

Synopsis
Successful treatment of unresectable and metastatic gastroenteropancreatic neuroendocrine tumors (GEP-NETs) requires the thoughtful choice of systemic therapy as a component of a multidisciplinary therapeutic approach. The role of somatostatin analogs is established in symptom relief, but there is less clarity in respect of the efficacy of interferon and radiopeptide targeted therapy. The utility of a variety of tyrosine kinase and anti-angiogenic agents is very variable and under investigation, while a role for cytotoxic chemotherapy in poorly differentiated GEP-NETs is accepted. Overall, the ideal treatment of more indolent tumors is less certain. Reassessments of the GEP-NET pathology classification has provided improved logic for the role of a variety of agents, while the precise positioning of many new agents that target molecular pathways of angiogenesis and proliferation are under examination. This paper describes the current options for systemic therapy for GEP-NETs within the framework of the current (World Health Organization) WHO classification system
Keywords: anti-neoplastic, biotherapy, carcinoid, chemotherapy, neuroendocrine tumor
Introduction
Gastroenteropancreatic neuroendocrine tumors (GEP-NETs) are more common than previously thought and second only to colorectal cancers as the most prevalent gastrointestinal cancers (1, 2). Systemic therapy for GEP-NETs is indicated to control symptoms of bioactive peptide and amine hypersecretion and to reduce tumor proliferation in the setting of unresectable or recurrent disease (3). Although the treatment of each individual tumor is often complex and requires multi-disciplinary expertise (4), there are stable themes that guide choice of systemic treatment of GEP-NETs. These are well described in recent consensus statements such as the European Neuroendocrine Tumor Society (ENETS), the UK and Ireland neuroendocrine tumor network (UKINET), the National Comprehensive Cancer Network (NCCN) and the European Society of Medical Oncology (ESMO) (5, 6, 7, 8). Broadly speaking, all systemic treatment is palliative: somatostatin (SST) analogs effectively reduce symptoms of neuroendocrine hypersecretion (e.g., carcinoid syndrome), systemic treatment does not markedly change the natural history of well-differentiated tumors, cytotoxic chemotherapy is indicated for poorly differentiated neuroendocrine carcinomas, and systemic therapy should be one part of a multi-modal intervention that considers surgery, local therapies and peptide receptor radio-labeled therapy. This paper describes recent developments in systemic pharmacotherapy, particularly in the more controversial indolent tumors, with a focus on novel treatments that target specific proteins in cellular proliferation pathways. The fact that there is a lack of globally accepted definitions of what precisely represents a particular grade or stage of tumor has hampered both delineation of treatment strategy and assessment of efficacy and outcome with different treatment regimes (4).
Classification
The terms used to describe NETs are bewildering in their diversity. NET nomenclature has been difficult since Obendorfer named the tumor as ‘karzinoide’ or ‘cancer-like’ before later observing that these tumors were truly malignant. A simplistic classification, based on embryological origin, classified carcinoids as of fore-, mid-, or hind-gut origin emerged in the 1960’s and is still used in some areas (9). In the current decade, two new classifications have emerged, although neither has gained universal acceptance. The WHO classification describes tumors by their degree of differentiation and site of origin (10), and the more recent ENETS classification includes TNM-type staging, augmented by the Ki-67 index of proliferation (11). However, broad agreement on whether Ki-67 should be assessed or the precise number (of positive cells) that represents a clinically or therapeutically relevant cipher remains controversial (12). The changing classification and most clinician’s natural reluctance to abandon terms associated with a known therapeutic approach have resulted in a variety of names remaining in the pathologist and clinician lexicon. For example, the term ‘neuroendocrine tumor’ is often used to describe the entire family of tumors, but is now a histological stage in the WHO classification, where it describes the well-differentiated and more indolent end of the GEP-NET spectrum. Similarly, the term ‘carcinoid’ is used interchangeably by clinicians to describe all NETs, only NETs with indolent biological behavior, or only NETs originating in the small bowel. However, in the WHO classification, it is now synonymous with ‘neuroendocrine tumor’ and either term describes grade 1 tumors (see Table 1). It is, however, generally agreed that the term ‘carcinoid’ should be phased out since it denotes an archaic concept that is no longer compatible with the modern biological understanding of neuroendocrine malignancy.
Table 1.
WHO Classification of Neuroendocrine tumors
| WHO Classification | WHO Synonym | Biological Behaviour |
|---|---|---|
| Well Differentiated Neuroendocrine Tumor | Carcinoid | Uncertain Malignant potential |
| Well Differentiated Neuroendocrine Carcinoma | Malignant Carcinoid | Low Grade Malignancy |
| Poorly Differentiated Neuroendocrine carcinoma | High Grade Malignancy |
In this paper, we group GEP-NETs along the lines of the WHO definition, but have adjusted this slightly to approximate the inclusion criteria of pharmaco-therapeutic trials, and to separate GEP-NETs by the appropriate therapeutic approaches. This should not imply that we believe this grouping has any inherent value for future classification but simply that it provides a more easily comprehensible system by which the logic of therapeutic strategies can be appreciated. We therefore combine the WHO grade 1 and 2 tumors and divide them by site of tumor origin, as either well differentiated carcinoids (i.e., of enterochromaffin cell origin,) or well differentiated pancreatic neuroendocrine tumors (Figure 1). The group of poorly differentiated tumors irrespective of site of origin is discussed separately.
Figure 1.
