

Abstract
p90 ribosomal protein S6 kinases (RSKs) integrate upstream signals through two catalytic domains. Autophosphorylation of Ser386 by the regulatory C-terminal kinase domain (CTD) is thought to be essential for activation of the N-terminal kinase domain (NTD), which phosphorylates multiple downstream targets1. We recently reported fmk, an irreversible inhibitor of the CTD of RSK1 and RSK22. Here we describe fmk-pa, a propargylamine variant that has improved cellular potency and a ‘clickable’ tag for assessing the extent and selectivity of covalent RSK modification. Copper-catalyzed conjugation of an azidoalkyl reporter (the click reaction) revealed that fmk-pa achieves selective and saturable modification of endogenous RSK1 and RSK2 in mammalian cells. Saturating concentrations of fmk-pa inhibited Ser386 phosphorylation and downstream signaling in response to phorbol ester stimulation, but had no effect on RSK activation by lipopolysaccharide. RSK autoactivation by the CTD is therefore context dependent, which suggests that NTD and CTD inhibitors should have distinct physiological effects.
RSKs are serine/threonine kinases that are activated by signaling inputs from extracellular-regulated kinase3,4 (ERK) and phosphoinositide-dependent kinase 1 (PDK1)5–7. ERK phosphorylates and activates the CTD, which can autophosphorylate Ser386 (human RSK2 amino acid numbering) in a hydrophobic motif, most likely via an intramolecular mechanism8–10. The phosphorylated hydrophobic motif serves as a docking site for PDK1 (ref. 5), which phosphorylates and activates the NTD (Fig. 1). The NTD phosphorylates all known RSK substrates8,9,11. CTD-mediated phosphorylation of the hydrophobic motif is thought to be essential for RSK function, as S386A (RSK2) and S381A (RSK1) mutants do not support NTD-mediated signaling6,9. However, overexpression of CTD kinase-inactive12 mutants or CTD-deletion5 mutants of RSK2 results in constitutive Ser386 phosphorylation (albeit with reduced NTD activity), which suggests that hydrophobic motif phosphorylation can occur in the absence of the CTD (Fig. 1). Whether a CTD-independent activation pathway exists for endogenous RSK remains unknown.
Figure 1.

Mechanism of RSK activation. fmk is a selective inhibitor of the RSK CTD, which is thought to mediate autophosphorylation of Ser386 in the RSK hydrophobic motif (HM).
Using a structural bioinformatics approach, we designed fmk (1) (Fig. 1), the first selective inhibitor of the CTD of RSK1 and RSK2 (ref. 2). fmk exploits two selectivity filters in the RSK ATP binding site: a cysteine, which is covalently modified by the fluoromethylketone electrophile, and a threonine gatekeeper, which accommodates the p-tolyl group. A biotinylated derivative of fmk selectively modified RSK1 and RSK2 in cell lysates, but this probe was inappropriate for targeting RSK in intact cells because of its reduced membrane permeability and because of the presence of endogenous biotinylated proteins. To extend the utility of fmk as a cellular probe of RSK CTD-mediated signaling, we sought a tagged membrane-permeable derivative. Such a probe would allow us to determine (i) the selectivity of covalent modification in intact cells and (ii) the relationship between CTD inactivation and hydrophobic motif phosphorylation in endogenous RSK.
