

Abstract
The present review aims to systematically and critically analyze the current knowledge on phospholipases and their role in physiological and pathological mineralization undertaken by mineralization competent cells. Cellular lipid metabolism plays an important role in biological mineralization. The physiological mechanisms of mineralization are likely to take place in tissues other than in bones and teeth under specific pathological conditions. For instance, vascular calcification in arteries of patients with renal failure, diabetes mellitus or atherosclerosis recapitulates the mechanisms of bone formation. Osteoporosis—a bone resorbing disease—and rheumatoid arthritis originating from the inflammation in the synovium are also affected by cellular lipid metabolism. The focus is on the lipid metabolism due to the effects of dietary lipids on bone health. These and other phenomena indicate that phospholipases may participate in bone remodelling as evidenced by their expression in smooth muscle cells, in bone forming osteoblasts, chondrocytes and in bone resorbing osteoclasts. Among various enzymes involved, phospholipases A1 or A2, phospholipase C, phospholipase D, autotaxin and sphingomyelinase are engaged in membrane lipid remodelling during early stages of mineralization and cell maturation in mineralization-competent cells. Numerous experimental evidences suggested that phospholipases exert their action at various stages of mineralization by affecting intracellular signaling and cell differentiation. The lipid metabolites—such as arachidonic acid, lysophospholipids, and sphingosine-1-phosphate are involved in cell signaling and inflammation reactions. Phospholipases are also important members of the cellular machinery engaged in matrix vesicle (MV) biogenesis and exocytosis. They may favour mineral formation inside MVs, may catalyse MV membrane breakdown necessary for the release of mineral deposits into extracellular matrix (ECM), or participate in hydrolysis of ECM. The biological functions of phospholipases are discussed from the perspective of animal and cellular knockout models, as well as disease implications, development of potent inhibitors and therapeutic interventions.
Keywords: bone, cartilage, osteoarthritis, osteoporosis, phospholipases, rheumatoid arthritis, sphingomyelinase, osteoblasts, osteoclasts, chondrocytes, Smooth muscle cells, matrix vesicle, mineralization, vascular calcification
Contents
-
1. Introduction 5040
1.1. Bone Biology and Physiological Mineralization 5040
1.2. Ectopic Calcifications and Defective Mineralizations 5042
1.3. Matrix Vesicles and Early Stages of Mineralization 5043
1.4. Dietary Lipids and Bone Health 5044
1.5. Groups of Phospholipases and Possible Roles during Mineralization 5044
-
2. Phospholipases A1 5047
2.1. Groups, Subgroups and Specificity 5047
-
3. Phospholipases A2 5048
3.1. Groups, Subgroups and Specificity 5048
3.2. Presence of PLA2s in Chondrocytes and Possible Roles 5049
3.3 Presence of PLA2s in Osteoblasts and Possible Roles 5050
3.4. Presence of PLA2s in osteoclasts and Possible Roles 5051
3.5. Presence of PLA2s in Smooth Muscle Cells and Possible Roles 5052
3.6. The Expressions of PLA2s under Pathological Conditions 5052
3.7. Transgenic Knockout Animal for PLA2 Enzymes as Models for Bone Formation and Mineralization Diseases 5053
3.8. Inhibitors of PLA2 as Drug Therapy 5053
-
3.9. Effects Mediated by Arachidonic Acid and Its Pathways at Cellular Level 5054
3.9.1. Effects Mediated by PGE2 5055
3.9.2. Effects Mediated by PGF2α and PGD2 5056
3.10. Effects Mediated by Lysophospholipids and Their Pathways at Cellular Level 5057
3.11. The Effects of PLA Metabolites at Matrix Vesicle Level 5060
-
4. Non-Specific Phospholipase C 5060
4.1. Groups, Subgroups and Specificity 5060
4.2. Presence of PC-PLC in Chondrocytes and in Osteoblasts and Its Possible Role 5061
4.3. Presence of PC-PLC in Osteoclasts and Possible Roles 5061
4.4. Presence of PC-PLC in Smooth Muscle Cells and Possible Roles 5061
4.5. The Effect of PLC Metabolites in Matrix Vesicles 5061
-
5. PI-Specific Phospholipase C 5062
5.1. Groups, Subgroups and Specificity 5062
5.2. PI-PLC in Tissues 5063
5.3. Presence of PI-PLC in Chondrocytes and Possible Roles. 5064
-
5.4. Presence of PI-PLC in Osteoblasts 5065
5.4.1. Endothelin-1 Induced Signaling Pathway 5065
5.4.2. Basic FGF Induced Signaling Pathway 5066
5.4.3. Platelet-Derived Growth Factor Induced Signaling Pathway 5066
5.4.4. Parathyroid Hormone Induced Signaling Pathway 5066
5.4.5. PGD2 Induced-Signaling Pathway 5066
5.4.6. PGE2 Induced-Signaling Pathway 5067
5.4.7. PGF2 Induced-Signaling Pathway 5067
5.4.8. Vitamin D-Induced Signaling Pathway 5067
5.4.9. Interleukin-1-Induced Signaling Pathway 5067
5.4.10. Miscelanous Ligand Binding Stimulated PI-PLC in Osteoblasts 5068
5.4.11. Purinergic and Serotonin-2 B Receptors 5068
-
5.5. Presence of PI-PLC in Osteoclasts 5068
5.5.1. Calcitonin Induced Signaling Pathway 5069
5.5.2. Intracellular Ca2+ Induced Signaling Pathway 5069
5.5.3. Osteoprotegrin Induced Signaling Pathway 5070
5.5.4. RANK Induced Signaling Pathways 5071
5.5.5. Parathyroid Hormone Induced Signaling Pathway 5071
5.6. Presence of PI-PLC in Smooth Muscle Cells and Possible Roles 5072
5.7. Presence of PI-PLC in Odontoblasts and Possible Roles 5072
5.8. Genetic Models 5073
6. PLC-Related but Catalytically Inactive Protein 5073
-
7. Sphingomyelinase 5074
7.1. Groups, Subgroups and Specificity 5074
7.2. Presence of Sphingomyelinase in Chondrocytes and Possible Roles 5074
7.3. Presence of Sphingomyelinase in Osteoblasts and Possible Roles 5074
7.4. Presence of Sphyngomyelinase in Osteoclasts and Possible Roles 5075
7.5. Genetic Models 5075
7.6. Effects of Sphyngomyelinase Metabolites at Matrix Vesicle Level 5075
-
8. Phospholipase D 5076
8.1. Groups, Subgroups and Specificity 5076
8.2. Presence of PLD in Chondrocytes and Possible Roles 5077
8.3. Presence of PLD in Osteoblasts and Possible Roles 5079
8.4. Presence of PLD in Osteoclasts and Possible Roles 5080
8.5. Genetic Models 5080
8.6. Effects of PLD Metabolite at Matrix Vesicle Level 5080
-
9. Non-HKD Enzymes—GPI-PLD 5081
9.1. Groups, Subgroups and Specificity 5081
9.2. Presence of GPI-PLD in Chondrocytes and Possible Roles 5081
9.3. Presence of GPI-PLD in Osteoblasts and Possible Roles 5082
-
10. Non-HKD Enzymes—Autotaxin 5082
10.1. Groups, Subgroups and Specificity 5082
10.2. Presence of ATX in Chondrocytes and Possible Roles. 5083
10.3. Presence of ATX in Osteoblasts and Possible Roles 5083
10.4. Presence of ATX in Osteoclasts and Possible Roles. 5084
10.5. Presence of ATX in Smooth Muscle Cells and Possible Roles 5084
11. Concluding Remarks 5084
Acknowledgments 5084
References 5084
Abbreviations
- 1α,25-(OH)2D3
1α,25-dihydroxyvitamin D3
- 24R,25(OH)2D3
24R,25-dihydroxyvitamin D3
- AA
arachidonic acid
- ATX
autotaxin
- BM
bone marrow
- Ca2+e
extracellular Ca2+
- Ca2+i
intracellular Ca2+
- CaR
calcium-sensing receptor
- CIA
collagen-induced arthritis
- cPLA2
cytosolic Ca2+-dependent PLA2
- COX
cyclooxygenase
- DAG
diacylglycerol
- DHT
5α-dihydrotestosterone
- ECM
extracellular matrix
- ERK
extracellular signal-regulated kinase
- ET
endothelin
- FGF
fibroblast growth factor
- GPCR
G-protein-coupled receptor
- GPI-PLD
glycosyl-PI specific PLD
- HA
hydroxyapatite
- IL
interleukin
- IP3
inositol 1,4,5-trisphosphate
- iPLA2
Ca2+-independent PLA2
- LOX
lipooxygenase
- LPA
lysophosphatidic acid
- LPC
lysophosphatidylcholine
- LPE
lysophosphatidylethanolamine
- LPG
lysophosphatidylglycerol
- LPI
lysophosphatidylinositol
- LPL
lysophospholipid
- LPS
lysophosphatidylserine
- LRRc17
leucine-rich repeat-containing 17
- MAP
mitogen activated protein
- MPP
metalloproteinase
- MV
matrix vesicle
- NF-κB
nuclear factor κB
- NFAT
nuclear factor of activated T cell
- NPP
ectonucleotide pyrophosphatase phosphodiesterase
- NSAID
non-steroidal anti-inflammatory drug
- OA
osteoarthritis
- OPG
osteoprotegerin
- PA
phosphatidic acid
- PAF
platelet-activating factor
- PAF-AH
PAF-acetylhydrolase
- PBMC
peripheral blood mononuclear cell
- PC
phosphatidylcholine
- PChol
phosphocholine
- PE
phosphatidylethanolamine
- PEA
phosphoethanolamine
- PGD2
prostaglandin D2
- PGE1
prostaglandin E1
- PGE2
prostaglandin E2
- PGF2
prostaglandin F2
- PH
pleckstrin homology
- PHOSPHO1
phosphatase orphan 1
- PI
phosphatidylinositol
- PI-PLC
PI-specific
- PIP2
PI 4,5-bisphosphate
- PIP3
PI 3,4,5-trisphosphate
- PKC
protein kinase C
- PLA1
phospholipase A1
- PLA2
phospholipase A2
- PLC
phospholipase C
- PLD
phospholipase D
- Pi
inorganic phosphate
- PPi
inorganic pyrophosphate
- PRIP
PLC-related but catalytically inactive protein
- PS
phosphatidylserine
- PS-PLA1
PS-specific PLA1
- PTH
parathyroid hormone
- PTX
pertussis toxin
- PUFA
polyunsaturated fatty acid
- RA
rheumatoid arthritis
- RANKL
receptor activator of nuclear factor κB ligand
- Runx2
runt-related transcription factor 2
- SH
src homology
- SM
sphingomyelin
- SMase
sphingomyelinase
- SMPD3
sphingomyeline phosphodiesterase-3
- sPLA2
secreted PLA2
- S1P
sphingosine-1-phosphate
- STAT
signal transducer and activator of transcription
- TNAP
tissue-non specific alkaline phosphatase
- TNF
tumor necrosis factor
- VSMC
vascular smooth muscle cell
1. Introduction
1.1. Bone Biology and Physiological Mineralization
The extracellular matrix (ECM) mineralization is a physiological process occurring in bone and teeth during skeletal growth in growth plate cartilage. In these tissues, this process is maintained by mineralization-competent cells, e.g., osteoblasts, odontoblasts, and hypertrophic chondrocytes. Bone formation begins when mesenchymal cells form condensations (Figure 1). During intramembranous ossification, which occurs in a few areas such as the flat bones of the skull, cells present in these condensations differentiate directly into bone-forming osteoblasts producing a matrix particulary rich in collagen I. At the end of the bone formation phase, osteoblasts may be subjected to apoptosis, become inactive osteoblasts, bone lining cells or osteocytes (Figure 1) [1]. The proportion of osteoblasts following each fate is not the same in all mammals and is not conserved among all types of bone [2]. In human cancellous bone, 65% of the osteoblasts undergo apoptosis and only about 30% transform into osteocytes [3], while in the antlers of the white-tailed deer (Odocoileus virginianus) 10% osteoblasts transform into osteocytes [4]. In advanced bony fishes with acellular bone, the number of osteoblasts that turn into osteocytes is zero [5,6]. During endochondral ossification, which occurs in most condensations, the cells become chondrocytes (Figure 1), i.e., the primary cell type of cartilage, which secretes a matrix rich in collagen II [7]. Subsequently, chondrocytes in the centre of cartilage mould stop proliferating, become hypertrophic and start to produce collagen X. The hypertrophic chondrocytes are the principal engine of bone growth [8]. Hypertrophic chondrocytes direct the mineralization of the surrounding matrix, attract blood vessels and attract chondroclasts (closely related or identical to osteoclasts) [7]. These cells direct adjacent perichondrial cells to become osteoblasts. Finally, hypertrophic chondrocytes undergo apoptotic cell death [7]. Osteoblasts, lining cells and osteoclasts on the surface of bone as well as osteocytes in the interior of the bone are the four different types of bone cells (Figure 1). Osteoblasts originate from local mesenchymal cells called osteoprogenitor cells, while osteoclasts originate from bone marrow (BM) hematopoietic stem cells. Bone is a dynamic tissue that is constantly being reshaped by osteoblasts, which are in charge of matrix and mineral production, and by osteoclasts, which have long been recognized as cells that resorb the bone in normal bone remodelling and in pathological conditions when bone resorption is increased [9]. In fact, the mechanisms of mineralization used by mineralization-competent cells are very similar to each other. First, mineralization-competent cells differentiate under the control of the runt-related transcription factor 2 (Runx2). Then, they are able to secrete ECM, principally composed of fibrillar collagen, in which the calcium phosphate crystals with the hydroxyapatite (HA) structure are deposited [10]. The initiation of formation of calcium phosphate deposits is likely to start at matrix vesicles (MVs) [11], which are then released by the mineralization-competent cells into ECM. MVs are spherical bodies in 50–200 nm in diameter [11], enriched in tissue-non specific alkaline phosphatase (TNAP), which is indispensable for mineralization [12]. It was established a long time ago that inorganic pyrophosphate (PPi) or polyphosphate must be removed from the sites of mineralization, before calcification can occur [13]. On the other hand, it was commonly thought until recently that main function of TNAP is to generate inorganic phosphate (Pi) to sustain mineral formation. This discrepancy has been solved by demonstrating that TNAP initiates mineralization by hydrolysing PPi to Pi, therefore removing PPi which is a strong mineralization inhibitor [14]. Furthermore, the results of elegant experiments have provided evidence that TNAP itself may be sufficient to induce mineralization in any tissue containing fibrillar collagen [15]. Later, other factors were also identified and shown to be involved in the mineralization process. For instance, fetuin protein identified in serum that limits the production of calcium phosphate crystals and their deposition in the collagen fibrils [16]. In addition to the role of MVs as TNAP carriers during mineralization, it is also believed that these extracellular organelles are able to accumulate calcium and phosphate ions, which result in the precipitation of calcium phosphate crystals, and that these crystals, by a still unknown mechanism, are transferred from MVs into ECM.
Figure 1.

Schematic representation of bone formation and resorption. Bone formation: During endochondral ossification: chondrocytes first synthesize a cartilage. Central chondrocytes undergo hypertrophic differentiation and then undergo apoptose inducing vascular invasion and formation of a primary ossification centre. During intramembranous ossification, mesenchyme condensations differentiate into osteoblasts, which synthesize and mineralize osteoid to form a new bone without the requirement for a cartilage intermediate. A large fraction of mature osteoblasts undergo to apoptosis while a small fraction of mature osteoblasts become osteocytes. Bone resorption: Hematopoietic stem cells of the monocyte/macrophage lineage differentiate to mature osteoclasts and resorb bone.
Phosphate homeostasis maintained by the gut, bone and kidney is regulated by many hormones such as the classical ones, parathyroid hormone (PTH) and 1α,25-dihydroxyvitamin D3 (1α,25-(OH)2D3), as well as the fibroblast growth factor 23 (FGF23) which was recently reported to have a role in phosphate homeostasis. FGF23—a circulating hormone synthesized by osteocytes and osteoblasts in bone [17]—reduces serum phosphate and 1,25-dihydroxyvitamin D levels by acting on kidney through some FGF receptor [18]. FGF23 null mice have soft tissue calcifications, severe growth retardation, abnormalities of bone mineralization, a markedly shortened lifespan, and abnormalities of glucose metabolism [19,20].
1.2. Ectopic Calcifications and Defective Mineralizations
The physiological mechanisms of mineralization described above are likely to take place also in tissues other than bones and teeth under specific pathological conditions. Vascular calcification for instance in arteries of patients with renal failure, diabetes mellitus or atherosclerosis recapitulates the mechanisms of bone formation [11,21–24]. Generalized artery calcification of infancy, a rare but severe autosomal recessive disorder characterized by calcification and stenosis of arteries [25], calcification in cartilage (osteoarthritis (OA)) [26] tendons and/or in ligaments (calcific tendonitis or ankylosing spondylitis) [27] result from pathologic HA deposition in soft tissues. In patients with end-stage renal disease, FGF23 may exert a toxic effect on the cardiovascular system in a Klotho-independent manner [18]. Elevated levels of FGF23 have been associated with risks of end-stage renal disease, cardiovascular disease and mortality [28]. On the other hand, disorders that are caused by high circulating level of FGF23 are associated with hypophosphatemia while those corresponding to low circulating levels of FGF23 are associated with hyperphosphemia [20]. Heterotopic ossification is a pathological condition where bone formation occurs in extra skeletal tissues (skin, soft tissues, muscle). The two known genetic forms of heterotopic ossification are fibrodysplasia ossificans progressiva and progressive osseous heteroplasia. Fibrodysplasia ossificans progressiva is a rare heritable disorder of connective tissue disease characterized by congenital malformations of the great toes [29] and recurrent episodes of painful soft-tissue swelling that lead to heterotopic ossification [30]. Fibrodysplasia ossificans progressiva is associated with overexpression of bone morphogenetic protein 4 [31,32]. Progressive osseous heteroplasia is a developmental disorder of mesenchymal differentiation characterized by dermal ossification during infancy and by progressive heterotopic ossification of cutaneous, subcutaneous, and deep connective tissue during childhood [33]. In contrast, other diseases such as hypophosphatasia [34,35], osteoporosis [36] and rheumatoid arthritis (RA) [37] result from defective bone mineralization. Hypophosphatasia is a rare inherited disorder caused by mutations in the gene-encoding TNAP that cause a decrease in enzyme activity leading to defective bone and teeth mineralizations [34,35]. Osteoporosis is a skeletal disease characterized by low bone mass and microarchitectural deterioration resulting in bone fragility and in increasing number of bone fractures [36]. RA is characterized by inflammation in the synovium and symmetric polyarthritis. Infiltrations of the synovial tissues by inflammatory cells such as macrophages and T cell occur. Following this, local cellular proliferation of synoviocytes results in an expansion of the synovium—so called pannus—which invades and destroys articular structures [38,39]. Cytokine- and cell contact- activation of synoviocytes and monocytic cells occurs and for some of them differentiate into bone-resorbing osteoclasts [37]. Therefore, understanding the mechanisms of mineralization and bone resorption is important not only in the context of bone formation and modelling, but also in the field of calcific diseases, affecting skeletal and no-skeletal tissues [40]. Given that vascular calcification significantly increases the mortality risk in patients with chronic kidney disease [41] as well as in the general population [42], deciphering the molecular mechanisms of mineralization deserves broad consideration.
1.3. Matrix Vesicles and Early Stages of Mineralization
As it has been already mentioned, early stages of mineralization occur in MVs [11]. These organelles are enriched in phosphatases, especially TNAP [43–45] which can hydrolyze any phosphoesters including PPi, phosphatase orphan 1 (PHOSPHO1) [46,47] which can hydrolyze phosphocholine (PChol) or phosphoethanol producing Pi. MVs also house various ion-motive ATPases [48] providing Pi, as well as progressive ankylosis protein [45], a PPi transporter from the lumen of MVs or from cells to the extracellular medium, and ectonucleotide pyrophosphatase phosphodiesterase 1 (NPP1) [45,49], which produce PPi from ATP or UTP. The Pi pool generated by TNAP, PHOSPHO1 and ATPases significantly affects the phosphate homeostasis and is indispensable for tissue mineralization. On the other hand, the PPi pool produced by ankylosis protein and NPP1 is inhibitory for mineralization [14]. Analysis of lipid composition of growth plate cartilage [50] and MVs [50–52] revealed significant phospholipase activity, although none of these enzymes have been isolated [53]. Other protein constituents of MVs and their possible roles in mineralization have been reviewed elsewhere [53,54].
1.4. Dietary Lipids and Bone Health
The fat and bone connection plays an important role in the pathophysiology of age-related bone loss [55]. Several reviews reported that dietary lipids such as α-linolenic acid [56], conjugated linoleic acid [57], n-3 fatty acid [58–60] could promote bone health. Long-chain n-3 polyunsaturated fatty acids (PUFA) such as eicosapentaenoic acid and docosahexaenoic acid are beneficial for bone health. They can increase bone formation, affect peak bone mass in adolescents and reduce bone loss [61]. Such beneficial effects may include the prevention or reduction of RA [57] and of osteoporosis [59,62]. However, bone mineral density is negatively associated with saturated fat intake, and men may be particularly vulnerable to these effects [63]. On the other hand, the nature or type of the diet fat were not assessed and may possibly mask the beneficial effects of some PUFAs. Nevertheless, chronic exposure to free fatty acids can be deleterious to some cell types and may contribute to lipotoxicity [64] and lead to cardiomyopathy, hepatohepatitis and diabetes [65]. Diets containing foods naturally rich in antioxidants and n-3 PUFAs could be used to treat patients with inflammatory periodontitis. However, the effect of nutritional approaches to periodontal management still need to be evaluated [66]. Skeletal lipidomics is just emerging and targeted lipidomics have not been applied to bone tissue. A partial profile of endocannabinoids and endocannabinoid-like compounds has demonstrated the presence of several long-chain fatty acid amides, some of which display potent effects on osteoblasts and osteoclasts [67]. Therefore, it became clear that an understanding of the role of phospholipases, which produce various lipids, including fatty acids, would provide additional insights into the physiological and pathological mechanisms of mineralization leading to calcification.
1.5. Groups of Phospholipases and Possible Roles during Mineralization
There are two families of phospholipase A (PLA), PLA1[68,69] and PLA2[70–74] that hydrolyze the acyl group attached to the sn-1 and sn-2 positions of glycerophospholipids, respectively. In both cases, free fatty acids as well as lysophospholipids (LPLs) are liberated (Figure 2).
Figure 2.