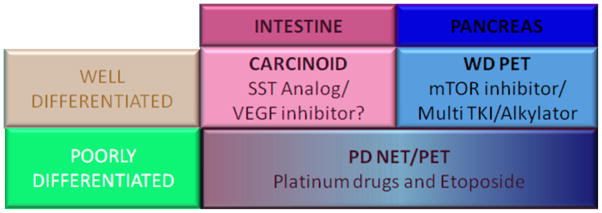
Schema of Gastroenteropancreatic neuroendocrine tumors and treatment options
Impediments to the development of new pharmacological therapy for GEP-NETs
Although some consensus has emerged for choice of systemic treatment, the trial literature used to make these decisions is problematic. The incidence of each sub-type of GEP-NET is low, and there is broad variability in biological behavior within each sub-type. The lack of a widely accepted classification and staging system hinders recruitment of homogenous trial participants. Single centers lack sufficient cases of GEP-NET sub-types for adequate recruitment. As a result, most studies are retrospective, include heterogeneous tumor types, often lack standardized entry criteria, reflect single centre experience and are underpowered (3). Most trials are uncontrolled, and the few controlled trials do not compare the intervention to best supportive care. Furthermore, the majority of studies are funded by pharmaceutical companies with the unavoidable implications inherent therein. Finally, the more indolent GEP-NETs are difficult to assess using traditional measures of efficacy in trial design. The traditional gold standard of therapeutic efficacy in a non-randomized trial is the objective response rate, but this measure has uncertain utility when the tumor is slow growing, and the action of the therapeutic agent is cytostatic. Nonetheless, this compromised dataset has been used to draw the conclusions that follow, based upon the somewhat flawed assertion of, “it’s the best we have.”
Well-differentiated (low grade) Carcinoid tumors
Only a minority of carcinoid tumors are completely resectable (13), meaning that most would benefit from systemic anti-proliferative therapy if an effective option existed. Disease symptoms related to secretion of bioactive peptides and amines were a dominant event in the past, when a far greater percentage of advanced NETs were undetected. Now, only approximately 10–20% of carcinoid tumors (enterochromaffin cell derived tumors of the small bowel), produce the flushing, diarrhea, bronchospasm, and cardiac fibrosis (previously referred to as “the carcinoid syndrome”). This group will benefit from agents that control hypersecretion, namely the somatostatin analog class of drugs (3) or more specific agents that actually interfere with serotonin secretion (14).
Somatostatin (SST) Analogs
SST analogs such as octreotide and lantreotide effectively control symptoms of carcinoid syndrome in most cases where the underlying tumor has a low proliferative index, although in some patients the effect tends to wane over time. This reflects either increase in tumor growth or tachyphylaxis. This can be temporarily overcome by incremental dose increases of the SST analog, or the addition of interferon. Octreotide LAR, for example, has been used above the usual dose range (20 – 30 mg monthly) without additional toxicity (15).
SST analogs may also have an anti-proliferative effect on GEP-NETs. Endogenous SST has a well-documented inhibitory effect on gastrointestinal cell physiology and has an anti-proliferative effect on type II and III gastric carcinoids (16). The PROMID study randomized 85 patients with well-differentiated mid-gut NETs to receive octreotide LAR or placebo (17). Almost all patients (95%) had tumors with Ki67-positive cells less than 2%, and 39% of patients had symptoms of the carcinoid syndrome. The time to progression was 6 months in the placebo group, compared to 14 months in the group given octreotide LAR. This effect was most marked in the subgroup with low volume liver metastases (<10% of liver volume), where time to progression was 29 months. This study has been criticized because, despite multi-center participation, it was slow to recruit (8 years), had a small sample size, and the intervention and placebo groups were not identical (18). The intervention group had a significantly longer time between diagnosis and commencing octreotide, raising a concern that the group who received octreotide included more indolent tumors. Furthermore, there was no difference in median overall survival, although all patients in the placebo group were allowed to cross over to octreotide LAR at progression. This study exemplifies the difficulty in conducting NET research, and we are left with an appealing but uncertain conclusion that has excited much debate. While some groups have adopted the PROMID data as providing the basis for a paradigm shift in therapeutic strategy (all NETs should be treated with an “effective” anti proliferative therapy), significant reservations have been expressed and it seems likely that more data (especially survival improvement) to validate SST as an anti-proliferative agent will be required before the matter is satisfactorily resolved.
The promising novel SST analog pasireotide, a pan-receptor SST agonist, has been proposed as possessing the “biological credentials” to outperform octreotide and lantreotide. There are 5 types of SST receptor (SSTR). Pasireotide has far higher affinity for SSTR1, 3 and 5 than octreotide and lantreotide. This broader specificity has been touted as possibly likely to provide an anti-proliferative advantage. For example, stimulation of SSTR1 is associated with reduced Vascular Endothelial Growth factor (VEGF) and VEGF Receptor 2 (VEGFR2) expression, which may prevent tumor growth by reducing effective blood supply to the tumor. Also, receptor subtypes vary between and within tumor types, so a broad receptor sub-type affinity might be an advantage. Pasireotide reduced symptoms of hyper-secretion in 27% of patients with carcinoid syndrome resistant to octreotide (19), and inhibits growth in carcinoid cell lines in vitro (20). To date, no convincing data have been presented to indicate a major advantage for this pan-receptor agonist and evidence of significant, as yet unresolved, issues relating to pasireotide-induced abnormalities in glucose metabolism continue to dominate clinical concerns (21).
Therapy targeting cellular pathways of proliferation
NETs exhibit some differences in molecular profile to most other tumors. In contrast to many adenocarcinomas, they seldom express microsatellite instability and mutations in the Ras-Raf-MAPKinase or the TGFβ pathway (22, 23). Molecular profiling of tumors instead shows over-expression of the phosphoinotiside-3 kinase - Akt - mammalian target of rapamycin (PI3K-Akt-mTOR) pathway, and the insulin-like growth factor-1 receptor (24, 25, 26). Administering a pharmacologic agent that inhibits one or more of these pro-proliferative proteins provides a rationale for patient- or tumor-specific therapy, and has been tested in early-stage clinical trials.
