Å. The region away from the surface is more hydrophobic and less deuterated as suggested by the lower protein hydration and lower protein SLD in this region and the previous Rosetta simulations of LRAP on HAP.
We found that LRAP monomers are the dominant species in the physiological solutions at pH 7.4. The mechanism for adsorption is the preferred adsorption of monomers onto the surfaces from solution. This mechanism is in contrast to the mechanism of the disassembly of adsorbed nanospheres we previously found for full length amelogenin.60,61 Amelogenin solutions consisting of rp(H)M180 (recombinantly synthesized, histidine tagged, mouse amelogenin) and rM179 (recombinantly synthesized mouse amelogenin) primarily contained nanospheres. Monomers and small oligomers shed from the nanospheres onto surfaces, reversing the process of amelogenin assembly. The presence of LRAP monomers in in-vitro suggests that monomers may also exist in-vivo and may be an important functional form of LRAP. The monomer quaternary structure may be critical for LRAP’s proposed role as a mesenchymal cell signaling molecule.62 For example, LRAP has been found to promote osteogenesis in rat muscle fibroblasts,63 mouse cementoblasts,64 and mouse stem cells.65 There is also evidence that LRAP is involved in enamel formation, promoting enamel crystal growth in amelogenin knock-out mice.66–68
In order to give a more complete picture of the structure of amelogenins in solution and on surfaces, further studies of the structure of LRAP at various solution conditions relevant to enamel forming will be of interest. Developing a detailed molecular-level description of the structure of amelogenin proteins on surfaces, from the secondary to the quaternary level, is experimentally challenging but is being addressed by the use of advanced surface analytical techniques such as NR and SSNMR. These approaches have lead to a better fundamental understanding of the structures of biomineralization proteins on surfaces and have provided insights on relationships between protein structure and function.
SUMMARY
We found that monomers are the dominant species for LRAP in pH 7.4, physiologically relevant solutions. The monomer has an experimentally measured hydrodynamic radius of 20.2 Å and a simulated radius of gyration of 18.5 Å and an extended, asymmetric structure. NR experiments show that LRAP adsorbs onto SAM surfaces as a monomer with a z dimension of ~32 Å. The NR modeling is consistent with the protein adsorbing with the more hydrophilic, deuterated domains (C-terminal and inner N-terminal) near the surface and more hydrophobic domains away from the surface. We have demonstrated that NR is a valuable technique for determining the tertiary and quaternary structure of adsorbed proteins and can also provide information on protein orientation. This work shows that monomeric species may be an important functional form of LRAP amelogenins in solution and at surfaces.
Supplementary Material
Acknowledgments
This work was supported by NIH-NIDCR Grant DE-015347 (PNNL) and was performed in part at Pacific Northwest National Laboratory, operated by Battelle for the US-DOE. This work was supported in part by the National Science Foundation under Agreement No. DMR-0454672 and was performed in part at NIST. We would like to thank Andrew Rockwell, who helped collect initial NR data. We thank Robert Latour and Nadeem Vellore for providing coordinates for the COOH SAM surface.
ABBREVIATIONS
- NR
Neutron Reflectivity
- SSNMR
Solid state NMR
- HAP
Hydroxyapatite
- LRAP
Leucine-Rich Amelogenin Peptide
- SLD
Scattering length density
- SAMs
Self-assembled monolayers
- NIST
National Institute of Standards and Technology
- NCNR
Center for Neutron Research
- SCP
saturated calcium phosphate solution with 0.15 M NaCl
- PBS
