

Abstract
The Alzheimer's disease (AD) process is understood to involve the accumulation of amyloid plaques and tau tangles in the brain. However, attempts at targeting the main culprits, neurotoxic Aβ peptides, have thus far proven unsuccessful for improving cognitive function. Recent clinical trials with passively administrated anti-Aβ antibodies failed to slow cognitive decline in mild to moderate AD patients, but suggest that an immunotherapeutic approach could be effective in patients with mild AD. Using an AD mouse model (Tg2576), we tested the immunogenicity (cellular and humoral immune responses) and efficacy (AD-like pathology) of clinical grade Lu AF20513 vaccine. We found that Lu AF20513 induces robust “non-self” T-cell responses and the production of anti-Aβ antibodies that reduce AD-like pathology in the brains of Tg2576 mice without inducing microglial activation and enhancing astrocytosis or cerebral amyloid angiopathy. A single immunization with Lu AF20513 induced strong humoral immunity in mice with preexisting memory T-helper cells. In addition, Lu AF20513 induced strong humoral responses in guinea pigs and monkeys. These data support the translation of Lu AF20513 to the clinical setting with the aims of: (1) inducing therapeutically potent anti-Aβ antibody responses in patients with mild AD, particularly if they have memory T-helper cells generated after immunizations with conventional tetanus toxoid vaccine, and (2) preventing pathological autoreactive T-cell responses.
Introduction
Neuropathological features of Alzheimer's disease (AD) include the deposition of the amyloid-β (Aβ) fragment of amyloid precursor protein (APP) in senile plaques, the accumulation of neurofibrillary tangles composed of tau protein, and the death of neurons (Hardy and Allsop, 1991; Pike et al., 1991; Price and Sisodia, 1994; Hardy and Selkoe, 2002; Nikolaev et al., 2009). For over two decades, Aβ peptides have been thought to play a central role in the onset and progression of AD (Selkoe, 1991, 1994), and it was from this idea that the “amyloid cascade hypothesis” emerged (Hardy and Higgins, 1992; Golde et al., 2006; Hardy, 2006). According to this hypothesis, in AD, the normally soluble Aβ molecule undergoes conformational changes and is deposited as insoluble fibrils and soluble oligomers and protofibrils. The amyloid cascade hypothesis has evolved to focus mainly on soluble oligomers and protofibrils of Aβ, which are now considered to be the most toxic forms of Aβ, responsible for causing synaptic destruction (Harper et al., 1997; Walsh et al., 1997; Lambert et al., 1998; Yong et al., 2002; Klein et al., 2004; Cleary et al., 2005; Haass and Selkoe, 2007). Accordingly, therapeutic interventions for AD have been directed toward decreasing Aβ production using β- and γ-secretase inhibitors/modulators or by immunotherapeutic strategies to enhance Aβ clearance and to block tau aggregation (Sigurdsson et al., 2004; Rafii and Aisen, 2009; Holtzman et al., 2011). The first clinical trial of the AN1792 vaccine in AD patients was unsuccessful due to a small but statistically significant incidence of meningoencephalitis (Orgogozo et al., 2003). However, vaccine strategies for AD treatment will remain highly promising if new-generation vaccines can avoid anti-Aβ T-cell responses, which may underlie the incidences of meningoencephalitis (Orgogozo et al., 2003), and T-cell tolerance, which might have accounted for the low antibody titers in many patients in the AN1792 trial (Nicoll et al., 2003; Ferrer et al., 2004; Gilman et al., 2005; Boche et al., 2008; Holmes et al., 2008). In this translational study, we have devised and validated a novel AD epitope vaccine, Lu AF20513, in which the T-helper (Th) cell epitopes of Aβ42 were replaced by two foreign Th epitopes from tetanus toxoid (TT), P2, and P30, and the immunodominant B-cell epitope of amyloid Aβ1–12. Our data reveal that the Lu AF20513: (1) overcomes T-cell tolerance induced by self-antigen, (2) greatly reduces the possibility of inducing harmful autoreactive T-cell responses that may explain the failure of the AN1792 vaccine, and (3) may improve the ability of the elderly to mount an effective immune response by stimulation of preexisting memory Th-cells. Our results on the immunogenicity, efficacy, and safety of GMP (Good Manufacturing Practice) grade Lu AF20513 in an APP/Tg mouse model of AD, in guinea pigs and in cynomolgus monkeys support the translation to Phase I/IIa clinical trials.
Materials and Methods
Animals, epitope vaccine, and experimental protocols
Mice.
Female and male, 4- to 6-month-old Tg2576 mice (H-2bxs haplotype) were bred at the animal facility of the University of California at Irvine. Female, 6- to 8-week-old B6SJL mice (H-2bxs haplotype) were obtained from The Jackson Laboratory. All animals were housed in a temperature- and light-cycle-controlled facility and cared for under the guidelines of the National Institutes of Health and an approved institutional animal care and use committee protocol at the University of California, Irvine.
Guinea pigs.
Female and male guinea pigs (albino Dunkin–Hartley) were obtained from Charles River Laboratories and were ∼8–10 weeks of age at the commencement of treatment. All animals were housed in a temperature- and light-cycle-controlled facility at TNO Triskelion (Zeist, The Netherlands) and cared for under the guidelines of the European Communities (Directive 86/609/EEC) and Dutch legislation (The Experiments on Animals Act, 1997).
Cynomolgus monkeys.
Female and male, purpose-bred cynomolgus monkeys (Macaca fascicularis), ∼3 years of age, were obtained from Biodia. All animals were housed in a temperature- and light-cycle-controlled facility at Ricerca Biosciences (Concord, OH) and their care was in compliance with the following: Guide for the Care and Use of Laboratory Animals by the National Research Council, 1996, Decree #2001-464 regarding experiments with laboratory animals, which was described in the Journal Officiel de la République Francaise on May 29, 2001, and Decree #2001-486 regarding the protection of animals used in scientific experiments, which was described in the Journal Officiel de la République Francaise on June 6, 2001.
Epitope vaccine.
The GMP grade recombinant protein vaccine (Lu AF20513) that was used in this study was composed of two foreign Th-cell epitopes from TT, P30, and P2 and three copies of the B-cell epitopes of Aβ42 and Aβ1–12 (Fig. 1A), avoiding known Th-cell epitopes in Aβ. Lu AF20513 is expressed in E. coli and is found primarily in the inclusion bodies. The inclusion bodies were solubilized and the protein was purified through a series of chromatographic methods and buffer exchanges to the final formulation.
Figure 1.

Lu AF20513 vaccine and design of immunization studies of Tg2576 mice. A, Schematic representation of multivalent clinical grade AD epitope vaccine (Lu AF20513). Three copies of B-cell epitope of Aβ42 (Aβ1–12) are attached to P30 and P2 Th epitopes from TT conventional vaccine. B, Four- to 6-month-old mice were immunized with 100 μg/mouse of Lu AF20513 formulated in CFA/IFA adjuvant. Mice were killed on day 7 after the third immunization and cellular responses were analyzed. C, Four- to 6-month-old mice were vaccinated four times with 100 μg/mouse of Lu AF20513 formulated in either CFA/IFA or Quil-A adjuvants. Two groups of control mice were injected with CFA/IFA and Quil-A. Mice were bled 10 d after each immunization and the concentration of anti-Aβ antibodies was measured in experimental and control sera. D, Mice from C were combined and additionally vaccinated 7 times with 100 μg/mouse of Lu AF20513 formulated in Quil-A to continue to monitor antibody responses and to measure neuropathological changes at an age of 15–17 months. Control mice were injected with Quil-A adjuvant.
Experimental protocols.
All Tg2576 mice were from the same breeding cohort and were allocated randomly to experimental and control groups. Four to 6-month-old mice were immunized with Lu AF20513 as described previously (Cribbs et al., 2003; Petrushina et al., 2003; Agadjanyan et al., 2005). Briefly, Lu AF20513 protein was formulated in complete (CFA)/incomplete (IFA) Freund's adjuvant or Quil-A adjuvants, as described previously (Cribbs et al., 2003), and mice were injected subcutaneously with the indicated concentrations of antigen (Fig. 1B–D). Control mice were injected with adjuvant only. All mice were boosted at monthly intervals. More detailed experimental protocols are provided in Figure 1B–D. The guinea pigs and cynomolgus monkeys were immunized subcutaneously every month with 20 and 50 μg per animal, respectively, of Lu AF20513 formulated in Alhydrogel. Control groups of guinea pigs and cynomolgus monkeys were injected with adjuvant only. Blood was collected before injections (prebleed) or 10–11 d after the second injection with Lu AF20513 formulated in Alhydrogel.