The most successful therapeutic agents in well-differentiated carcinoid tumors have targeted tumor angiogenesis. VEGF and VEGFR appear to be excellent targets based on clinical phenotype and cell line data. Carcinoid tumors are macroscopically vascular, as evidenced by enhancement in the arterial phase of CT scanning, a characteristic often used to diagnose GEP-NETs radiologically. Carcinoid tumors express high levels of VEGF in surgical specimens (27). Elevated expression of VEGF correlates with increased angiogenesis and decreased progression free survival among patients with low grade NETs (28). Bevacizumab is a monoclonal antibody that binds to VEGF and prevents activation of the VEGF receptor (Figure 2). Bevacizumab has been evaluated in a phase II trial of 44 patients with unresectable or metastatic carcinoid tumors on a stable dose of SST analog. Patients were randomized to 18 weeks of Bevacizumab or pegylated-interferon-α2b therapy before going on to receive both drugs. The Bevacizumab group achieved radiological reduction in tumor vascularity, tumor shrinkage in 18%, and a better 18-week progression free survival of 95% versus 67% (29). Because of planned cross over at 18 weeks, no viable overall survival data was generated. Despite the small numbers, this promising finding warrants further investigation.
Figure 2. Overview of the novel therapeutic targets for GEP-NETs.
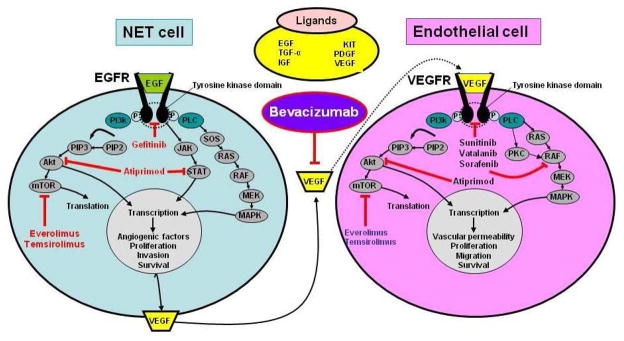
Neuroendocrine tumors are highly angiogenic and dependent on EGF/EGFR and VEGF/VEGFR activation to initiate and maintain, proliferation, survival and invasion. Intracellular targeting of the mTor pathway (Everolimus, Temsirolimus), AKT/STAT (Atiprimod) or growth factor receptors (e.g. Gefitinib or Sorafenib) have “some” therapeutic efficacy in individual tumors. Targeting VEGF with Bevacizumab inhibits”cross-talk” between tumor cells and endothelial cells in the tumor microenvironment. This is a critical relationship in metastatic development.
Two other tyrosine kinase inhibitors (TKI’s) (Sorafenib and Sunitinib) have anti-VEGF or VEGFR properties as part of their targeting arsenal, however, they exhibit only modest in vivo activity in carcinoid tumors. The oral multi-TKI Sorafenib inhibits VEGFR 2 and 3, PDGFR β, RAF, FLT-3 and c-kit. Four of 41 patients with carcinoid tumor had a radiological response by RECIST criteria, and another 3 had a smaller non-RECIST response (30). Of concern is that ~40% of patients experienced grade 3 toxicity. Sunitinib is an oral multi-TKI targeting VEGFR 1, 2 and 3, Platelet derived growth factor receptor (PDGFR) α and β, stem cell factor receptor, glial cell line derived neurotrophic factor receptor, and FMS-like tyrosine kinase 3 (FLT-3). Forty-one patients with well differentiated metastatic carcinoid tumor received Sunitinib as part of a non-randomized phase II study (31). Only 1 of 41 patients achieved a radiological response, so that further accrual was abandoned. Tumor differentiation of participants is not described, although small cell carcinoma was excluded. Sunitinib exhibited significant adverse events with over 50% of patients needing a dose reduction or treatment delay.
Valatinib, an oral TKI that targets multiple VEGFRs and PDGFRs, did not achieve any radiological response in two small studies, and the drug was moderately toxic (32, 33). Given the failure of the VEGFR inhibitors Sunitinib and Valatinib, and the relative success of Bevacizumab and Sorafenib, it seems likely that targeting VEGF is more effective than targeting its receptor, VEGFR in well differentiated carcinoid tumors.
Two other drugs with anti-angiogeneic effects have had minimal impact on carcinoid tumors. Thalidomide has an anti-angiogenic and immune modulating effect by inhibiting TNFα. No radiologic responses were seen in 18 patients with carcinoid, and grade 3 toxicities were common, with 4 patients stopping treatment due to toxicity (34). Endostatin is an endogenous inhibitor of vascular endothelial migration and proliferation. No patients (0/40) achieved a measurable tumor response when given a recombinant version of endostatin (35). The experience with these five drugs represents the practical clinical difficulties encountered when seeking to inhibit a rational biochemical selected target.
Although the P13K-Akt-mTOR pathway is up-regulated in carcinoid cell lines and mTOR inhibitors reduce cell-lines proliferation (20), the in vivo response has been disappointing. The intravenous mTOR inhibitor Temsirolimus achieved only 5% radiological response rate in progressive carcinoid tumors in a small phase II trial (36). Similarly, experience with the small molecule EGFR tyrosine kinase inhibitor (TKI) Gefitinib is an example of thwarted rational target choice. EGFR is over-expressed in NETs, and EGFR inhibitors reduce growth in carcinoid cell lines (37). A phase II study of Gefitinib including 57 patients with carcinoid tumors reported that only 1 of 40 evaluable patients achieved a radiological response, although 32% had a longer period of stability (time to progression - TTP) than they had experienced prior to study entry (determined if TTP on study was 4 months longer than TTP prior to study) (38). This overall poor response might reflect the observation that NETs lack either of the two facilitating EGFR tyrosine kinase mutations associated with Gefitinib activity in non-small cell lung cancer (39). Therefore, although EGFR remains a rational target, the current agents might not be able to adequately inhibit the receptor.
Other targeted agents have been disappointing. A trial of the cKit and the PDGFR inhibitor imatinib showed minimal activity in a group of endocrine tumors that included 2 patients with carcinoid and one with a pancreatic NET (40). There were no objective responses in these three patients.