phosphate buffered saline
- TEM
transmission electron microscopy
- and AFM
atomic force microscopy
Footnotes
SUPPORTING INFORMATION AVAILABLE
Supporting information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lowenstam HA, Weiner S. On Biomineralization. Oxford University Press; New York: 1989. [Google Scholar]
- 2.Addadi L, Moradian-Oldak J, Furedi-Milhofer H, Weiner S, Veis A. In: Proceedings of the fourth International Conference on the Chemistry and Biology of Mineralized Tissues. Slavkin H, Price P, editors. Excerpta Medica; Coronado, CA: 1992. pp. 153–162. [Google Scholar]
- 3.Berman A, Addadi L, Weiner S. Interactions of Sea-Urchin Skeleton Macromolecules with Growing Calcite Crystals--a Study of Intracrystalline Proteins. Nature. 1988;331:546–548. [Google Scholar]
- 4.Gibson CW, Yuan ZA, Hall B, Longenecker G, Chen EH, Thyagarajan T, Sreenath T, Wright JT, Decker S, Piddington R, et al. Amelogenin-deficient mice display an amelognesis imperfecta phenotype. Journal of Biological Chemistry. 2001;276:31871–31875. doi: 10.1074/jbc.M104624200. [DOI] [PubMed] [Google Scholar]
- 5.Hunter G. Interfacial Aspects of Biomineralization. Current Opinion in Solid State and Materials Science. 1996;1:430–435. [Google Scholar]
- 6.Simmer JP, Fincham AG. Molecular Mechanisms of Dental Enamel Formation. Critical Reviews in Oral Biology and Medicine. 1995;6:84–108. doi: 10.1177/10454411950060020701. [DOI] [PubMed] [Google Scholar]
- 7.Addadi L, Weiner S. Interactions between acidic proteins and crystals: Stereochemical requirements in biomineralization. Proceedings of the National Academy of Science. 1985;82:4110–4114. doi: 10.1073/pnas.82.12.4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeOliveira, Laursen RA. Control of Calcite Crystal Morphology by a Peptide Designed To Bind to a Specific Surface. Journal of the American Chemical Society. 1997;119:10627–10631. [Google Scholar]
- 9.Hunter GK, O’Young J, Grohe B, Karttunen M, Goldberg HA. The flexible polyelectrolyte hypothesis of protein-biomineral interaction. Langmuir. 2010;26:18639–18646. doi: 10.1021/la100401r. [DOI] [PubMed] [Google Scholar]
- 10.Hunter GK, Curtis HA, Grynpas MD, Simmer JP, Fincham AG. Effects of Recombinant Amelogenin on Hydroxyapatite Growth. Calcified Tissue International. 1999;65:226–231. doi: 10.1007/s002239900688. [DOI] [PubMed] [Google Scholar]
- 11.Termine JD, Belcourt AB, Christner PJ, Conn KM, Nylen MU. Properties of Dissociatively Extracted Fetal Tooth Matrix Proteins. The Journal of Biological Chemistry. 1980;255:9760–9768. [PubMed] [Google Scholar]
- 12.Gibson CW, Golub E, Ding W, Shimokawa H, Young M, Termine J, Rosenbloom J. Identification of the Leucine-Rich Amelogenin Peptide (LRAP) as the Translation Product of an Alternatively Spliced Transcript. Biochemical and Biophysical Research Communications. 1991;174:1306–1312. doi: 10.1016/0006-291x(91)91564-s. [DOI] [PubMed] [Google Scholar]
- 13.Fincham AG, Moradian-Oldak J, Diekwisch TGH, Lyaruu DM, Wright JT, Jr, PB, Slavkin HC. Evidence for Amelogenein”Nanospheres” as Functional Components of Secretory-Stage Enamel Matrix. Journal of Structural Biology. 1995;115:50–59. doi: 10.1006/jsbi.1995.1029. [DOI] [PubMed] [Google Scholar]
- 14.Moradian-Oldak J, Bouropoulos N, Wang L, Gharakhanian N. Analysis of self-assembly and apatite binding propertied of amelogenin protein lacking the hydrophilic C-terminal. Matrix Biology. 2002;21:197–205. doi: 10.1016/s0945-053x(01)00190-1. [DOI] [PubMed] [Google Scholar]
- 15.Chen X, Wang Q, Shen J, Pan H, Wu T. Adsorption of Leucine-Rich Amelogenin Protein on Hydroxyapatite (001) Surface through -COO− Claws. Journal of Physical Chemistry C. 2007;111:1284–1290. [Google Scholar]