We initially synthesized fmk-BODIPY (2), a derivative of fmk containing a membrane-permeable fluorescent tag (Fig. 2a). The primary hydroxyl group of fmk was chosen as the attachment site for the BODIPY tag because a homology model of fmk bound to the CTD of RSK2 suggested that this position would be solvent-exposed. fmk-BODIPY irreversibly modified recombinant RSK2 CTD, as shown by denaturing gel electrophoresis followed by in-gel fluorescence detection (Fig. 2b). Unfortunately, fmk-BODIPY was ~100-fold less potent than fmk at inhibiting RSK2 CTD kinase activity in vitro (Supplementary Table 1 online), and it had similarly reduced potency in cellular assays. Compared to fmk, which inhibited phorbol myristate acetate (PMA)-induced Ser386 phosphorylation with an effector concentration for half-maximum response (EC50) of ~150 nM (Supplementary Fig. 1 online), fmk-BODIPY was much less effective, with an EC50 of ~10 μM (Fig. 2c). Fluorescent bands corresponding to RSK were detected in lysates from cells treated with fmk-BODIPY, but saturable labeling was not achieved. Moreover, we detected extensive off-target modification at concentrations of fmk-BODIPY above 1 μM (Fig. 2d). We hypothesized that the large, hydrophobic BODIPY tag not only interfered with RSK binding but also promoted nonspecific protein modification. We therefore sought a less intrusive tag that could be conjugated to a fluorescent reporter after covalent modification of RSK.
Figure 2.

fmk-BODIPY irreversibly targets RSK but has only modest selectivity and potency in cells. (a) Chemical structure of fmk-BODIPY, a fluorescent fmk derivative. (b) Covalent labeling of RSK2 CTD by fmk-BODIPY. RSK2 CTD was treated with the indicated concentrations of unlabeled fmk for 1 h and then with 3 μM fmk-BODIPY for 1 h. Proteins were resolved by 10% SDS-PAGE and detected by in-gel fluorescence scanning, followed by western blot with an antibody to RSK2. (c) Effect of fmk-BODIPY on PMA-stimulated RSK Ser386 phosphorylation. HEK 293 cells were deprived of serum for 2 h and then treated with the indicated concentrations of fmk-BODIPY for 1 h. Cells were stimulated with PMA (0.1 μg ml−1) for 30 min and harvested in PBS. Proteins were resolved by 10% SDS-PAGE and detected by western blot with antibodies to phospho-Ser386 RSK (pS386) and total RSK2. (d) Selectivity of protein modification by fmk-BODIPY in intact cells. Proteins in cell lysates from c were resolved by 10% SDS-PAGE and detected by in-gel fluorescence scanning (left) and Coomassie blue staining (right). The asterisk marks the position of RSK1 and RSK2.
Bioorthogonal conjugation methods have been used to identify protein targets of irreversible inhibitors added to cell lysates13–15, intact cells14–18 and animals14,15,17. The click reaction, in which copper(I) catalyzes a [3+2] azide/alkyne cycloaddition to yield a stable triazole, is particularly effective. We therefore synthesized a clickable RSK inhibitor by replacing the primary hydroxyl of fmk with propargylamine to yield fmk-pa (3) (Fig. 3a). We treated recombinant RSK2 CTD with fmk-pa and then conjugated it to a tetramethylrhodamine-azide reporter (TAMRA-N3) using click chemistry (see Methods). Saturable labeling of RSK2 CTD was achieved by fmk-pa, as determined by in-gel fluorescence scanning (Fig. 3b). fmk-pa prevented labeling of RSK2 CTD by fmk-BODIPY (Fig. 3b) and inhibited CTD kinase activity in vitro with potency similar to that of fmk (Supplementary Table 1).
Figure 3.