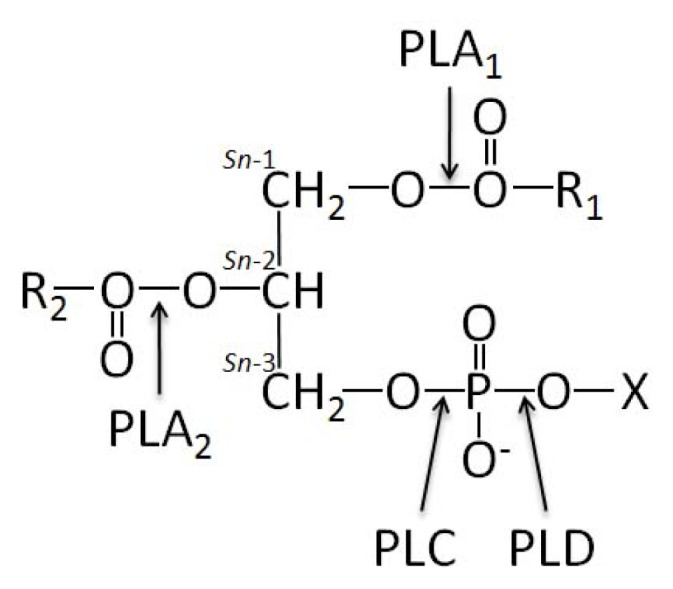
Glycerophospholipid structure and the site of action of phospholipases. The glycerophospholipid molecule consists of a glycerol-3-phosphate esterified at its sn-1 and sn-2 positions to non-polar fatty acids (R1 and R2, respectively) and, at its phosphoryl group to a polar head group, containing alcohol, X. Arrows indicate the sites of phospholipase-catalyzed hydrolysis. The carbon atoms of the glycerol backbone of the glycerophospholipid are indicated according to the stereochemical numbering (sn-1, sn-2 and sn-3).
Phospholipase C (PLC) cleaves the polar head phosphate from glycerophospholipids, producing diacylglycerol (DAG) [75,76], while phospholipase D (PLD) catalyzes the hydrolysis of the terminal phosphodiester bond of membrane glycerophospholipids, producing phosphatidic acid (PA) and free polar head group (Figure 2) [77,78]. Before discussing the potential roles of phospholipases during mineralization, it is necessary to discuss the general basis of their actions and their consequences during mineralization. Phospholipases are not only localized on cellular or organelle membranes but they can be secreted or reside in the cytoplasm. The secreted phospholipases can regulate in an autocrine or paracrine manner the osseous cells, osteoclasts and chondrocytes. Their catalytic products (Figure 3) can be involved in lipid-mediated signaling, in membrane remodelling, in endocytosis or in exocytosis of MVs. In addition, phospholipids contain phosphate, a precursor of HA formation. Their hydrolytic products may serve as a phosphate reservoir to sustain mineralization in MVs. There are at least two distinct types of targets for these lipolytic enzymes, namely those in mineralization competent cells and those in MVs (Figure 4). One can suppose that in a cell, the effects of phospholipase activities shall initiate membrane modelling, intracellular signaling events and exocytosis of MVs. In MVs, phospholipases shall break the membranous structure facilitating the release of HA crystals into ECM [11,79]. In addtion PLC and SMase will provide PChol or phosphoethanolamine (PEA) which are phosphate precursors, that could be further hydrolysed by phosphatase such as PHOSPHO1 to yield Pi[46,47]. To illustrate the potential roles of phospholipases in MVs, it is worth looking into the lipid comparison of MVs and chondrocyte membrane fractions, which reveals small but significant differences [53]. Among them one may notice an enrichment of MV membrane in phosphatidylserine (PS) (2.3–3.5 fold), sphingomyelin (SM) (1.9–2.8 fold) and total LPLs (1.3–3.6 fold), with concomitant depletion in phosphatidylcholine (PC) content (0.8–0.9 fold) in comparison to the membrane fractions isolated from proliferating chondrocytes ([53] and Table 1). An enrichment in SM but not in LPLs was observed in membrane fractions isolated from hypertrophic cells as compared with proliferating cells (Table 1) suggesting that SMases may be silent during hypertrophy, a phenomenon that precedes MV formation [53]. This suggests that the differences in lipid compositions in membrane MVs and in plasma membranes could be not fortuitous but may have a functional significance.
Figure 3.

Phospholipase metabolites with biological activity at various levels of mineralization process. (A) Production of eicosanoids by phospholipase A2 (PLA2); (B) phospholipase D (PLD)-catalyzed hydrolysis of phosphatidylcholine (PC) to phosphatidic acid (PA) and Choline; (C) phosphatidylinositol-specific phospholipase C (PI-PLC)-catalyzed hydrolysis of PIP2 generating membrane-associated second messengers (inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG)). PC-PLC hydrolyzes PC to DAG and phosphocholine (PChol). PE-PC hydrolyzes phosphatidylethanolamine (PE) to DAG and phosphoethanolamine (PEA); (D) sphingomyelinase (SMase)-catalyzed hydrolysis of sphingomyelin (SM) to ceramide and PChol.
Figure 4.

Initial steps of mineralization in which particular phospholipases can be involved.
Table 1.
Total lipid composition of growth plate tissues: Chondrocytes, membranes, and matrix vesicles (MVs). The cell membrane fraction represents more dense membranous material that sediments more rapidly than MVs; it probably contains some MVs that are partially calcified. It does not specifically represent the plasma membrane. Adapted from [53].
| % of Total lipid | |||||
|---|---|---|---|---|---|
|
| |||||
| Chondrocytes | |||||
| Lipid | Whole cartilage | Proliferating | Hypertrophic | Cell membranes | MVs |
| SM | 8.6 ± 0.7 | 5.8 ± 0.4 | 8.0 ± 0.8 | 8.1 ± 0.8 | 13.4 ± 1.8 |
| PC | 45.2 ± 1.9 | 47.6 ± 1.5 | 38.0 ± 1.5 | 53.2 ± 2.2 | 41.8 ± 2.5 |
| LPC | 2.0 ± 0.6 | 1.9 ± 0.4 | 1.8 ± 0.4 | 3.5 ± 0.8 | 3.4 ± 0.8 |
| PE | 17.6 ± 1.0 | 16.9 ± 0.7 | 14.7 ± 0.8 | 14.6 ± 1.8 | 14.9 ± 1.8 |
| LPE | 2.0 ± 0.4 | 3.3 ± 0.7 | 2.4 ± 0.4 | 4.9 ± 1.2 | 6.5 ± 1.2 |
| PS | 5.1 ± 0.8 | 3.3 ± 0.3 | 5.0 ± 1.0 | 5.4 ± 0.7 | 9.3 ± 1.1 |
| LPS | 0.5 ± 0.2 | 0.2 ± 0.1 | 0.3 ± 0.2 | 2.2 ± 0.7 | 2.4 ± 0.8 |
| PI | 7.2 ± 0.8 | 6.2 ± 0.8 | 6.4 ± 0.8 | 6.1 ± 0.8 | 6.6 ± 0.6 |
| LPI | 1.1 ± 0.7 | 1.0 ± 0.6 | 0.5 ± 0.4 | 0.3 ± 0.2 | 1.1 ± 0.3 |
| PA | 2.0 ± 0.5 | 0.8 ± 0.3 | 1.6 ± 0.5 | 1.1 ± 0.2 | 0.9 ± 0.3 |
| PG | 1.2 ± 0.6 | 0.7 ± 0.3 | 1.2 ± 0.6 | 0.9 ± 0.2 | 1.3 ± 0.3 |
| di-PG | 3.0 ± 0.6 | 2.5 ± 0.4 | 2.9 ± 0.6 | 1.7 ± 0.3 | 1.5 ± 1.4 |
2. Phospholipases A1
2.1. Groups, Subgroups and Specificity
There are at least nine known PLA1 molecules in mammals; at least six are extracellular enzymes— belonging to the pancreatic lipase gene family—and the other three are intracellular enzymes [69] (Table 2). The extracellular PLA1 comprise PS-specific PLA1 (PS-PLA1) [80], membrane-associated PA-selective PLA1 (mPA-PLA1α and mPA-PLA1β) [81,82], hepatic lipase, endothelial lipase and pancreatic lipase-related protein 2. PS-PLA1 is specific to PS and gives a rise to lyso-PS (LPS), while mPA-PLA1α and mPA-PLA1β are specific to PA and form lyso-PAs (LPAs). Hepatic lipase, endothelial lipase and pancreatic lipase-related protein 2, in addition to PLA1 activity, can hydrolyze triacylglycerols [69,83,84]. In mammals, there are three intracellular PLA1, a PA-preferential PLA1, (iPLA1α) [85–87]; a p125 (iPLA1β) [88] and KIAA0725 (iPLA1γ) [89] (Table 2). The physiological functions of PLA1 remain largely unknown in bone cells and chondrocytes in contrast to those of PLA2 and other phospholipases [69].
Table 2.
PLA1 family (adapted from [69]).
| Types of PLA1 | Groups | Origin |
|---|---|---|
| Extracellular PLA1 | PS-PLA1 | Human |
| mPLA1α | Human | |
| mPLA1β | Human | |
| Hepatic lipase | Human | |
| Endothelial lipase | Human | |
| Pancreatic lipase-related protein 2 | Human | |
| Intracellular PLA1 | iPLA1α | Human |
| iPLA1β | Human | |
| iPLA1γ | Human |
3. Phospholipases A2
3.1. Groups, Subgroups and Specificity
To date there are more than 30 enzymes identified in mammals that possess PLA2 or related activity [90,91] (Table 3). There are six types of PLA2: the secreted small molecular weight extracellular enzymes (sPLA2s) [90,92–96]; the larger cytosolic Ca2+-dependent enzymes (cPLA2s) [97–102]; the Ca2+-independent enzymes (iPLA2s) [103–107], the platelet-activating factor (PAF) acetylhydrolases (PAF-AH) [108–116]; the lysosomal PLA2 (LPLA2) [117] and the adipose-tissue PLA2 (AdPLA2) [118,119] (Table 3). Among the subgroups of secreted PLA2 (sPLA2): IB, IIA, IID, IIE, IIF, III, V, XIIA, XIIB are of human origin. Among them, the group II subfamily (IIA, IID, IIE and V) is thought to play a role in the production of several lipid mediators especially in the delayed phase of the cell activation process, because their expression levels are up-regulated under various inflammatory conditions. In contrast, sPLA2-IB has long been thought to be a digestive enzyme, given its abundance in the pancreas. However, the discovery of the PLA2 receptor (PLA2R) which can bind sPLA2-IB suggests that IB sPLA2 could exerts various biological responses in addition to its digestive function [120]. sPLA2-IA is found in cobras and kraits, -IIB is evidenced in the Gaboon viper and –IX originates from the snail venom. sPLA2-IIC is found in rat/murine testis. sPLA2-XIA and -XIB are evidenced in green rice shoots. sPLA2-XIII has been evidenced in parvovirus and XIV was found in symbiotis fungus and bacteria [72]. Among the cPLA2 -subgroups, cPLA2α, cPLA2β, cPLA2γ are from human origin while the three others—cPLA2δ, cPLA2ɛ, cPLA2η—are of murine origin (Table 3). All the six calcium independent PLA2—group VI iPLA2—have been identified in humans (iPLA2 A,B,C,D,E,F) as well as all the PAF PLA2-VIIA, -VIIB, -VIIIA and -VIIIB (Table 3). Among PLA2s, so far only sPLA2-II, -V and -X as well as cPLA-IVA [91] and iPLA2β [121] have been evidenced to be involved in osseous diseases. Since AA is a precursor of prostaglandins, prostacyclins and thromboxanes, as well as leukotrienes and lipoxins (Figure 3A), PLA2 especially cPLA2-α is involved in cellular signaling affecting bone formation and resorption. cPLA2-α is constitutively expressed in most tissues although its level of expression can be increased in response to growth factors and proinflammatory cytokines. cPLA2-α is unique among the PLA2 enzymes in having a preference for phospholipids with arachidonic acid (AA) at the sn-2 position [122].
Table 3.
PLA2 family (adapted from [91]).
| Type | Group | Subgroup | Origin or commun source |
|---|---|---|---|
| sPLA2 | I | A | Cobras and kraits |
| I | B | Human/porcine pancreas | |
| II | A | Rattlesnake/human synovial | |
| II | B | Gaboon viper | |
| II | C | Rat/murine testis | |
| II | D | Human/murine pancreas/spleen | |
| II | E | Human/murine brain/heart/uterus | |
| II | F | Human/murine testis/embryo | |
| III | Lizard/bee | ||
| V | Human/murine heart/lung/macrophage | ||
| IX | Snail venom | ||
| X | Human spleen/thymus/leucocyte | ||
| XI | A | Green rice shoots (PLA2-I) | |
| XI | B | Green rice shoots (PLA2-II) | |
| XII | A | Human/murine | |
| XII | B | Human/murine | |
| XIII | Parvovirus | ||
| XIV | Symbiotic fungus/bacteria | ||
| cPLA2 | IV | A(α) | Human macrophage-like U937 cells/Platelets/Raw 264.7/rat kidney, ubiquitous |
| IV | B(β) | Human pancreas/liver/heart/brain/ubiquitous | |
| IV | C(γ) | Human heart/skeletal muscle | |
| IV | D(δ) | Murine placenta | |
| IV | E(ɛ) | Murine heart/skeletal muscle/testis/thyroid | |
| IV | F(η) | Murine thyroid/stomach | |
| iPLA2 | VI | A(β) | Human/murine |
| VI | B(γ) | Human/murine | |
| VI | C(δ) | Human/murine | |
| VI | D(ɛ) | Human | |
| VI | E(ζ) | Human | |
| VI | F(η) | Human | |
| PAF-AH | VII | A(lipoprotein-associated-PLA2) | Human, murine, porcine, bovine |
| VII | B(PAF-AH II) | Human, bovine | |
| VIII | A(α1) | Human | |
| VIII | B(α2) | Human | |
| Lysosomal PLA2 | XV | Human, murine, bovine | |
| Adipose PLA | XVI | Human,mouse |
3.2. Presence of PLA2s in Chondrocytes and Possible Roles
Experimental evidence of the presence of specific types of PLA2s such as sPLA2-IIA, sPLA2-V and sPLA2-X, cPLA-IVA and iPLA2β in chondrocytes arise from analysis of human synovial fluid, especially from RA or OA patients or from the effects of cell stimulation with interleukin-1 (IL-1) and tumor necrosis factor (TNF). sPLA2-IIA was found in human synovial fluid of arthritic knee [123,124]. PLA2s were evidenced in chondrocytes stimulated with IL-1 [125]. Indeed, the expression of sPLA2-IIA and -V is cytokine-dependent [124]. Immunohistochemistry of RA sections revealed that sPLA2-IIA was generally located in synovial lining and sublining cells and cartilage chondrocytes [124]. In healthy and in OA patients, sPLA2-IIA is predominantly located in blood vessel endothelium and in vascular smooth muscle [126]. sPLA2 in the inflammation joint may originate from chondrocytes [127–130]. Indeed, cultured chondrocytes synthetise and release sPLA2[131]. IL-1 and TNF can stimulate the expression of mRNA encoding sPLA2 in chondrocytes [132,133]. IL-1 induces the secretion of PLA2 from chrondrocytes [134–137]. IL-1 and TNF can activate sPLA2 gene expression not only in chondrocytes, but also in fibroblasts, smooth muscle cells and endothelial cells [126,138]. sPLA2-IIA, -IID, -V as well as cPLA2-IVA expressions were upregulated in human-OA chondrocytes upon IL-1, TNF, IL-6 or IL-8 stimulations [139]. Usually, among the members of cPLA2-IVA (Table 3), cPLA2-α is the most ubiquitously expressed enzyme [102].
3.3 Presence of PLA2s in Osteoblasts and Possible Roles
Inflammatory processes are characterized by increased levels of extracellular PLA2, IL-1 and TNF. Stimulated Fetal rat calvarial bone forming cells, treated with recombinant human IL-1 and TNF stimulated extracellular sPLA2[140] and the PLA2 activity in osteosarcoma cell lines is stimulated [141]. Fetal rat calvaria osteoblastic cells are emblematic since they continuously synthesize and release sPLA. 1α,25-(OH)2D3)—a regulator of bone biology—stimulates PLA2 activity in three osteoblastic cell lines: ROS 17/2.8 cells, MC-3T3-E1 cells, and MG-63 cells. 1α,25-(OH)2D3-dependent alkaline phosphatase and PLA2 activities were correlated with production of prostaglandin E1 (PGE1) and prostaglandin E2 (PGE2) in the MC-3T3-E1 cells [142]. PLA2 inhibitors (such as quinacrine or mepacrine) [143–145] and PLA2 activators (such as melittin) [146,147] served to evaluate PLA2 in osteoblasts. In MC3T3-E1 cells, quinacrine showed partial inhibitory effect on prostaglandin F2 (PGF2) induced AA release [143] while it suppressed the thrombin-induced AA release [145]. Mepacrine, significantly inhibited the bradykinin-induced AA release [144] suggesting the presence of PLA2 in osteoblasts. Microtubule depolymerizing agents inhibit the expression and release of sPLA2 by fetal rat calvarial osteoblasts [148]. MC3T3-E1 cells originating from IIA sPLA2-deficient C57BL/6J mouse had delayed PGE2 generation but introduction of type IIA sPLA2 augmented PGE2 production. This was accompanied by increased expression of both cPLA2 and cyclooxygenase-2 (COX-2) [149] revealing a particular cross-talk between the two PLA2 enzymes and COX-2. sPLA2 augments cPLA2 and COX-2 expression in mouse osteoblasts via endogenous PGE1[150]. IL-1α treatment induced an augmentation of PGE2 production by mineralizing osteoblasts involving cPLA2, sPLA2, COX-2 and PGE synthase activities [151]. However, the crosstalk between sPLA2 and cPLA2 may not hold in other cellular responses. In mouse osteoblastic cells, cPLA2 mRNA and protein were constitutively expressed and increased approximately 2-fold by IL-1α treatment, but secretory sPLA2 mRNA was not detected [152]. Using arachidonoyltrifluoromethyl ketone—a cPLA2 inhibitor—it was found that Cd increased cPLA2 activity followed by COX-2 induction, which resulted in PGE2 production in primary mouse osteoblastic cells [153,154]. However, the results obtained with arachidonoyltrifluoromethyl ketone should be analyzed with some caution since it is not a selective cPLA2 inhibitor and it may inhibit other enzymes, such as COX [155]. So far, most of the reports were focused on sPLA2 (among them IIA sPLA2) and a few were concentrated on cPLA2. Only recently, the presence of other PLA2 types in osteoblasts was evidenced. MC3T3-E1 cells possess high levels of native PLA2R and sPLA2-X is one of its high-affinity ligands. PLA2-VIA or iPLA2β is expressed in normal bone. It was suggested that iPLA2β mRNA is more abundant in bone forming osteoblast cells than in osteoclast cells [121]. From the findings based on knockout mice lacking iPLA2β, an unrecognized role of iPLA2β in bone formation is yet to be found. The absence of iPLA2β causes abnormalities in osteoblast function and BM stromal cells differentiation [121].
3.4. Presence of PLA2s in osteoclasts and Possible Roles
In BM cultures, IL-1 stimulated PGE2 production and osteoclast formation in cells from wild-type mice but not from those taken from cPLA2α−/− mice [156] indicating that cPLA2-α is essential for PGE2 production. PGE2 may act to enhance osteoclast formation and action (Figure 5). In response to IL-1 (or other agents such as TNF-α), cPLA2-α is activated and PGE2 is produced and secreted. PGE2 may act in autocrine manner modulating the stromal cell response or in a paracrine manner on the osteoclast precursor cells. Following this, the newly formed osteoclast activates bone resorbtion [122] (Figure 5). The Enzymatic activity of cytosolic PLA2 was detected in human osteoclasts extracted from human fetuses and in human osteoclast-like cells differentiated from peripheral blood mononuclear cells [157]. Human osteoclasts actively produced prostaglandin, and the COX-1 pathway was implicated in the control of bone resorption. COX-2 and sPLA2-IIA are also implicated in osteoclastogenesis as suggested by the results obtained with the use of DFU—an inhibitor of COX-2—and KH064—an inhibitor of sPLA2-IIA—in ovariectomized Wistar rats. KH064 suppressed increases in osteoclast surface induced by ovariectomy while the effect of COX-2 inhibition was less marked [158].
Figure 5.

Possible role of cPLA2α in bone resorption. In response to lysophosphatidylserine (LPS), interleukin (IL)-1 or tumor necrosis factor-α (TNF-α), cPLA2α is activated and releases arachidonic acid (AA). AA is then transformed into prostaglandin E2 (PGE2) which may bind in an autocrine manner to a PGE receptor on the stromal cell or, in a paracrine manner, on the osteoclast precursor cell. Then osteoclasts derived from osteoclast precursor cells undertake bone resorption. Adapted from [122] (PL, phospholipid).
3.5. Presence of PLA2s in Smooth Muscle Cells and Possible Roles
PLA2 was evidenced in vascular smooth muscle cells [159]. The isoenzyme sPLA2-IIA has been localized in smooth muscle cell [160–164] and has a close spatial relationship with collagen fibers [161].
3.6. The Expressions of PLA2s under Pathological Conditions
sPLA2-IIA is highly expressed in synovial fluid [123,126,165,166], in chrondrocytes [131], in the joints of patients with RA and to a lesser extent in synovium of OA patients but not in healthy patients [126] (Table 4). Increased catalytic activity of group II sPLA2 was observed in synovial fluid of OA patients [167,168]. Circulating sPLA2 activity correlates with juvenile RA activity [169]. The sPLA2-IIA activity in the serum of 212 RA patients was determined and appeared to be correlated with the Lansbury index, number of effusions, number of damaged joints, erythrocyte sedimentation rate, platelet count and low hemoglobin [170]. However, the enzyme activity is not always correlated with the severity of the RA disease [171]. sPLA2-IIA, sPLA2-IID, sPLA2-IIE sPLA2-V are more often detected in active RA than in inactive RA synovial tissues, while sPLA2-X is diversely expressed in both active and inactive RA tissues [124] (Table 4). This suggests that transcriptional regulation of the groups -V and -X as well as group II subfamilies are distinct. Exogenous addition of sPLA2-IIA, dose-dependently amplified TNF-α stimulated PGE2 production accompanied by increased expression of COX-2 and cPLA2-IIA in cultured synovial cells [172]. Exogenous addition of Crotalus adamantus sPLA2-II, as well as continuous exposure to IL-1α inhibited mineralization of the osteoid formed by fetal rat calvaria cells [173]. In normal heart, sPLA2-IIA was detected in coronary vascular smooth muscle cells (VSMCs) and sPLA2-V in cardiomyocytes beneath the endocardium. In infarcted hearts, expression of sPLA2-IIA and sPLA2-V increased in damaged cardiomyocytes and VSMCs. Expression of sPLA2-IID and -IIE, which were indetectable in normal heart, was elevated in damaged cardiomyocytes and VSMCs, respectively [164] (Table 4).