T-cell proliferation and production of cytokines by immune splenocytes
Analysis of T-cell proliferation was performed in splenocyte cultures from individual animals using a [3H]-thymidine incorporation assay, as described previously (Cribbs et al., 2003; Agadjanyan et al., 2005; Davtyan et al., 2010). The same splenocytes used to assess T-cell proliferation were used for detection of T-cells producing IFNγ (Th1) or IL-4 (Th2) cytokines by ELISPOT (BD Pharmingen), as described previously (Cribbs et al., 2003; Agadjanyan et al., 2005; Petrushina et al., 2007). The cultures of splenocytes from experimental and control mice were restimulated in vitro with P30, P2, Lu AF20513, Aβ40, or an irrelevant peptide (25 μg/ml of each peptide) for 18 h.
Detection of anti-Aβ antibodies by ELISA
Total anti-Aβ antibodies were detected using ELISA, as described previously (Ghochikyan et al., 2006; Petrushina et al., 2007; Davtyan et al., 2010). Anti-Aβ antibody concentrations in mice were calculated using a calibration curve generated with 6E10 monoclonal antibody (Signet). HRP-conjugated anti-IgG1, IgG2ab, IgG2b, and IgM-specific antibodies (Bethyl Laboratories) were used to characterize the isotype profiles of antibodies in pooled sera at a dilution of 1:200.
Anti-Aβ antibodies in guinea pigs and monkeys were detected using 96-well ELISA plates coated with Aβ1–28 (American Peptide). For guinea pigs, sheep anti-guinea pig IgG-biotin (Nordic Immunology) coupled to streptavidin-europium (PerkinElmer) was used as a secondary antibody; in monkeys, goat anti-monkey IgG antibodies conjugated with HRP (Nordic Immunology) was used. The individual samples were diluted to 1:100, 1:200, 1:400, 1:800, 1:2400, 1:7200, and 1:21600, and the titer was determined by back-calculating the OD of the assay cut point on a 4-parameter logistic curve using Prism software (GraphPad).
Purification of anti-Aβ1–12 antibody
Anti-Aβ1–12 antibodies were purified from sera of mice immunized with Lu AF20513 by an affinity column (SulfoLink; Pierce) as described previously (Mamikonyan et al., 2007). The column was immobilized with Aβ18-C peptide (GenScript). Purified antibodies were analyzed with 10% Bis-Tris gel (Invitrogen) and the concentration was determined using a BCA protein assay kit (Pierce).
Surface plasmon resonance analysis
Binding studies were performed on the BIAcore 3000 surface plasmon resonance (SPR) platform (Biacore) as described previously (Mamikonyan et al., 2007). Monomeric, oligomeric, and fibrillar forms of Aβ42 peptides were prepared as described previously and confirmed by electron microscopy, and binding to the 6E10, A11, and OC antibodies was confirmed by dot blot (Kayed et al., 2007; Mamikonyan et al., 2007; Kayed et al., 2010). These peptides were immobilized to the surface of biosensor chip CM5 (GE Healthcare) via an amine coupling of primary amino groups of the appropriate peptide to carboxyl groups in the dextran matrix of the chip. Serial dilutions of anti-Aβ1–12 antibody, irrelevant mouse IgG (SouthernBiotech), or 6E10 antibody (666.6, 222.2, 74.1, 24.7, 8.23, and 2.74 nm) in the running buffer containing 10 mm HEPES, 150 mm NaCl, 0.05% surfactant P20, pH 7.4, were injected at 20 μl/min over each immobilized form of peptide, and the kinetics of binding/dissociation was measured as change of the SPR signal in resonance units. Each injection was followed by a regeneration step of a 25 s pulse of 1 M NaCl and 50 mm NaOH. Fitting of experimental data was done with BIAevaluation 4.1.1 software (GE Healthcare) using a 1:1 interaction model to determine apparent binding constants.
Detection of Aβ plaques in human brain tissues
Anti-Aβ1–12 antibodies (1.25 μg/ml) were screened for their ability to bind to human Aβ plaques using 50 μm brain sections of formalin-fixed cortical tissue from a severe AD case (received from the Brain Bank and Tissue Repository, MIND, at University of California, Irvine) using immunohistochemistry as described previously (Ghochikyan et al., 2003; Agadjanyan et al., 2005; Davtyan et al., 2011). A digital camera (Olympus) was used to capture images of the plaques at a 10× magnification.
Neurotoxicity assay
Cell culture MTT assay was performed as described previously with minor modifications (Davtyan et al., 2011). Aβ42 oligomers and fibrils were incubated alone or with purified anti-Aβ1–12 antibodies or irrelevant antibodies (BD Biosciences) for 2 h at room temperature with occasional mixing to ensure maximal interaction. After incubation, the peptide/immune sera mixtures were diluted into culture media so that the final concentration of peptide and antibodies was 2 and 0.2 μm, respectively. This medium was then added (100 μl) to SH-SY5Y cells (ATCC). The treatment time was 48 h. Untreated controls were run in parallel. After incubation, neurotoxicity was assayed using the MTT assay according to the manufacturer's instructions (Promega). Cell viability was calculated by dividing the absorbance of wells containing samples by the absorbance of wells containing medium alone.
Brain collection, immunohistochemistry, histostaining, quantitative image analysis, and biochemical analysis
Brain collection.
At 15–17 months of age, vaccinated and control mice were killed for further neuropathological analysis as described previously (Movsesyan et al., 2008b). The left hemisphere was snap-frozen and reserved for the measurement of both soluble and insoluble Aβ levels by ELISA. The right hemisphere was fixed in 4% paraformaldehyde in PBS at +4°C for 24 h for further sectioning.
Immunohistochemistry.
Fifty-micrometer-thick coronal sections of fixed hemibrains were cut using a Vibratome. To assess the extent of neuropathology, several sets of free-floating equally spaced sections were selected for each mouse brain, and immunostained using the antibodies described below. Aβ cored and defused deposits were detected with 6E10 (1:1000, Signet), as described previously (Petrushina et al., 2007; Movsesyan et al., 2008b). Activated microglia were stained with the anti-I-A/I-E antibody, a marker of MHC II alloantigens (1:100; BD Pharmingen), and astrocytes were labeled with anti-GFAP antibody (1:500; Dako), both as described previously (Petrushina et al., 2007; Movsesyan et al., 2008b).
Histostaining.
Fibrillar Aβ deposits were visualized using thioflavin S (ThS; Sigma) and hemosiderin deposits were detected with Prussian blue staining. Both assays were performed as described previously (Petrushina et al., 2007; Movsesyan et al., 2008b).
Quantitative image analysis.
The number of 6E10- or ThS-positive plaques was analyzed through the whole hemisphere and in cortical and hippocampal regions. The number of blood vessels showing amyloid depositions characteristic of cerebral amyloid angiopathy (CAA) was determined in 6E10-stained brain sections; activated microglial and hemosiderin-positive profiles were also counted through the whole hemisphere. The mean semiquantitative scores per hemisphere or per neuroanatomical region in these assays were determined based on visual microscopic inspection of sets of five coronal sections equally spaced between −0.80 and −2.92 mm with respect to bregma by three independent observers blinded to the treatment conditions.
NIH ImageJ 1.45s software was used to analyze the number of GFAP-positive astrocytes. For every animal, 19 images (802 × 650 μm each) of cortex, striatum, and hippocampus were selected at approximately the same plane (3 sections between −0.82 and −2.75 mm with respect to bregma). The images were captured using a MD700 video camera (AmScope) and a 10× objective.
Biochemical analysis.
To determine the levels of both soluble Aβ40 and Aβ42 and insoluble total Aβ, a 10% (w/v) of brain homogenate was prepared from each mouse brain at Amorfix Life Sciences using 2% NP-40 containing proprietary protease inhibitors in PBS. Each 10% brain homogenate was centrifuged at 100,000 × g for 1 h at 4°C. The supernatant was collected as the source of soluble aggregates for analysis of soluble Aβ40 and Aβ42 using human β-amyloid ELISA kits (Invitrogen) according to the manufacturer's recommendations. The final values of soluble Aβ were expressed as nanograms per gram wet weight of hemibrain. The remaining pellet was solubilized in 70% formic acid by sonication and centrifuged at 100,000 × g for 1 h at 4°C. Avoiding the upper lipid layer, the lower aqueous layer was collected and stored at −70°C. Quantification of the insoluble Aβ level was done using human β-amyloid ELISA kit (Invitrogen) according to the manufacturer's recommendations. The final values of insoluble Aβ were expressed as micrograms per gram wet weight of hemibrain.