Cytotoxic Chemotherapy
A maxim of the medical oncologist is that slow growing tumors seldom respond to traditional cytotoxic chemotherapy. The clinical determination of slow growth can be quantified using indices of proliferation such as the number of mitoses per high power field, or the percent of cells expressing Ki-67. Higher indices of proliferation (Ki-67 > 20%) are associated with significant but short-lived responses to chemotherapy, while very low indices of proliferation (Ki-67<2%) tend to infer resistance to cytotoxic chemotherapy (10). In carcinoid tumors, it is now accepted that there is virtually no role for cytotoxics in well differentiated carcinoid tumors, and response rates are less than 15% (5, 41, 42). Similarly, adjuvant chemotherapy after resection of liver metastases does not reduce the rate of relapse (43). Former enthusiasm for agents such as streptozotocin, 5-fluorouracil, doxorubicin and cyclophosphamide has abated given their toxicity and limited efficacy. The real advance in cytotoxic chemotherapy is the realization that it has no role in carcinoid tumors with low indices of proliferation. Cytotoxic chemotherapy takes primacy in the aggressive poorly differentiated carcinoid tumors and will be described later.
Summary
Overall, therapy targeting cellular pathways of proliferation has been ineffective for the majority of patients with well differentiated carcinoid tumors. Radiological responses were seen in only 2 to 18% of patients in each series. There might be a role in well differentiated carcinoid tumors for agents that target VEGF directly. Although small, most drugs had activity in some tumors. The real challenge is to identify and predict which tumors will respond to a specific therapy. For example, although VEGF inhibitors were successful in a few partientss, it is well recognized that the level of VEGF expression is highly variable between different carcinoid tumors (27). Delineation of specific levels would identify patients likely to be responsive to VEGF inhibition. Similarly, VEGF and VEGF receptor expression is lower in liver metastases than in the primary tumor (44), suggesting that VEGFR inhibition might be a better strategy when disease is unresectable but not yet metastatic. And finally, if a cytostatic response is achieved in uncontrolled phase II trials, the activity of such agents may be undetected by measuring response rate, unless a randomized controlled trial is undertaken.
Well differentiated Pancreatic Neuroendocrine Tumors
Therapy targeting cellular pathways of proliferation has been more effective in pancreatic NETs than carcinoid tumors. Pancreatic NETs are a heterogenous group of tumors that includes the functioning tumors insulinoma, gastrinoma, glucagonoma, VIPoma, and rarer cases of pancreatic NETs secreting somatostatin, adrenocorticotrophic hormone, and parathyroid hormone related hormone (3). They differ by the type of endocrine cell that undergoes neoplastic transformation, for example the α, β and δ cells are progenitors of glucagonoma, insulinoma, and somatostatinoma respectively. A group of “so called” non-functioning pancreatic NETs, although biologically indistinguishable, secrete neurotensin, chromogranin A, or several other hormones but do not produce clinically evident symptomatology. The symptoms associated with bioactive peptide hypersecretion can be controlled by tumor resection, SST analogs, or occasionally exogenous counter-hormones (e.g. insulin in glucagonomas). Overall, the somatostatin analog class of agents, particularly the long acting moieties (Octreotide LAR and Lantreotide autogel), have been extremely effective (45). SST analogs have even shown some paradoxical efficacy in somatostatinomas (46).
Therapy targeting cellular pathways of proliferation
Inherited diseases that carry a predisposition to pancreatic NETs such as neurofibromatosis, tuberose Sclerosis, and Cowden’s syndrome are associated with loss of the tumor suppressor genes NF-1, TSC2 and PTEN respectively. Since these genes usually act as tumor suppressor genes by inhibiting the PI3K-AKT-mTOR pathway, there is considerable rationale for exploring therapy directed at the AKT/mTOR pathway in pancreatic NETs. Expression profiling of 72 primary pancreatic NETs showed under-expression of PTEN and TSC2 (47). The subsequent over activity in this pathway results in angiogenesis and tumor cell proliferation. Inhibiting the downstream protein in this pathway, mTOR, therefore appears a rational strategy to control growth of pancreatic NETs.
Everolimus is an oral mTOR inhibitor and had an anti-tumor effect in the RADIANT-1 non-randomized phase II trial of patients with well to moderately differentiated metastatic pancreatic NETs (48). A stratum of patients given single agent Everolimus achieved a radiological response rate of 10% and a progression free survival (PFS) of about 10 months. A stratum of patients already on octreotide when Everolimus was started, achieved a radiological response of 17% and a PFS of about 17 months. This might be explained by a synergistic effect of mTOR inhibition and somatostatin analogs, which has been observed in NET cell lines (20). Alternatively the patients already on octreotide might have represented a more indolent group.
In contrast to NETs of the gut (carcinoid), the multi-TKI Sunitinib has clear activity in pancreatic NETs. Sunitinib achieved a radiological response of 17% and a TTP of 8 months when given to 66 patients with pancreatic NETs (31). A subsequent multi-center randomized placebo-controlled double-blind trial in advanced pancreatic NETs using sunitinib resulted in early study cessation due to differences in efficacy after 171 patients had been randomized (49). Despite a radiological response rate of only 9%, the median PFS in the Sunitinib arm of 11 months was significantly longer than 6 months in the group receiving placebo. This illustrates the difficulty in phase II trial design to detect activity in potentially cytostatic agents, and in this case, the real benefit was only evident when Sunitinib moved to controlled trial.
The multi-TKI Sorafenib also demonstrated some activity against pancreatic NETs. Sorafenib achieved a 10% radiological response, rising to 32% if smaller non-RECIST (20–29% reduction) responses were included (26). Toxicity was an issue as previously described. The EGFR inhibitor Gefitinib may have had even less effect on pancreatic NETs than gut neuroendocrine (carcinoid) tumors, with only a 6% response rate and 10% achieving a longer TTP than their previous TTP (38). Atiprimod exhibits an anti-angiogenic and pro-apoptotic effect by deactivating the AKT and STAT3 signaling pathways. The agent was well tolerated, and may have stabilized low to intermediate grade neuroendocrine carcinomas in 23 patients in phase II trial (50).