Table 4.
Diseases and affected PLA2 expressions in human patients and in knockout mice.
| Types of PLA2 | Expression levels | Diseases | References |
|---|---|---|---|
| sPLA2-IIA | Highly expressed in synovial fluid | RA | [123,126,165,166,169] |
| sPLA2-IIA | Highly expressed in chondrocytes | RA | [126] |
| sPLA2-IID | Overexpressed in synovial fluid | RA | [124] |
| sPLA2-IIE | Overexpressed in synovial fluid | RA | [124] |
| sPLA2-V | Overexpressed in synovial fluid | RA | [124] |
| sPLA2-X | More or less expressed in synovial fluid | Active and inactive RA | [124] |
| sPLA2-IIA | Overexpressed in synovial fluid | OA | [126,167,168] |
| sPLA2-IIA | Overexpressed in VSMC | Infarctus heart | [164] |
| sPLA2-V | Overexpressed in VSMC | Infarctus Heart | [164] |
| cPLA2-α | cPLA2-α−/− mice loss in function | Prevention in collagen-induced arthritis | [174] |
| iPLA2β | iPLA2-β−/− mice loss in function | Low bone mass | [121] |
3.7. Transgenic Knockout Animal for PLA2 Enzymes as Models for Bone Formation and Mineralization Diseases
There are at least five knockout mice for for sPLA2 isoforms (-IB, -IIA, -III, -V and -X) [74,90,95,175] as well as one for the cPLA2α isoform [74,176–178] and at least two for iPLA2 isoforms -β [179,180] and -γ [181–183]. The PLA2γ2a gene coding for sPLA2-IIA has been knocked out in BALB/c mice by breeding them with C57BL/6 mice and then backcrossing with BALB/c mice [184]. The PLA2γ2a−/− BALB/c mice displayed a reduced degree of arthritic inflammation in K/BxN autoantibody-induced mouse arthritis model as compared with wild-type BALB/c mice. This suggests that sPLA2-IIA is playing a pro-inflammatory role in this mouse arthritis model [184]. In contrast with PLA2γ2a−/− BALB/c mice, the PLA2γ5a−/− BALB/c mice lacking the sPLA2-V gene product exacerbated the K/BxN autoantibody-induced arthritis. Indeed, supplementation of sPLA2-V slowed down the K/BxN autoantibody-induced arthritis in PLA2γ5a−/− BALB/c mice by facilitating phagocytic uptake of the immune complex by macrophages. This suggests that sPLA2-V has an anti-inflammatory effect, while sPLA2-IIA has a pro-inflammatory effect [184]. cPLA2α−/− mice were characterized by reduced severity and incidence in collagen-induced arthritis (CIA) indicating that cPLA2α, plays a key role in the development of CIA [174]. A patient having a heterozygous mutation of cPLA2α (PLA2γ4a) with loss of cPLA2α function develops small intestinal ulcers, presented platelet dysfunction, and globally decreased eicosanoid production [185]. It has been previously reported that cPLA2α−/− mice have developed intestinal ulcerative lesions [186]. Therefore, pharmacologic inhibition of the cPLA2α enzyme may induce non-steroidal anti-inflammatory drug (NSAID)-like-induced gastric and intestinal lesions. Knockout mouse models [176] indicated that cPLA2 is important for macrophage production of inflammatory mediators, fertility, and in the pathophysiology of neuronal death after transient focal cerebral ischaemia. iPLA2β-null mice exhibit defective spermatozoa mobility [179], pancreatic islet insulin secretion [180] and lower bone mass associated with a decrease in bone strength [121]. It was concluded that iPLA2β may be an important factor of bone formation and BM stromal cell differentiation [121].
3.8. Inhibitors of PLA2 as Drug Therapy
It was previously recognized that PLA2 may be an attractive therapeutic target since PLA2 inhibition may lead to suppression of prostaglandins, leukotrienes, and PAFs (Figure 3A) [187]. Sulfasalazine—widely used in the therapy of RA, spondyloarthropathies and inflammatory level diseases—inhibited extracellular release of sPLA2 from fetal rat calvaria osteoblasts suggesting that the anti-inflammatory activity may be related, in part, to the selective inhibition of the extracellular release of proinflammatory sPLA2[188]. sPLA2-IIA-inhibitor, LY333013, was administrated to 251 RA patients. Although 12-week treatment with LY333013 or methyl Varespladib—a prodrug that is rapidly converted in vivo to Varespladib was well tolerated, it did not significantly affect RA activity [189]. One explanation is that sPLA2-V has an anti-inflammatory effect, while sPLA2-IIA has a pro-inflammatory effect [184]. Since the inhibitor could block both enzymes [90], its action may cancel the beneficial effect. Alternatively, the regulation of TNF-dependent prostaglandin production by exogenous sPLA2-IIA does not depend on its enzymatic activity. Indeed, sPLA2-IIA mutant H48Q having only 1% of the sPLA2-IIA enzyme activity is as effective as the fully functional enzyme in up-regulating PGE2 production and in over inducing TNF-mediated COX-2 production [190]. sPLA2-IB, sPLA2-IIA, sPLA2-V and sPLA2-X can produce proinflammatory cytokines and chemokines independently of the hydrolytic activity [191]. Nevertheless, methyl Varespladib is in phase III trials for the treatment of cardiovascular diseases [192]. There is an evidence that sPLA2-IIA is involved in the development of atherosclerosis [193]. One possible mechanism of atherogenesis may rely on the ability of sPLA2 to hydrolyze the phospholipids on LDL particles promoting lipid accumulation and leading to enhanced macrophage uptake [91]. On the other hand, inhibitor of cPLA2α could serve as drug to treat human RA [194]. Oral administration of pyrroxyphene in a CIA in mice results in anti-arthritic activity probably due to inhibition of cPLA2α activity and subsequent reduction in eicosanoid levels as well as suppression of metalloproteinase (MMP) and COX-2 mRNA expression [194].
3.9. Effects Mediated by Arachidonic Acid and Its Pathways at Cellular Level
The fatty acid moieties that are incorporated into phospholipids vary, generating a broad range of molecular species. One of the most important fatty acids that can be released from phospholipids by PLA2 is AA which is converted via the COX- and lipooxygenase (LOX)-mediated pathways to eicosanoids, including prostaglandins, thromboxanes, prostacyclins, leukotrienes and lipoxins (eicosatetraenoic acid) (Figure 3A) [74,195]. sPLA2 does not show distinct preference for the sn-2 position fatty acyl chains [91]. In general, most of the sPLA2s have higher activity towards anionic phospholipids such as phosphatidylglycerol (PG), phosphatidylethanolamine (PE) and PS. sPLA2V and sPLA2-X can hydrolyze both PC and anionic phospholipids vesicles at comparable rates [196,197], while sPLA2-IA and sPLA2-XIV are more active against PC [91]. sPLA2 can release AA intracellularly prior to secretion [198], or after secretion into extracellular space. The latter is especially true for sPLA2-V and sPLA2-XV, which have high affinity for PC and act at the outer plasma membrane [199–207] or through a heparan sulfate proteoglycan (HSPG) shuttling pathway [205,207–211]. For example, sPLA2-IIA, -IID and -V often bind to HSPGs, internalized through caveolae/raft-dependent endocytosis, and then exert their function [205,207–211]. In addition, sPLA2 acts as a ligands for a M-type transmembrane sPLA2 receptor, which in turn activates cPLA2-IV [212]. cPLA2-IV (or cPLA2α) have a marked specificity for AA at the sn-2 position of phospholipids [97,99]. Most phospholipids such as PC, PE and phosphatidylinositol (PI) are substrates for cPLA2-IV. PC is a good substrate, especially radiolabeled PC, and has been often used to determine cPLA2-IV activity [97,98]. cPLA2-IV has a calcium-dependent phospholipase activity, as well as a transacylcyclase activity and a calcium-independent lysophospholipase activity [213]. Table 5 contains an overview of the overall beneficial or pathological effects of the AA pathway - including enzymes, their products and their metabolites - on bone formation or bone resorption. Most of information concerning the effects of AA metabolites comes from osteoblasts, osteoclasts and from RA chondrocytes. Several of the cytokines and growth factors implicated in the inflammatory processes in rheumatic diseases have also been demonstrated to impact osteoclast differentiation and function either directly, by acting on cells of the osteoclast-lineage, or indirectly, by acting on other cell types to modulate expression of the key osteoclastogenic factor receptor activator of nuclear factor κB ligand (RANKL) and/or its inhibitor, osteoprotegerin (OPG) [38]. In RA, synovial cells behave like osteoblasts in the induction of RANKL which is an essential ligand for differentiation of bone-resorbing osteoclasts from macrophage precursors [38,214]. It was proposed that by targeting the pathways involved in osteoclast differentiation and function, focal articular bone erosion may be attenuated in the setting of inflammatory arthritis [38].
Table 5.
Beneficial or pathological effects of AA pathways on bone formation.
| Enzymes or products or animal models | Expression level or concentration | Physiological effects | Pathological effects | References |
|---|---|---|---|---|
| COX-2 | Increase in synovial fluid | RA | [38] | |
| mPGES-1 | Increase in synovial fluid | RA | [38] | |
| Mice deficient in COX2 | Null-COX | CIA reduction | [215] | |
| Mice deficient in mPGES-1 | Null-PGES-1 | CIA reduction | [217] | |
| PGE2 | High level in synovial fluid | RA | [239,240] | |
| Prostaglandin | Stimulate bone formation | [233] | ||
| Prostaglandin | Activate bone resorption in osteoporosis, RA, OA or in periodontis | [238] | ||
| PGD2 | Stimulate osteoblast calcification | [249] | ||
| PGF2α | Promote osteogenic differentiation | [251] | ||
| 15-Deoxy-Δ12,14- prostaglandin J2 | Prostaglandin D2 metabolite | Activates PPARγ and TNAP expression | [249] | |
| n-3 PUFA or conjugated linoleic acid | Exogenous addition | Beneficial effects due to modulation of COX-2 | [59] |
3.9.1. Effects Mediated by PGE2
RA synovial cells have high COX-2 and microsomal prostaglandin E synthase-1 expression suggesting that both enzymes are mediatiators of PGE2 production in RA inflamed joints [38]. Mice deficient in COX-2 were protected from CIA [215] while selective inhibitors to COX-2 significantly reduced the severity of arthritis in murine CIA [216]. Mice deficient in microsomal prostaglandin E synthase also exhibited significant reduction in CIA [217] or collagen antibody-induced arthritis inflammation and joint destruction [218]. On the other hand, numerous data support the notion that COX enzymes play an important role in bone formation. COX-1 can be considered as a housekeeping enzyme and is present in healthy tissues, while COX-2 is activated by tissue damage. Both enzymes have a similar Km and Vmax values for their reaction with AA [219]. NSAIDs, ibuprofen [220,221] or indomethacin [220–223] that inhibit COX-1 and COX-2 activity have been reported to inhibit fracture healing in animals (Table 6). NSAIDS can decrease heterotopic calcification in humans [224–226]. Indomethacin was shown to decrease spinal fusion rate in an animal model [227], while keterolac significantly reduces the rate of spinal fusion in humans [228]. In cultured mouse cells, indomethacin decreased TNAP activity and mineralization without significantly affecting Runx2, collagen type I and osteocalcin levels [229]. In the skeleton, prostaglandins (Figure 3A), mainly PGE2 produced by osteoblasts under COX-2 stimulation, play either a stimulatory or an inhibitory role in bone metabolism, depending on the physiological or pathological conditions. PGE2 mediates anti-inflammatory [230] as well as inflammatory effects [38,231,232]. Prostaglandins are potent multifunctional regulators in bone [233] having both stimulatory and inhibitory effects in bone metabolism [234,235]. Under physiological conditions, prostaglandins can stimulate bone formation by increasing proliferation and differentiation of osteoblasts [236]. iPLA2β−/− null mice showed an age-related bone loss that was not accompanied by an increase in osteoclast abundance/activity. However, it was linked to increased adipogenesis from BM stromal cells and decreased osteoblastogenesis associated with higher PPARγ and lower Runx2 levels [121]. These findings suggest that iPLA2β is involved in bone formation and BM stromal cells differentiation. On the other hand, under pathological conditions like osteoporosis, RA, OA and periodontis, prostaglandins can activate bone resorption by increasing the activity of osteoclasts [237,238]. PGE2 is detected at high level in the synovial fluid of RA patients [239,240]. PGE2 mediates pain and inflammatory responses [38,231,235]. Indeed COX-2 inhibitors are effective for decreasing pain in RA [231,241]. cPLA2 in osteoblasts, upon stimulation by IL-1, TNFα or lipopolysaccharide, produces PGE2 which may acts through one or more receptors in autocrine manner as well as in a paracrine manner on the osteoclast precursor cells. Then the number of osteoclast cells increases leading to bone resorption (Figure 5). Results obtained using MG63 osteoblast-like cells cultured on commercially pure titanium surfaces of two different roughnesses in the presence of media containing 1α,25-(OH)2D3 without or with quinacrine (a PLA2 inhibitor) indicated that surface roughness and 1α,25-(OH)2D3 transmit their effects through PLA2, which catalyzes one of the rate-limiting steps in PGE2 production [242]. PGE2 exerts indirect effects on osteoclastic differentiation through osteoblasts but can have direct effects on osteoclast precursor cells and mature osteoclasts [122,243]. A cPLA2α-deficient mice had reduced CIA symptoms which suggests that AA, as a precursor of prostaglandins (Figure 3A) and leukotrienes is the inflammatory mediator in the development of CIA [174]. Indeed, it was suggested that cPLA2 antagonists might be effective in reducing inflammatory bone resorption associated with RA and periodontal diseases [122]. Therefore, analysis of AA metabolites should become an obvious target for further investigations with potential pathophysiological, therapeutic and regenerative outcomes [244]. Beneficial effects of n-3 PUFAs and conjugated linoleic acids on bone formation rate are likely due the modulation of COX-2 mediated biosynthesis of PGE2[244]. It was proposed that increased consumption of foods providing n-3 PUFA and conjugated linoleic acid will balance eicosanoid biosynthesis by decreasing pro-inflammatory AA concentration and will help to prevent or relieve diseases associated with increased n-6 fatty acid derived eicosanoid production [244].
Table 6.
Effect of cyclooxygenase (COX) inhibitors on bone formation.
| Cox inhibitors | Physiological effects | Pathological effects | References |
|---|---|---|---|
| NSAIDS Ibuprofen Indomethacin | Inhibit fracture healing | [221–223] | |
| Indomethacin | Decrease TNAP activity | [222] | |
| NSAIDS | Decrease heterotopic calcification | [224–226] | |
| Keterolac | Decrease in spinal fusion | [228] | |
| COX-2 inhibitor | Decrease pain in RA | [231,241] |
3.9.2. Effects Mediated by PGF2α and PGD2
Other prostanoids and leukotrienes may also play a role in bone metabolism and bone remodeling but they are less defined. Prostaglandin F2α (PGF2α) contributes to osteoblast functions. Locally produced PGF2α might be beneficial in promoting osteogenic differentiation of adipose tissue-derived mesenchymal stem cells [245]. It has been reported [246] that PGF2 stimulates Na+-dependant phosphate transport (Pi transport) activity [247], playing important role in the mineralization of osteoblast-like cells. 15-deoxy-Δ12,14-prostaglandin J2, a prostaglandin D2 (PGD2) metabolite, by binding to and activating PPARγ, may also modulate TNAP expression and mineralization [248]. PGD2 has a stimulatory activity on osteoblast calcification [249]. In addition to COX-metabolized prostaglandins, LOX-mediated generation of leukotrien B4 may be involved in TNAP post-translational activation during osteoblastic differentiation [250].
3.10. Effects Mediated by Lysophospholipids and Their Pathways at Cellular Level
LPLs are produced either by PLA1 or PLA2 and are potential lipid mediators [252]. Among various LPLs detected lyso-PC (LPC) is the most abundant with a plasma concentration of several hundred micromoles per liter [252]. Other LPLs such as lysophosphatidylglycerol (LPG), lyso-PE (LPE), lyso-PI (LPI) and LPS are present at low concentration [252]. GPR55—a G protein-coupled receptor and cannabinoid-sensitive receptor—is activated by 1-acyl LPI [253–256]. GPR55 has only a distant phylogenetic relationship to CB1 and CB2 cannabinoid receptors, but clusters with the LPA receptor LPA4[257]. Since LPI and LPA are similar, LPI is a good agonist of GPR55. The most active species is 2-arachidonyl-sn-glycero-3-phosphoinositol which can be considered as one of the possible natural substrates of GPR55 [258]. GPR55 expression was found to be 8-fold higher in osteoclasts than in monocytes from human healty donors. The GPR55-agonist LPI inhibited mouse osteoclast formation but it stimulated mouse and human osteoclast polarization and resorption in vitro[259]. Although GPR55-agonist LPI induced a decrease in osteoclast number it stimulated osteoclast function. Treatment of human osteoclasts with LPI caused activation of RhoA and ERK1/2 [259] suggesting that activation of GPR55—via inducing a signaling pathway—promotes a regulatory mechanism in osteoclasts. Male GPR55−/− mice but not female GPR55−/− mice presented a decrease in bone resorption and an osteopetrotic phenotype with an increasing osteoclast number [259]. Although osteoclast number increased, osteoclast function was impaired, consistent with the role of GPR55 in stimulating osteoclast function [259]. GPR55−/− mice are resistant to neuropathic and inflammatory pain suggesting that GPR55 antagonists may serve to treat arthritic pain [260]. GPR55 has the potential to influence bone resorption and could be a target to treat bone diseases, such as osteoporosis, by inhibitory drugs, or calcification diseases (osteopetrosis, etc.) by activatory drugs. The role of LPA in skeletal biology has been reviewed [261] and only selected findings will be discussed here. LPA is a stimulator of resting zone chondrocyte proliferation and maturation and an inhibitor of chondrocyte apoptosis. LPA decreases the abundance of p53 to alter p53-target gene expression resulting in the inhibition of caspase activity [262]. Different LPAs vary according to acyl chain length and degree of saturation. Typically, 1-oleoyl LPA is the specimen used in describing its effects on cell and tissues [261]. LPAs are present in plasma at around high nM to low μM concentrations [263–268] and at elevated levels at sites of tissue injury or inflammation [267]. LPA species with saturated fatty acids (16:0, 18:0) and unsaturated fatty acids (16:1, 18:1, 18:2 and 20:4) have been detected in serum, plasma, and activated platelets [269–271]. LPA is an important intermediate product of the synthetic pathway for phospholipids and triacyglycerols in many cell types from various species. In this pathway, LPA is produced by acylation of glycerol-3-phosphate. However, so far, there is no evidence of LPA release into the extracellular fluid or accumulation in plasma membrane and this pathway in not considered to be involved in extracelular LPA signaling [272]. LPA is produced both in the cells and in biological fluids, such as serum and plasma, by distinct pathways [273,274]. (1) In serum and in plasma, LPA is mainly converted from LPLs by a lyso-PLD [275]—later identified as autotaxin (ATX) [276,277]. LPL in plasma and serum are produced by PLA1-like enzymes [266,272] and by a lecithin: cholesterol acyltransferase [278] (Figure 6). Another source of LPLs (about half part of the total amount) in serum are activated platelets. In platelets, PS-PLA1[279] is involved in the production of 2-acyl-LPL while sPLA2-IIA could be involved in the production of 1-acyl-LPL [70,280] (Figure 6). The PS-PLA1 and PE substrate-preferred sPLA2-IIA are extracellular enzymes. Since their substrates are in the inner leaflet of the lipid bilayer, the phospholipids asymmetry should be disrupted and indeed such phenomenon is known to occur in activated platelets, as well as in apoptotic cells and cytokine-stimulated cells [272]. The exposed phospholipids could be targets for PS-PLA1 and sPLA2-IIA enzymes. LPA production by this pathway could be involved in wound healing and inflammation [272]. LPAs are not only produced and released by activated platelets [281] but also by activated inflammatory cells such as erythrocytes and white blood cells [280]. In addition LPA may originate from cancer cells, fibroblasts or adipocytes. Lipoproteins are also a source of LPA. Therefore, the concept of local (autocrine, paracrine) action of LPA which has been demonstrated in platelet aggregation should be extended to other cellular responses. Moreover, several pathological conditions such as cancer, injuries, hematoma, renal failure, are associated with a relatively high production of LPA, thus constituting an interesting marker of cell aggression [282]. In contrast to LPA in plasma and serum, LPA in cells may originate from PA [273]. In this pathway, a PLD or DAG kinase convert lipids in PA and then PA is hydrolyzed by PLA1 or PLA2. There was no direct evidence of LPA production from PA by PLA2 (such as the secretory type or cytosolic type), however a PA-selective PLA1 (PA-PLA1a/LIPH or mPA-PLA1a) could be involved in the production of LPA [81]. As reviewed [273], very little is known about this pathway. For example, production of LPA by mPA-PLA1a has been evidenced in hair follicles. Then, the secreted LPA can activate the P2Y5 receptor which is the closest homolog of LPA4, a G-protein-coupled receptors (GPCR) for LPAs [283] (Figure 6). Albumin binds with high affinity to LPAs and may serve as LPA carrier [284]. Synovial fluid of patients with RA contains a significant amount of LPA. To evaluate its effects, the synovial fluid from RA patients was used in fibroblast-like synovial cells and was found to stimulate COX-2 induction in these cells [285]. Lyso-PLD, LPC and LPA-producing enzymes are present in synovial fluid [285]. Platelets can contribute to LPA production in bone tissue [286,287]. LPA biosynthesis can occur in response to purinergic signaling via P2X7 receptors [288,289]. There are distinct mechanisms involved in the control of the paracrine secretion of LPA [280,286,290]. LPA can affect neighbouring cells in an autocrine/paracrine manner via interactions with a subset of six GPCRs for LPAs so called LPA1-LPA6[291]. Addition of LPAs stimulated the proliferation of primary rat osteoblasts [292], osteoblast-like MC3T3-E1 cells [293], primary human osteoblasts [294], human osteosarcoma cell line G92 and MG-63 [294,295] as well as proliferation of rat primary chondrocytes [296]. LPA addition induced chemotaxis, elevated resistance to apoptosis, activated mitogen-activated protein (MAP) kinases, and elevated Ca2+ in osteoblasts, the precursors of osteocytes [292,294,297–299]. LPA stimulated osteoblast fibronectin assembly and binding with concomitant changes to the cytoskeleton [300–302]. It was predicted that osteocytes could be a target for LPA and indeed LPA stimulated dentrite outgrowth in MLO-Y4 osteocytes [303]. LPA induced osteogenic differentiation of human mesenchymal stem cells hMSC-TERT by interacting with LPA1 and LPA2 receptors. LPA1 receptor activation—coupled to a rise of Ca2+—promotes osteogenic differentiation while LPA4 receptor activation, coupled to cAMP, inhibits differentiation [304]. LPA1−/− mice exhibits impaired suckling behavior and neurological abnormalities [305,306] as well as defects in bone formation leading to osteoporosis [306]. Since LPA1 and LPA4 displays completely opposite effects, as expected LPA4−/− mice had, as expected, an increased bone volume, trabecular thickness and trabecular number [304]. The potential effects of LPA on osteogenesesis or osteoclasis may depend on the respective expressions of LPA receptors [306]. In osteoclasts, LPA acts through multiple receptor subtypes to elevate intracellular Ca2+ (Ca2+i), induce cellular retraction, activates nuclear factor of activated T cell 1 (NFAT1) and prolongs osteoclast survival [307]. Undifferentiated and differentiated ATDC5 chondroprogenitor cells were found to express LPA [308].