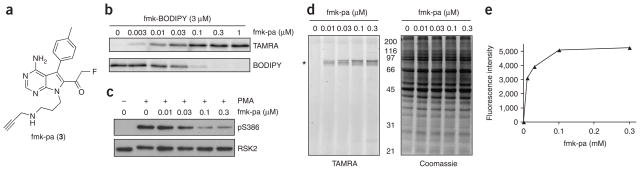
Irreversible, selective and potent inhibition of RSK by fmk-pa. (a) Chemical structure of fmk-pa, a clickable variant of fmk. (b) Click chemistry–mediated detection of RSK2 CTD. RSK2 CTD was treated with the indicated concentrations of fmk-pa for 1 h followed by 3 μM fmk-BODIPY for 1 h. After click conjugation with TAMRA-N3 (see Methods), proteins were resolved by 10% SDS-PAGE and detected by sequential in-gel fluorescence scanning using filters specific for TAMRA and BODIPY. (c) Effect of fmk-pa on PMA-stimulated Ser386 RSK phosphorylation. HEK 293 cells were deprived of serum for 2 h and then treated with the indicated concentrations of fmk-pa for 1 h. Cells were stimulated with PMA (0.1 μg ml−1) for 30 min and processed as described for Figure 2c. (d) Selectivity of protein modification by fmk-pa in intact cells. Cell lysates from c were subjected to click conjugation with TAMRA-N3. Proteins were resolved by 10% SDS-PAGE and detected by in-gel fluorescence scanning (left) and Coomassie blue staining (right). The asterisk marks the position of RSK1 and RSK2. (e) Quantification of fluorescent bands in d.
In contrast to fmk-BODIPY, fmk-pa was even more potent than fmk at inhibiting RSK Ser386 phosphorylation in intact cells (EC50 of ~30 nM, Fig. 3c). The five-fold increase in cellular potency of fmk-pa relative to fmk (EC50 of ~150 nM, Supplementary Fig. 1) may be due to higher cell permeability or potency toward full-length endogenous RSK, given that the inhibitors were equipotent against RSK2 CTD in vitro (Supplementary Table 1). We examined the extent and selectivity of covalent modification of RSK by fmk-pa by performing the click reaction in lysates prepared from the above-treated cells. Labeling of RSK1 and RSK2 by fmk-pa (up to 300 nM) was remarkably specific, as indicated by the prominent fluorescent bands at ~90 kDa (Fig. 3d). We confirmed the identities of these bands as RSK1 and RSK2 by immunoprecipitation with specific antibodies (Supplementary Fig. 2 online). Importantly, the extent of covalent modification correlated with inhibition of Ser386 phosphorylation. Maximum labeling and inhibition were achieved at 100 nM fmk-pa (Fig. 3c–e), although even saturating concentrations of fmk-pa failed to block Ser386 phosphorylation completely. In contrast to fmk-BODIPY, inhibition of RSK by fmk-pa in intact cells was unaffected by the presence of 10% serum in the incubation medium (Supplementary Fig. 3online). Together, these results demonstrate the advantage of click chemistry as an alternative labeling strategy for cases in which direct conjugation of a reporter tag to an irreversible inhibitor reduces potency and promotes nonspecific protein modification.
Having demonstrated selective and saturable modification of endogenous RSK by fmk-pa, we next tested whether RSK CTD activity is required for phosphorylation of ribosomal protein S6 (rpS6), which is involved in translation initiation and regulation of cell size19. Until recently, phosphorylation of rpS6 at Ser235 and Ser236 was thought to be mediated exclusively by p70 S6 kinases 1 and 2 (S6K1 and S6K2), the activation of which requires another protein kinase, the mammalian target of rapamycin (mTOR)20. However, a recent study showed that in cells derived from S6K1/S6K2 double knockout mice, rpS6 phosphorylation is still observed and is sensitive to pharmacological inhibition of ERK21. These results led to the proposal that p90 RSK might promote phosphorylation of rpS6 under certain conditions21.