Generation and testing of memory T-cells
Six- to 8-week-old B6SJL mice were immunized 3 times with P30 peptides formulated in Quil-A (50 μg/mouse, n = 30). Control mice were injected with adjuvant only (n = 25). After a resting period of 6 months, 10 mice from the P30-primed and control groups were killed and the memory T-cell response was analyzed before and after a single booster injection using the ELISPOT assay. Recall responses of CD4+ T-cells were detected after isolation of this subpopulation of cells from splenocytes using a specific isolation kit and as suggested by the manufacturer (Miltenyi Biotec). Isolated CD4+ T-cells from splenocytes of primed (n = 4) and nonimmune mice (n = 4) were used for enrichment of naive splenocytes (5 × 104 CD4+ cells were added into 15 × 104 splenocytes). The purity of isolated CD4+ cells and the efficacy of depletion were analyzed by flow cytometry (Miltenyi Biotec). For booster injection, 100 μg/mouse of Lu AF20513 was used, and both cellular and humoral immune responses were analyzed in experimental and control mice, as indicated in Figure 7A.
Figure 7.

Testing the effect of preexisting memory T-cells on the generation of the anti-Aβ antibody response: simulation of vaccination in humans. A, Two groups of B6SJL mice (8 weeks of age) were injected three times biweekly with P30 peptide formulated in Quil-A adjuvant or with Quil-A alone. Cellular immune responses were detected in splenocytes of mice (n = 10/group) after a 6-month resting period. The rest of the primed (n = 20) and control (n = 15) mice were immunized with 100 μg/mouse of Lu AF20513 and cellular (on day 7) as well as humoral (on day 10) responses were analyzed. B, Experimental animals primed with the Th epitope P30, but not control animals (n = 10 per group), generated memory Th-cells that after a 6-month resting period vigorously responded to in vitro restimulation with P30 by the production of IFNγ. Lines indicate averages. ***p ≤ 0.001. C, Depletion of CD4+ T-cells from immune splenocytes significantly decreases the number of IFNγ+ spots formulating cells (SFCs). D, Enrichment of nonimmune splenocytes from control mice with CD4+ T-cells purified from P30-primed mice significantly enhances the number of IFNγ+ SFCs. Error bars indicate average ± SD (n = 4). ***p ≤ 0.001.
Statistical analysis
All statistical parameters (means, SD, significant differences, etc.) used in our experiments were calculated using Prism 6 software (GraphPad). Statistically significant differences were examined using ANOVA and post hoc comparisons were done using Tukey's test (p < 0.05 was considered statistically different).
Results
Lu AF20513 induces strong Th-cell responses specific to foreign Th epitopes from TT
One challenge associated with the clinical use of the Aβ “self” T-cell epitopes as part of a vaccine to treat AD patients is the development of undesirable anti-Aβ or anti-APP-specific T-cell responses (Cribbs et al., 2003; Orgogozo et al., 2003; Ferrer et al., 2004; Agadjanyan et al., 2005; Lemere, 2009). With this in mind, we synthesized the Lu AF20513 vaccine, which was designed to activate non-self CD4+ T-lymphocytes (Th-cells) specific to TT that are preexistent in most subjects due to prior TT immunization to enhance antibody production by Aβ-specific B-cells (Fig. 1A).
We first tested the cellular responses to Lu AF20513 vaccination in Tg2576 mice (Fig. 2A,B), an APP-overexpressing model of AD (Hsiao et al., 1996). Control groups of mice were injected with adjuvant only (Fig. 1B). Vaccinations of Tg2576 mice with Lu AF20513 led to the production of IFNγ and IL-4 after restimulation with P30, but not Aβ40 (Fig. 2A). In proliferation assays, Th-cells were activated by Lu AF20513 and P30, but not by Aβ40 peptide (Fig. 2B). T-cell responses were not detected in mice injected with adjuvant only (Fig. 2C,D). We tested Th-cell responses to only one TT epitope, P30, based on our previous experiments performed in wild-type B6SJL mice demonstrating that two immunizations with Lu AF20513 induced equally effective Th-cell responses specific to both the P2 and P30 epitopes of TT and Lu AF20513 (Fig. 2E,F). Restimulation of immune splenocytes with Lu AF20513 induced significantly stronger Th-cell proliferation (p ≤ 0.001) than when stimulated by P2 and P30 peptides individually (Fig. 2E,F). Therefore, splenocytes from Lu AF20513-immunized wild-type and Tg2576 mice did not induce autoreactive anti-Aβ specific Th-cell responses (Fig. 2).
Figure 2.

Efficacy of Lu AF20513 vaccine: induction of cellular immune responses in Tg2576 and wild-type mice. A, C, Number of IFNγ- and IL-4-producing cells detected by ELISPOT in splenocyte cultures obtained from Lu AF20513-immunized or nonimmunized Tg2576 mice. B, D, Proliferation of cells detected by [3H]-thymidine incorporation assay in splenocyte cultures obtained from experimental and control animals and expressed as the stimulation index. E, F, Immunization of 6- to 8-week-old B6SJL mice (2× s.c.) with 100 μg/mouse of Lu AF20513 formulated in Quil-A adjuvant induces T-cell responses specific to both non-self epitopes, P30 and P2, but not to self-Aβ40 peptide. E, F, T-cells producing IFNγ and IL-4 cytokines detected by ELISPOT (E) and T-cell proliferation assessed by [3H]-thymidine incorporation assay (F). Cellular immune responses in the control group were at the background level (IFNγ+ and IL-4+ spots formulating cells (SFCs) were < 15 and the stimulation index was < 1.6). Error bars represent average ± SD (n = 8 for Tg2576 and n = 5 for B6SJL mice). *p ≤ 0.05;**p ≤ 0.01; ***p ≤ 0.001.
Lu AF20513 immunization induces therapeutically potent anti-Aβ antibody production in Tg2576 mice with very early AD-like pathology
To demonstrate that activation of P30- and P2-specific Th-cells culminates in the activation of anti-Aβ-specific B-cells, we measured the anti-Aβ42 antibody concentrations in Tg2576 mice vaccinated with Lu AF20513 formulated in two different adjuvants, CFA/IFA and Quil-A (Fig. 1C). These animals were in the very early stage of AD-like pathology at the start of immunization (Lesne et al., 2006). Control mice injected with CFA/IFA and Quil-A adjuvants did not generate anti-Aβ42 antibodies, whereas Lu AF20513 formulated in either adjuvant induced robust antibody production after two to three immunizations. No differences were seen in the level or kinetics of antibody production in mice immunized with Lu AF20513 formulated either in CFA/IFA or in Quil-A (Fig. 3A). To characterize the type of humoral immune response, we measured the production of IgG1, IgG2ab, IgG2b, and IgM isotypes of anti-Aβ antibodies in Tg2576 mice immunized with Lu AF20513 formulated in CFA/IFA or Quil-A. We did not observe any differences in the magnitude or type of antibody responses: mice from both vaccinated groups that received either CFA/IFA or Quil-A generated mostly IgG antibodies (IgG2b > IgG2ab > IgG1; Fig. 3B). Because CFA/IFA adjuvant is used only for animal studies and the equivalent Quil-A for humans, QS21, has already been used in many clinical trials, we combined mice from groups immunized with Lu AF20513/Quil-A and Lu AF20513/CFA/IFA as indicated in Figure 1D and performed subsequent immunizations with Lu AF20513 formulated in Quil-A only. These data demonstrated that the anti-Aβ antibody concentration in vaccinated mice reached the maximum titers after three immunizations (Fig. 3A), declined slightly after that, and remained at steady-state during subsequent immunizations until the end of the experiment (Fig. 3C). The levels of IgG1, IgG2ab, and IgG2b immune responses were robust and stable, whereas the level of IgM was low during the whole vaccination process (Fig. 3B,D). These results demonstrate that Th-cells specific to P30 and P2 epitopes from TT provide the support necessary for a therapeutically potent anti-Aβ antibody response in Tg2576 mice vaccinated with the Lu AF20513.
Figure 3.

Efficacy of Lu AF20513 vaccine: induction of humoral immune responses specific to amyloid. A, CFA/IFA and Quil-A adjuvants are equally effective in the stimulation of anti-Aβ antibody responses to immunizations with Lu AF20513. B, Lu AF20513 induced mostly IgG anti-Aβ antibody of the IgG2b, IgG2ab, and IgG1 isotypes. C, D, Multiple immunizations of Tg2576 mice with Lu AF20513 formulated in Quil-A changes neither the strength of antibody production (C) nor the isotype profile (D) of humoral immune responses. Error bars indicate the average ± SD of individual animals for A and pooled sera from 3 different ELISA for B, C, D.