Figure 6.

Production of lysophosphatidic acid (LPA) via two main pathways: Half of serum LPA is formed through the generation of lysophospholipids (LPLs), such as lyso-PC (LPC), lyso-PE (LPE), and LPS, by secreted PLA2 (sPLA2-IIA) or PS-PLA1 from membrane phospholipids of activated platelets, followed by conversion of the LPLs to LPA by autotaxin (ATX). The other half of serum LPA can be generated by sequential action of lecithin: cholesterol acyltransferase (LCAT) or PLA1 and ATX. LPA produced by ATX has various roles mediated by LPA receptors. LPA in cells may originate from PA. In this pathway, a PLD or DAG kinase convert lipids in PA and then PA is hydrolyzed by PLA1 or PLA2.
3.11. The Effects of PLA Metabolites at Matrix Vesicle Level
In vivo, increasing content of LPLs in the growth plate is associated with the onset of mineralization events and participates in the breakdown of MV membrane. Depletion of PC and increase in LPC are characteristic of MVs and are already observed in the microvilli from which MVs derive [309]. Indeed, 10%–15% of the total phospholipids in MVs from chicken growth plate cartilage are LPLs [52,310–312]. LPLs may destabilize the MV membrane, leading eventually to the loss of membrane integrity and release of MMP-3 into ECM, stimulating the ECM components digestion [294] as well as release of calcium phosphate crystals from MVs to ECM [50]. LPLs could also serve as a reservoir for generation of pro-mineralizing Pi, as TNAP was reported to hydrolyse LPLs [313]. This enzyme, purified from the osseous plate, displayed broad substrate specificity. In fact, TNAP from osseous tissues as well as hyperthrophic chondrocytes is a multifunctional enzyme capable of hydrolyzing phosphate monoesters, PPi and phosphodiesters [314,315]. In addition, NPPs present in MVs share the same capability to hydrolyze phosphodiester bonds, acting on distinct substrates that include nucleosides triphosphates, LPLs and choline phosphate esters [49]. The composition of lipids in chicken growth plate cartilage MVs has been described to change significantly during the process of mineralization and it is accompanied by a rise in free fatty acid and LPLs produced by PLAs identified in MVs [50]. By analysing chicken cartilage MV phospholipid content and composition, it was suggested that LPE, detected by UV absorption at 205 nm which indicates the presence of unsaturated fatty acids found in the MV membrane, must have arisen from action of PLA1[50]. On the other hand, PLA in MVs displayed a PLA2 activity, which was Ca2+-dependent, highly selective for intramembranous, as opposed to externally added phospholipids substrates, had optimal activity at pH 8 and hydrolyzed PC in preference of PE or other membrane phospholipids [53]. It has been observed that MVs released by hypertrophic chondrocytes contain higher levels of TNAP and PLA2 specific activities than MVs released by the resting zone cells [316,317]. Addition of 10−8 to 10−9 M of 1α,25-(OH)D3 significantly increased PLA2 activity in MV but not in plasma membrane.
4. Non-Specific Phospholipase C
4.1. Groups, Subgroups and Specificity
PLC cleaves the polar head phosphate from phospholipids, producing DAG (Figures 2,3C). The polar head phosphate is released into the cytoplasm, whereas DAG remains as an integral component of the membrane. The observation that certain activators of protein kinase C (PKC) function as DAG or potent tumor promoters [318] suggests the possibility that uncontrolled activation of PLC may trigger a mitogenic response. In mammalian cells, PLC has been implicated in intracellular signal transduction, vesicle transport, endocytosis, exocytosis, ion channel function, mitosis, cytoskeletal reorganization, and neuronal signal transduction [319]. On the basis of their substrate specificity, two major classes of PLC, namely PI-specific PLC (PI-PLC), with specificity towards PI and non specific PLC, PC-PLC, hydrolysing PE or PC, were identified. PC-PLC has been found in many organisms, from bacteria to mammals [319]. PC-PLC catalyzes the hydrolysis of PC, generating PChol and DAG. PC-PLC was found in the cytoplasm, plasma membrane, and the nucleus. PC-PLC is a potential target for therapy in inflammation-associated diseases such as atherosclerosis [320,321]. PC-PLC is implicated in cytokine signaling pathways, such those of interferon-γ [322], transforming growth factor-β [323] and TNF-α [324]. D609, an inhibitor of PC-PLC, blocked the progression of atherosclerotic lesions in ApoE−/− mice. The lesions contained less lipid and matrix MMPs, fewer macrophages and more smooth muscle cells and collagen [325]. In the future, PC-PLC might serve as a marker in diagnosis of atherosclerosis in the future and as a new target for atherosclerosis therapy.
4.2. Presence of PC-PLC in Chondrocytes and in Osteoblasts and Its Possible Role
To the best of our knowledge, there are almost no reports on the presence and role of PC-PLC in chondrocytes. A pathway for the liberation of AA in osteoblasts involves the non specific hydrolysis of PI and PE by PLC followed by the deesterification of DAG. This pathway can be activated by a phorbol ester through a protein kinase C-independent mechanism [326]. Little information is available on the roles of PC-PLC in osteoblasts, especially during intracellular cell signaling in osteoblast-like MCT3T3-E1 cells. IL-6 stimulates bone resorption and induces osteoclast formation [327]. IL-6 synthesis is induced by IL-1 which is a potent resorptive agent and osteoblasts, rather than osteoclasts, have receptors for many resorptive molecules [328]. The regulatory mechanism of IL-1action in MCT3T3-E1 cells consists of activation of PKC via PC-PLC [329]. Inhibitors of PKC (staurosporine and calphostin) or of PC-PLC tricylodecan-9-yl xanthogenate (D609) enhanced the IL-6 secretion by IL-1 [329] suggesting that IL-1, by activating PKC via PC-PLC limits IL-6 synthesis, while possibly another pathway induced by IL-1 stimulates IL-6 synthesis. IL-6 synthesis is also regulated by TNF which activates PKC via PC-PLC in MCT3-E1 cells [330]. Hydrolysis of sphyngomyelin and PC are activated upon TNF stimulation [330]. PGF2α-induced proliferation in osteoblast-like MC3T3-E1 cells was accompanied by sustained increase in DAG which, in turn, was mediated mainly via tyrosine kinase(s)-dependent PC hydrolysis by a PC-PLC [331]. One of the mechanisms leading to lead-induced apoptosis in cultured rat primary osteoblasts may involve a PC-PLC activity [332].
4.3. Presence of PC-PLC in Osteoclasts and Possible Roles
A possible role of a PC-PLC in BM cells has been evidenced using a PC-PLC specific inhibitor D609. It was found that TNF-α promoted RANKL-induced osteoclastogenesis, at least partially, through the PC-PLC/inositol-1,4,5-trisphosphate (IP3) receptors/NFAT1 pathway [333].
4.4. Presence of PC-PLC in Smooth Muscle Cells and Possible Roles
IL-4 induced OPG mRNA levels and protein secretion by 5-fold in a dose- and time-dependent fashion in human coronary artery smooth muscle cells. Addition of inhibitor D609 decreased OPG expression controlled by IL-4-induced signal transducer and activator of transcription (STAT6) activation suggesting that PLC may participate in the transformation of smooth muscle cells towards an osteoblastic phenotype [334].
4.5. The Effect of PLC Metabolites in Matrix Vesicles
It has been described [50] that the PE and PC content of the MV membrane decreases during mineralization with some accumulation of DAG in MVs, suggesting the involvement of a non-specific PLC activity. In addition there was some accumulation of monoacylglycerol during MV mineralization indicating a lyso-PLC activity [50]. The MV membranes are rich in both PE and PC and may act as a reservoir for both phospholipid classes during early stages of mineralization. Among different enzymes involved in further metabolism of PLC metabolites (Figure 3C), PHOSPHO1, a soluble cytosolic phosphatase entrapped inside MVs [47,53,54], was found to be implicated in generation of Pi for mineralization [46,47]. PHOSPHO1 is capable to hydrolyse PEA and PChol to generate Pi[46,47]. The enzyme activity is highly Mg2+-dependent, with optimal concentration of Mg2+ between 1 and 10 mM. The apparent Km values amounted to 3.0 μM for PEA and 11.4 μM for PChol [46,47]. It has also been hypothesized that PEA is a natural substrate for TNAP since an increase in its urinary excretion in patients diagnosed with hypophosphatasia has been observed [335]. Kinetic and biochemical analysis of TNAP isolated from human Saos 2 cells revealed that this enzyme possesses also phosphatase activity towards PEA [336]. One possible role of PLC in MVs is to produce PEA and PChol which can be further hydrolyzed by PHOSPHO1 and TNAP producing Pi necessary for MV-mediated mineralization [53]. So far, there are no reports that point out experimental evidences of PLC activity toward PC or PE in chondrocytes [53]. Alternatively, a sphyngomyelinase activity could contribute to the production of PEA and PChol.
5. PI-Specific Phospholipase C
5.1. Groups, Subgroups and Specificity
PI hydrolysis by PLC results in the production of two second messengers, IP3 (Figures 2,3C) which mobilizes calcium ions from intracellular calcium stores, and DAG, a physiological activator of PKC isoforms [337]. PI-PLC is a soluble protein that is localized mainly in the cytoplasm and is translocated to the plasma membrane where it hydrolyzes PI 4,5-bisphosphate (PIP2) in response to cell activation [75]. PIP2 is a precursor not only of IP3 but also of PI 3,4,5-trsiphosphate (PIP3), which is produced by the action of PI-3 kinase. Strict regulation of the levels of PIP2 and PIP3 is very important for maintaining homeostasis of the body. PIP2 regulates a variety of cells functions, among them cytoskeletal rearrangement [338], membrane trafficking such as endocytosis of the EGF receptor [339], ion channel activity [340]. PIP3 transduces intracellular signals related to cell proliferation or motility. Therefore defects in the generation/degradation of PIP3 cause cancer, diabetes and inflammation [341,342]. Thirteen different mammalian PI-PLC isoforms have been described, and divided into six subclasses namely β [343–345], γ [343,346,347], δ [343], ɛ [348–351], ζ [352] and η [353–355] each of which comprising different isoenzymes: PI-PLC-β1–β4, γ1 and γ2, δ1–δ4, ɛ, ζ and η. The activation mechanisms of several PLCs have been clarified [356] (Table 7). The isoforms can be divided on the basis of amino-acid sequence and their ways of activation. All the PI-PLC isoforms contain catalytic X and Y domains. Outside of the core conserved regions, there is diversity in protein structure that reflects the range of mechanisms utilized for regulation of these enzymes. Except for PI-PLC-ζ, all PLC isoenzymes have a pleckstrin domain (PH) which binds membrane phosphoinositides or regulatory proteins [356,357]. PI-PLC β1–β4 isoenzymes are composed of subtype specific domains and conserved domains, such as catalytic core regions designated as the X and Y domains, which are located between the EF-hand motif and two phospholipid-binding regions the PH and C2 domains [356]. The catalytic core of PI-PLC-γ isozymes comprises a split PH domain flanking two tandem src homology 2 (SH2) domain inserts of the catalytic core of PLC-γ and SH3 domain between the two halves of the TIM Barrel catalytic domain [356]. Members of the PLCγ class are regulated by receptors that are coupled to tyrosine kinases [357]. PI-PLC-δ, comprising of a PH domain, EF hand motif, X and Y domains and a C2 domain is considered as the most basic isozyme due to its simple structure [356]. PI-PLCɛ is unique in relation to other PLC isoforms in terms of its ability to be regulated by multiple signaling inputs from both Rho family GTPases and heteromeric G protein. [358,359]. Two forms of PI-PLC-ɛ differing in size by 25 kDa were found and are designated as PI-PLC-ɛ1a and PI-PLC-ɛ1b [360]. No functional differences between the two splice variants have as yet been identified. The identification of an independent nuclear polyphosphoinositides signaling machinery has paved the way to find new roles for these molecules. Although several distinct isozymes of PI-PLC have been detected in the nucleus, the isoform that has been most consistently highlighted as being nuclear is PI-PLC-β1 where signaling occurs not only at the plasma membrane but also in the nucleus [356]. Indeed, all the four members of PI-PLC-β contain a high proportion of basic residues at their C-terminal domain, critical for nuclear localization [361,362]. Nuclear PI-PLC-β1 has been linked with either cell proliferation or cell differentiation [363]. The presence of other PI-PLC isozymes, such as PI-PLC-γ1, -δ1, -δ4, and PLC-ζ, in the nucleus have been reported [362]. PLC-γ1 is essential for cell proliferation and cell differentiation [364–367].
Table 7.
| Type | Group | Origin |
|---|---|---|
| Non-specific PLC | Mammalian | |
| PI-specific | PLC-β PLCB1, PLCB2, PLCB3, PLCB4 | Mammalian |
| PI-specific | PLC-γ PLCG1, PLCG2 | Mammalian |
| PI-specific | PLC-δ PLCD1, PLCD3, PLCD4 | Mammalian |
| PI-specific | PLC-ɛ PLCE1 | Mammalian |
| PI-specific | PLC-η PLCH1, PLCH2 | Mammalian |
| PI-specific | PLC-ζ PLCZ1 | Mammalian |
| Phospholipase C-like | PLCL1, PLCL2 | Mammalian |
| Zinc-dependent prokaryotic PLC | Bacterial | |
| PI-DAG-lyase | Trypanosome | |
| SMase | Neutral SMase1 | Mammalian |
| Neutral SMase2 (SMPD3) | Mammalian | |
| Neutral SMase3 | Mammalian | |
| Lysosmal acid SMase | Mammalian | |
| Secreted zinc-dependent acid SMase | Mammalian | |
| Alkaline SMase | Mammalian |
5.2. PI-PLC in Tissues
The distribution of PI-PLC isozymes is tissue and organ specific [356,359,362,369]. PI-PLC-β1 is highly expressed in the cerebral cortex and hippocampus [370] compared to limited expression of PI-PLC-β2 in hematopoietic cells [369,371]; PI-PLC-β3 is found in brain, liver, and parotid gland [372]; PI-PLC-β4 is present at the highest level in cerebellum and retina [373]. Two mammalian subtypes of PI-PLC-γ isozymes have been identified. PI-PLC-γ1 is abundantly expressed in embryonal cortical structures, neurons, oligodendrocytes and astrocytes [374]. The expression of PI-PLC-γ2 is primarily limited to cells of hematopoietic lineage. PI-PLCδ1 is present at high abundance in brain, heart, lung, skeletal muscle and testis. PI-PLC-δ3 is detected abundantly in brain, skeletal muscle and heart [375]. PI-PLC-δ4 mRNA is expressed in various tissues with the highest levels detected selectively in brain, skeletal muscle, testis and kidney [376]. PI-PLC-ɛ mRNA expression has been detected in brain, lung, and colon, with the highest expression detected in heart. PI-PLC-ζ expression within the testis is sperm-specific. Two PI-PLC-η isozymes, PI-PLC-η1 and PI-PLC-η2, were identified in humans and mice. The highest level of PI-PLC-η1 mRNA was observed in brain and kidney and smaller levels were detected in lung, spleen, intestine, thymus and pancreas [353]. As evaluated from the EST database in NCBI Unigene (http:/www.ncbi.nlm.nih.gov/sites/entrez?db=unigene), PI-PLC-β2, PI-PLC-γ1, PI-PLC-γ2, PI-PLC-δ1 and PI-PLC-ɛ genes are expressed in bone tissues but to a limited degree compared with their expressions in other tissues [356].
5.3. Presence of PI-PLC in Chondrocytes and Possible Roles
Chondrocytes treated with PLC from Clostridium welchii divided repeatedly but failed to synthesize metachromatic matrix [377] suggesting that PLC may have a functional role in chondrocytes. Most of the experimental evidence of PI-PLC in chondrocytes comes from the use of U-73122, a PLC inhibitor. Earlier experimental evidence of PLC activity derived from articular chondrocytes upon fluid-induced shear. The shear-induced change in matrix molecule metabolism was influenced by NO synthesis, G protein activation and PLC activation [378]. The molecular mechanisms on the effects induced by mechanical stress indicate the participation of PI-PLC. Mechanical stress induced Ca2+ influx in primary cultures of rabbit articular chondrocytes and stimulated PI-PLC activity [379]. Periodic mechanical stress of rat chondrocytes—leading to chondrocyte area expansion and migration—implies phophorylation of tyrosine kinase protein Src, which in turn activates PI-PLC that regulates ERK1/2 activity [380]. Since the later activity was inhibited by U73122, it was assumed that a PI-PLC-γ1 was involved in this pathway [380] probably due to its activation by tyrosine protein kinase [381]. The use of calcium blockers or ionophores served to identify the presence of Ca2+-sensitive PI-PLC. Insulin-like growth factor-1 induced an increase in Ca2+i that was pertussis toxin (PTX) dependent in articular chondrocytes from 21-day-old rabbits. Treatment with U-73122 [382] partially blocked the Ca2+i increase suggesting that PLC coupled to a PTX-sensitive G protein was present in chondrocytes [383]. In HIG-82 synovial cells, the ionophore ionomycin stimulated phosphoinositide hydrolysis indicating the expression of a Ca2+-sensitive PI-PLC activity in these cells [384,385]. PI-PLC (very often abbreviated as PLC) activity has been evidenced during determination of signaling pathways induced by vitamin D metabolites, sex hormornes, 17β-estradiol, adrenocorticotrpin, eotaxin-1, FGF-3, etc. The resting zone chondrocytes from costochondral cartilage responds preferentially to the vitamin D metabolite 24R,25-dihydroxyvitamin D3 (24R,25(OH)2D3), whereas hypertrophic chondrocytes respond preferentially to 1α,25(OH)2D3[386,387]. In chondrocytes from the costochondral cartilage growth zone, 1α,25(OH2)D3 causes a rapid increase in PLA2 activity [388] producing AA and LPL. AA can stimulate PKC activity [389] and can serve as a substrate for COX-1. LPL activates PI-PLC (possibly PI-PLC-β) since it is a G protein sensitive (Gαq) pathway- resulting in IP3 and DAG production which contributes to PKCα activation and downstream activation of ERK1/2 [388,390–393]. PI-PLC-β1 and PI-PLC-β3 were proposed to be involved in LPL-activation of PI-PLC [393] but they are not expressed in bone or in BM [356]. Growth-plate chondrocytes respond to17β-estradiol in a sex-specific manner by inducing an increase in IP3 which suggests the involvement of PI-PLC activity [394] as reported earlier [395,396]. It was suggested that in resting zone chondrocytes, 24R,25(OH)2D3 was initiating LPA mediated stimulation of G-induced PI-PLC activity [397]. Resting chondrocytes treated with melanocortin peptide or/and ACTH showed elevated basal Ca2+ level that was decreased [398] by U-73122 [382]. The sex hormorne, testosterone, and its metabolite, 5α-dihydrotestosterone (DHT), play an important role in skeletal development in males during adolescence [399]. The effects of DHT were observed in resting-zone chondrocytes from rats in a sex-specific manner i.e., only in males. PI-PLC was required for the DHT–dependent activation of PKC [400] as evidenced by the effect of U-73122. The eotaxin-dependent matrix metalloproteinase secretion in human chondrosarcoma cell line SW1353 is regulated by a PI-PLC-PKC cascade and c-Jun N-terminal kinase/MAP kinase pathways [401]. PI-PLC-γ mediates FGF-3-induced a STAT1 in ATDC5 chondrogenic cells [402].
5.4. Presence of PI-PLC in Osteoblasts
Osteoblast-like osteosarcoma UMR-106 cells possess at least two distinct PLC activities, one predominant in the cytosol and activated by increasing cytosolic Ca2+ with PI as the substrate. The second enzyme, a GTP-activated PI-4, 5-bisphosphate (PIP2)-specific PLC is found in the plasma membranes [403]. PI-PLC-β1, -β3, -γ1, -γ2, and -δ1 were detected by Western blot in osteosarcoma MG-63, MNNG/HOS, OST, U-2/OS, and SaOS-2 cell lines, while PI-PLC-β2 was only expressed in MG-63 and MNNG/HOS cells [404]. PI-PLC-β2 is involved in the mechanotransduction in primary osteoblasts [404]. PLC-γ1 plays an important role in the regulation of cell proliferation and differentiation by generation of the second messengers, DAG, and IP3. PLC-γ1, ERK1/2, and nuclear factor κB (NF-κB) signaling pathways are stimulated while p38 MAP kinase is inhibited by H2O2-induced oxidative stress during rabbit BM stromal cell differentiation. [405]. Elevated extracellular Ca2+ (Ca2+e) stimulates both chemotaxis and mitogenesis of MC3T3-E1 osteoblasts via a calcium-sensing receptor (CaR). Ca2+e-mediated chemotaxis of these bone-forming cells is dependent on PLC [406]. Alternatively, most of the evidence of the presence of PI-PLC in osteoblasts originates from ligand-induced signaling pathways involving Ca2+i increase with the production of DAG and IP3 as reported below.
5.4.1. Endothelin-1 Induced Signaling Pathway
Endothelin-1 (ET-1)—a vasoactive peptide derived from endothelial cells—by binding to high-affinity receptors in MC3T3-E1 osteoblast cells, induces PLC activation with the production of two second messengers, IP3 and DAG and a biphasic increase in Ca2+i, as measured with a fluorescent indicator, fura-2 [407,408]. It has been shown that ET-1 inhibits osteoclast bone resorption by a direct effect on cell motility and that it can also activate PLC in the osteoblast [409]. In MC3T3-E1 osteoblast-like cells, ET-1 acting through ET receptor, links to a stimulation of Pi transport via activation of PKC through both phosphoinositide and PC hydrolyses [410]. In osteoblast-like MC3T3-E1 cells, various ET peptides and their homologous sarafotoxins generate PGE2 release through an ET(A) receptor subtype. PLC-dependent calcium activation mechanisms seem to be involved [411].