To test this hypothesis, we examined the effects of rapamycin and fmk-pa on rpS6 phosphorylation in human embryonic kidney (HEK) 293 cells. Increasing concentrations of rapamycin, which prevents S6K phosphorylation by mTOR, inhibited PMA-stimulated rpS6 phosphorylation at Ser235 and Ser236 to a maximum extent of 76 ± 8% (mean ± s.e.m.) (Fig. 4a,b). By contrast, fmk-pa (300 nM) inhibited rpS6 phosphorylation by 28 ± 6% (Fig. 4b), without affecting S6K phosphorylation (Supplementary Fig. 4 online). Combining fmk-pa and rapamycin produced additive effects on rpS6 phosphorylation (Fig. 4a,b); phosphorylation was abolished by 300 nM fmk-pa and 30 nM rapamycin. Similar results were obtained in MDA-MB-231 breast cancer cells growing in serum (Supplementary Fig. 5 online). It is possible that fmk-pa binds reversibly to off-target kinases that mediate rpS6 phosphorylation. To test this, we pretreated cells with fmk-pa (300 nM) for 1 h and then washed the cells with compound-free medium and incubated them with rapamycin (30 nM) for 1 h in the absence of fmk-pa. Using this washout protocol, RSK and rpS6 phosphorylation were blocked to the same extent as when cells were treated continuously with fmk-pa and rapamycin (Fig. 4c). Together, these results demonstrate that the RSK CTD contributes to rpS6 phosphorylation, most likely by activating the NTD, which phosphorylates rpS6 at Ser235 and Ser236 in vitro8. RSK NTDs are highly homologous to S6K1 and S6K2; they also phosphorylate a similar consensus motif22 and may share multiple substrates. Consistent with this idea, recent studies have shown that RSK mediates phosphorylation of two additional S6K substrates: eEF2K23 and eIF4B24.
Figure 4.
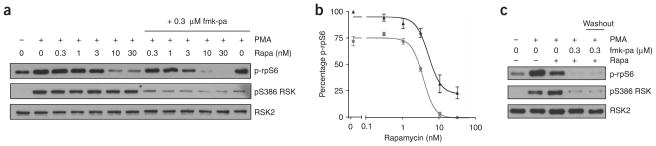
fmk-pa reveals a RSK CTD–dependent pathway for rpS6 phosphorylation. (a) Effect of rapamycin and fmk-pa on rpS6 phosphorylation. Serum-starved HEK 293 cells were treated with the indicated concentrations of rapamycin in the absence (left lanes) or presence (right lanes) of 300 nM fmk-pa for 1 h and stimulated with PMA (0.1 μg ml−1) for 30 min. Proteins were resolved by 10% SDS-PAGE and detected by western blot with antibodies to phospho-Ser235/236 rpS6 (p-rpS6), phospho-Ser386 RSK and total RSK2. (b) Dose-response curves showing additive inhibition of rpS6 phosphorylation: rapamycin alone, black triangles; rapamycin plus 300 nM fmk-pa, gray circles. Percentage rpS6 Ser235/236 phosphorylation was determined by quantification of western blots relative to DMSO controls using ImageJ (NIH). Data represent mean ± s.e.m. from three independent experiments. (c) Irreversible inhibition of rpS6 and RSK phosphorylation by fmk-pa. Serum-starved HEK 293 cells were treated with either fmk-pa (300 nM) or DMSO for 1 h. In the washout sample, fmk-pa was removed and cells were washed with compound-free medium and incubated with rapamycin (30 nM) for 1 h. Cells were stimulated with PMA (0.1 μg ml−1) for 30 min and processed as described for Figure 4a.
Saturating concentrations of fmk-pa (as revealed by click chemistry) failed to inhibit Ser386 phosphorylation completely (Figs. 3c and 4a), raising the possibility that Ser386 phosphorylation occurs in a CTD-independent manner in certain cellular contexts. To test this possibility more rigorously, we compared the inhibitory effects of fmk-pa on Ser386 phosphorylation induced by two different stimuli in the same cell type. In addition, we used click chemistry to approximate the extent of RSK modification with varying concentrations of fmk-pa. In primary bone marrow–derived macrophages, PMA-stimulated Ser386 phosphorylation was maximally inhibited by 67 ± 10% in the presence of 300 nM fmk-pa (Fig. 5a,b); by contrast, Ser386 phosphorylation induced by lipopolysaccharide (LPS) was unaffected by up to 1 μM fmk-pa (Fig. 5a,b), despite saturation of RSK labeling at this concentration (Fig. 5c). Click conjugation of a biotin-azide reporter in lysates derived from LPS-stimulated cells was followed by affinity purification of biotinylated proteins with avidin-agarose beads (see Methods). Western blot analysis of the avidin-agarose eluates revealed biotinylated RSK1 and RSK2, phosphorylated at Ser386 (Fig. 5d). Taken together, our results provide strong evidence for the existence of an alternative kinase (or kinases) that mediates phosphorylation of the RSK hydrophobic motif (Ser386) in the absence of a functional CTD. In primary macrophages, use of the alternative kinase is context dependent, occurring maximally in response to LPS and, to a lesser extent, in response to PMA.