- 16.Shaw WJ, Ferris KF, Krueger S, Perez-Salas U, Silin V, McGillivray DJ, Campbell AA, Paine ML, Snead ML. The Orientation of an Amelogenin on Hydroxyapatite Determined Using Neutron Scattering, Solid State NMR and Computational Methods. Proceedings of the International Conference on the Chemistry and Biology of Mineralized Tissues; 2004. pp. 150–153. [Google Scholar]
- 17.Shaw W, Campbell A, Paine M, Snead M. The COOH terminus of the amelogenin, LRAP, is oriented next to the hydroxyapatite surface. Journal Biological Chemistry. 2004;279:40263–40266. doi: 10.1074/jbc.C400322200. [DOI] [PubMed] [Google Scholar]
- 18.Shaw WJ, Ferris KF, Tarasevich BJ, Larson JL. The Structure and Orientation of the C-terminus of LRAP. Biophysical Journal. 2008;94:3247–3257. doi: 10.1529/biophysj.107.119636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw WJ, Ferris K. Structure, Orientation, and Dynamics of the C-Terminal Hexapeptide of LRAP Determined Using Solid-State NMR. Journal of Physical Chemistry B. 2008;112:16975–16981. doi: 10.1021/jp808012g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masica DL, Gray JJ, Shaw WJ. Partial High-Resolution Structure of Phosphorylated and Non-phosphorylated Leucine-Rich Amelogenin Protein Adsorbed to Hydroxyapatite. Journal of Physical Chemistry C. 2011;115:13775–13785. doi: 10.1021/jp202965h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fincham AG, Moradian-Oldak J, Simmer JP. The Structural Biology of the Developing Dental Enamel Matrix. Journal of Structural Biology. 1999;136:270–299. doi: 10.1006/jsbi.1999.4130. [DOI] [PubMed] [Google Scholar]
- 22.Lu JR, Zhao X, Yaseen M. Protein adsorption studied by neutron reflection. Current Opinion in Colloid & Interface Science. 2007;12:9–16. [Google Scholar]
- 23.Fragneto G, Graner F, Charitat T, Dubos P, Bellet-Amalric E. Interaction of the Third Helix of Antennapedia Homeodomain with a Deposited Phospholipid Bilayer: A Neutron Reflectivity Structural Study. Langmuir. 2000;16:4581–4588. [Google Scholar]
- 24.Su TJ, Lu JR, Thomas RK, Cui ZF, Penfold J. The Conformational Structure of Bovine Serum Albumin Layers Adsorbed at the Silica-Water Interface. Journal of Physical Chemistry B. 1998;102:8100–8108. [Google Scholar]
- 25.Vaknin D, Kjaer K, Ringsdorf H, Blankenburg R, Piepenstock M, Diederich A, Losche M. X-ray and Neutron Reflectivity Studies of a Protein Monolayer Adsorbed to a Functionalized Aqueous Surface. Langmuir. 1993;9:1171–1174. [Google Scholar]
- 26.Skoda MWA, Schreiber F, Jacobs RMJ, Webster JRP, Wolff M, Dahint RM. Protein Density Profile at the Interface of Water with Oligo(ethyleneglycol) Self-Assembled Monolayers. Langmuir 2009. 2009;25:4056–4064. doi: 10.1021/la8028534. [DOI] [PubMed] [Google Scholar]
- 27.Carpino LA, Han GY. The 9-Fluorenylmethoxycarbonyl Amino-Protecting Group. Journal of Organic Chemistry. 1972;37:3404–3409. [Google Scholar]
- 28.Tarasevich BJ, Lea S, Shaw WJ. The leucine rich amelogenin protein (LRAP) adsorbs as monomers or dimers onto surfaces. Journal of Structural Biology. 2010;169:266–276. doi: 10.1016/j.jsb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lebowitz J, Lewis MS, Schuck P. Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Science. 2002:11. doi: 10.1110/ps.0207702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Philo JS. A method for directly fitting the time derivative of sedimentation velocity data and an alternative algorithm for calculating sedimentation coefficient distribution functions. Anal Biochem. 2000;279:151–163. doi: 10.1006/abio.2000.4480. [DOI] [PubMed] [Google Scholar]
- 32.Philo JS. Improved methods for fitting sedimentation coefficient distributions drived by time-derivative techniques. Anal Biochem. 2006;354:238–246. doi: 10.1016/j.ab.2006.04.053. [DOI] [PubMed] [Google Scholar]
- 33.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. In: Analytical ultracentrifugation in biochemistry and polymer science. Harding SE, Rowe AJ, Horton JC, editors. Royal Society of Chemistry; Cambridge: 1992. pp. 90–125. [Google Scholar]