Cytotoxic chemotherapy
Well differentiated advanced pancreatic NETs sometimes respond to cytotoxic chemotherapy, however, radiological response rates vary widely. For the past two decades, the cytotoxic standard of care for well differentiated pancreatic NETs has been Streptozotocin with 5FU, doxorubicin or both (51). Trials without placebo control suggested response rates as high as 69% (e.g., 52, 53), but subsequent clinical experience failed to substantiate this level of benefit (54). The positive studies did not describe indices of proliferation in their inclusion criteria, and the high response rates may relate to a high proportion of patients with more aggressive tumors. There is now consensus that cytotoxic chemotherapy has moderate utility in pancreatic NETs with higher indices of proliferation (Ki-67 and mitoses per high power field) more aligned with the WHO grade 2 class of well differentiated neuroendocrine carcinoma (55).
The most recent combination of an alkylator and a flouropyrimidine utilizes the oral agents Temozolamide (oral equivalent of dacarbazine) and Capecitabine (the oral equivalent of 5-fluorouracil). The oral administration of cytotoxic agents enhances the convenience of drug delivery, especially in a slow-growing tumor where administration is prolonged. Temozolamide might be effective in combination with Capecitabine. In a small retrospective review, 10 of 17 patients achieved a radiological response (PR or CR) despite progression on at least one previous line of chemotherapy (56). It is too early to be certain as to whether this oral combination can supplant the intravenous equivalent, but there is a biological rational in support of the increased efficacy of Temozolamide as opposed to the IV alkylators Dacarbazine and Streptozotocin. Temozolamide is more effective in tumors with lower levels of the DNA repair enzyme MGMT, and as a group, pancreatic NETs are often MGMT deficient on immunohistochemical staining (57).
Combination of targeted and cytotoxic agents
Temozolamide has also been combined with the mTOR inhibitor Everolimus in patients with advanced pancreatic NETs (58). There was a radiological response in 6 (35%) of the 17 evaluable patients in this phase I/II trial. Tolerability was not obviously different when using single cytotoxic agents. Temozolamide has also been combined with Thalidomide with a 45% radiological response in a small group of pancreatic NETs, but had little activity in patients with gut neuroendocrine (carcinoid) tumors in the same trial (59). Based upon current data, it appears that a combination of cytotoxic and targeted agent has synergistic activity in pancreatic NETs.
Summary
The response to targeted agents and cytotoxic chemotherapy exhibited by pancreatic NETs differs from gut neuroendocrine (carcinoid) tumors. As a class, pancreatic NETs are more responsive to pharmaco-therapy, but particularly to mTOR and multi-TKI inhibition. The activity of cytotoxic chemotherapy might be limited to the WHO grade 2 tumors with slightly higher indices of proliferation, and trials of these agents in therapeutic doublets will need to carefully describe the proliferative indices of tumors of trial participants.
Poorly differentiated carcinomas
Cytotoxic chemotherapy is the only active therapy for poorly differentiated GEP-NETs (corresponding to WHO grade 3), but chemotherapy rarely achieves prolonged remission. The mainstay of chemotherapy is a platinum agent e.g., Cisplatin coupled with the topoisomerase II inhibitor, Etoposide. Typical response rates range from 42 to 67% but only lasted for a median of around 6 months (e.g., 60, 61).
Recent trials of new cytotoxic combinations in high grade NETs do not appear likely to improve this prognosis. One phase II study substituted the topoisiomerase I inhibitor Irinotecan in place of Etopside. A similar trial had suggested parity in small cell lung cancer (62), the equivalent poorly differentiated NET in the lung. Noting the danger of inter-trial comparisons the Cisplatin, Irinotecan regimen achieved a radiological response of 58%, and the median TTP among responders was 6 months (63). The study was closed early due to slow accrual. A phase II study of the Oxaliplatin, Capecitabine and Bevacizumab regimen commonly used in colorectal cancer was recruiting in 2008 with an early signal of activity, but it is not possible to determine the histological group from the abstract (64). There is little role for SST analogs in poorly differentiated tumors since a high percentage of these lesions exhibit diminished SST receptor expression with the aggressive phenotype (65). Nevertheless, if severe symptomatology is evident, a SST analog may have some utility.
Summary
GEP-NETS are heterogeneous tumors but their pharmacological treatment is governed by several unifying themes (Figure 1). SST analogs are effective in ameliorating symptoms but their role in reducing tumor proliferation of low grade GEP-NETs is felt by many to lack robust data support. Similarly, combinations of SST analogs and kinase inhibitors, while biologically appealing, are unsupported by rigorous long term data. Novel agents that target pro-proliferative cellular proteins, appear to have some promise in well differentiated pancreatic NETs, but to date have been disappointing in well differentiated carcinoid tumors. The newer oral cytotoxics Temozolamide and capecitabine may prove to be more effective than traditional agents in pancreatic NETs, and could conceivably be used in combination with targeted therapies. There have been no recent advances in systemic therapy for poorly differentiated GEP-NETs. All trials continue to be limited by small sample sizes, poor delineation of tumor types and grade description of tumors, the lack of a globally acceptable classification and staging system, and paucity of control group data. Future strategies should focus on not only developing new drugs but identifying patient/tumor specific molecular profiles that provide predictive information necessary to align a targeted agent with a specific tumor in an individual patient.
Footnotes
The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Ben Lawrence, Email: benjaminla@adhb.govt.nz.
Bjorn I. Gustafsson, Email: bjorn.gustafsson@ntnu.no.
Mark Kidd, Email: mark.kidd@yale.edu.
Irvin Modlin, Email: imodlin@yale.edu.