Figure 5.
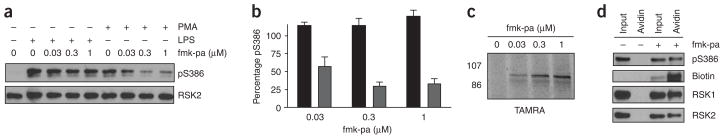
fmk-pa and click chemistry reveal a CTD-independent pathway for RSK Ser386 phosphorylation. (a) Effect of fmk-pa on Ser386 phosphorylation in primary bone marrow–derived macrophages stimulated with LPS or PMA. Macrophages were cultured in 0.5% serum for 20 h and then treated with the indicated concentrations of fmk-pa for 1 h. Cells were stimulated with either PMA (0.1 μg ml−1) or LPS (1 μg ml−1) for 30 min and harvested in PBS. Proteins were resolved by 10% SDS-PAGE and detected by western blot with antibodies to phospho-Ser386 RSK and total RSK2. (b) Percentage Ser386 phosphorylation was determined by quantification of western blots relative to DMSO controls (PMA-treated, gray bars; LPS-treated, black bars). Data represent mean ± s.e.m. from three independent experiments. (c) Saturable RSK modification by pretreatment of intact macrophages with fmk-pa. Lysates from LPS-stimulated macrophages (pretreated for 1 h with the indicated concentrations of fmk-pa) were incubated with TAMRA-N3 under the click reaction conditions. Proteins were resolved by 10% SDS-PAGE and detected by in-gel fluorescence scanning. (d) Use of click chemistry to demonstrate that fmk-pa–modified RSK1 and RSK2 are phosphorylated at Ser386 in response to LPS stimulation. Lysates from LPS-stimulated cells (pretreated with 1 μM fmk-pa or DMSO control) were incubated with biotin-N3 under the click reaction conditions for 1 h. Biotinylated proteins were affinity purified with avidin-agarose beads. Eluted proteins were resolved by 10% SDS-PAGE and detected by western blot with streptavidin-HRP or antibodies to RSK1, RSK2 and phospho-Ser386 RSK.
fmk-pa selectively and irreversibly modifies endogenous RSK in intact mammalian cells and can be conjugated to a fluorescent or biotin tag via the click reaction. These properties allowed us to monitor RSK CTD modification by fmk-pa and (in parallel) its effects on Ser386 phosphorylation and downstream signaling. In certain cellular contexts (for example, LPS-stimulated macrophages, Fig. 5) but not others (for example, MDA-MB-231 breast cancer cells growing in serum, Supplementary Fig. 5), an unidentified kinase bypassed the CTD requirement and phosphorylated RSK at Ser386, which is essential for activation of the NTD5,6,9. Thus, CTD-targeted inhibitors, in contrast to NTD-targeted inhibitors25,26, should block a subset of RSK-mediated signaling events in vivo, depending on the cellular context. Recent studies of transgenic mice with cardiac-specific over-expression of active or dominant-negative RSK suggest that RSK hyperactivation may play a role in heart disease27,28. Clickable inhibitors such as fmk-pa should facilitate determination of the specific roles played by the RSK CTD in cellular and animal models relevant to heart failure and other human diseases.