Ex vivo and in vitro characterization of anti-Aβ antibodies generated by Lu AF20513 epitope vaccine
To characterize functionally the anti-Aβ1–12 antibodies generated in response to Lu AF20513, we measured the binding of these antibodies to various forms of β-amyloid, including plaques in the brains from a clinical AD patient, as well as their neuroprotective ability (Fig. 4).
Figure 4.

In vitro assays suggest that Lu AF20513 induces the production of functionally potent anti-Aβ antibody. A, Purified anti-Aβ antibody binds to monomeric, oligomeric, and fibrillar forms of Aβ42 with high affinity. The black lines represent individual data points and the orange lines represent fitted curves B. Binding kinetics (KD is the affinity constant, ka the association rate, and kd the dissociation rate) of purified anti-Aβ and 6E10 antibodies to different forms of Aβ42. C, Anti-Aβ antibody, but not irrelevant mouse IgG, binds to cortical plaques in AD brain. Original magnification is 10×. Scale bar, 100 μm. D, Purified anti-Aβ antibody inhibits neurotoxicity mediated by 2 μm Aβ42 (fibrillar or oligomeric forms). Data were collected from four replicates and are expressed as a percentage of control ± SD. ****p ≤ 0.0001.
One of the important features of anti-Aβ antibodies is their capacity to bind the toxic forms of β-amyloid (Glabe, 2008). Our results from the SPR assay showed that polyclonal anti-Aβ antibodies bound with high average affinity to immobilized monomers (KD = 2.83 × 10−8 M), oligomers (KD = 9.9 × 10−9 M), and fibrils (KD = 2.8 × 10−8 M) (Fig. 4A,B), whereas irrelevant mouse IgG antibody purified from normal mouse serum bound to all these forms of Aβ42 on the baseline level. The binding affinity of purified antibodies to oligomers was higher than the binding affinity to fibrils and monomers. The binding affinity for the control monoclonal antibody, 6E10, was higher for the monomeric form of Aβ42 (KD = 1.21 × 10−10 M) compared with that of the oligomeric (KD = 9.37 × 10−9 M) and fibrillar (KD = 2.97 × 10−9 M) forms (Fig. 4B). Nevertheless, the affinity of binding to oligomeric forms was similar for anti-Aβ1–12 and 6E10 antibodies (KD = 9.9 × 10−9 M vs KD = 9.37 × 10−9 M). In addition, the anti-Aβ antibody generated in mice immunized with Lu AF20513 also bound to Aβ plaques in the brain tissues from an AD patient, whereas irrelevant mouse IgG did not bind to AD plaques (Fig. 4C).
We also tested the protective effect of anti-Aβ antibodies against neurotoxicity induced by Aβ42 oligomers and fibrils in an in vitro model. Culture of SH-SY5Y neuroblastoma cells with Aβ42 oligomers and fibrils had cytotoxic effects, reducing cell viability to 50% and 63.1%, respectively, whereas preincubation with anti-Aβ antibodies rescued the cell viability to 79.5% and 91.6%, respectively (Fig. 4D). Binding to amyloid plaques in the brain sections and the neuroprotective effect of anti-Aβ1–12 antibodies suggest that the Lu AF20513 vaccination strategy is functionally active.
Vaccination with Lu AF20513 significantly reduces AD-like pathology in aged Tg2576 mice
As a preclinical test of the therapeutic efficacy of Lu AF20513 vaccination, we studied neurological changes in the brains of aged Tg2576 mice (15–17 months old) vaccinated with Lu AF20513 (Fig. 1D). Figure 5A shows a significant decrease in hemibrain plaque burden in mice immunized with Lu AF20513 compared with the control, adjuvant-only-injected group. We detected significantly less 6E10-positive diffuse and cored plaques in both cortical and hippocampal regions of brains of Lu AF20513-vaccinated mice compared with the same brain regions of control animals (Fig. 5B). In addition, the results showed a significant reduction of cored ThS-positive Aβ plaques in the hemibrains and as in cortical and hippocampal regions of these brains obtained from vaccinated mice versus brains isolated from control Tg2576 mice (Fig. 5C,D). These data were confirmed by the detection of insoluble Aβ in the brains of vaccinated and control mice. As demonstrated in Figure 5E, vaccination with Lu AF20513 induced a significant reduction (p < 0.001) of insoluble total Aβ in the brains of immunized mice compared with control animals.
Figure 5.
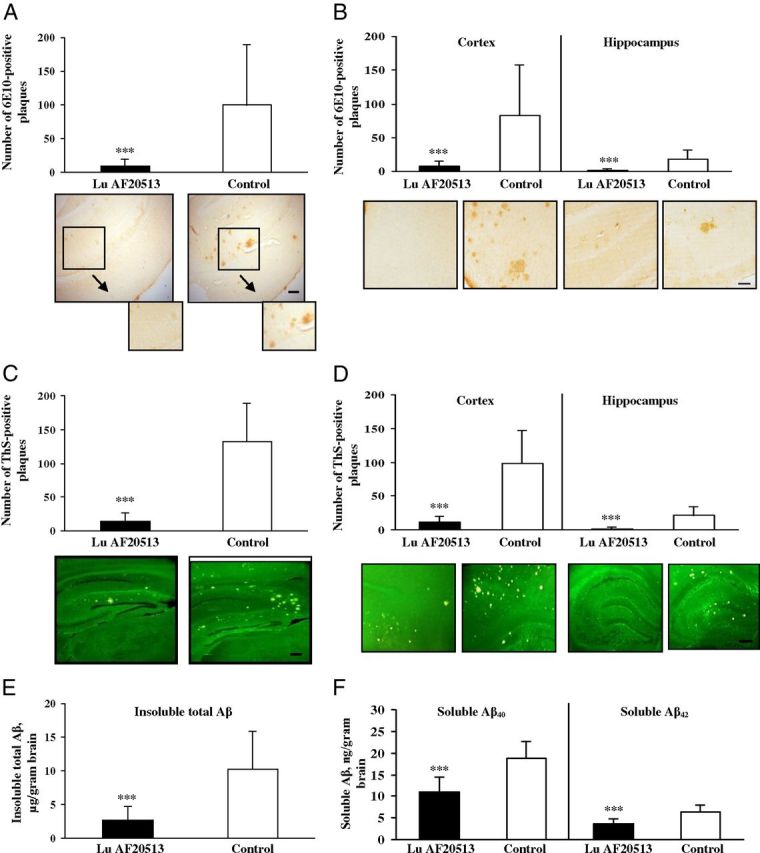
Anti-Aβ antibody inhibits AD-like neuropathology in Tg2576 mice vaccinated with Lu AF20513. A–D, Significant inhibition of numbers of 6E10-immunoreactive cored and diffused (A, B) and ThS-positive cored amyloid-β plaques (C, D) was detected in the brains of 15- to 17-month-old vaccinated Tg2576 mice (n = 20) compared with control animals (n = 17) of the same age. Aβ pathology was analyzed through the whole hemibrains (A, C) and compared in cortical versus hippocampal brain regions (B, D). Photomicrographs show representative images of hemibrain regions of immunized and control mice stained with 6E10 or ThS at a 4× magnification. Scale bar, 200 μm in A, C, D, and 10×. Scale bar (in B), 100 μm (boxed brain areas are shown in detail using 10× magnification). E, As a result of Lu AF20513 vaccination (n = 20), significant reduction of insoluble total β-amyloid levels in brain homogenates was observed compared with control mice (n = 17). F, Significant inhibition of soluble Aβ40 and Aβ42 levels detected in brain homogenates of immune mice (n = 20) compared with control animals (n = 17). Error bars represent average ± SD. ***p < 0.001.
Increasing evidence suggests that soluble (or diffusible) Aβ oligomers mediate different toxic pathways in AD, such as tau hyperphosphorylation (De Felice et al., 2008), impairment of memory, and neuronal death (Lesne et al., 2006; Haass and Selkoe, 2007; Shankar et al., 2008). A potential problem of immunotherapy is that a reduction of insoluble Aβ may lead to increased levels of soluble forms of this peptide (Patton et al., 2006). We observed that vaccination significantly (p < 0.001) reduced the levels of potentially toxic forms of amyloid, soluble Aβ42, and Aβ40 peptides in the brains of Lu AF20513-immunized Tg2576 mice compared with control (adjuvant-injected) animals (Fig. 5F). Therefore, Lu AF20513 vaccination reduces the levels of pathologic forms of Aβ in the brains of immunized mice.