References
- 1.Hauso O, Gustafsson BI, Kidd M, et al. Neuroendocrine tumor epidemiology: contrasting Norway and North America. Cancer. 2008;113(10):2655–64. doi: 10.1002/cncr.23883. [DOI] [PubMed] [Google Scholar]
- 2.Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–72. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 3.Modlin IM, Oberg K, Chung DC, et al. Gastroenteropancreatic neuroendocrine tumors. Lancet Oncol. 2008;9:61–72. doi: 10.1016/S1470-2045(07)70410-2. [DOI] [PubMed] [Google Scholar]
- 4.Modlin IM, Moss SF, Chung DC, et al. Priorities for Improving the Management of Gastroenteropancreatic Neuroendocrine Tumors. J Nat Cancer Instit. 2008;100(18):1282–1289. doi: 10.1093/jnci/djn275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eriksson B, Kloppel G, Krenning E, et al. Consensus guidelines for the management of patients with digestive neuroendocrine tumors – well-differentiated jejuna-ileal tumor/carcinoma. Neuroendocrinology. 2008;87:8–19. doi: 10.1159/000111034. [DOI] [PubMed] [Google Scholar]
- 6.Ramage JK, Davies AHG, Ardill J, et al. Guidelines for the management of Gastroenteropancreatic neuroendocrine (including carcinoid) tumours. Gut. 2005;54:1–16. doi: 10.1136/gut.2004.053314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark OH, Benson AB, 3rd, Berlin JD, et al. NCCN Clinical Practice Guidelines in Oncology: neuroendocrine tumors. Journal of the National Comprehensive Cancer Network. 2009;7(7):712–47. doi: 10.6004/jnccn.2009.0050. [DOI] [PubMed] [Google Scholar]
- 8.Oberg K, Jelic S ESMO Guidelines Working Group. Neuroendocrine gastroenteropancreatic tumors: ESMO clinical recommendation for diagnosis, treatment and follow-up. Annals of Oncology. 2009;20 (Suppl 4):150–3. doi: 10.1093/annonc/mdp158. [DOI] [PubMed] [Google Scholar]
- 9.Williams E, Sandler M. The classification of carcinoid tumors. Lancet. 1963 Feb 2;1:238–9. doi: 10.1016/s0140-6736(63)90951-6. [DOI] [PubMed] [Google Scholar]
- 10.DeLellis RA, Lloyd RV, Heitz PU, et al. World Health Organization Classification of Tumors, Pathology and Genetics of Tumors of Endocrine Organs. Lyon: IARC Press; 2004. [Google Scholar]
- 11.Rindi G, Kloppel G, Couvelard A, et al. TNM staging of midgut and hindgut (neuro) endocrine tumors: a consensus proposal including a grading system. Virchows Arch. 2007;451:757–62. doi: 10.1007/s00428-007-0452-1. [DOI] [PubMed] [Google Scholar]
- 12.Klimstra DS, Modlin IR, Adsay NV, et al. Pathology reporting of neuroendocrine tumors: application of the Delphic consensus process to the development of a minimum pathology data set. American Journal of Surgical Pathology. 2010;34(3):300–13. doi: 10.1097/PAS.0b013e3181ce1447. [DOI] [PubMed] [Google Scholar]
- 13.Modlin IM, Kidd M, Latich I, et al. Current status of gastrointestinal carcinoids. Gastroenterology. 2005;128:1717–51. doi: 10.1053/j.gastro.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 14.Moertel CG, Kvols LK, Rubin J. A study of cyproheptadine in the treatment of metastatic carcinoid tumor and the malignant carcinoidsyndrome. Cancer. 1991;67:33–36. doi: 10.1002/1097-0142(19910101)67:1<33::aid-cncr2820670107>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 15.Chadha MK, Lombardo J, Mashtare T, et al. High-dose Octreotide Acetate for Management of Gastroenteropancreatic Neuroendocrine Tumors. Anticancer Research. 2009;29(10):4127–30. [PubMed] [Google Scholar]
- 16.Tomasetti P, Migliori M, Caletti GC, et al. Treatment of type II gastric carcinoid tumors with somatostatin analogues. N Eng J Med. 2000;343(8):551–4. doi: 10.1056/NEJM200008243430805. [DOI] [PubMed] [Google Scholar]
- 17.Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656–63. doi: 10.1200/JCO.2009.22.8510. [DOI] [PubMed] [Google Scholar]
- 18.Chua YJ, Michael M, Zalcberg JR, et al. Anti-tumor effects of somatostatin analogues in neuroendocrine tumors. J Clin Oncol. 2010;28(3):e41–2. doi: 10.1200/JCO.2009.26.0612. (letter) [DOI] [PubMed] [Google Scholar]
- 19.Kvols L, Wiedenmann B, Oberg K, et al. Safety and efficacy of pasireotide (SOM230) in patients with metastatic carcinoid tumors refractory or resistant to octreotide LAR: results of a phase II study. J Clin Oncol. 2006;24:198s. doi: 10.1530/ERC-11-0367. [DOI] [PubMed] [Google Scholar]
- 20.Kidd M, Svejda B, Giovinazzo F, et al. Effective inhibition of human neuroendocrine tumor cell proliferation by a TOR kinase inhibitor and a novel somatostatin receptor agonist, pasireotide. Proc Am Soc Clin Oncol Gast Intest. 2010:Abstract 174. [Google Scholar]
- 21.Kvols L, Wiedenmann B, Oberg K, et al. Safety and efficacy of pasireotide (SOM230) in patients with metastatic carcinoid tumors refractory or resistant to octreotide LAR: Results of a phase II study. J Clin Oncol; ASCO Annual Meeting Proceedings; 2006. p. 4082. [Google Scholar]
- 22.Tannapfel A, Vomschloss S, Karhoff D, et al. BRAF gene mutations are rare events in gastroenteropancreatic neuroendocrine tumors. Am J Clin Pathol. 2005 Feb;123(2):256–60. [PubMed] [Google Scholar]
- 23.Kidd M, Eick G, Shapiro MD, et al. Microsatellite instability and gene mutations in transforming growth factor-beta type II receptor are absent in small bowel carcinoid tumors. Cancer. 2005 Jan 15;103(2):229–36. doi: 10.1002/cncr.20750. [DOI] [PubMed] [Google Scholar]