METHODS
Synthesis of fmk, fmk-BODIPY and fmk-pa
Experimental procedures are provided in Supplementary Methods online.
Proteins, reagents and antibodies
RSK2 CTD with a hexahistidine tag was expressed in Escherichia coli and purified by immobilized nickel affinity chromatography as previously described2. PMA and LPS were purchased from Sigma. Rapamycin was purchased from Cell Signaling Technology. Tris-(benzyltriazolylmethyl)amine (TBTA), TAMRA-N3 and biotin-N3 were synthesized as described15,27. Primary antibodies were purchased from Santa Cruz Biotechnology (RSK1 and RSK2) and Cell Signaling Technology (phospho-Ser386 RSK, phospho-Ser235/236 rpS6 and phospho-Thr389 S6K).
Cell culture
HEK 293 and MDA-MB-231 cells were cultured in DMEM supplemented with 10% FBS. Primary mouse bone marrow–derived macrophages were maintained in DMEM supplemented with 20 mM HEPES (pH 7.4), 10% FBS and 10% CMG14-12 cell supernatant (source of macrophage-colony-stimulating factor)28.
Western blot
Cells were washed with cold (4 °C) phosphate-buffered saline (PBS) and lysed by freezing and thawing in either PBS or hypertonic lysis buffer (20 mM HEPES (pH 7.9), 450 mM NaCl, 25% glycerol, 3 mM MgCl2 and 0.5 mM EDTA), both of which contained protease (Roche) and phosphatase (Sigma) inhibitor cocktails. Clarified lysates were normalized for protein content with the Bradford assay (Bio-Rad). Proteins were resolved by 10% SDS-PAGE, transferred to a nitrocellulose membrane, blocked with 5% (w/v) nonfat milk, probed with primary and secondary antibodies, and detected by enhanced chemiluminescence.
Labeling of RSK2 CTD with fmk-BODIPY
RSK2 CTD (30 nM) in PBS (pH 7.4) was treated with increasing concentrations of fmk or fmk-pa for 1 h and then with fmk-BODIPY (3 μM) for 1 h. Proteins were resolved by 10% SDS-PAGE and detected by in-gel fluorescence scanning with a Typhoon 9400 flatbed laser-induced scanner (Amersham Biosciences).
Click chemistry
Click reactions were performed as previously described15. Briefly, RSK2 CTD (30 nM) pretreated with the indicated concentrations of fmk-pa, or lysates from fmk-pa–pretreated cells, were incubated with TAMRA-N3 or biotin-N3 (50 μM), tris(carboxyethyl)phosphine (TCEP) (1 mM), CuSO4 (1 mM), and TBTA (100 μM) in PBS (pH 7.4) containing 1% SDS for 1 h at room temperature 20–25 °C. Proteins were resolved by 10% SDS-PAGE and detected by in-gel fluorescence scanning (TAMRA-N3) or were affinity purified with avidin-agarose and detected by western blot (biotin-N3).
PMA stimulation of HEK 293 cells
Confluent HEK 293 cells in 12-well plates were deprived of serum for 2 h and then treated with the indicated concentrations of inhibitors for 1 h at 37 °C. After stimulation with PMA (0.1 μg ml−1) for 30 min, cells were washed with cold PBS and harvested in either PBS (Figs. 2c and 3c) or hypertonic lysis buffer (Fig. 4). Click chemistry was performed with TAMRA-N3 in lysates from cells treated with fmk-pa. Proteins were analyzed by in-gel fluorescence scanning or western blot.
LPS and PMA stimulation of primary mouse macrophages
Confluent macrophages in 12-well plates were incubated in DMEM supplemented with 20 mM HEPES (pH 7.4) and 0.5% FBS for 12 h before treatment with the indicated concentrations of fmk-pa for 1 h at 37 °C. After stimulation with either PMA (0.1 μg ml−1) or LPS (1 μg ml−1) for 30 min, cells were washed with cold PBS and harvested in PBS lysis buffer. Click chemistry was performed with TAMRA-N3 and proteins were then analyzed by in-gel fluorescence scanning or western blot.