Vaccination with Lu AF20513 reduces glial activation without increasing cerebral amyloid angiopathy and microhemorrhages
To detect inflammation-related pathology in the brains of animals immunized with the Lu AF20513 vaccine, microglial (MHC class II) and astrocyte (GFAP) activation were examined. Semiquantitative image analysis of MHC class II-positive cells (I-Ab/I-Eb) demonstrated significantly fewer activated (MHC class II positive) microglial cells in Tg2576 mice vaccinated with Lu AF20513 (Fig. 6A, representative MHC class II staining). An astrocyte-specific marker in the brains of Lu AF20513-immunized mice revealed a significant reduction in GFAP-positive cells via quantitative image analysis indicative of the presence of fewer astrocytes compared with the control group (Fig. 6B, representative GFAP staining).
Figure 6.
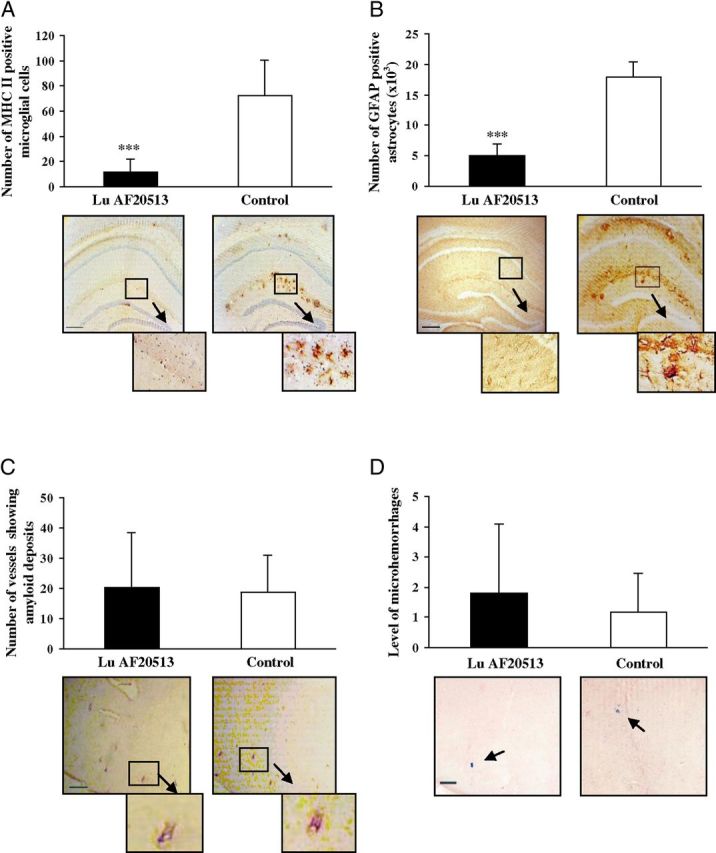
Less activation of microglia and astrocytes in Lu AF20513-vaccinated Tg2576 mice. A, B, Anti-Aβ antibody reduced glial activation (A) and astrocytosis (B) without changing CAA (C) in the brains of 15- to 17-month-old Tg2576 mice vaccinated with Lu AF20513. Error bars represent average ± SD for vaccinated (n = 20) and control nonvaccinated mice (n = 17). ***p < 0.001. Image analysis of hemibrains stained with anti-MHC II (A) and anti-GFAP (B) antibodies demonstrated significantly less microglia activation and astrocytosis, respectively, in vaccinated mice compared with control animals. ***p < 0.001. C, Number of vessels positive for amyloid deposits was calculated in brain sections of experimental and control mice. D, Number of microhemorrhages in the hemibrains of vaccinated mice did not differ from that in control animals. Image analysis of hemibrains from vaccinated or control mice was performed after Prussian blue staining. Photomicrographs represent hemibrains of immunized and control mice at an original magnification of 4×. Scale bars, 200 μm in A–C and 10×. Scale bar (in D), 100 μm (boxed brain areas are shown in detail using a 10× magnification).
One possible side effect of the disruption of amyloid plaques could be the increase of CAA, as was observed in preclinical trials (Wilcock et al., 2007) and the AN1792 trial (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005). To assess the effect of vaccination with Lu AF20513 on vascular deposition of Aβ, we calculated 6E10-positive vessels in the brain sections of immunized and control Tg2576 mice. No differences in the number of amyloid-containing blood vessels were observed in brains of vaccinated or control mice (Fig. 6C). However, the ratio of amyloid-containing blood vessels to parenchymal plaques was increased in vaccinated mice compared with control animals (5.07 ± 5.6 vs 0.24 ± 0.15, respectively), suggesting that amyloid or amyloid–antibody immune complexes may be redistributed to the vessels for clearance from the brain.
We also analyzed microhemorrhages in brains of Lu AF20513-immunized and control mice. It was reported previously that active immunizations with Aβ42 formulated in strong adjuvants (CFA/IFA; Wilcock et al., 2007) may induce cerebral microhemorrhages in ∼20-month-old APP/Tg mice. However, microhemorrhages have not been reported in 15- to 17-month-old mice. Characteristic blue hemosiderin-positive profiles were not increased in Lu AF20513-immunized mice compared with control animals (Fig. 6D). Therefore, immunization with Lu AF20513 did not exacerbate cerebral amyloid angiopathy or increase microhemorrhages and had the positive impact of lowering microglial activation and astrocytosis.
Lu AF20513 induces high titers of anti-Aβ antibodies in natural immunotolerant animal models: guinea pigs and cynomolgus monkeys
Encouraged by the results generated in APP/Tg mice, we tested the Lu AF20513 vaccine in guinea pigs and cynomolgus monkeys after formulation in the widely used mild adjuvant Alhydrogel to strengthen the link to a clinical situation. Fifteen of 16 guinea pigs (94%) responded to two immunizations with Lu AF20513 by producing anti-Aβ antibodies with the average titer = 1:7954 ± 7676 (Table 1). Data from the first clinical trial with the AN1792 vaccine demonstrated that anti-Aβ antibody titers ≥ 1:2200 showed some slowing of cognitive decline and localized reduction of plaques (Hock et al., 2003; Gilman et al., 2005; Masliah et al., 2005; Nicoll et al., 2006). Lu AF20513 vaccine generated therapeutically relevant titers of anti-Aβ antibody (>1:2200) in 80% of responding guinea pigs (Table 1). More importantly, all 14 monkeys immunized twice with Lu AF20513 formulated in Alhydrogel responded to the vaccine (average titer = 1:6491 ± 5338) and 71.5% of these animals generated therapeutically relevant titers of anti-Aβ IgG antibody (Table 2). In prebleed sera (Tables 1 and 2), we detected only a background level of anti-Aβ antibodies.
Table 1.
Humoral immune responses in guinea pigs immunized with Lu AF20513 (20 μg/guinea pig) formulated in Alhydrogel
| Guinea pig # | Anti-Aβ IgG titers |
|
|---|---|---|
| Prebleed | After two immunizations | |
| 1 | <1:100 | 1:5260 |
| 2 | <1:100 | 1:8640 |
| 3 | <1:100 | 1:219 |
| 4 | <1:100 | <1:100 |
| 5 | <1:100 | 1:4748 |
| 6 | <1:100 | 1:3905 |
| 7 | <1:100 | 1:14032 |
| 8 | <1:100 | >1:21600 |
| 9 | <1:100 | >1:21600 |
| 10 | <1:100 | 1:6236 |
| 11 | <1:100 | >1:21600 |
| 12 | <1:100 | 1:1227 |
| 13 | <1:100 | 1:2751 |
| 14 | <1:100 | 1:9606 |
| 15 | <1:100 | 1:1759 |
| 16 | <1:100 | 1:3974 |
Table 2.