- 34.Masica DL, Gray JJ. Solution- and Adsorbed-State Structural Ensembles Predicted for the Statherin-Hydroxyapatite System Biophysical. Journal Volume. 2009;96:3082–3091. doi: 10.1016/j.bpj.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;1:2577. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 36.Kortemme T, Morozov AV, Baker DJ. Mol. Biol. 2003;326:1239–1259. doi: 10.1016/s0022-2836(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 37.Majkrzak CF. Polarized Neutron Reflectometry. Physica B. 1991;173:75–88. doi: 10.1103/PhysRevLett.88.067201. [DOI] [PubMed] [Google Scholar]
- 38.Perez-Salas UA, Faucher KM, Majkrzak CF, Berk NF, Krueger S, Chaikof EL. Characterization of a Biomimetic Polymeric Lipid Bilayer by Phase Sensitive Neutron Reflectometry. Langmuir. 2003;19:7688–7694. [Google Scholar]
- 39.Vrugt JA, ter Braak CJF, Diks CGH, Higdon D, Robinson BA, Hyman JM. Accelerating Markov chain Monte Carlo simulation by differential evolution with self-adaptive randomized subspace sampling. International Journal of Nonlinear Sciences and Numerical Simulation. 2009;10:273–290. [Google Scholar]
- 40.Hamilton WC. Acta Crystallogr. 1965;18:502–510. [Google Scholar]
- 41.Li Y, Williams TD, Schowen RL, Topp EM. Characterizing protein structure in amorphous solids using hydrogen/deuterium exchange with mass spectrometry. Analytical Biochemistry. 2007;366:18–28. doi: 10.1016/j.ab.2007.03.041. [DOI] [PubMed] [Google Scholar]
- 42.Dobson CM, Evans PA, Radford SE. Understanding how proteins fold: the lysozyme story so far. Trends in Biochemical Science. 1994;19:31–37. doi: 10.1016/0968-0004(94)90171-6. [DOI] [PubMed] [Google Scholar]
- 43.Hoofnagle AN, Resing KA, Ahn NG. Protein analysis by hydrogen exchange mass spectrometry. Annual Rev Biophys Biomol Struct. 2003;32:1–25. doi: 10.1146/annurev.biophys.32.110601.142417. [DOI] [PubMed] [Google Scholar]
- 44.Efimova YM, Well AAv, Hanefeld U, Wierczinski B, Bouwman WG. On the scattering length density of proteins in H2O/D2O:Determination of HD exchange using ES+I-MS. Journal of Radioanalytical and Nuclear Chemistry. 2005;264:271–275. [Google Scholar]
- 45.Efimova YM, Wella AAv, Hanefeldb U, Wierczinskia B, Bouwman WG. On the neutron scattering length density of proteins in H2O/D2O. Physica B. 2004;350:877–880. [Google Scholar]
- 46.Bull HB, Breese K. Protein Hydration: I. Binding Sites. Archives of Biochemistry and Biophysics. 1968;128:488–496. doi: 10.1016/0003-9861(68)90055-6. [DOI] [PubMed] [Google Scholar]
- 47.Moradian-Oldak J, Paine ML, Lei YP, Fincham AG, Snead ML. Self-assembly properties of recombinant engineered amelogenin proteins analyzed by dynamic light scattering and atomic force microscopy. Journal of Structural Biology. 2000;131:27–37. doi: 10.1006/jsbi.2000.4237. [DOI] [PubMed] [Google Scholar]
- 48.Moradian-Oldak J, Simmer JP, Lau EC, Sarte PE, Slavkin HC, Fincham AG. Detection of monodisperse aggregates of a recombinant amelogenin by dynamic light scattering. Biopolymers. 1994;34:1339–1347. doi: 10.1002/bip.360341006. [DOI] [PubMed] [Google Scholar]
- 49.Fincham AG, Moradian-Oldak J, Simmer JP, Sarte P, Lau EC, Diekwisch T, Slavkin HC. Self-assembly of a recombinant amelogenin protein generates supramolecular structures. Journal of Structural Biology. 1994;112:103–109. doi: 10.1006/jsbi.1994.1011. [DOI] [PubMed] [Google Scholar]
- 50.Le Norcy E, Kwak SY, Wiedemann-Bidlack FB, Beniash E, Yamakoshi Y, Simmer JP, Margolis HC. Leucine-rich amelogenin peptides regulate mineralization in vitro. J Dent Res. 2011;90:1091–1097. doi: 10.1177/0022034511411301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scott DJ, Harding SE, Rowe AJ. Analytical Ultracentrifugation: Techniques and Methods. Royal Society of Chemistry; 2005. [Google Scholar]