5.4.2. Basic FGF Induced Signaling Pathway
Basic FGF selectively stimulates sodium coupled Pi transport activity in osteoblast-like cells. Signaling mechanisms responsible for this effect involve mainly activation of PI-PLC-γ and PKC, with some possible contribution of the p38 MAP kinase [412]. Basic FGF, which is able to increase the rate of bone formation, stimulates fibronectin expression by binding to FGF-2 receptor and activation of PI-PLCγ2, PKCα, c-Src in rat osteoblasts [413].
5.4.3. Platelet-Derived Growth Factor Induced Signaling Pathway
Platelet-derived growth factor (PDGF) is a potent and selective stimulator of Pi transport in osteoblastic cells. The mechanism responsible for this effect is not mediated by MAP kinases but involves tyrosine phosphorylation-dependent activation of PLCγ and PI-3-kinase [414]. PDGF-mediated chemotaxis of MC3T3-E1 osteoblast-like cells is dependent on both PLC and PI-3-kinase [415]. Moennings et al.[416] showed for the first time that PDGFRα signaling stimulates osteogenesis of neural crest cells-derived osteoblasts by activating the PI-PLC-γ pathway. This may suggest an involvement of this pathway in the etiology of human craniosynostosis.
5.4.4. Parathyroid Hormone Induced Signaling Pathway
PTH is known to have both catabolic and anabolic effects on bone. The dual functionality of PTH may stem from its ability to activate two signal transduction mechanisms: adenylate cyclase and PLC [417,418]. In osteoblasts, PI-PLC-β2 transduces the signals from PTH, PGE2, and other prostanoids [419,420]. Several reports suggest that PTH activation of PKC, via the stimulation of PLC, plays a role in stimulating the synthesis and release of transforming growth factor-b1 (TGF-b1) [421] as well as in the PTH-stimulated synthesis of insulin-like growth factor binding protein-5 [422]. Both of these factors stimulate bone deposition by activating osteoblast growth and differentiation and may, therefore, play a role in the coupling of bone resorption to bone deposition. Regulation of the PLC pathway through the PTH1R can be significantly increased by elevating the expression of G(11)α in osteoblastic cells. This leads to increased PTH stimulation of MMP-13 expression by activation of AP-1 factors: c-jun and c-fos [423].
5.4.5. PGD2 Induced-Signaling Pathway
PGD2 stimulates Ca2+ influx from the extracellular space and activates phosphoinositide (PI)-hydrolyzing PLC and PC-hydrolyzing PLD independently of PGE2 or PGF2α in osteoblast-like MC3T3-E1 cells [251,424]. Thrombin raises Ca2+i, in UMR 106-H5 rat osteoblast-like osteosarcoma cells by activating PI-PLC [425]. Exogenous PA appears to increase IP3 accumulation in osteoblast-like cell line MOB 3-4 by activating PI-PLC [426].
5.4.6. PGE2 Induced-Signaling Pathway
U-73122 and calphostin C reduced the PGE2-induced phosphorylation of p44/p42 MAP kinase and p38 MAP kinase. These results indicate that PGE2 stimulates the induction of HSP27 through PKC-dependent activations of both p44/p42 MAP kinase and p38 MAP kinase in osteoblasts [427]. Bradykinin increased both IL-6 and PGE2 synthesis in osteoblastic cells via B2R. PLC, IP3-induced Ca2+i, and MAP kinases were involved in signal transduction in these cells [428]. PGE2 evoked a Ca2+i, rise via a PI-PLC pathway in MC3T3-E1 osteoblasts, particularly in the growing phase [429]. The proton induced COX-2 expression and PGE2 production were mediated through the ovarian cancer G protein–coupled receptor/Gq/11/PLC pathway in human osteoblastic cells [430].
5.4.7. PGF2 Induced-Signaling Pathway
PGF2α induces phosphoinositide hydrolysis by PLC and PC hydrolysis by PLD through heterotrimeric GTP-binding protein, resulting in the activation of PKC in osteoblast-like MC3T3-E1 cells. PGF2α can also stimulate the synthesis of DNA [431,432]. Zinc reduces PGF2α-induced IL-6 synthesis via suppression of phosphoinositide-hydrolyzing PLC and PC-hydrolyzing PLD in osteoblasts [433]. It is well known that osteoporosis is a common complication in patients with glucocorticoid excess. Glucocorticoid inhibits PGF2α-induced PGE2 synthesis through the inhibition of PI hydrolysis by PLC as well as PLA2 in osteoblast-like cells [434]. Contrary to sphingosine 1-phosphate (S1P), sphingosine inhibits PGF2α-induced phosphoinositide hydrolysis by PLC via p38 MAP kinase in osteoblasts. [435,436].
5.4.8. Vitamin D-Induced Signaling Pathway
Only PI-PLC-β1 linked to a PTX-insensitive G-protein and PLC-β2 coupled to a PTX-sensitive G protein are involved in the effects of calcitriol and 17β estradiol (the hormonally active form of vitamin D,) respectively, on the mobilization of Ca2+ from Ca2+i stores. [437]. PLC-β1 is the target effector of Gα(q/11), whereas PI-PLC-β2 is only activated by βγ subunits; this specificity may help to generate membrane receptor-specific responses in vivo[438]. When osteoblasts are cultured on surfaces of increasing micro roughness, they exhibit decreases in proliferation, increases in differentiation and local factor production, and enhanced response to 1α,25(OH)2D3. The cells interact with surfaces through integrins, which signal by the same pathways used by 1α,25(OH)2D3, i.e., they activate PKC via PLC and protein kinase A via PLA2. This provides opportunities for crosstalk that may contribute to the synergistic effects of surface roughness and the vitamin D metabolite [439].
5.4.9. Interleukin-1-Induced Signaling Pathway
PI-PLC-β1 is specifically localized in the nucleus of Saos-2 osteoblast, where it is activated when cells are stimulated with IL-1 [440,441]. Saos-2 cells are characterized by the expression of high affinity receptors for IL-lα, which is one of the most potent stimulators of bone resorption [442]. A recent report [443] demonstrated that nuclear activation of PI-PLCβ1 was dependent on its phosphorylation by the MAP kinase. The MAP kinase pathway is implicated in the pathogenesis of RA because it is activated by proinflammatory cytokines, such as TNF-α and IL-1β [444].
5.4.10. Miscelanous Ligand Binding Stimulated PI-PLC in Osteoblasts
Pasteurella multocida toxin, a mitogenic toxin, acts to inhibit differentiation, in particular of bone cells. In vitro, it prevents the formation of mineralized bone nodules. P. multocida toxin action stimulates PLC leading to activation of protein kinase C, an increase in inositol phosphates, and a rise in Ca2+i[445]. In human osteosarcoma MG63 cells, thymol causes a Ca2+i rise by inducing PLC-dependent Ca2+release from the endoplasmic reticulum and Ca2+ entry via protein kinase C-sensitive store-operated Ca2+channels [446]. CGRP a peptide produced locally in bone, and that may act as a cytokine on bone cells, is not coupled to adenylate cyclase but increases Ca2+i levels in CGRP receptor-positive OHS-4 osteosarcoma cells, suggesting that in these cells CGRP induces downstream events driven by PLC in these cells [446]. N-formyl-methionyl-leucyl-phenylalanine -stimulated osteogenic differentiation of human mesenchymal stem cell which was mediated via the N-formyl peptide receptor-PLC/PLD-Ca2+-calmodulin-dependent kinase II-ERK-CREB signaling pathways [447]. The pretreatment of human osteoblast SaM-1 cells with U-73122, a PLC inhibitor, stopped IL-6 and IL-8 synthesis in response to extracellular LPA. The proposed mechanism may involve activation of PLC and IP3-mediated Ca2+i release in human SaM-1 cells [448].
5.4.11. Purinergic and Serotonin-2 B Receptors
Under certain stress conditions that lead to release of nucleotides from the rat osteoblastic cell line ROS-A 17/2.8, the stimulation of specific purinergic receptors such as P2Y2, sensitizes mechanical stress activated Ca2+ channel through a mechanism that involves PI-PLC activation. [449]. U-73122 and thapsigargin, a calcium-pump inhibitor, both significantly inhibited the increase in Ca2+i induced by extracellular ATP in TBR31-2 cells. This suggests that the increase in Ca2+i is due to Ca2+i release from the calcium store following activation of PLC [450]. Mice knocked out for the serotonin-2B receptor (5-HT2BR) show defects in bone homeostasis. In C1 osteogenic cells the positive action of 5-HT2BR on TNAP downstream from eicosanoids requires the activity of the glycosyl-PI-solubilizing enzyme PI-PLC [250].
5.5. Presence of PI-PLC in Osteoclasts
During bone resorption osteoclasts remove large amounts of ECM. At the beginning of the resorption cycle, the plasma membrane in contact with the bone expands through fusion of lysosomes and intracellular vesicles into the convoluted ruffled borders. Consistent with the function of osteoclasts, some of the PI-PLC identified in osteoclasts are involved in membrane trafficking and in cytoskeletal rearrangement. PI-PLCγ2 modulates bone homeostasis by affecting osteoclast recruitment and function. PI-PLCγ2 is implicated in actin cytoskeletal reorganisation in osteoclasts and neutrophils. It is an important regulator of α(v)β(3) integrin-mediated bone osteoclast cell adhesion, migration, and in bone resorption [346,451,452]. Although PI-PLCγ1 is expressed in osteoclasts, it cannot compensate for the absence of PI-PLCγ2 [346], suggesting that PI-PLCγ1 and PI-PLCγ2 are implicated in distinct signaling pathways. Experimental evidence of the presence of PI-PLC in osteoclasts arises from the analysis of the mechanisms induced by endocrine and paracrine factors that regulate osteoclast formation and activity. Such factors include calcitonin, RANK ligand, Ca2+, H+, nucleotides regulating osteoclast activity in several ways. Very often, the PLC activity in osteoclasts was evidenced by the use of PLC inhibitors such as U-73122.
5.5.1. Calcitonin Induced Signaling Pathway
Calcitonin inhibits the activity and changes the morphology of osteoclasts by interfering with trafficking to and from the ruffled border [453,454]. Such action makes it a possible therapeutic target for the treatment of osteoporosis [455]. Using U-73122, PLC was shown to be implicated in the inhibition of endocytosis from the ruffled borders of rabbit osteoclasts induced by a calcitonin treatment. The PLC inhibitor reversed the calcitonin effect and restored endocytic trafficking to the level equivalent to 75% of that in untreated controls [456].
5.5.2. Intracellular Ca2+ Induced Signaling Pathway
It is essential to make a distinction between the effect of Ca2+e in osteoclasts that may stimulate the rise in Ca2+i in osteoclasts and the effect of other ligands that may induce an increase in Ca2+i concentration. The latter may affect osteoclast differentiation and function differently although both may involve PLC activity. Increasing Ca2+e to levels comparable to those resulting from local bone resorption inhibits osteoclast differentiation and osteoclastic bone resorption [457]. Osteoclasts can sense increasing levels of Ca2+e, which in turn trigger a rapid rise in the cytosolic calcium concentration, disassembly of podosomes, and osteoclast apoptosis [458–461]. RANKL induced an increase in Ca2+i of extracellular origin resulting of the opening of calcium channels—possibly transient receptor potential vanilloid channels 5—on the surface of human osteoclasts [462]. Ca2+i oscillation could be triggered by a RANKL-dependent receptor potential vanilloid channels 2 calcium channel in preosteoclast RAW264.7 cells [463]. Similarly to RANKL, Ca2+e (20 mM) appeared to trigger rapid and significant nuclear translocation of NF-κB in a CaR- and PLC-dependent manner [464,465] (Figure 7). The CaR is coupled to PLC activity that induces an increase in Ca2+i concentration. Sre2+—which exerts both an anti-catabolic and an anabolic effect on bone cells—acts through CaR and induces osteoclast apoptosis through a signaling pathway similar to but different in certain respects from that of Cae2+ (Figure 7) [465]. The cation sensing by osteoclast-like GCT23 cells is mediated by a PLC-coupled receptor [466]. Osteoclast activity is inhibited by elevated Ca2+e that induce a PLC-dependent rise in Ca2+i Lyn [467,468] as well as leucine-rich repeat-containing 17 (LRRc17) [469]—by interfering with the PI-PLC-γ1—both down-regulated Ca2+e signaling and inhibited osteoclast differentiation [467]. In constrast to the inhibition effects on osteoclasts induced by Lyn or LRRc17, calcitonin increases Ca2+i via a PLC-PKC-calcium signaling pathway, stimulating endocytosis in osteoclasts [456]. Several reports confirmed the involvement of PLC in the increase in Ca2+i. U-73122 blocks the calcium sensitive component in avian osteoclasts [470]. Extracellular nucleotides caused elevation of Ca2+i in osteoclasts—isolated from femora and tibiae of rat or rabbit pups—by activating P2Y receptors [471]. Inhibition of PLC with U-73122 or inhibition of endoplasmic reticulum Ca2+-ATPase with cyclopiazonic acid or thapsigargin abolished the rise of Ca2+i induced by the binding of nucleotides to P2Y receptors [471]. RANKL acting on osteoclasts isolated from the long bones of neonatal Wistar rats or or New Zealand white rabbits elevated Ca2+i in Ca2+-containing and Ca2+-free media. The increase in Ca2+ was prevented by U-73122 [472,473]. It was suggested that PI-PLC-γ could regulate Ca2+ channel during RANKL-signaling for terminal differentiation of osteoclasts and that RGS12 was essential for the terminal differentiation of osteoclasts induced by RANKL [474]. It was proposed that PI-PLC-γ2 mediates RANKL-induced osteoclastogenesis and is a potential candidate for the antiresorptive therapy [475]. Other proteins such as RGS10 can mediate PLC activation and Ca2+i oscillations [474]. The Ca2+i calmodulin complex competes for the PIP3-binding site on RGS10 and frees the bound PIP3. Once the Ca2+i concentration reaches its peak, Ca2+i begins to reload into the endoplasmic reticulum and the Ca2+i calmodulin complex dissociates from RGS10 at the low Ca2+i concentration. Free PIP3 activates PLC and then binds RGS10 again. PLC activation triggers a release of Ca2+i from intracellular stores by generating IP3 to induce a second peak. This process continues to repeat itself, causing Ca2+i oscillations. RGS10 mediates PLC activation and Ca2+i oscillations through its Ca2+i -dependent dual interaction with Ca2+i calmodulin and PIP3[474].
Figure 7.

Calcium and strontium induce distinct intracelular calcium signaling. Upon stimulation by extracellular calcium, calcium-sensing receptor (CaR) activates PLC, which is responsible for the translocation of nuclear factor κB (NF-κB) from the cytoplasm to the nucleus in mature osteoclasts, in an IP3-dependent manner. Upon stimulation by extracellular strontium, CaR also activates PLC inducing the DAG-PKCβII signaling pathway and then promoting translocation of NF-κB from the cytoplasm to the nucleus in an IP3-independent manner. Taken from [465].
5.5.3. Osteoprotegrin Induced Signaling Pathway
OPG-inhibitor of osteoclast differentiation—directly binds to RANKL, whereas LRRc17 acts as a negative regulator of RANKL-induced murine osteoclast differentiation by blocking PI-PLCγ signaling [469]. PLC exerts indirect effect in endothelial cells of the bone vasculature, modulating development, remodeling, and bone repair by secreting OPG which acts on osteoblastic and osteoclastic lineage cells. For example, IL-4 and IL-13 induced OPG expression through activation of a PLC-sensitive STAT6 pathway in human umbilical vein endothelial cells [476].
5.5.4. RANK Induced Signaling Pathways
Mice lacking the tyrosine kinases Btk and Tec show severe osteopetrosis caused by a defect in bone resorption indicating that Btk and Tec kinases are crucial in the regulation of osteoclast differentiation [477]. RANKL induced tyrosine phosphorylation of PI-PLCγ1 and PI-PLCγ2 was markedly suppressed in Tec−/−Btk−/− cells. Taken together the findings suggest that RANKL binding to RANK results in activation of classical pathways involving TRAF6 and c-Fos. In addition, Tec kinases are phosphorylated by RANK (Figure 8). ITAM phosphorylation results in the recruitment of Syk, leading to activation of adaptor proteins such as BLNK and SLP-76, which function as scaffolds that recruit both Tec kinases and PI-PLCγ to form the osteoclastogenic signaling complex (Figure 8). This complex is required for calcium signaling to activate NFAT1, the key transcription factor for osteoclast differentiation [477]. A molecular mechanism for the long-term link between RANK and ITAM signals has been proposed. A domain in RANK is dispensable for the early phase of RANK and ITAM signaling but is essential for the late-phase signaling, which involves PI-PLC-γ2 binding to RANK [478] (Figure 8). A linker for activation of T cells (LAT), regulates RANKL-induced osteoclast differentiation and is involved in RANKL-induced PI-PLC-γ activation and NFAT1 induction [479]. PI-PLCγ1 is also involved in RANKL-induced Ca2+ oscillations as shown by marked inhibition of the oscillations in BM-derived monocyte/macrophage precursor cells in which PI-PLCγ1 was knocked down with PLCγ1 siRNA [480]. The absence of PI-PLC-γ1 is not compensated by the presence of PI-PLCγ2, suggesting that both PI-PLC-γ1 and PI-PLC-γ2 participate in RANKL-induced Ca2+ oscillations. RANKL induced a significant increase in Ca2+i of extracellular origin, probably as a result of the opening of TRPV-5 calcium channels on the surface of human osteoclasts. Mutant forms of SH3BP2—occuring in patients with cherubism- potentiate RANKL-induced phosphorylation of PI-PLC-γ isoforms, suggesting that SH3BP2, as well as PLC-γ2, are potential targets in the treatment of disorders characterized by excessive osteoclastic development [481,482].
Figure 8.

Integration of the RANK and ITAM Signals by Tec Kinases. RANKL binding to RANK results in activation of pathways involving TRAF6 and c-Fos. Tec kinases are phosphorylated by RANK. ITAM phosphorylation results in the recruitment of Syk, leading to activation of adaptor proteins such as BLNK and SLP-76, which function as scaffolds that recruit both Tec kinases and PLCγ to form the osteoclastogenic signaling complex. This complex induces calcium signaling required for the induction and activation of NFATc1, the transcription factor for osteoclast differentiation. Adapted from [477].
5.5.5. Parathyroid Hormone Induced Signaling Pathway
As in most cells expressing the PTH/PTH-related peptide receptor in cells, stimulation with with PTH agonists results in the activation of two G protein-dependent signaling pathways, the Gαs/adenylyl cyclase/cAMP/protein kinase A pathway and the Gαq/11/PLC/IP3/Ca2+/PKC pathway, as many other GPCRs, activates several signaling pathways, including the Gq/11-linked PI-PLC-PKC signaling pathway as determined in cell cultures. However, there are only few reports investigating whether such signaling occurs in vivo[483]. Estrogens modulate the catabolic effects of PTH on bone in vivo and in vitro. Estrogens suppress PTH-stimulated osteoclast-like cell formation by blocking both the cAMP-dependent protein kinase A pathway and the PLC-coupled calcium/PKC pathway [304,484].
5.6. Presence of PI-PLC in Smooth Muscle Cells and Possible Roles
Most of experimental evidence on the presence of PI-PLC in smooth muscle cells originates from analysis of signaling pathways [485–487] and the use of general PLC inhibitor U73122 or PLC isotype inhibitors D609 and ET-18-OCH3. No evidence was obtained by Western blotting for the presence of PLC-β, PLC-γ and PLC-δ in bovine mesenteric lymphatic smooth muscle cells. However, a PLC activity was concentration-dependently stimulated by Ca2+[488]. In rat thoracic aortic smooth muscle cell, vasopressin induces V1 receptors to release AA, DAG and PChol via activation of both a PI- and PC-PLC [489]. Both isotypes of PLC were involved during VSMC proliferation [490]. S-1-P in Rat arterial VSMC induced time-dependent activation of PI-PLC-β as evidenced by the use of U-73122 [491].
5.7. Presence of PI-PLC in Odontoblasts and Possible Roles
Odontoblasts—extracted from dental pulp of newborn Wistar rats—demonstrated an IP3-induced Ca2+ release activated by PLC-coupled receptors [492]. Calcium ions and PLC were required for the capsaicin-induced expression of OPG in human periodontal ligament, which is known to play an important role in the bone-remodeling process [493]. U73122 was able to ablate the basic FGF-induced neuronal differentiation of dental pulp stem cell (DPSC) and the authors suggested that basic FGF-induced neuronal differentiation of DSPC could involve a PI-PLC-γ pathway [494].
5.8. Genetic Models
Genetically manipulated mice revealed that the lack of some PLC isozymes causes defects in fertilization and development of the circulatory, hematopoietic, immune, and skin systems [495]. PI-PLC-ζ and PI-PLC-δ4 are critical in fertilization [495]. Liao et al.[496] reported that vasculogenesis is impaired in PLC-1 knockout embryos. PI-PLC-δ1/-δ3 knockout mice show mainly placental vascular defects [497]. PI-PLC-β3 plays some role in angiogenesis [498]. PI-PLC-γ1 is essential for renal development and for the development of hematopoietic stem cells [499]. PLC-γ1 and PLC-γ2 play important roles in the immune system, in development of B cells [500] as well in in bone homeostasis [501–506]. PI-PLC-γ2 null mice (PLC-γ2−/−) are osteopetrotic, i.e., shox features of a hereditary disease marked by abnormally dense bone, and by the common occurrence of fractures of the affected bone. [452]. PI-PLC-γ2 knockout mice have less osteoclasts due to defective upregulation of NFAT2, which is a critical transcription factor activated by RANKL that controls osteoclast differentiation [507]. These findings indicate that PLC-γ2 regulates osteoclastogenesis as a downstream effector of RANKL in mice. PI-PLC-γ2 is essential for RANK signaling, and its deficiency leads to defective lymph node organogenesis and osteoclast differentiation [508]. PI-PLCγ2 activation/function may provide opportunities to develop targeted therapeutic approaches for treatment of inflammatory and osteolytic diseases. However, PLC-γ2−/− mice proved that PI-PLC-γ2 is not a major player in ovariectomy-induced bone loss, indicating that PLC-γ2 may not be a suitable therapeutic target in postmenopausal osteoporosis [509].