Humoral immune responses in cynomolgus monkeys immunized with Lu AF20513 (50 μg/monkey) formulated in Alhydrogel
| Monkey # | Anti-Aβ IgG titers |
|
|---|---|---|
| Prebleed | After two immunizations | |
| 1 | <1:100 | 1:1810 |
| 2 | <1:100 | 1:1135 |
| 3 | <1:100 | 1:7400 |
| 4 | <1:100 | 1:1857 |
| 5 | <1:100 | 1:6590 |
| 6 | <1:100 | 1:1775 |
| 7 | <1:100 | 1:3362 |
| 8 | <1:100 | 1:12085 |
| 9 | <1:100 | 1:19028 |
| 10 | <1:100 | 1:8233 |
| 11 | <1:100 | 1:6515 |
| 12 | <1:100 | 1:2766 |
| 13 | <1:100 | 1:13951 |
| 14 | <1:100 | 1:4368 |
Analysis of immune mechanism associated with Lu AF20513 vaccinations
One important aspect of the design of the Lu AF20513 is that immunization with this vaccine should quickly induce a robust anti-Aβ antibody production in elderly subjects with preexisting TT-reactive memory Th-cells. To test this hypothesis, we immunized two groups of B6SJL mice with either P30 peptide formulated in Quil-A or Quil-A only, and first analyzed the memory Th-cells specific to P30 after a 6-month of resting period (Fig. 7A). After in vitro restimulation of immune, but not control, splenocytes with P30, we detected high numbers of cells producing IFNγ (Fig. 7B). Depletion of almost 95% of CD4+ cells from primed splenocytes completely abrogated detection of IFNγ-producing cells (Fig. 7C). Moreover, after enrichment of splenocytes isolated from naive (nonimmunized) mice with CD4+ T-cells isolated from P30-primed splenocytes (∼75% nonimmune splenocytes mixed with ∼25% of P30 primed CD4+ T-cells), immune responses to P30 were restored (Fig. 7D). Therefore, these data reveal that after a 6-month resting period, mice had functional memory CD4+ Th-cells specific to the P30 epitope of TT vaccine.
We also tested whether a single booster injection of Lu AF20513 formulated in Quil-A adjuvant could activate preexisting anti-P30 specific memory Th-cells and lead to a quicker and stronger anti-TT cellular responses and anti-Aβ antibody responses. After a 6-month resting period, we boosted the P30-primed mice with Lu AF20513 formulated in Quil-A and analyzed both cellular and humoral immune responses (Fig. 8). Boosting of experimental mice with epitope vaccine induced strong Th-cell responses specific to P30: a very large number of cells producing IFNγ was detected in this group of mice with preexisting memory Th-cells compared with control mice (Fig. 8A). Most importantly, the single injection with Lu AF20513 formulated in the strong Th1 adjuvant Quil-A led to the induction of robust anti-Aβ antibody responses only in mice with preexisting memory Th-cells: concentrations of anti-Aβ antibodies were significantly higher (p ≤ 0.001) than that in control mice (Fig. 8B). Therefore, a single immunization with Lu AF20513 strongly activated preexisting memory CD4+ T-cells specific to the Th epitopes of this vaccine and rapidly led to the robust production of antibodies specific to the B-cell epitope (Aβ1–12) of the same vaccine.
Figure 8.

The generation of strong cellular and humoral immune responses to Lu AF20513 in mice with preexisting memory T-cells. A, Single boost of Lu AF20513 in experimental, but not control, animals leads to robust activation of these preexisting memory CD4+ Th-cells specific to the TT epitope P30 (Fig. 7C,D). The number of IFNγ-producing cells is significantly higher in splenocytes cultures obtained from experimental mice primed with P30 plus Quil-A (n = 8) than in control animals injected with adjuvant only (n = 4). B, Experimental animals (n = 12) possessing anti-P30 specific preexisting memory Th-cells, but not control animals injected with adjuvant (n = 11), rapidly and robustly responded to a single injection with Lu AF20513 by producing high titers of anti-Aβ antibody. Lines indicate averages. ***p ≤ 0.001.
Discussion
According to the amyloid cascade hypothesis (Hardy and Higgins, 1992), the accumulation of Aβ peptide is a primary pathological event in the development of AD, preceding tau accumulation and leading to neurodegeneration and dementia. The identification of the Aβ peptide as a target for therapeutic interventions for AD has led to many preclinical and clinical studies. Despite the fact that recent clinical data from Pfizer and Eli-Lilly with passively administrated anti-Aβ antibodies have been disappointing, there is a consensus in the field that the removal or lowering of Aβ in patients with very early AD pathology or even in presymptomatic subjects could be an effective measure. Obviously, for such preventive treatments, passive vaccination is not practical but a safe active vaccine might be beneficial. Although the first clinical trial with AN1792 was unsuccessful due to meningoencephalitis (possibly caused by the activation of autoreactive T-cells; Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005), various epitope-based vaccines similar to one that we suggested previously (Agadjanyan et al., 2005; Petrushina et al., 2007; Movsesyan et al., 2008b) are currently in clinical trials (e.g., UBITh, V950, CAD106, and ACC-001 on http://www.clinicaltrials.gov; Wang et al., 2007; Lemere and Masliah, 2010; Wiessner et al., 2011; Winblad et al., 2012). Designs of these vaccines aim to circumvent the problem of T-cell autoreactivity and overcome tolerance induction to self-antigen (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005). Published data are currently available only for a clinical trial with CAD106 composed of the bacteriophage Qβ-expressing Aβ1–6 B-cell epitope. Although the investigators reported that anti-Aβ antibodies were detected in 62% of low-dose and 82% of high-dose subjects, the exact titers of antibodies were not presented, so the actual magnitude of the humoral immune responses is not clear (Winblad et al., 2012). No data have been presented in this report on antigen-specific cellular immune responses generated after vaccinations of AD patients. Likewise, humoral and cellular immune responses to CAD106 have not been properly reported in preclinical mouse and monkey studies (Wiessner et al., 2011).
In the study, we tested a novel clinical grade epitope vaccine, Lu AF20513, which is composed of the same immunodominant B-cell epitope of Aβ42 (Aβ1–12) fused with two Th epitopes from a conventional TT vaccine that are recognized by different human MHC-class II molecules (James et al., 2007). We used an active immunization approach in Tg2576 mice with very early-stage AD-like pathology. In our previous studies, we tested several protein-, peptide-, and DNA-based AD epitope vaccines containing a synthetic Th-cell epitope, PADRE, and showed that they were immunogenic in mice and did not induce T-cell responses against self-Aβ molecules (Mamikonyan et al., 2007; Petrushina et al., 2007; Movsesyan et al., 2008a; Movsesyan et al., 2008b; Ghochikyan, 2009; Davtyan et al., 2010; Davtyan et al., 2012). In the design of the Lu AF20513 vaccine, we replaced PADRE with P2 and P30 epitopes (Fig. 1A) with the goal of activating non-self preexisting memory Th-cells in the general human population, who are immune to TT due to the public health vaccination program. Data from this study demonstrated that Lu AF20513 induced therapeutically potent Aβ-specific humoral immune responses in mice (Fig. 3) and strong anti-P2 and anti-P30 Th-cell responses without the activation of autoreactive Th-cells (Fig. 2). These anti-Aβ antibodies bound to oligomeric and fibrillar forms of Aβ (Fig. 4A,B) and pathological plaques in the brain sections from an AD patient (Fig. 4C) and also protected neuronal cells from Aβ42 oligomer- and fibril-mediated toxicity (Fig. 4D). The most striking observation from our study was that Lu AF20513 vaccination offered protection from the development of cored and diffuse plaques in the brains of Tg2576 mice (Fig. 5A–D).
One potential problem with immunotherapy is that a reduction of insoluble Aβ plaques may lead to increased levels of soluble forms of Aβ, which are toxic to neurons. Two major mechanisms for antibody-mediated clearance of Aβ have been suggested: sequestration of Aβ from the CNS into the periphery (“peripheral sink”; DeMattos et al., 2001) and entry of anti-Aβ antibodies into the CNS and clearance of antigen-antibody complexes by microglial cells (Bard et al., 2000). Regardless of the mechanism of action, we observed that anti-Aβ antibodies generated by Lu AF20513 vaccination inhibited the accumulation of not only insoluble, but also the soluble, forms of Aβ42 and Aβ40 peptides in the brains of experimental compared with control Tg2576 mice (Fig. 5E,F). Despite the significant reduction in plaque burden and soluble Aβ42, no differences in CAA were observed in the brains of vaccinated mice compared with control animals (Fig. 6C). An increased ratio of amyloid-containing blood vessels to the numbers of parenchymal plaques in vaccinated mice suggests that anti-Aβ antibodies were not as effective at inhibiting cerebral vascular deposition of Aβ as they were at blocking amyloid plaque formation. In addition, we have demonstrated that the inhibition of Aβ42 and Aβ40 depositions in the brains of Lu AF20513-vaccinated Tg2576 mice was associated with a reduction in the frequencies of activated microglia and astrocytes (Fig. 6A,B).