- 24.Stilling GA, Zhang H, Ruebel KH, et al. Characterization of the functional and growth properties of cell lines established from ileal and rectal carcinoid tumors. Endocr Pathol. 2007;18(4):223–32. doi: 10.1007/s12022-007-9001-3. [DOI] [PubMed] [Google Scholar]
- 25.Pitt SC, Davis R, Kunnimalaiyaan M, Chen H. AKT and PTEN expression in human gastrointestinal carcinoid tumors. Am J Transl Res. 2009 Feb 28;1(3):291–9. [PMC free article] [PubMed] [Google Scholar]
- 26.Bowen KA, Silva SR, Johnson JN, et al. An analysis of trends and growth factor receptor expression of GI carcinoid tumors. J Gastrointest Surg. 2009 Oct;13(10):1773–80. doi: 10.1007/s11605-009-0958-8. Epub 2009 Jul 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oxboel J, Binderup T, Knigge U, et al. Quantitative gene-expression of the tumor angiogenesis markers vascular endothelial growth factor, integrin alphaV and integrin beta3 in human neuroendocrine tumors. Oncology Reports. 2009;21(3):769–75. [PubMed] [Google Scholar]
- 28.Zhang J, Jia Z, Li Q, et al. Elevated expression of vascular endothelial growth factor correlates with increased angiogenesis and decreased progression-free survival among patients with low-grade neuroendocrine tumors. Cancer. 2007;109:1478–86. doi: 10.1002/cncr.22554. [DOI] [PubMed] [Google Scholar]
- 29.Yao JC, Ng C, Hoff PM, et al. Improved progression free survival (PFS), and rapid, sustained decrease in tumor perfusion among patients with advanced carcinoid treated with Bevacizumab. Proc Am Soc Clin Oncol. 2005;23:309. [Google Scholar]
- 30.Hobday TJ, Rubin J, Holen K, et al. MC044h, a phase II trial of sorafenib in patients (pts) with metastatic neuroendocrine tumors (NET): a Phase II Consortium (P2C) study. J Clin Oncol. 2007;25 (18S):4504. [Google Scholar]
- 31.Kulke MH, Lenz H, Meropol NJ, et al. Activity of Sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008;26(20):3403–10. doi: 10.1200/JCO.2007.15.9020. [DOI] [PubMed] [Google Scholar]
- 32.Anthony LB, McCall J, Nunez J, et al. An open-label phase II clinical trial of PTK787 in patients with progressive neuroendocrine cancer. J Clin Oncol. 2007;25 (18S):14127. [Google Scholar]
- 33.Pavel ME, Bartel C, Heuck F, et al. Open-label, non-randomized, multicenter phase II study evaluating the angiogenesis inhibitor PTK787/ZK222584 (PTK/ZK) in patients with advanced neuroendocrine carcinomas (NEC) J Clin Oncol. 2008 May 20;26(suppl):abstr 14684. [Google Scholar]
- 34.Varker KA, Campbell J, Shah MH. Phase II study of thalidomide in patients with metastatic carcinoid and islet cell tumors. Cancer Chemother & Pharmacol. 2008;61(4):661–8. doi: 10.1007/s00280-007-0521-9. [DOI] [PubMed] [Google Scholar]
- 35.Kulke MH, Bergsland EK, Ryan DP, et al. Phase II study of recombinant human endostatin in patients with advanced neuroendocrine tumors. J Clin Oncol. 2006;24(22):3555–61. doi: 10.1200/JCO.2006.05.6762. [DOI] [PubMed] [Google Scholar]
- 36.Duran I, Kortmansky J, Singh D, et al. A phase II clinical and pharmacodynamic study of temsirolimus in advanced neuroendocrine carcinomas. Br J Cancer. 2006;95:1148–54. doi: 10.1038/sj.bjc.6603419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stilling GA, Zhang H, Ruebel KH, et al. Characterization of the functional and growth properties of cell lines established from ileal and rectal carcinoid tumors. Endocr Pathol. 2007;18(4):223–32. doi: 10.1007/s12022-007-9001-3. [DOI] [PubMed] [Google Scholar]
- 38.Hobday T, Holen K, Donehower R, et al. A phase II trial of gefitinib in patients (pts) with progressive metastatic neuroendocrine tumors (NET): A Phase II Consortium (P2C) study. J Clin Oncol ASCO Annual Meeting Proceedings. 2006;24(18s):189s. [Google Scholar]
- 39.Gilbert JA, Lloyd RV, Ames MM. Lack of mutations in EGFR in gastroenteropancreatic neuroendocrine tumors. N Engl J Med. 2005;353(2):209–10. doi: 10.1056/NEJM200507143530219. [DOI] [PubMed] [Google Scholar]
- 40.Gross DJ, Munter G, Bitan M, et al. The role of imatinib mesylate (Glivec) for treatment of patients with malignant endocrine tumors positive for c-kit or PDGF-R. Endocr Relat Cancer. 2006 Jun;13(2):535–40. doi: 10.1677/erc.1.01124. [DOI] [PubMed] [Google Scholar]
- 41.Sun W, Lipsitz S, Catalano P, et al. Phase II/III study of doxorubicin with fluorouracil compared with streptozotocin with fluorouracil or dacarbazine in the treatment of advanced carcinoid tumors: Eastern Cooperative Oncology Group Study E1281. J Clin Oncol. 2005;23(22):4897–904. doi: 10.1200/JCO.2005.03.616. [DOI] [PubMed] [Google Scholar]
- 42.Kulke MH, Wu B, Ryan DP, et al. A phase II trial of irinotecan and cisplatin in patients with metastatic neuroendocrine tumors. Dig Dis Sci. 2006 Jun;51(6):1033–8. doi: 10.1007/s10620-006-8001-3. [DOI] [PubMed] [Google Scholar]
- 43.Maire F, Hammel P, Kianmanesh R, et al. Is adjuvant therapy with streptozotocin and 5-fluorouracil useful after resection of liver metastases from digestive endocrine tumors? Surgery. 2009;145(1):69–75. doi: 10.1016/j.surg.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 44.Besig S, Voland P, Baur DM, et al. Vascular endothelial growth factors, angiogenesis, and survival in human ileal enterochromaffin cell carcinoids. Neuroendocrinology. 2009;90(4):402–15. doi: 10.1159/000245900. [DOI] [PubMed] [Google Scholar]