6. PLC-Related but Catalytically Inactive Protein
PLC related but catalytically inactive protein (PRIP) is a novel molecule in bone biology research and was originally identified as IP3-binding protein. This protein is similar to PLC-δ1 but is catalytically inactive [510,511]. The PRIP family consists of at least two types of proteins (PRIP-1 and PRIP-2 subfamilies). PRIP has a number of binding partners, including the catalytic subunit of protein phosphatase 1α (PP1α) and PP2A [512,513] and the phosphorylated (active) form of Akt [514] in addition to IP3 and PIP2[515]. PRIP gene-deficient mice (prip−/−), genetically deficient in type 1 or type 2 isoforms or both, brought light the physiological functions of PRIP: modulation of GABAA receptor signaling [516], dysfunction of reproduction, negative regulation of multiple-hormone secretion [517] and bone properties [518]. PRIP is involved in the phosphorylation-dependent modulation of exocytosis in PC-12 cells [519]. Exocytosis of various peptide hormones, such as gonadotropins and insulin, was up-regulated in prip−/− mice, indicating that PRIP is likely to be involved in dense core vesicle exocytosis in a negative manner. PRIP is implicated in the regulation of bone formation in a negative manner, partly through the regulation of SMAD phosphorylation [518]. Since PC-PLC contributes to the progression of atherosclerosis, it was proposed that pharmacological blockade of PC-PLC is a possible approach to atherosclerosis therapy.
7. Sphingomyelinase
7.1. Groups, Subgroups and Specificity
Sphingomyelinase (SMase), which may be considered as a subtype of PLC, cleaves SM (ceramide phosphorylcholine) to yield ceramide and PChol (Figure 3D). Ceramide, subsequently metabolized by ceramidase and sphingosine kinase to sphingosine and S1P, respectively, appeared to be a lipid second messenger in programmed cell death, cell differentiation and cell proliferation [368,520,521]. There are at least five isoforms of acidic, neutral, and basic SMases differing mainly on pH profiles, cation requirements, and cellular localization. Smpd3 encodes neutral SMase 2, a membrane-bound enzyme, and is highly expressed in bone. A local neutral SMase2 (also called sphingomyeline phosphodiesterase 3 (SMPD3)) activity is required for a normal bone mineralization and for physiological apoptosis of hypertrophic chondrocytes in the cartilage during early skeletal development [522].
7.2. Presence of Sphingomyelinase in Chondrocytes and Possible Roles
Six years ago it has been shown for the first time [523], that articular chondrocytes express both acidic and neutral SMases and are able, in response to the appropriate external signal, to raise levels of endogenous ceramide; depending on which SMase is activated, an inflammatory (neutral SMase) or apoptotic (acidic SMase) response is observed. SMase is implicated in both chondrocyte apoptosis and ECM degradation during cartilage degeneration [524]. Ceramide stimulated synthesis of specific MMPs which in turn induced degradation of ECM and cell death in cartilage. This suggests that the SMase pathway could participate in vascular invasion of the growth plate by disrupting cartilage-ECM homeostasis, resulting in down-regulation of the type II collagen [524]. In addition, some ceramide metabolites have been implicated in the cartilage degradation and arthritic disease. For example, in Farber’s disease, the lack of ceramidase causes excessive accumulation of ceramide within the cartilage and bone, and is associated with joint pain and arthritis-like joint degeneration [525]. SMase down-regulates type II collagen in articular chondrocytes via activation of the ERK signaling cascade, redistribution of SOX9, and recruitment of c-Fos [526]. These findings provide direct evidence for a role of SMase metabolites in human arthritic disease. This enzyme could represent a target for pharmacological intervention against cartilage loss in arthritic diseases.
7.3. Presence of Sphingomyelinase in Osteoblasts and Possible Roles
Information regarding the effects of ceramide in cells of skeletal origin is limited and conflicting. Ceramide in vitro may be either a pro-death agent or it may protect cells, depending on the experimental conditions. Moreover, TNF-α, IL-1, platelet-derived growth factor and vitamin D3 as potent regulators of bone remodelling may utilize sphingosine metabolites such as ceramide or S1P as second messengers in their respective signal transduction pathways via activation of SMase. This suggests that sphingosine metabolites are operating as intracellular signaling molecules in osteoblasts and osteoclasts. In fact, these metabolites are able to mimic the biological actions of the above cytokines in osteoblasts. The proapoptotic agent TNF-α has been reported to induce osteoblast cell death in a process involving ceramide [527]. However, conversely, ceramide was shown to be mitogenic in MC3T3-E1 osteoblast cells [528]. Among SM metabolites, ceramide enhances the BMP stimulated osteocalcin synthesis in osteoblasts and its effect is exerted at a point upstream from p44/p42 MAP kinase [529]. Scyphostatin—a neutral SMase inhibitor—revealed that neutral SMase-induced release of ceramide directly activated the intrinsic mitochondrial apoptotic pathway [530]. Ceramide signals osteoblast survival and apoptosis through different intracellular pathways, and alteration in the intracellular levels of ceramide may play an important role in bone remodeling [531]. S1P acts as a second messenger for tumor necrosis factor-α-induced synthesis of IL-6 in MC3T3-E1 cells and the p44/p42 MAP kinase is involved in the signaling [330,435,532,533]. It has been shown that not ceramide but sphingosine and S1P transiently mobilize Ca2+i from intracellular stores in osteoblast-like MC3T3-E1 cells [534].
7.4. Presence of Sphyngomyelinase in Osteoclasts and Possible Roles
Sphingolipid metabolism is implicated in osteoclastogenesis. Acid SMase gene was identified as a gene induced by NFAT2 during the late stages of osteoclastogenesis [535]. SMase and C2 ceramide inhibited bone resorption by suppressing osteoclast activity through suppression of F-actin ring formation essential for ruffled border formation [536].
7.5. Genetic Models
Bone deformities in mouse models lacking a functional Smpd3 gene underscore the importance of sphingolipid metabolism in skeletal tissues [522,537,538]. Stoffel et al.[538,539] characterized the skeletal phenotypes of the Smpd3−/− mice as chondrodysplasia and speculated a systemic role for neuronal SMPD3 in the regulation of the skeletal development.
7.6. Effects of Sphyngomyelinase Metabolites at Matrix Vesicle Level
SM is a structural component of MVs released by pre-hypertrophic/upper hypertrophic chondrocytes. SM is specifically enriched in MVs as compared to plasma membrane of growth plate chondrocytes from which they derive, Table 1[53]. During cartilage MV-induced mineralization, there is a progressive disappearance of SM [53]. It was suggested that a neutral SMase-2 could be a possible candidate for the SM hydrolysis [53]. Indeed, SMPD3 has been identified in MVs isolated from osteoblast-like Saos-2 cells [540]. The hydrolytic activity of SMPD3 may serve as additional source of Pi, since SMase produces PChol which is hydrolysed by PHOSPHO1 present in MVs [47,523,541]. This gives rise to the possibility of a novel mechanism by which phosphate may be unleashed through the action of PHOSPHO1 and SMase such as SMPD3 to contribute to the changes in Pi concentration inside MV lumen. Exosome formation in multivesicular bodies is triggered by hydrolysis of sphingolipids and release of ceramide. This reaction is catalyzed by SMPD3 and an inhibitor of SMPD3, GW4869, efficiently abrogates exosome release in the oligodendroglial cell line OLI-neu.
8. Phospholipase D
8.1. Groups, Subgroups and Specificity
PLD belongs to a large superfamily of enzymes which hydrolyzes the phosphodiester bonds of membrane phospholipids, producing PA and polar head group (Figures 2,3B). A large subset of enzymes with PLD activity share a conserved HxKx4Dx6GSxN motif (HKD) [542] or a variation of thereof, which is responsible for catalytic activity [77]. Non-HKD enzymes—such as glycosyl-PI specific PLD (GPI-PLD), N-acyl PE-PLD, cytochrome P450 1A2 and 2E1 as well as ATX- have a PLD activity but have divergent structures and catalytic mechanisms [77] (Table 8). PA is also produced by a DAG kinase from DAG or by LPA acyltransferase from LPA. Alternatively, PA can be also be synthesized by sequential enzyme catalyzed alcylation from glycerol-2-phosphate [77]. PA—due to its small negatively charged group—binds to protein and facilitates changes in lipid bilayer and therefore is implicated in vesicular trafficking, exocytosis and endocytosis [543,544]. PA is also precursor to other lipid signaling molecules such as DAG and LPA. DAG is a well known activator of PKC [545–548]. To date 10 isotypes of PKC have been identified, which are subgrouped in three categories: classical PKC (PKC-α, -β and -γ) require Ca2+, DAG and phospholipids; novel PKC (PKC-δ, -ɛ, -η and -θ) are Ca2+-independent but DAG and phospholipid dependent; and atypical PKC (PKC-ζ, -1/λ and -μ) are insensitive to Ca2+ and DAG [549]. DAG can be converted to AA, a precursor of eicosanoids [550]. In addition, PA—as a lipid messager, can interact with several signaling proteins including Raf-1 [551,552] and the mammalian target of rapamycin (m-TOR) [553]. PA is involved in signaling cascades affecting cell- growth, proliferation and survival [77]. PLD catalyzes the reaction of transphosphatidylation using water or primary alcohols (ethanol or 1-butanol) as nucleophiles to generate PA, phosphatidylethanol or phosphatidylbutanol, respectively [554]. PLD activity has been evidenced in various organisms, including plants, mammals, bacteria and yeast. In humans, two genes, pld1 and pld2, encoding the PLD enzyme were found. pld1 encodes the 124 kDa protein PLD1a (1074 amino acids), and an alternatively spliced form PLD1b (1036 amino acids), which lacks 38 amino acid residues, the most studied variants. There are two other PLD1 splice variants PLD1c and PLD1d. PLD2 encodes a 106 kDa protein with 50% homology to PLD1 [77,554]. PLD2 has three variants PLD2a, 2b and 2c (Table 8). The PLD2b variant lacks 11 amino acids in its C-terminus compared to PLD2a, but it is still functional [555]. Both isoforms are capable of hydrolyzing PC, PE, PS but are not capable of hydrolyzing PI, PG or cardiolipin [77]. In addition, PLD can hydrolyze LPC and LPS and produce LPA. Two other mammalian PLD enzymes have been identified with significant sequence homology to viral PLD: PLD3 or Hu-K4 [556] and an endonuclease-like mitochondrial PLD enzyme [557]. PLD3 activity has not been detected [77], while mitochondrial PLD hydrolyzes cardiolipin to generate PA [557]. It is usually stated that PLD1 and PLD2 are expressed in nearly all mammalian tissues [77,558] and that PLD plays an important role in modulating cellular function [559]. However, very little is known about the presence and function of PLD in osseous tissues. Although both isoforms catalyze the same reactions and utilize similar substrates to generate PA or transphophatidylation species, they have usually distinct subcellular localizations [77]. As reviewed [77], it is generally accepted that PLD1 is localized to perinuclear membranes, including early endosomes and Golgi, under basal conditions [560]. Upon stimulation, PLD1 translocates to the plasma membranes or late endosomes [77]. PLD2 is usually located in the plasma membrane under basal conditions and translocates to the recycling vesicles [77]. PLD2 also binds to β-actin [561]. PLD1 [562] and PLD2 [563] are palmitoylated at two cystein residues and both contain PH and phox homology lipid binding domains. The palmitoylation and the two lipid binding domains contribute to the association of PLD with membrane lipids [554]. PLD activity is regulated by many factors, including phosphoinositides. PLD1 has a low basal activity and is extensively regulated by PKC and members of the ARF and Rho (RhoA, Rac1, Cdc42) families of small GTPases. PLD2 has a higher basal activity than PLD1 but has been shown to respond to ARF and PKC [564]. It has been reported that PLD/PA can directly activate regulatory proteins playing key roles in cell physiology, such as PI-4-phosphate 5-kinase, PKC, PLCγ, Raf-1 kinase and MAP kinase [565]. These proteins are also considered as candidates mediating cellular signaling during osteoblast proliferation and differentiation [566] but also during osteoclast differentiation [567]. PLD and its enzymatic product, PA, regulate the actin cytoskeleton, vesicle trafficking for secretion and endocytosis, and receptor signaling [568]. Free choline is not thought to fulfil any intracellular signaling roles [568]. Although PLD is important for many physiological processes, its function in bone metabolism is unclear. Their presences in chondrocytes and in osteoblasts have been reported.
Table 8.
PLD family (according to [77]).
| Type | Variants | Origin |
|---|---|---|
| PLDs with HKD motif | ||
| PLD1 | PLD1a, PLD1b, PLD1c, PLD1d | Mammalian |
| PLD2 | PLD2a, PLD2b, PLD2c | Mammalian |
| PLD3 | Mammalian | |
| Endonuclease-like mitochondrial PLD | Mammalian | |
| Non-HKD PLDs | ||
| GPI-PLD | Mammalian | |
| N-acyl PE-PLD | Mammalian | |
| cytochrome P450 1A2 | Mammalian | |
| cytochtome P450 2E1 | Mammalian | |
| ATX | Mammalian |
8.2. Presence of PLD in Chondrocytes and Possible Roles
A PKC-regulated PLD activity stimulated by phorbol 12-myristate 13-acetate (a known PLD stimulator) has been evidenced in chondrocytes. This activity could be inhibited with staurosporine—a PKC inhibitor [569]. Other experimental evidence of PLD presence in chondrocytes, which provided more insight into the possible roles of PLD in biomineralization, originated from the determination of growth plate chondrocyte regulation by vitamin D3 metabolites [570]. 1α,25-(OH)2D3 and 24R,25-(OH)2D3 vitamine D3 metabolites are found in growth plate cartilage, indicating that they are implicated in regulation mechanisms of growth plate cartilage. Indeed, in the absence of vitamin D3, the growth plate fails to mineralize and the hypertrophic zone becomes enlarged [571,572]. Not only chondrocytes but osteoblasts produce 1α,25-(OH)2D3 and 24R,25-(OH)2D3 which may function as autocrine regulators of matrix events, including MV formation, enzyme activity and matrix protein remodelling during longitudinal growth, calcification, and growth factor activation [573]. The growth plate is an ideal model since the lack of a vasculature ensures that only one cell type, the chondrocyte, is present in the growth plate. The cells can be subdivided into maturation zones (post-proliferative, pre-hypertrophic and upper hypertrophic zones so-called “growth zone”) and resting zones [387]. Using rat costochondral growth zone and resting zone chondrocytes cultures, it has been shown that resting zone chondrocytes respond preferentially to 24R,25-(OH)2D3[574,575], while hypertrophic chondrocytes respond preferentially to 1α,25-(OH)2D3[317,576]. Vitamin D3 metabolite, 1α, 25-(OH2)D3, caused stimulation of PKC activity via a PI-PLC in growth zone chondrocytes [390,391]. In resting cells, 24R,25-(OH)2D3 caused also a rapid increase of PKC activity but the mechanism involved was independent of PI-PLC [577]. 24R,25-(OH)2D3 exerts its effect through a vitamin D receptor [578] resulting in activation of PLD2 (based on G-protein-independent property) [391,570] and production of LPA [262]. Both pathways produce DAG and cause PKC activation but their time course differs [579]. The mechanisms that render the 1α,25-(OH2)D3 pathway silent in resting zone chondrocytes and the 24R,25-(OH)2D3 pathway silent in growth zone chondrocytes is controlled by a PLA2 activity. Inhibition of PLA2 blocks the effect of 1α,25-(OH2)D3 on growth zone cells while activation of PLA2 with melitin mimics the effects of 1α, 25-(OH2)D3 on growth zone cells [580]. Inhibition of PLA2 activates PKC and mimics the effect of 24R,25-(OH)2D3 on resting zone cells whereas activation of PLA2 blocks the effect of 24R,25-(OH)2D3 on PKC [581]. RT-PCR and Northern blot analysis revealed the presence of PLD1a, PLD1b and PLD2 mRNAs in both resting zone and growth zone chondrocytes. PLD activity was detected in both resting zone and growth zone chondrocytes and could be inhibited by wortmannnin—a known PLD inhibitor [570]. PLD activity stimulated by 24R,25-(OH)2D3 in resting chondrocytes may have two functional roles. The first one is an indirect increase of DAG (which is not obtained via PLC) which activates PKC, increases MV production [582], maturation and cell survival [583]. So far it is not clear how DAG is produced in this pathway. The second hypothetical role of the PLD stimulation by 24R,25-(OH)2D3 in resting chondrocytes could be evoked is an increase of LPA that could then bind in an autocrine manner to the LPA1 or LPA3 receptor [262,397]. Although there is no dispute that resting zone chondrocytes contain intracellular and secrete extracellular LPA (among them, 1-oleoyl-2-hydroxy-sn-glycero-3-phosphate), which can be activated through a membrane-associated vitamin D receptor [578], the possible pathway of LPA production from PA needs to be ascertained. So far, there is no experimental evidence that the production of PA catalyzed by PLD in the resting chondrocytes is the only source of secreted LPA. Indeed no information on the type of PLA2 implicated in the hydrolysis of PA forming LPA in resting zone chondrocytes is reported for this pathway. Alternate pathways for the 24R,25-(OH)2D3 induced LPA production need to be considered. The actin cytoskeleton plays an essential role in adhesion and PLD is physically and functionally linked to actin cytoskeleton [584]. It has been reported that the release of MVs from cultured epiphyseal chondrocytes was correlated with changes in cellular actin distribution [309]. PA—the product of hydrolysis of phospholipids by PLD is a fusogenic lipid [585], implicated in different steps of vesicular trafficking and intracellular membrane fusion events [586–588]. Laulagnier et al., 2004 [589] have observed the enrichment of active PLD2 on exosomes secreted by RBL-2H3 cells and shown that PLD2 was necessary to obtain maximal exosome secretion. Taken together the overall findings may suggest that PLD-dependent remodelling of actin cytoskeleton could participate in promoting MV formation from chondrocytes as well as from osteoblasts.
8.3. Presence of PLD in Osteoblasts and Possible Roles
The earliest experimental evidence of PLD activity in osteoblasts and its regulation originated from osteoblast-like MC3T-E1 cells. Despite that the fact that a lot of information on the activation of PLD is available, little is known about the possible role of PLD in osteoblasts. In these cells, PLD can be activated by PKC or in a Ca2+ dependent manner. PLD is activated in a PKC dependent manner by the platelet-derived growth factor [590] and by thromboxane A2 [591], while PLD is activated Ca2+ dependently by PGD2[424,592], PGE2[593], extracellular ATP [559] and thrombin [594]. PLD is activated Ca2+ dependently by PGF2[595] and independently of the activation of PKC [596], while retinoic acid suppresses the PLD activity activated by PGF2[597]. Tyrosine kinase may regulate PLD activity in these cells [598,599]. Other factors such as ET-1 [600,601] and basic FGF [602] activate PLD activity in osteoblast-like MC3T-E1 cells independently of PKC. NaF activated PLD and induced Arf/Rhoa translocation in osteoblast-like Saos-2 cells [603]. More information on possible functional roles of PLD is gained from osteoblast-like UMR-106 cells. A phorbol 12-myristate 13-acetate treatment of osteoblast-like UMR-106 cells activated PLD and lead to the production of PGE2 but not PGF2α [604] confirming for the first time that in osteoblasts, PA can be converted in PGE2 via a PLD/phosphatidate phosphohydrolase/DAG lipase/COX pathway [605–607]. Arachidonate metabolites such as PGE2 were found to play an important role in bone and cartilage metabolism [608]. These findings reveal a new aspect of PLD action [604] as a possible mediator in bone metabolism. Other factors also revealed the functional roles of PLD in osteoblasts. For example, PTH stimulates bone formation by preventing osteoblast apoptosis [609] and by activating diverse signaling pathways. PTH can stimulate PLD activity in UMR-106 cells [605–607]. Another example is provided by epidermal growth factor (EGF) which participates in the regulation of bone resorption in mice and mouse calvaria in vitro organ cultures [610]. EGF activates PLD signaling cascade in osteoblasts from Sprague-Dawley 21-day fetal rat calvaria, suggesting a general mechanism of PLD signaling pathway in osteoblasts [611]. MG63 osteoblast-like cells showed increased PLD activity, phosphatase alkaline activity and osteocalcin production on sandblasted titanium surface suggesting that PLD regulates osteoblast differentiation [612]. PLD1 activity may promote adhesion-dependent osteoblast differentiation response [612]. It was reported that both PLD1 and PLD2 can mediate the response of osteoblasts to surface microstructure although they did so in a different manner [613]. PLD, by virtue of producing PA, could turn up the mineralization process by affecting Pi concentration because human alkaline phosphatase isoenzymes are able to hydrolyze phosphatidates with various fatty acyl chains (e.g., phosphatidate and dioleoyl, distearoyl, dipalmitoyl, dimyristoyl and dilauroyl phosphatidates) [614]. On the other hand it has been shown [426] that long-term incubation with PA increased TNAP activity in osteoblast-like cell line, MOB 3-4. It has been proposed that LPA, acting via its LPA1 cell surface receptor, is able to induce cell membrane bleb [288], the process that may be related to MV formation, mineralization and apoptosis. In addition, it has been reported that LPA production in response to ATP is necessary to trigger osteogenesis [289]. LPA1 deficient mice showed craniofacial dysmorphism attributed to abnormal development of the facial bones [305]. Moreover, LPA1-deficient osteoblasts were characterized by lower differentiation potency in vitro[306].
8.4. Presence of PLD in Osteoclasts and Possible Roles
The role of PLD in osteoclasts is best exemplified under pathological conditions such as lung cancer metastasis and RA. Therefore, PLD signaling in osteoclasts is proposed as possible therapeutic strategies to prevent bone destruction. Bone is a frequent target of lung cancer metastasis that has a significant impact on morbidity [615,616]. Elevated levels of IL-8 and/or its receptors have been evidenced in cancer cells, endothelial cells, infiltrating neurotrophils and tumor-associated macrophages [617,618]. After activation of heterotrimeric small G proteins, IL-8 signaling promotes activation of the PI-3-kinase, PLC and PLD [619,620]. Exposure of human peripheral blood mononuclear cells (PBMC) to conditioned medium derived from lung cancer lines A549 and NCI-H460 as well as to sera from invasive lung cancer patients increased osteoclastogenesis in PBMC that was associated with augmented PLD activity. Depletion of IL-8 in CM derived from lung cancer lines A549 and NCI-H460 reversed the induction of osteoclastogenesis in PBMC [621]. Taken together these findings suggest that IL-8 secreted by human lung cancer cells—by increasing PLD activation—can promote osteoclast differentiation of PBMC and that PLD is involved in bone resorption by stimulating osteoclast differentiation [621]. IL-8 or IL-8-mediated PLD signaling may constitute an attractive therapeutic target for osteolytic bone metastases in lung cancer patients [621]. Under normal conditions, RANKL is produced mainly by osteoblasts and BM stromal cells. However, under pathological conditions such as RA, RANKL is also produced by T and B lymphocytes, macrophages/monocytes and synovial fibroblasts. RA synovial tissue seems be a suitable microenvironment for osteoclastogenesis since activated synovial cells and fibroblast express RANKL in situ[622,623]. The proinflammatory cytokine interleulin-15 (IL-15) can induce multinucleation of osteoclast-like cells in rat BM cultures [624]. IL-15 produced by RA T cells can induce osteoclastogenesis in cocultured autologous monocytes [625]. This suggests that IL-15 can mediate inflammatory bone destruction and stimulate osteoclastogenesis. IL-15 stimulation of human RA synovial fibroblasts induces simultaneous the expression of RANKL and PLD1 but not PLD2 [567]. Synovial fibroblasts treated with IL-15 induced osteoclastogenesis and PLD1 activation through the MAP kinases and NF-κB signaling pathways [567]. PLD1 may be an efficient therapeutic strategy for preventing bone destruction in RA [567].