Because our data on glial activation was collected at the end of 11 months of active immunotherapy, we cannot exclude the possibility that glial cells were activated acutely after the active immunization protocol due to Fc-mediated microglial activation in response to antibody-Aβ immune complex formation in the CNS, because it was observed in old (19–20 months) APP Tg mice with substantial preexisting parenchymal and CAA in response to immunotherapy (Wilcock et al., 2001). However, because immunization was initiated before the onset of amyloid deposition (Tg2576 mice were 4–6 months of age), we believe that any acute inflammatory response would be rather mild under these experimental conditions. Moreover, we believe that early immunotherapy intervention may be critical to reducing the incidence of adverse cerebrovascular events such as microhemorrhages and vasogenic edema.
Data generated in Tg2576 mice vaccinated with Lu AF20513 suggest that this epitope vaccine may be safe for translation to human clinical studies. In this protective study, the behavioral changes in the cohort of vaccinated and control Tg2576 mice were not analyzed because there are many reports demonstrating that immunization with Aβ42 generates antibodies specific to the N terminus of Aβ (Janus et al., 2000; Lemere et al., 2000; Morgan et al., 2000) that improve cognitive functions in various APP mice, including Tg2576, especially if vaccination is started before the onset of amyloid-like pathology. In addition, results from previous active and passive vaccination clinical trials suggest that early intervention with immunotherapeutic approaches are likely to be the most clinically effective (Lemere and Masliah, 2010; Delrieu et al., 2012; Eli Lilly and Company Announcement, 2012; Johnson & Johnson Announcement, 2012).
We have also included analysis of anti-Aβ antibody titers in guinea pigs and monkeys, which demonstrated the immunogenic potential of the Lu AF20513 vaccine in animals that are naturally immune tolerant to Aβ42 and exhibit diverse genetic backgrounds for MHC class II molecules compared with mice. Two immunizations of guinea pigs were sufficient for the generation of anti-Aβ IgG antibody responses in 15 of 16 immunized animals, and 80% of responders generated anti-Aβ antibody titers > 1:2200 (Table 1). These represent therapeutically relevant titers, as was previously reported in the AN1792 trials (Gilman et al., 2005). Perhaps most importantly, Lu AF20513 vaccination induced robust humoral immune responses in 100% of cynomolgus monkeys, and in 71.5% of monkeys, the anti-Aβ antibody titers were therapeutically relevant (Table 2). These findings may translate into adequate clinical responses to vaccination in the elderly human population, in whom the generation of robust immunity is more difficult due to immunological senescence.
Although several different epitope vaccines are currently in clinical trials (Wang et al., 2007; Lemere and Masliah, 2010; Wiessner et al., 2011; Ghochikyan and Agadjanyan, 2012; and http://www.clinicaltrials.gov), the immune responses generated by these vaccines have not been fully characterized. To our knowledge, there are no published data on the exact specificity of Th-cells that could be activated by these vaccines either in animal models of AD or in humans. Because the antigen-specific T-cell responses were never defined, our challenge was to design an active vaccine against AD in humans with polymorphic MHC genes that overcomes the safety and efficacy barriers of previous approaches. In the present study, we have revealed the immunological mechanism of action of Lu AF20513 by demonstrating that this vaccine does indeed activate Th-cells specific to TT, leading to enhanced anti-Aβ antibody production. The most remarkable finding in this study is that a single injection with Lu AF20513 activated preexisting memory CD4+ T-cells specific to foreign Th epitopes and quickly induced strong anti-Aβ antibody responses (Fig. 8). Therefore, Lu AF20513 may represent an effective and safe form of active immunotherapy that may overcome the limited ability of the elderly to respond to vaccinations by activating preexisting anti-P30/P2 memory Th-cells (Fig. 9) in the general human population vaccinated with a conventional TT vaccine.
Figure 9.

Proposed model of early therapeutic or preventive Lu AF20513 vaccination. This is a model of vaccination of patients in the early stage of AD (early therapeutic) or asymptomatic subjects at risk of AD (preventive) possessing preexisting memory Th-cells specific to TT. Lu AF20513 vaccine should stimulate preexisting memory Th-cells generated in response to conventional TT vaccinations (the TT vaccine, given in 5 immunizations, is used for priming in childhood and is followed by a booster shot every 10 years).
Footnotes
This work was supported by funding from the NIH (Grants NS-50895, NS-065518, AG-20241, and NS 057395) and the Alzheimer's Association (Grant IIRG-0728314). H.D. and N.M. were supported by an NIA T32 training grant (AG000096). Additional support for AD case tissues was provided by University of California-Irvine Alzheimer's Disease Research Center (Grant P50 AG16573). We thank Jasja Wolthoorn (TNO Triskelion), Robert Pels Rijcken (TNO Triskelion), and Hervé Giorgi (Ricerca Biosciences) for their assistance in conducting the guinea pig and monkey studies.
A.K.L., P.J.M., and D.K.D. are employees of H. Lundbeck A/S. K.M.W. and L.O.P. are employees and shareholders of H. Lundbeck A/S.
References
- Agadjanyan MG, Ghochikyan A, Petrushina I, Vasilevko V, Movsesyan N, Mkrtichyan M, Saing T, Cribbs DH. Prototype Alzheimer's disease vaccine using the immunodominant B cell epitope from beta-amyloid and promiscuous T cell epitope pan HLA DR-binding peptide. J Immunol. 2005;174:1580–1586. doi: 10.4049/jimmunol.174.3.1580. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM, Wilkinson D, Holmes C, Nicoll JA. Consequence of Abeta immunization on the vasculature of human Alzheimer's disease brain. Brain. 2008;131:3299–3310. doi: 10.1093/brain/awn261. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Cribbs DH, Ghochikyan A, Tran M, Vasilevko V, Petrushina I, Sadzikava N, Babikyan D, Kesslak P, Kieber-Emmons T, Cotman CW, Agadjanyan MG. Adjuvant-dependent modulation of Th1 and Th2 responses to immunization with beta-amyloid. Int Immunol. 2003;15:505–514. doi: 10.1093/intimm/dxg049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davtyan H, Mkrtichyan M, Movsesyan N, Petrushina I, Mamikonyan G, Cribbs DH, Agadjanyan MG, Ghochikyan A. DNA prime-protein boost increased the titer, avidity and persistence of anti-Abeta antibodies in wild-type mice. Gene Ther. 2010;17:261–271. doi: 10.1038/gt.2009.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davtyan H, Ghochikyan A, Cadagan R, Zamarin D, Petrushina I, Movsesyan N, Martinez-Sobrido L, Albrecht RA, García-Sastre A, Agadjanyan MG. The immunological potency and therapeutic potential of a prototype dual vaccine against influenza and Alzheimer's disease. J Transl Med. 2011;9:127. doi: 10.1186/1479-5876-9-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davtyan H, Ghochikyan A, Movsesyan N, Ellefsen B, Petrushina I, Cribbs DH, Hannaman D, Evans CF, Agadjanyan MG. Delivery of a DNA Vaccine for Alzheimer's Disease by Electroporation versus Gene Gun Generates Potent and Similar Immune Responses. Neurodegener Dis. 2012;10:261–264. doi: 10.1159/000333359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, Chen-Dodson E, Kinney GG, Klein WL. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delrieu J, Ousset PJ, Caillaud C, Vellas B. ‘Clinical trials in Alzheimer's disease’: immunotherapy approaches. J Neurochem. 2012;120(Suppl 1):186–193. doi: 10.1111/j.1471-4159.2011.07458.x. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eli Lilly and Company Announces Top-Line Results on Solanezumab Phase 3 Clinical Trials in Patients with Alzheimer's Disease. 2012. Available at http://newsroom.lilly.com/releasedetail.cfm?releaseid=702211.
- Ferrer I, Boada Rovira M, Sánchez Guerra ML, Rey MJ, Costa-Jussá F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer's disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghochikyan A. Rationale for peptide and DNA based epitope vaccines for Alzheimer's disease immunotherapy. CNS Neurol Disord Drug Targets. 2009;8:128–143. doi: 10.2174/187152709787847298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghochikyan A, Agadjanyan MG. CAD-106, a beta-amyloid-based immunotherapeutic for Alzheimer's disease. 2012. Thompson Reuter https://partnering.thomson-pharma.com.