- 45.Modlin IM, Pavel M, Kidd M, Gustafsson BI. Review article: somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine (carcinoid) tumours. Alimentary Pharmacology & Therapeutics. 2010;31(2):169–88. doi: 10.1111/j.1365-2036.2009.04174.x. [DOI] [PubMed] [Google Scholar]
- 46.Arnold R, Trautmann ME, Creutzfeld W, et al. Somatostatin analogue octreotide and inhibition of tumor growth in metastatic endocrine gastroenteropancreatic tumors. Gut. 1996;38:430–8. doi: 10.1136/gut.38.3.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Missiaglia E, Dalai I, Barbi S, et al. Pancreatic endocrine tumors: Expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28(2):245–55. doi: 10.1200/JCO.2008.21.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yao JC, Lombard-Bohas C, Baudin E, et al. Daily everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. J Clin Oncol. 2010;28(1):69–76. doi: 10.1200/JCO.2009.24.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raymond E, Niccoli-Sire P, Bang Y, et al. Updated results of the phase III trial of sunitib (SU) versus placebo (PBO) for treatment of advanced pancreatic neuroendocrine tumors. Proc Am Soc Clin Oncol Gast Int. 2010:abstr 127. [Google Scholar]
- 50.Sung M, Kvois L, Wolin E, et al. Phase II proof-of-concept study of atiprimod in patients with advanced low- to intermediate-grade neuroendocrine carcinoma. J Clin Oncol. 2008 May 20;26(suppl):abstr 4611. [Google Scholar]
- 51.Granberg D. Investigational drugs for neuroendocrine tumors. Expert Opin Investigat Drugs. 2009;18(5):601–8. doi: 10.1517/13543780902850432. [DOI] [PubMed] [Google Scholar]
- 52.Kouvaraki M, Ajani JA, Hoff P, et al. Fluorouracil, Doxorubicin, and Streptozotocin in the Treatment of Patients With Locally Advanced and Metastatic Pancreatic Endocrine Carcinomas. J Clin Oncol. 2004;22(23):4762–71. doi: 10.1200/JCO.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 53.Moertel CG, Lefkopoulo M, Lipsitz S, et al. Streptozotocin-doxorubicin, streptozotocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Eng J Med. 1992;326(8):519–23. doi: 10.1056/NEJM199202203260804. [DOI] [PubMed] [Google Scholar]
- 54.Cheng PN, Saltz LB. Failure to Confirm Major Objective Antitumor Activity for Streptozotocin and Doxorubicin in the Treatment of Patients with Advanced Islet Cell Carcinoma. Cancer. 1999;86(6):944–8. [PubMed] [Google Scholar]
- 55.Ahlman H, Nilsson O, McNicol AM, et al. Poorly differentiated endocrine carcinomas of midgut and hindgut origin. Neuroendocrinology. 2008;87:40–6. doi: 10.1159/000109976. [DOI] [PubMed] [Google Scholar]
- 56.Isacoff WH, Moss RA, Pecora AL, et al. Temozolomide/Capecitabine therapy for metastatic neuroendocrine tumors of the pancreas. A retrospective review. J Clin Oncol. 2006;247(18S):14023. [Google Scholar]
- 57.Kulke MH, Hornick JL, Frauenhoffer C, et al. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clinical Cancer Research. 2009;15(1):338–45. doi: 10.1158/1078-0432.CCR-08-1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kulke M, Blaszkowsky LS, Zhu AX, Flortio S, Regan E, et al. Phase I/II study of everolimus (RAD001) in combination with Temozolamide (TMZ) in patients (pts) with advanced pancreatic neuroendocrine tumors (NET) Proc Am Soc Clin Oncol Gast Int. 2010:Abstr 223. [Google Scholar]
- 59.Kulke MH, Stuart K, Enzinger PC, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol. 2006;24(3):401–6. doi: 10.1200/JCO.2005.03.6046. [DOI] [PubMed] [Google Scholar]
- 60.Moertel CG, Kvols LK, O’Connell MJ, Rubin J. Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin. Evidence of major therapeutic activity in the anaplastic variants of these neoplasms. Cancer. 1991;68(2):227–32. doi: 10.1002/1097-0142(19910715)68:2<227::aid-cncr2820680202>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 61.Mitry E, Baudin E, Ducreux M, et al. Treatment of poorly differentiated neuroendocrine tumors with etoposide and Cisplatin. Brit J Cancer. 1999;81(8):1351–1355. doi: 10.1038/sj.bjc.6690325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lara PN, Jr, Natale R, Crowley J, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol. 27(15):2530–5. doi: 10.1200/JCO.2008.20.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mani MA, Shroff RT, Jacobs C, et al. A phase II study of irinotecan and cisplatin for metastatic or unresectable high grade neuroendocrine carcinoma. J Clin Oncol. 2008 May 20;26(suppl):abstr15550. [Google Scholar]
- 64.Kunz PL, Kuo T, Kaiser HL, et al. A phase II study of capecitabine, oxaliplatin, and bevicizumab for metastatic or unresectable neuroendocrine tumors: Preliminary results. J Clin Oncol. 2008 May 20;26(suppl):abstr15502. [Google Scholar]
- 65.Reubi JC, Kvols LK, Waser B, et al. Detection of somatostatin receptors in surgical and percutaneous needle biopsy samples of carcinoids and islet cell carcinomas. Cancer Res. 1990 Sep 15;50(18):5969–77. [PubMed] [Google Scholar]