8.5. Genetic Models
A recent generation of transgenic mice that do not express PLD1 [626] or PLD2 [626,627] indicated that platelets lacking PLD1 activity displayed impaired integrin activation under high shear conditions [626]. However, the skeletal formation in transgenic mice was not evaluated. The effects of silencing PLD genes on bone formation and on mineralization process were not determined.
8.6. Effects of PLD Metabolite at Matrix Vesicle Level
By using a fluorescence coupled-enzyme assay a phosphorylation-dependent PLD activity in MVs has been shown [628]. It is not yet known which type of PLD (PLD1 or PLD2) is predominant in MVs or what function PL has in MV. Since MV main function is to initiate HA formation, the hydrolytic activity of PLD leading to the production of PA may contribute to the mineralization process. Indeed, human alkaline phosphatase isoenzymes are able to hydrolyze phosphatidates with various fatty acyl chains (e.g., phosphatidate and dioleoyl, distearoyl, dipalmitoyl, dimyristoyl and dilauroyl phosphatidates) [614] forming Pi. Alternatively, PA can itself alter membrane curvature and contributes to the breaking of MV membrane.
9. Non-HKD Enzymes—GPI-PLD
9.1. Groups, Subgroups and Specificity
The glycosyl-PI specific PLD (GPI-PLD) activity has been characterized and implicated in the regulation of anchoring, thereby influencing the dispersal of anchored proteins or their maintenance on the cell surface, and in this way, possibly, cell signaling [629]. The only enzyme known to date that has specificity for cleavage of the GPI anchor is GPI-PLD, which cleaves the GPI structure to generate PA and the soluble protein. Although only one GPI-PLD cDNA has been identified in mouse [630] and ox [631], two have been described in human [632]. GPI-PLD is likely to be accessible to all cells of the body due to its abundance in serum. GPI-PLD expression has been detected in several tissue or cell types such as BM, liver and islets [633–637]. A potential role of GPI-PLD during bone formation certainly depends on the presence of suitable substrates. One GPI anchored protein that has a defined role in bone mineralization is TNAP. It is possible that TNAP found in bone tissue represents a substrate for endogenous GPI-PLD, which converts it from a membrane-bound to a soluble form [634]. Other GPI-anchored molecules that may be involved in bone formation include a subset of proteoglycans, as well as glypicans, which are part of collagen framework of the highly specialized ECM of cartilaginous tissue [638].
9.2. Presence of GPI-PLD in Chondrocytes and Possible Roles
Glypicans expressed by chondrocytes can act as cellular modulators of responses to bone morphogenetic factors [639] and defects in the glypican-3 gene cause an overgrowth and dysmorphic syndrome, the Simpson–Golabi–Behmel syndrome [640]. In addition, the GPI-anchored urokinase plasminogen activator receptor has been detected on the surface of chondrocytes [641], and it has been suggested that the plasminogen system may play a role in bone development by mediating effective degradation of the bone matrix. Deficiencies in this system can lead to bone overgrowth and malformations [642]. In addition, an endogenous GPI-PLD releases basic FGF-heparan sulfate proteoglycan complexes from human BM stromal cells. This mechanism of GP1 anchor cleavage could be relevant for mobilizing biologically active basic FGF in BM [633]. Gregory et al.[634] describe the first evidence of GPI-PLD expression during mouse embryonic ossification. GPI-PLD expression was detected predominantly at sites of skeletal development, increasing during the course of gestation. GPI-PLD was observed during both intramembraneous and endochondral ossification and localized predominantly to the ECM of chondrocytes and to primary trabeculae of the skeleton. In addition, the mouse chondrocyte cell line ATDC5 expressed GPI-PLD after experimental induction of differentiation.
9.3. Presence of GPI-PLD in Osteoblasts and Possible Roles
Decreasing GPI-anchored proteins by overexpressing GPI-PLD in MC3T3-E1 osteoblastic cells inhibits fluid flow induced Ca2+i mobilization and ERK1/2 phosphorylation, suggesting that GPI-anchored proteins in cell membranes may serve as transducer to transmit fluid shear stress to biochemical responses [643].
10. Non-HKD Enzymes—Autotaxin
10.1. Groups, Subgroups and Specificity
Autotaxin (ATX, NPP2) is an ecto-nucleotide pyrophosphatase/phosphodiesterase which hydrolyzes phosphodiester bonds of various nucleotides and nucleotide derivatives [644–647]. ATX hydrolyzes various LPL including LPC [277], LPE [266] and LPS [266] leading to the formation of LPA. ATX is encoded by a single gene on human chromosome 8 whose transcription, is regulated by diverse transcription factors, results in three alternatively spliced isoforms (α, β and γ) [648]. The expression of ATX is ubiquitous. Relatively high levels of ATX are expressed in brain, kidney and lymphoid organs [648]. As a lipid mediator LPA participates in many physiological processes. It promotes platelet aggregation and thrombosis, smooth muscle contraction, anti-apoptosis wound-healing, angiogenesis, development of the nervous systems through the cell surface G protein-coupled receptor pathways [268,646,647,649]. Thus LPLs and LPA may have a significant regulatory impact on the function of cells which are primarily involved in bone formation. On the other hand, LPLs are precursors of S1P which is a potential target for RA therapies [650]. Although the major source of S1P originates from the phosphorylation of sphingosine by sphingosine kinase, a part of S1P is hydrolyzed from sphingophosphorylcholine by ATX [651] or by S1P phosphatase and S1P lyase [650]. It has been suggested that ATX may be a potential target for the treatment of patients with RA [652]. In a CIA model, treatment with type-1 SphK siRNA suppressed articular inflammation and joint destruction and down regulated S1P, IL-6, TNF-α, and IFN-γ levels [653]. S1P level in synovial fluid from RA patients is higher than that from OA patients. S1P level in serum is about 2.5 times lower than that in RA synovial fluid [654]. S1P in serum is around 600–1000 nM [655]. S1P functions frequently in inflammatory processes, but is also implicated in autoimmune diseases as well as in cellular survival, proliferation and transformation, prevention of apoptosis and stimulation of angiogenesis [650]. S1P exerts its action via two distinct pathways: 1) intracellularly as a second messager; 2) extracellularly via activating specific GPCR [650]. So far, intracellular targets of S1P have not been found although they are implicated in the regulation of cellular proliferation, suppression of apoptosis and calcium homeostasis [650]. Five GPCR have been found on the cell surface: S1P(1–5) [656]. The S1P1 was markedly expressed in synovial lining cells, vascular endothelial cells and inflammatory mononuclear cells from RA synovial tissues when compared to those from OA synovial tissues, as determined by immunostaining [654]. S1P/S1P1 signaling enhanced synovial cell proliferation and COX-2 induced PGE2 production [654] and may enhance osteoclastogenesis via RANKL expression in RA synoviocytes and CD4+ cells [657]. Since the inflammation in RA is related to COX-2 induced PGE2 production by synoviocytes and since S1P/S1P1 signaling may induce synovial hyperplasia and inflammation in RA, S1P/S1P1 signaling could be a therapeutic target in RA [657].
10.2. Presence of ATX in Chondrocytes and Possible Roles
The presence of ATX in chondrocytes has been ascertained for the first time during the differentiation of cells. In C3H10T1/2—a multipotential cell line with the ability to differentiate into the major mesenchymal cell types such as myoblasts, adipocytes, osteoblasts, or chondrocytes the atx gene was expressed during BMP2-mediated osteo-/chondrogenic differentiation in vitro[658]. Atx expression in chondrocytes has been ascertained during murine embryogenesis [659]. The involvement of an α5β1 integrin in either the cartilage differentiation program or the joint formation program has been checked by blocking α5β1 integrin. Blocking α5β1 integrin resulted in the joint formation as indicated by the induction of an ectopic joint that expressed ATX as well as Wnt14, the earliest joint inducer and other specific markers of joints such as Gdf5, chordin and CD44 [660].
10.3. Presence of ATX in Osteoblasts and Possible Roles
So far the presence of ATX in osteoblasts has not been documented by immunoblot analysis. However, atx expression in preosteoblasts and osteoblasts has been ascertained during murine embryogenesis [659]. Possible roles of ATX have been proposed from the findings based on the involvement of LPA during bone metastases. Bone metastases are frequent complications in patients suffering from different types of cancers such as breast, kidney, lung, prostate and tyroid cancers [661]. Bone metastases have two distinct features, excessive bone loss involving osteoclasts and excess bone formation involving osteoblasts. Both types of bone lesions can occur in patients with metastatic prostate cancers [662]. Most of the information on the role of ATX during bone metastases comes from the synthesis of LPA and expression of its receptors LPA1, LPA2, LPA3 and LPA4[662]. LPA can be produced as a result of tumor-cell-induced platelet aggregation, by ATX-dependent or independent expression or following by P2X7 activation in osteoblasts. [662]. Then LPA can act directly in bone cells [299,303,663]. LPA can stimulate osteoblast proliferation and differentiation as well as can stimulate platelet aggregation which may initiate an amplification loop. LPA can stimulate osteocyte dentrite outgrow that thus could contribute to inhibition of bone formation [662]. Although it has been reported that atx expression is elevated in cancers compared to normal tissues [277], atx expression in primary tumors was not correlated with the occurrence of bone-metastases over a five-year period in a cohort of 167 breast cancer patients [664]. It was proposed that stratification of patients following the breast cancer intrinsic subtype’s classification should be carried to better evaluate the relationship between ATX and bone metastases [662]. Altough the atx expression levels in tumor cells have been reported [277,664], the expression changes of atx in osteoblasts during osteosclerosis were never determined. Silencing atx expression in 4T1 cells impaired their capacity to form osteolytic bone metastases in immunocompetent Balb/C mice [664] suggesting that LPA—the secreted product of 4T1-cell ATX activity—may act on osteoblasts. Taken together the findings tend to suggest that LPA production is a better marker for bone-metastases than the atx expression level. In this respect, LPA prevents PI3K-dependent apoptosis of osteoblasts [665], promotes cytoskeletal rearrangement and cell migration [299], induces differentiation of osteoblastic MG63 cells synergically with 1α,25-(OH)2D3[295] and the osteoblastogenesis of BM stem cells [666]. Based on Lpa1-4-deficient mice, it was concluded that LPA may induce osteoblast differentiation through LPA1 and LPA4 receptors [304].
10.4. Presence of ATX in Osteoclasts and Possible Roles
Incubation of BM cells with recombinant ATX in increased significantly M-CSF/RANK-L-induced osteoclast differentiation, suggesting that LPA generated by ATX in the presence of serum might directly control osteoclast differentiation [664]. LPA can act directly on osteoclast precursors to induce their differentiation and/or on mature osteoclasts to promote survival and bone resorption activity [662].
10.5. Presence of ATX in Smooth Muscle Cells and Possible Roles
Atx expression in smooth muscle cells has been ascertained during murine embryogenesis [659]. A time-dependent increase of around 2.5 fold of ATX in the vessels has been determined by immunoblotting after ligation injury of the carotid artery in mice. Mice deficient in LPA1 and LPA2, were protected from intimal hyperplasia in response to vascular injury. This indicates that LPA may regulate vascular development and function [667].
11. Concluding Remarks
Critical analysis of available reports revealed that phospholipases are implicated in the mineralization process at various levels of organization of the living matter. At the molecular level, phospholipases can provide precursors of Pi such as phosphatidates, PChols and PEAs, that can be further hydrolysed by other enzymes in mineralizing tissues and cells. At the membrane level, the degradation of phospholipids by phospholipases can affect the structural integrity and curvature of the plasma membrane from which MVs are released as well as membranes of mature MVs in ECM, favouring deposition of calcium phosphate complex formed inside MV into ECM. Last but not least, at cellular level, phospholipases, by producing signal molecules, may modulate cellular responses of mineralization-competent cells to the signals for mineralization. Gaining further knowledge about involvement of phospholipases in the mineralization process at distinct levels can contribute to our better understanding of the molecular mechanisms of lipid degradation during physiological and pathological mineralization, and can also help to create new targets for the cure of mineralization-related human diseases.
Acknowledgments
The work in the authors’ laboratories is supported by CNRS, by Polonium, by PICS, by the Polish National Science Center (grant NN401 642740) and by the Polish Ministry of Science and Higher Education (grant NN401 140639).
References
- 1.Del Fattore A., Teti A., Rucci N. Bone cells and the mechanisms of bone remodelling. Front. Biosci. 2012;4:2302–2321. doi: 10.2741/543. [DOI] [PubMed] [Google Scholar]
- 2.Franz-Odendaal T.A., Hall B.K., Witten P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 2006;235:176–190. doi: 10.1002/dvdy.20603. [DOI] [PubMed] [Google Scholar]
- 3.Parfitt A.M. Bone-forming cells in clinical conditions. In: Hall B.K., editor. Bone. Vol. 1. Telford Press; Calwell, NJ, USA: 1990. pp. 351–429. [Google Scholar]
- 4.Banks W.J. The ossification process of the developing antler in the white-tailed deer (Odocoileus virginianus) Calcif. Tissue Res. 1974;14:257–274. doi: 10.1007/BF02060300. [DOI] [PubMed] [Google Scholar]
- 5.Ekanayake S., Hall B.K. The development of acellularity of the vertebral bone of the Japanese medaka, Oryzias latipes (Teleostei; Cyprinidontidae) J. Morphol. 1987;193:253–261. doi: 10.1002/jmor.1051930304. [DOI] [PubMed] [Google Scholar]
- 6.Witten P.E., Huysseune A., Franz-Odendaal T.A., Fedak T., Vickaryous M., Cole A., Hall B.K. Acellular teleost bone: Primitive or derived, dead or alive? Palaeontol. Newslett. 2004;55:37–41. [Google Scholar]
- 7.Kronenberg H.M. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 8.Noonan K.J., Hunziker E.B., Nessler J., Buckwalter J.A. Changes in cell, matrix compartment, and fibrillar collagen volumes between growth-plate zones. J. Orthop. Res. 1998;16:500–508. doi: 10.1002/jor.1100160416. [DOI] [PubMed] [Google Scholar]
- 9.Pawelek J.M., Chakraborty A.K. The cancer cell—Leukocyte fusion theory of metastasis. Adv. Cancer Res. 2008;101:397–444. doi: 10.1016/S0065-230X(08)00410-7. [DOI] [PubMed] [Google Scholar]
- 10.Orimo H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J. Nihon Med. Sch. 2010;77:4–12. doi: 10.1272/jnms.77.4. [DOI] [PubMed] [Google Scholar]
- 11.Anderson H.C. The role of matrix vesicles in physiological and pathological calcification. Curr. Opin. Orthop. 2007;18:428–433. [Google Scholar]
- 12.Whyte M.P. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann. N. Y. Acad. Sci. 2010;1192:190–200. doi: 10.1111/j.1749-6632.2010.05387.x. [DOI] [PubMed] [Google Scholar]
- 13.Fleish H., Neuman W. Mechanisms of calcification: Role of collagen, polyphosphates, and phosphatase. Am. J. Physiol. 1961;200:1296–1300. doi: 10.1152/ajplegacy.1961.200.6.1296. [DOI] [PubMed] [Google Scholar]
- 14.Hessle L., Johnson K.A., Anderson H.C., Narisawa S., Sali A., Goding J.W., Terkeltaub R., Millan J.L. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc. Natl. Acad. Sci. USA. 2002;99:9445–9449. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murshed M., Harmey D., Millán J.L., McKee M.D., Karsenty G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005;19:1093–1104. doi: 10.1101/gad.1276205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Price P.A., Toroian D., Lim J.E. Mineralization by inhibitor exclusion: The calcification of collagen with fetuin. J. Biol. Chem. 2009;284:17092–17101. doi: 10.1074/jbc.M109.007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka H., Fukagawa M. Hormonal regulation of phosphate balance. Clin. Calcium. 2012;22:1477–1485. [PubMed] [Google Scholar]
- 18.Komaba H., Fukagawa M. The role of FGF23 in CKD—With or without Klotho. Nat. Rev. Nephrol. 2012;8:484–490. doi: 10.1038/nrneph.2012.116. [DOI] [PubMed] [Google Scholar]
- 19.Fukumoto S. The role of bone in phosphate metabolism. Mol. Cell. Endocrinol. 2009;310:63–70. doi: 10.1016/j.mce.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 20.Liu S., Quarles L.D. How fibroblast growth factor 23 works. J. Am. Soc. Nephrol. 2007;18:1637–1647. doi: 10.1681/ASN.2007010068. [DOI] [PubMed] [Google Scholar]
- 21.Anderson H.C., Mulhall D., Garimella R. Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis, and arthritis. Lab. Invest. 2010;90:1549–1557. doi: 10.1038/labinvest.2010.152. [DOI] [PubMed] [Google Scholar]
- 22.Doherty T.M., Fitzpatrick L.A., Inoue D., Qiao J.H., Fishbein M.C., Detrano R.C., Shah P.K., Rajavashisth T.B. Molecular, endocrine, and genetic mechanisms of arterial calcification. Endocr. Rev. 2004;25:629–672. doi: 10.1210/er.2003-0015. [DOI] [PubMed] [Google Scholar]
- 23.Magne D., Julien M., Vinatier C., Merhi-Soussi F., Weiss P., Guicheux J. Cartilage formation in growth plate and arteries: From physiology to pathology. Bioessays. 2005;27:708–716. doi: 10.1002/bies.20254. [DOI] [PubMed] [Google Scholar]
- 24.Shao J.S., Cai J., Towler D.A. Molecular mechanisms of vascular calcification: Lessons learned from the aorta. Arterioscler. Thromb. Vasc. Biol. 2006;26:1423–1430. doi: 10.1161/01.ATV.0000220441.42041.20. [DOI] [PubMed] [Google Scholar]
- 25.Rutsch F., Böyer P., Nitschke Y., Ruf N., Lorenz-Depierieux B., Wittkampf T., Weissen-Plenz G., Fischer R.J., Mughal Z., Gregory J.W., et al. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ. Cardiovasc. Genet. 2008;1:133–140. doi: 10.1161/CIRCGENETICS.108.797704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ali S.Y. Apatite crystal nodules in arthritic cartilage. Eur. J. Rheumatol. Inflamm. 1978;14:115–119. [Google Scholar]
- 27.Sampson H.W., Davis R.W., Dufner D.C. Spondyloarthropathy in progressive ankylosis mice: Ultrastructural features of the intervertebral disk. Acta Anat. 1991;141:36–41. doi: 10.1159/000147096. [DOI] [PubMed] [Google Scholar]
- 28.Isakova T. Fibroblast growth factor 23 and adverse clinical outcomes in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2012;21:334–340. doi: 10.1097/MNH.0b013e328351a391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connor J.M., Evans D.A. Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J. Bone Joint Surg. Br. 1982;64:76–83. doi: 10.1302/0301-620X.64B1.7068725. [DOI] [PubMed] [Google Scholar]
- 30.Kaplan F.S., Shore E.M., Connor J.M. Fibrodysplasia ossificans progressiva. In: Royce P.M., Steinmann B., editors. Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. John Wiley & Sons; New York, NY, USA: 2002. pp. 827–840. [Google Scholar]
- 31.Gannon F.H., Kaplan F.S., Olmsted E., Finkel G.C., Zasloff M.A., Shore E. Bone morphogenetic protein 2/4 in early fibromatous lesions of fibrodysplasia ossificans progressiva. Hum. Pathol. 1997;28:339–343. doi: 10.1016/s0046-8177(97)90133-7. [DOI] [PubMed] [Google Scholar]
- 32.Shafritz A.B., Shore E.M., Gannon F.H., Zasloff M.A., Taub R., Muenke M., Kaplan F.S. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N. Engl. J. Med. 1996;335:555–561. doi: 10.1056/NEJM199608223350804. [DOI] [PubMed] [Google Scholar]
- 33.Kaplan F.S., Shore E.M. Progressive osseous heteroplasia. J. Bone Miner. Res. 2000;15:2084–2094. doi: 10.1359/jbmr.2000.15.11.2084. [DOI] [PubMed] [Google Scholar]
- 34.Mornet E. Hypophosphatasia. Best Pract. Res. Clin. Rheumatol. 2008;22:113–127. doi: 10.1016/j.berh.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Whyte M.P. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr. Rev. 1994;15:439–461. doi: 10.1210/edrv-15-4-439. [DOI] [PubMed] [Google Scholar]
- 36.Sambrook P., Cooper C. Osteoporosis. Lancet. 2006;367:2010–2018. doi: 10.1016/S0140-6736(06)68891-0. [DOI] [PubMed] [Google Scholar]