- Ghochikyan A, Vasilevko V, Petrushina I, Tran M, Sadzikava N, Babikyan D, Movsesyan N, Tian W, Ross TM, Head E, Cribbs DH, Agadjanyan MG. Generation and characterization of the humoral immune response to DNA immunization with a chimeric β-amyloid-interleukin-4 minigene. Eur J Immunol. 2003;33:3232–3241. doi: 10.1002/eji.200324000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghochikyan A, Mkrtichyan M, Petrushina I, Movsesyan N, Karapetyan A, Cribbs DH, Agadjanyan MG. Prototype Alzheimer's disease epitope vaccine induced strong Th2-type anti-Abeta antibody response with Alum to Quil A adjuvant switch. Vaccine. 2006;24:2275–2282. doi: 10.1016/j.vaccine.2005.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM AN1792(QS-21)-201 Study Team. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde TE, Dickson D, Hutton M. Filling the gaps in the abeta cascade hypothesis of Alzheimer's disease. Curr Alzheimer Res. 2006;3:421–430. doi: 10.2174/156720506779025189. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hardy J. Has the amyloid cascade hypothesis for Alzheimer's disease been proved? Curr Alzheimer Res. 2006;3:71–73. doi: 10.2174/156720506775697098. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harper JD, Wong SS, Lieber CM, Lansbury PT. Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Müller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Antibodies against beta-amyloid slow cognitive decline in Alzheimer's disease. Neuron. 2003;38:547–554. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunization in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM. Alzheimer's disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr71. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- James EA, Bui J, Berger D, Huston L, Roti M, Kwok WW. Tetramer-guided epitope mapping reveals broad, individualized repertoires of tetanus toxin-specific CD4+ T cells and suggests HLA-based differences in epitope recognition. Int Immunol. 2007;19:1291–1301. doi: 10.1093/intimm/dxm099. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Johnson and Johnson Announces Discontinuation Of Phase 3 Development of Bapineuzumab Intravenous (IV) In Mild-To-Moderate Alzheimer's Disease. 2012. Found at http://www.jnj.com/connect/news/all/johnson-and-johnson-announces-discontinuation-of-phase-3-development-of-bapineuzumab-intravenous-iv-in-mild-to-moderate-alzheimers-disease.
- Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Canto I, Breydo L, Rasool S, Lukacsovich T, Wu J, Albay R, 3rd, Pensalfini A, Yeung S, Head E, Marsh JL, Glabe C. Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Abeta oligomers. Mol Neurodegener. 2010;5:57. doi: 10.1186/1750-1326-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein WL, Stine WB, Jr, Teplow DB. Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol Aging. 2004;25:569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere CA. Developing novel immunogens for a safe and effective Alzheimer's disease vaccine. Prog Brain Res. 2009;175:83–93. doi: 10.1016/S0079-6123(09)17506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010;6:108–119. doi: 10.1038/nrneurol.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere CA, Maron R, Spooner ET, Grenfell TJ, Mori C, Desai R, Hancock WW, Weiner HL, Selkoe DJ. Nasal Aβ treatment induces anti-Aβ antibody production and decreases cerebral amyloid burden in PD-APP mice. Ann N Y Acad Sci. 2000;920:328–331. doi: 10.1111/j.1749-6632.2000.tb06943.x. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Mamikonyan G, Necula M, Mkrtichyan M, Ghochikyan A, Petrushina I, Movsesyan N, Mina E, Kiyatkin A, Glabe C, Cribbs DH, Agadjanyan MG. Anti-Abeta 1–11 antibody binds to different beta-amyloid species, inhibits fibril formation, and disaggregates preformed fibrils, but not the most toxic oligomers. J Biol Chem. 2007;282:22376–22386. doi: 10.1074/jbc.M700088200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Movsesyan N, Mkrtichyan M, Petrushina I, Ross TM, Cribbs DH, Agadjanyan MG, Ghochikyan A. DNA epitope vaccine containing complement component C3d enhances anti-amyloid-beta antibody production and polarizes the immune response towards a Th2 phenotype. J Neuroimmunol. 2008a;205:57–63. doi: 10.1016/j.jneuroim.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movsesyan N, Ghochikyan A, Mkrtichyan M, Petrushina I, Davtyan H, Olkhanud PB, Head E, Biragyn A, Cribbs DH, Agadjanyan MG. Reducing AD-like pathology in 3xTg-AD mouse model by DNA epitope vaccine-a novel immunotherapeutic strategy. PLos One. 2008b;3:e21–24. doi: 10.1371/journal.pone.0002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–1048. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC, Kuo YM, Lopez J, Brune D, Ferrer I, Masliah E, Newel AJ, Beach TG, Castaño EM, Roher AE. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer's disease patients: a biochemical analysis. Am J Pathol. 2006;169:1048–1063. doi: 10.2353/ajpath.2006.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrushina I, Tran M, Sadzikava N, Ghochikyan A, Vasilevko V, Agadjanyan MG, Cribbs DH. Importance of IgG2c isotype in the immune response to b-amyloid in APP/Tg mice. Neurosci Lett. 2003;338:5–8. doi: 10.1016/s0304-3940(02)01357-5. [DOI] [PubMed] [Google Scholar]
- Petrushina I, Ghochikyan A, Mktrichyan M, Mamikonyan G, Movsesyan N, Davtyan H, Patel A, Head E, Cribbs DH, Agadjanyan MG. Alzheimer's disease peptide epitope vaccine reduces insoluble but not soluble/oligomeric A{beta} species in amyloid precursor protein transgenic mice. J Neurosci. 2007;27:12721–12731. doi: 10.1523/JNEUROSCI.3201-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. In vitro aging of beta-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- Price DL, Sisodia SS. Cellular and molecular biology of Alzheimer's disease and animal models. Annu Rev Med. 1994;45:435–446. doi: 10.1146/annurev.med.45.1.435. [DOI] [PubMed] [Google Scholar]
- Rafii MS, Aisen PS. Recent developments in Alzheimer's disease therapeutics. BMC Med. 2009;7:7. doi: 10.1186/1741-7015-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. The molecular pathology of Alzheimer's disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: a central role for amyloid. J Neuropathol Exp Neurol. 1994;53:438–447. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM, Knudsen E, Asuni A, Fitzer-Attas C, Sage D, Quartermain D, Goni F, Frangione B, Wisniewski T. An attenuated immune response is sufficient to enhance cognition in an Alzheimer's disease mouse model immunized with amyloid-beta derivatives. J Neurosci. 2004;24:6277–6282. doi: 10.1523/JNEUROSCI.1344-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- Wang CY, Finstad CL, Walfield AM, Sia C, Sokoll KK, Chang TY, Fang XD, Hung CH, Hutter-Paier B, Windisch M. Site-specific UBITh amyloid-beta vaccine for immunotherapy of Alzheimer's disease. Vaccine. 2007;25:3041–3052. doi: 10.1016/j.vaccine.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Wiessner C, Wiederhold KH, Tissot AC, Frey P, Danner S, Jacobson LH, Jennings GT, Lüönd R, Ortmann R, Reichwald J, Zurini M, Mir A, Bachmann MF, Staufenbiel M. The second-generation active Abeta immunotherapy CAD106 reduces amyloid accumulation in APP transgenic mice while minimizing potential side effects. J Neurosci. 2011;31:9323–9331. doi: 10.1523/JNEUROSCI.0293-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Jantzen PT, Li Q, Morgan D, Gordon MN. Amyloid-b vaccination, but not nitro-NSAID treatment, increases vascular amyloid and microhemorrhage while both reduce parenchymal amyloid. Neuroscience. 2007;144:950–960. doi: 10.1016/j.neuroscience.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Gordon MN, Ugen KE, Gottschall PE, DiCarlo G, Dickey C, Boyett KW, Jantzen PT, Connor KE, Melachrino J, Hardy J, Morgan D. Number of Ab inoculations in APP+PS1 transgenic mice influences antibody titers, microglia activation, and congophilic plaque levels. DNA Cell Biol. 2001;20:731–736. doi: 10.1089/10445490152717596. [DOI] [PubMed] [Google Scholar]
- Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, Dumortier T, Maguire RP, Blennow K, Lundmark J, Staufenbiel M, Orgogozo JM, Graf A. Safety, tolerability, and antibody response of active Abeta immunotherapy with CAD106 in patients with Alzheimer's disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012;11:597–604. doi: 10.1016/S1474-4422(12)70140-0. [DOI] [PubMed] [Google Scholar]
- Yong W, Lomakin A, Kirkitadze MD, Teplow DB, Chen SH, Benedek GB. Structure determination of micelle-like intermediates in amyloid beta-protein fibril assembly by using small angle neutron scattering. Proc Natl Acad Sci U S A. 2002;99:150–154. doi: 10.1073/pnas.012584899. [DOI] [PMC free article] [PubMed] [Google Scholar]