Avidin pulldown from LPS-stimulated primary macrophages
Confluent primary macrophages in 15-cm plates were incubated in DMEM supplemented with 20 mM HEPES (pH 7.4) and 0.5% FBS for 12 h before treatment with either DMSO or 1 μM fmk-pa for 1 h at 37 °C. Lysates prepared in PBS (200 μl, precleared with streptavidin-agarose beads) were subjected to click chemistry to conjugate biotin-N3 to fmk-pa–modified RSK. After the click reaction, proteins were precipitated using 15% (v/v) trichloroacetic acid and pelleted by centrifugation (13,000 r.p.m., 5 min, 4 °C). The supernatant was removed and the protein pellet was washed twice with 0 °C acetone (2 × 200 μl). The resulting acetone pellet was solubilized in PBS (40 μl) containing 2% (w/v) SDS by heating and sonication. Samples were diluted ten-fold in PBS containing 0.5% (v/v) Nonidet P-40 (Aldrich). Avidin-agarose (30 μl of 50% slurry in PBS) was added and samples were rotated overnight at 4 °C. The supernatant was removed and the beads were washed with PBS containing 0.5% Nonidet P-40 and 0.1% SDS (2 × 1 ml). Affinity-purified proteins were eluted from the beads with 2× Laemmli sample buffer containing 1 mM biotin (100 μl), resolved by 10% SDS-PAGE, and detected by western blot.
Supplementary Material
Acknowledgments
This work was supported by the US National Institutes of Health (GM71434) and a University of California at San Francisco President’s Dissertation-Year Fellowship to M.S.C. We thank M. Pak and E. Brown (University of California at San Francisco) for the primary mouse macrophages. We thank H. Luecke, M. Simon and members of the Taunton laboratory for many helpful discussions and for critical reading of the manuscript.
Footnotes
AUTHOR CONTRIBUTIONS
J.T. and M.S.C. designed the experiments; M.S.C. and H.H. executed the experiments; J.T. and M.S.C. analyzed the data and wrote the manuscript.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
Note: Supplementary information and chemical compound information is available on the Nature Chemical Biology website.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Hauge C, Frodin M. RSK and MSK in MAP kinase signalling. J Cell Sci. 2006;119:3021–3023. doi: 10.1242/jcs.02950. [DOI] [PubMed] [Google Scholar]
- 2.Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith JA, Poteet-Smith CE, Malarkey K, Sturgill TW. Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J Biol Chem. 1999;274:2893–2898. doi: 10.1074/jbc.274.5.2893. [DOI] [PubMed] [Google Scholar]
- 4.Gavin AC, Nebreda ARA. MAP kinase docking site is required for phosphorylation and activation of p90(rsk)/MAPKAP kinase-1. Curr Biol. 1999;9:281–284. doi: 10.1016/s0960-9822(99)80120-1. [DOI] [PubMed] [Google Scholar]
- 5.Frodin M, Jensen CJ, Merienne K, Gammeltoft S. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J. 2000;19:2924–2934. doi: 10.1093/emboj/19.12.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jensen CJ, et al. 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3-phosphoinositide-dependent protein kinase-1. J Biol Chem. 1999;274:27168–27176. doi: 10.1074/jbc.274.38.27168. [DOI] [PubMed] [Google Scholar]
- 7.Richards SA, Fu J, Romanelli A, Shimamura A, Blenis J. Ribosomal S6 kinase 1 (RSK1) activation requires signals dependent on and independent of the MAP kinase ERK. Curr Biol. 1999;9:810–820. doi: 10.1016/s0960-9822(99)80364-9. [DOI] [PubMed] [Google Scholar]
- 8.Fisher TL, Blenis J. Evidence for two catalytically active kinase domains in pp90rsk. Mol Cell Biol. 1996;16:1212–1219. doi: 10.1128/mcb.16.3.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dalby KN, Morrice N, Caudwell FB, Avruch J, Cohen P. Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J Biol Chem. 1998;273:1496–1505. doi: 10.1074/jbc.273.3.1496. [DOI] [PubMed] [Google Scholar]