- 52.Yang D, Xiong Y, Guo Y, Da D, Lu W. Sizes correction on AFM images of nanometer spherical particles. J Material Science. 2001;36:263–267. [Google Scholar]
- 53.San Paulo A, Garcia R. Unifying theory of tapping-mode atomic-force microscopy. Phys Rev B. 2002;66:041406(R). [Google Scholar]
- 54.Pauling L. The adsorption of water by proteins. JACS. 1945;67:555–557. [Google Scholar]
- 55.Towns JK. Moisture content in proteins: its effects and measurement. J Chromatography A. 1995;705:115–127. doi: 10.1016/0021-9673(94)01257-f. [DOI] [PubMed] [Google Scholar]
- 56.Halle B, Andersson T, Forsen S, Lindman B. Protein hydration from water oxygen-17 magnetic relaxation. J of the American Chemical Society. 1981;103:500–508. [Google Scholar]
- 57.Kuntz ID. Hydration of macromolecules III. Hydration of polypeptides. J of the American Chemical Society. 1971;93:514–516. doi: 10.1021/ja00731a037. [DOI] [PubMed] [Google Scholar]
- 58.Kuntz ID, Kauzmann W. In: Adv Protein Chem. Anfinsen CB, Edsall JT, Richards FM, editors. Vol. 28. Academic Press; NY: 1974. pp. 239–345. [DOI] [PubMed] [Google Scholar]
- 59.Liebmann-Vinson A, Lander LM, Foster MD, Brittain WJ, Vogler EA, Majkrzak CF, Satija S. A Neutron Reflectometry Study of Human Serum Albumin Adsorption in Situ. Langmuir. 1996;12:2256–2262. [Google Scholar]
- 60.Tarasevich BJ, Lea S, Bernt W, Engelhard M, Shaw WJ. Adsorption of Amelogenin onto Self-Assembled and Fluoroapatite Surfaces. Journal of Physical Chemistry B. 2009;113:1833–1842. doi: 10.1021/jp804548x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tarasevich BJ, Lea S, Bernt W, Engelhard MH, Shaw WJ. Rapid Communication Changes in the Quaternary Structure of Amelogenin When Adsorbed onto Surfaces. Biopolymers. 2009;91:103–107. doi: 10.1002/bip.21095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haruyama N, Hatakeyama J, Moriyama K, Kulkarni AB. Amelogenins: multi-functional enamel matrix proteins and their binding partners. J Oral Biosci. 2011;53:257–266. [PMC free article] [PubMed] [Google Scholar]
- 63.Veis A, Tompkins K, Alvares K, Wei K, Wang L, Wang XS, Browneil AG, Jengh SM, Healy KE. Specific amelogenin gene splice products have signaling effects on cells in culture and in implants in vitro. Journal of Biological Chemistry. 2000;275:41263–41272. doi: 10.1074/jbc.M002308200. [DOI] [PubMed] [Google Scholar]
- 64.Boabaid F, Gibson CW, Kuehl MA, Berry JE, Snead ML, Nocitl FH, Katchburian E, Somerman MJ. Leucine-rich amelogenin peptide: a candidate signaling molecule during cementogenesis. Journal of Periodontology. 2004;75:1126–1136. doi: 10.1902/jop.2004.75.8.1126. [DOI] [PubMed] [Google Scholar]
- 65.Warotayanont R, Zhu D, Snead ML, Zhou Y. Leucine-rich amelogenin peptide induces osteogenesis in mouse embryonic stem cells. Biochemical and Biophysical Res Comm. 2008;367:2008. doi: 10.1016/j.bbrc.2007.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ravindranath RMH, Devarajan A, Bringas P., Jr Enamel formation in vitro in mouse molar explants exposed to amelogenin polypeptides: ATMP, LRAP on enamel development. Arch Oral Biol. 2007;52:1161–1171. doi: 10.1016/j.archoralbio.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 67.Gibson CW, Li Y, Suggs C, Kuehl MA, Pugach MK, Kulkarni AB, Wright JT. Rescue of the murine amelogenin null phenotype with two amelogenin transgenes. Eur J Oral Sci. 2011;119:70–74. doi: 10.1111/j.1600-0722.2011.00882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gibson CW, Li Y, Daly B, Suggs C, Yuan ZA, Fong H, Simmons D, Aragon M, Kulkarni AB, Wright JT. The leucine-rich amelogenin peptide alters the amelogenin null enamel phenotype. Cells Tissues Organs. 2008;189:169–174. doi: 10.1159/000151384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.