- 10.Vik TA, Ryder JW. Identification of serine 380 as the major site of auto-phosphorylation of Xenopus pp90rsk. Biochem Biophys Res Commun. 1997;235:398–402. doi: 10.1006/bbrc.1997.6794. [DOI] [PubMed] [Google Scholar]
- 11.Bjorbaek C, Zhao Y, Moller DE. Divergent functional roles for p90rsk kinase domains. J Biol Chem. 1995;270:18848–18852. doi: 10.1074/jbc.270.32.18848. [DOI] [PubMed] [Google Scholar]
- 12.Chrestensen CA, Sturgill TW. Characterization of the p90 ribosomal S6 kinase 2 carboxyl-terminal domain as a protein kinase. J Biol Chem. 2002;277:27733–27741. doi: 10.1074/jbc.M202663200. [DOI] [PubMed] [Google Scholar]
- 13.Vocadlo DJ, Bertozzi CR. A strategy for functional proteomic analysis of glycosidase activity from cell lysates. Angew Chem Int Edn Engl. 2004;43:5338–5342. doi: 10.1002/anie.200454235. [DOI] [PubMed] [Google Scholar]
- 14.Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 15.Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chem Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 16.Ovaa H, et al. Chemistry in living cells: detection of active proteasomes by a two-step labeling strategy. Angew Chem Int Edn Engl. 2003;42:3626–3629. doi: 10.1002/anie.200351314. [DOI] [PubMed] [Google Scholar]
- 17.Alexander JP, Cravatt BF. Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem Biol. 2005;12:1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans MJ, Saghatelian A, Sorensen EJ, Cravatt BF. Target discovery in small-molecule cell-based screens by in situ proteome reactivity profiling. Nat Biotechnol. 2005;23:1303–1307. doi: 10.1038/nbt1149. [DOI] [PubMed] [Google Scholar]
- 19.Ruvinsky I, et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005;19:2199–2211. doi: 10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 21.Pende M, et al. S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–3124. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leighton IA, Dalby KN, Caudwell FB, Cohen PT, Cohen P. Comparison of the specificities of p70 S6 kinase and MAPKAP kinase-1 identifies a relatively specific substrate for p70 S6 kinase: the N-terminal kinase domain of MAPKAP kinase-1 is essential for peptide phosphorylation. FEBS Lett. 1995;375:289–293. doi: 10.1016/0014-5793(95)01170-j. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, et al. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001;20:4370–4379. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shahbazian D, et al. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006;25:2781–2791. doi: 10.1038/sj.emboj.7601166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sapkota GP, et al. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem J. 2007;401:29–38. doi: 10.1042/BJ20061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith JA, et al. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 2005;65:1027–1034. [PubMed] [Google Scholar]
- 27.Itoh S, et al. Role of p90 ribosomal S6 kinase-mediated prorenin-converting enzyme in ischemic and diabetic myocardium. Circulation. 2006;113:1787–1798. doi: 10.1161/CIRCULATIONAHA.105.578278. [DOI] [PubMed] [Google Scholar]
- 28.Maekawa N, et al. Inhibiting p90 ribosomal S6 kinase prevents (Na+)-H+ exchanger-mediated cardiac ischemia-reperfusion injury. Circulation. 2006;113:2516–2523. doi: 10.1161/CIRCULATIONAHA.105.563486. [DOI] [PubMed] [Google Scholar]
- 29.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 30.Stamm LM, et al. Mycobacterium marinum escapes from phagosomes and is propelled by actin-based motility. J Exp Med. 2003;198:1361–1368. doi: 10.1084/jem.20031072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.