- 37.Goronzy J.J., Weyand C.M. Developments in the scientific understanding of rheumatoid arthritis. Arthritis Res. Ther. 2009;11:249. doi: 10.1186/ar2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walsh N.C., Crotti T.N., Goldring S.R., Gravallese E.M. Rheumatic diseases: The effects of inflammation on bone. Immunol. Rev. 2005;208:228–251. doi: 10.1111/j.0105-2896.2005.00338.x. [DOI] [PubMed] [Google Scholar]
- 39.Karouzakis E., Neidhart M., Gay R.E., Gay S. Molecular and cellular basis of rheumatoid joint destruction. Immunol. Lett. 2006;106:8–13. doi: 10.1016/j.imlet.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 40.Mebarek S., Hamade E., Thouverey C., Bandorowicz-Pikula J., Pikula S., Magne D., Buchet R. Ankylosing spondylitis, late osteoarthritis, vascular calcification, chondrocalcinosis and pseudo gout: Toward a possible drug therapy. Curr. Med. Chem. 2011;18:2196–2203. doi: 10.2174/092986711795656153. [DOI] [PubMed] [Google Scholar]
- 41.Blacher J., Guerin A.P., Pannier B., Marchais S.J., London G.M. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension. 2001;38:938–942. doi: 10.1161/hy1001.096358. [DOI] [PubMed] [Google Scholar]
- 42.Detrano R., Guerci A.D., Carr J.J., Bild D.E., Burke G., Folsom A.R., Liu K., Shea S., Szklo M., Bluemke D.A., et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N. Engl. J. Med. 2008;358:1336–1345. doi: 10.1056/NEJMoa072100. [DOI] [PubMed] [Google Scholar]
- 43.Ali S.Y., Sajdera S.W., Anderson H.C. Isolation and characterization of calcifying matrix vesicles from epiphyseal cartilage. Proc. Natl. Acad. Sci. USA. 1970;67:1513–1520. doi: 10.1073/pnas.67.3.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ali S.Y., Anderson H.C., Sajdera S.W. Enzymic and electron-microscopic analysis of extracellular matrix vesicles associated with calcification in cartilage. Biochem. J. 1971;122:56P–57P. doi: 10.1042/bj1220056pa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaji H. Pyrophosphate and mineralization (TNSALP, PC-1, ANK) Clin. Calcium. 2007;17:1574–1579. [PubMed] [Google Scholar]
- 46.Roberts S.J., Stewart A.J., Schmid R., Blindauer C.A., Bond S.R., Sadler P.J., Farquharson C. Probing the substrate specificities of human PHOSPHO1 and PHOSPHO2. Biochim. Biophys. Acta. 2005;1752:73–82. doi: 10.1016/j.bbapap.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 47.Roberts S., Narisawa S., Harmey D., Millán J.L., Farquharson C. Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization. J. Bone Miner. Res. 2007;22:617–627. doi: 10.1359/jbmr.070108. [DOI] [PubMed] [Google Scholar]
- 48.Hsu H.H., Anderson H.C. A role for ATPase in the mechanisms of ATP-dependent Ca and phosphate deposition by isolated rachitic matrix vesicles. Int. J. Biochem. Cell. Biol. 1995;27:1349–1356. doi: 10.1016/1357-2725(95)00103-v. [DOI] [PubMed] [Google Scholar]
- 49.Terkeltaub R. Physiologic and pathologic functions of the NPP nucleotide pyrophosphatase/phosphodiesterase family focusing on NPP1 in calcification. Purinergic Signal. 2006;2:371–377. doi: 10.1007/s11302-005-5304-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu L.N., Genge B.R., Kang M.W., Arsenault A.L., Wuthier R.E. Changes in phospholipid extractability and composition accompany mineralization of chicken growth plate cartilage matrix vesicles. J. Biol. Chem. 2002;277:5126–5133. doi: 10.1074/jbc.M107899200. [DOI] [PubMed] [Google Scholar]
- 51.Wuthier R.E. Effect of phospholipids on the transformation of amorphous calcium phosphate to hydroxapatite in vitro. Calcif. Tissue Res. 1975;19:197–210. doi: 10.1007/BF02564004. [DOI] [PubMed] [Google Scholar]
- 52.Wuthier R.E. Lipid composition of isolated epiphyseal cartilage cells, membranes and matrix vesicles. Biochim. Biophys. Acta. 1975;409:128–143. doi: 10.1016/0005-2760(75)90087-9. [DOI] [PubMed] [Google Scholar]
- 53.Wuthier R.E., Lipscomb G.F. Matrix vesicles: Structure, composition, formation and function in calcification. Front. Biosci. 2011;16:2812–2902. doi: 10.2741/3887. [DOI] [PubMed] [Google Scholar]
- 54.Golub E.E. Biomineralization and matrix vesicles in biology and pathology. Semin. Immunopathol. 2011;33:409–417. doi: 10.1007/s00281-010-0230-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duque G. Bone and fat connection in aging bone. Curr. Opin. Rheumatol. 2008;20:429–434. doi: 10.1097/BOR.0b013e3283025e9c. [DOI] [PubMed] [Google Scholar]
- 56.Kim Y., Ilich J.Z. Implications of dietary α-linolenic acid in bone health. Nutrition. 2011;27:1101–1107. doi: 10.1016/j.nut.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 57.Hur S.J., Park Y. Effect of conjugated linoleic acid on bone formation and rheumatoid arthritis. Eur. J. Pharmacol. 2007;568:16–24. doi: 10.1016/j.ejphar.2007.04.056. [DOI] [PubMed] [Google Scholar]
- 58.Fernandes G., Bhattacharya A., Rahman M., Zaman K., Banu J. Effects of n-3 fatty acids on autoimmunity and osteoporosis. Front. Biosci. 2008;13:4015–4020. doi: 10.2741/2989. [DOI] [PubMed] [Google Scholar]
- 59.Salari P., Rezaie A., Larijani B., Abdollahi M. A systematic review of the impact of n-3 fatty acids in bone health and osteoporosis. Med. Sci. Monit. 2008;14:RA37–44. [PubMed] [Google Scholar]
- 60.Poulsen R.C., Moughan P.J., Kruger M.C. Long-chain polyunsaturated fatty acids and the regulation of bone metabolism. Exp. Biol. Med. 2007;232:1275–1288. doi: 10.3181/0704-MR-100. [DOI] [PubMed] [Google Scholar]
- 61.Kruger M.C., Coetzee M., Haag M., Weiler H. Long-chain polyunsaturated fatty acids: Selected mechanisms of action on bone. Prog. Lipid Res. 2010;49:438–449. doi: 10.1016/j.plipres.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 62.Genuis S.J., Schwalfenberg G.K. Picking a bone with contemporary osteoporosis management: Nutrient strategies to enhance skeletal integrity. Clin. Nutr. 2007;26:193–207. doi: 10.1016/j.clnu.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 63.Corwin R.L., Hartman T.J., Maczuga S.A., Graubard B.I. Dietary saturated fat intake is inversely associated with bone density in humans: Analysis of NHANES III. J. Nutr. 2006;136:159–165. doi: 10.1093/jn/136.1.159. [DOI] [PubMed] [Google Scholar]
- 64.Weinberg J.M. Lipotoxicity. Kidney Int. 2006;70:1560–1566. doi: 10.1038/sj.ki.5001834. [DOI] [PubMed] [Google Scholar]
- 65.Unger R.H., Orci L. Lipoapoptosis: Its mechanism and its diseases. Biochim. Biophys. Acta. 2002;1585:202–212. doi: 10.1016/s1388-1981(02)00342-6. [DOI] [PubMed] [Google Scholar]
- 66.Chapple I.L. Potential mechanisms underpinning the nutritional modulation of periodontal inflammation. J. Am. Dent. Assoc. 2009;140:178–184. doi: 10.14219/jada.archive.2009.0131. [DOI] [PubMed] [Google Scholar]
- 67.Bab I., Smoum R., Bradshaw H., Mechoulam R. Skeletal lipidomics: Regulation of bone metabolism by fatty acid amide family. Br. J. Pharmacol. 2011;163:1441–1446. doi: 10.1111/j.1476-5381.2011.01474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wong H., Schotz M.C. The lipase gene family. J. Lipid Res. 2002;43:993–999. doi: 10.1194/jlr.r200007-jlr200. [DOI] [PubMed] [Google Scholar]
- 69.Aoki J., Inoue A., Makide K., Saiki N., Arai H. Structure and function of extracellular phospholipase A1 belonging to the pancreatic lipase gene family. Biochimie. 2007;89:197–204. doi: 10.1016/j.biochi.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 70.Kudo I., Murakami M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002;68–69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 71.Burke J.E., Dennis E.A. Phospholipase A2 biochemistry. Cardiovasc. Drugs Ther. 2009;23:49–59. doi: 10.1007/s10557-008-6132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schaloske R.H., Dennis E.A. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 73.Scott K.F., Sajinovic M., Hein J., Nixdorf S., Galettis P., Liauw W., de Souza P., Dong Q., Graham G.G., Russell P.J. Emerging roles for phospholipase A2 enzymes in cancer. Biochimie. 2010;92:601–610. doi: 10.1016/j.biochi.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 74.Murakami M., Taketomi Y., Miki Y., Sato H., Hirabayashi T., Yamamoto K. Recent progress in phospholipase A2 research: From cells to animals to humans. Prog. Lipid Res. 2011;50:152–192. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 75.Fukami K., Inanobe S., Kanemaru K., Nakamura Y. Phospholipase C is a key enzyme regulating intracellular calcium and modulating the phosphoinositide balance. Prog. Lipid Res. 2010;49:429–437. doi: 10.1016/j.plipres.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 76.Bunney T.D., Katan M. PLC regulation: Emerging pictures for molecular mechanisms. Trends Biochem. Sci. 2011;36:88–96. doi: 10.1016/j.tibs.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 77.Selvy P.E., Lavieri R.R., Lindsley C.W., Brown H.A. Phospholipase D: Enzymology, functionality, and chemical modulation. Chem. Rev. 2011;111:6064–6119. doi: 10.1021/cr200296t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peng X., Frohman M.A. Mammalian phospholipase D physiological and pathological roles. Acta Physiol. 2012;204:219–226. doi: 10.1111/j.1748-1716.2011.02298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kirsch T. Physiological and pathological mineralization: A complex multifactorial process. Curr. Opin. Orthop. 2007;18:425–427. [Google Scholar]
- 80.Aoki J., Nagai Y., Hosono H., Inoue K., Arai H. Structure and function of phosphatidylserine-specific phospholipase A1. Biochim. Biophys. Acta. 2002;1582:26–32. doi: 10.1016/s1388-1981(02)00134-8. [DOI] [PubMed] [Google Scholar]
- 81.Sonoda H., Aoki J., Hiramatsu T., Ishida M., Bandoh K., Nagai Y., Taguchi R., Inoue K., Arai H. A novel phosphatidic acid-selective phospholipase A1 that produces lysophosphatidic acid. J. Biol. Chem. 2002;277:34254–34263. doi: 10.1074/jbc.M201659200. [DOI] [PubMed] [Google Scholar]
- 82.Hiramatsu T., Sonoda H., Takanezawa Y., Morikawa R., Ishida M., Kasahara K., Sanai Y., Taguchi R., Aoki J., Arai H. Biochemical and molecular characterization of two phosphatidic acid-selective phospholipase A1s, mPA-PLA1alpha and mPA-PLA1beta. J. Biol. Chem. 2003;278:49438–49447. doi: 10.1074/jbc.M213018200. [DOI] [PubMed] [Google Scholar]
- 83.Carrière F., Withers-Martinez C., van Tilbeurgh H., Roussel A., Cambillau C., Verger R. Structural basis for the substrate selectivity of pancreatic lipases and some related proteins. Biochim. Biophys. Acta. 1998;1376:417–432. doi: 10.1016/s0304-4157(98)00016-1. [DOI] [PubMed] [Google Scholar]
- 84.Hide W.A., Chan L., Li W.H. Structure and evolution of the lipase superfamily. J. Lipid Res. 1992;33:167–178. [PubMed] [Google Scholar]
- 85.Pete M.J., Ross A.H., Exton J.H. Purification and properties of phospholipase A1 from bovine brain. J. Biol. Chem. 1994;269:19494–19500. [PubMed] [Google Scholar]
- 86.Higgs H.N., Glomset J.A. Identification of a phosphatidic acid-preferring phospholipase A1 from bovine brain and testis. Proc. Natl. Acad. Sci. USA. 1994;91:9574–9578. doi: 10.1073/pnas.91.20.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Higgs H.N., Han M.H., Johnson G.E., Glomset J.A. Cloning of a phosphatidic acid-preferring phospholipase A1 from bovine testis. J. Biol. Chem. 1998;273:5468–5477. doi: 10.1074/jbc.273.10.5468. [DOI] [PubMed] [Google Scholar]
- 88.Tani K., Mizoguchi T., Iwamatsu A., Hatsuzawa K., Tagaya M. p125 is a novel mammalian Sec23p-interacting protein with structural similarity to phospholipid-modifying proteins. J. Biol. Chem. 1999;274:20505–20512. doi: 10.1074/jbc.274.29.20505. [DOI] [PubMed] [Google Scholar]
- 89.Nakajima K., Sonoda H., Mizoguchi T., Aoki J., Arai H., Nagahama M., Tagaya M., Tani K. A novel phospholipase A1 with sequence homology to a mammalian Sec23p-interacting protein, p125. J. Biol. Chem. 2002;277:11329–11335. doi: 10.1074/jbc.M111092200. [DOI] [PubMed] [Google Scholar]
- 90.Murakami M., Taketomi Y., Sato H., Yamamoto K. Secreted phospholipase A2 revisited. J. Biochem. 2011;150:233–255. doi: 10.1093/jb/mvr088. [DOI] [PubMed] [Google Scholar]
- 91.Dennis E.A., Cao J., Hsu Y.H., Magrioti V., Kokotos G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011;111:6130–6185. doi: 10.1021/cr200085w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Menschikowski M., Hagelgans A., Siegert G. Secretory phospholipase A2 of group IIA: Is it an offensive or a defensive player during atherosclerosis and other inflammatory diseases? Prostaglandins Other Lipid Mediat. 2006;79:1–33. doi: 10.1016/j.prostaglandins.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 93.Rosenson R.S., Gelb M.H. Secretory phospholipase A2: A multifaceted family of proatherogenic enzymes. Curr. Cardiol. Rep. 2009;11:445–451. doi: 10.1007/s11886-009-0064-2. [DOI] [PubMed] [Google Scholar]
- 94.Nevalainen T.J., Graham G.G., Scott K.F. Antibacterial actions of secreted phospholipases A2. Review. Biochim. Biophys. Acta. 2008;1781:1–9. doi: 10.1016/j.bbalip.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 95.Lambeau G., Gelb M.H. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem. 2008;77:495–520. doi: 10.1146/annurev.biochem.76.062405.154007. [DOI] [PubMed] [Google Scholar]
- 96.Boyanovsky B.B., Webb N.R. Biology of secretory phospholipase A2. Cardiovasc. Drugs Ther. 2009;23:61–72. doi: 10.1007/s10557-008-6134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Clark J.D., Lin L.L., Kriz R.W., Ramesha C.S., Sultzman L.A., Lin A.Y., Milona N., Knopf J.L. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell. 1991;65:1043–1051. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- 98.Kramer R.M., Roberts E.F., Manetta J., Putnam J.E. The Ca2(+)-sensitive cytosolic phospholipase A2 is a 100-kDa protein in human monoblast U937 cells. J. Biol. Chem. 1991;266:5268–5272. [PubMed] [Google Scholar]
- 99.Ghosh M., Tucker D.E., Burchett S.A., Leslie C.C. Properties of the Group IV phospholipase A2 family. Prog. Lipid Res. 2006;45:487–510. doi: 10.1016/j.plipres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 100.Leslie C.C., Gangelhoff T.A., Gelb M.H. Localization and function of cytosolic phospholipase A2alpha at the Golgi. Biochimie. 2010;92:620–626. doi: 10.1016/j.biochi.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kita Y., Ohto T., Uozumi N., Shimizu T. Biochemical properties and pathophysiological roles of cytosolic phospholipase A2s. Biochim. Biophys. Acta. 2006;1761:1317–1322. doi: 10.1016/j.bbalip.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 102.Linkous A., Yazlovitskaya E. Cytosolic phospholipase A2 as a mediator of disease pathogenesis. Cell. Microbiol. 2010;12:1369–1377. doi: 10.1111/j.1462-5822.2010.01505.x. [DOI] [PubMed] [Google Scholar]
- 103.Balsinde J., Pérez R., Balboa M.A. Calcium-independent phospholipase A2 and apoptosis. Biochim. Biophys. Acta. 2006;1761:1344–1350. doi: 10.1016/j.bbalip.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 104.Green J.T., Orr S.K., Bazinet R.P. The emerging role of group VI calcium-independent phospholipase A2 in releasing docosahexaenoic acid from brain phospholipids. J. Lipid Res. 2008;49:939–944. doi: 10.1194/jlr.R700017-JLR200. [DOI] [PubMed] [Google Scholar]
- 105.Hooks S.B., Cummings B.S. Role of Ca2+-independent phospholipase A2 in cell growth and signaling. Biochem. Pharmacol. 2008;76:1059–1067. doi: 10.1016/j.bcp.2008.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cedars A., Jenkins C.M., Mancuso D.J., Gross R.W. Calcium-independent phospholipases in the heart: Mediators of cellular signaling, bioenergetics, and ischemia-induced electrophysiologic dysfunction. J. Cardiovasc. Pharmacol. 2009;53:277–289. [PMC free article] [PubMed] [Google Scholar]
- 107.Lei X., Barbour S.E., Ramanadham S. Group VIA Ca2+-independent phospholipase A2 (iPLA2beta) and its role in beta-cell programmed cell death. Biochimie. 2010;92:627–637. doi: 10.1016/j.biochi.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Arai H., Koizumi H., Aoki J., Inoue K. Platelet-activating factor acetylhydrolase (PAF-AH) J. Biochem. 2002;131:635–640. doi: 10.1093/oxfordjournals.jbchem.a003145. [DOI] [PubMed] [Google Scholar]
- 109.Karasawa K., Harada A., Satoh N., Inoue K., Setaka M. Plasma platelet activating factor-acetylhydrolase (PAF-AH) Prog. Lipid Res. 2003;42:93–114. doi: 10.1016/s0163-7827(02)00049-8. [DOI] [PubMed] [Google Scholar]
- 110.Mallat Z., Lambeau G., Tedgui A. Lipoprotein-associated and secreted phospholipases A2 in cardiovascular disease: Roles as biological effectors and biomarkers. Circulation. 2010;122:2183–2200. doi: 10.1161/CIRCULATIONAHA.110.936393. [DOI] [PubMed] [Google Scholar]
- 111.McIntyre T.M., Prescott S.M., Stafforini D.M. The emerging roles of PAF acetylhydrolase. J. Lipid Res. 2009;50:S255–S259. doi: 10.1194/jlr.R800024-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stafforini D.M. Biology of platelet-activating factor acetylhydrolase (PAF-AH, lipoprotein associated phospholipase A2) Cardiovasc. Drugs Ther. 2009;23:73–83. doi: 10.1007/s10557-008-6133-8. [DOI] [PubMed] [Google Scholar]
- 113.Stafforini D.M., Elstad M.R., McIntyre T.M., Zimmerman G.A., Prescott S.M. Human macrophages secret platelet-activating factor acetylhydrolase. J. Biol. Chem. 1990;265:9682–9687. [PubMed] [Google Scholar]
- 114.Stafforini D.M., McIntyre T.M., Zimmerman G.A., Prescott S.M. Platelet-activating factor, a pleiotrophic mediator of physiological and pathological processes. Crit. Rev. Clin. Lab. Sci. 2003;40:643–672. doi: 10.1080/714037693. [DOI] [PubMed] [Google Scholar]
- 115.Tjoelker L.W., Wilder C., Eberhardt C., Stafforini D.M., Dietsch G., Schimpf B., Hooper S., Le Trong H., Cousens L.S., Zimmerman G.A., et al. Anti-inflammatory properties of a platelet-activating factor acetylhydrolase. Nature. 1995;374:549–553. doi: 10.1038/374549a0. [DOI] [PubMed] [Google Scholar]
- 116.Wilensky R.L., Macphee C.H. Lipoprotein-associated phospholipase A2 and atherosclerosis. Curr. Opin. Lipidol. 2009;20:415–420. doi: 10.1097/MOL.0b013e3283307c16. [DOI] [PubMed] [Google Scholar]
- 117.Shayman J.A., Kelly R., Kollmeyer J., He Y., Abe A. Group XV phospholipase A2, a lysosomal phospholipase A2. Prog. Lipid Res. 2011;50:1–13. doi: 10.1016/j.plipres.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Duncan R.E., Sarkadi-Nagy E., Jaworski K., Ahmadian M., Sul H.S. Identification and functional characterization of adipose-specific phospholipase A2 (AdPLA) J. Biol. Chem. 2008;283:25428–25436. doi: 10.1074/jbc.M804146200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jaworski K., Ahmadian M., Duncan R.E., Sarkadi-Nagy E., Varady K.A., Hellerstein M.K., Lee H.Y., Samuel V.T., Shulman G.I., Kim K.H., et al. AdPLA ablation increases lipolysis and prevents obesity induced by high-fat feeding or leptin deficiency. Nat. Med. 2009;15:159–168. doi: 10.1038/nm.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yokota Y., Higashino K., Nakano K., Arita H., Hanasaki K. Identification of group X secretory phospholipase A2 as a natural ligand for mouse phospholipase A2 receptor. FEBS Lett. 2000;478:187–191. doi: 10.1016/s0014-5793(00)01848-2. [DOI] [PubMed] [Google Scholar]
- 121.Ramanadham S., Yarasheski K.E., Silva M.J., Wohltmann M., Novack D.V., Christiansen B., Tu X., Zhang S., Lei X., Turk J. Age-related changes in bone morphology are accelerated in group VIA phospholipase A2 (iPLA2beta)-null mice. Am. J. Pathol. 2008;172:868–881. doi: 10.2353/ajpath.2008.070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bonventre J. Cytosolic phospholipase A2alpha reigns supreme in arthritis and bone resorption. Trends Immunol. 2004;25:116–119. doi: 10.1016/j.it.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 123.Seilhamer J.J., Pruzanski W., Vadas P., Plant S., Miller J.A., Kloss J., Johnson L.K. Cloning and recombinant expression of phospholipase A2 present in rheumatoid arthritic synovial fluid. J. Biol. Chem. 1989;264:5335–5338. [PubMed] [Google Scholar]
- 124.Masuda S., Murakami M., Komiyama K., Ishihara M., Ishikawa Y., Ishii T., Kudo I. Various secretory phospholipase A2 enzymes are expressed in rheumatoid arthritis and augment prostaglandin production in cultured synovial cells. FEBS J. 2005;272:655–672. doi: 10.1111/j.1742-4658.2004.04489.x. [DOI] [PubMed] [Google Scholar]
- 125.Chang J., Gilman S.C., Lewis A.J. Interleukin 1 activates phospholipase A2 in rabbit chondrocytes: A possible signal for IL 1 action. J. Immunol. 1986;136:1283–1287. [PubMed] [Google Scholar]
- 126.Jamal O.S., Conaghan P.G., Cunningham A.M., Brooks P.M., Munro V.F., Scott K.F. Increased expression of human type IIa secretory phospholipase A2 antigen in arthritic synovium. Ann. Rheum. Dis. 1998;57:550–558. doi: 10.1136/ard.57.9.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nevalainen T.J., Haapanen T.J. Distribution of pancreatic (group I) and synovial-type (group II) phospholipases A2 in human tissues. Inflammation. 1993;17:453–464. doi: 10.1007/BF00916585. [DOI] [PubMed] [Google Scholar]
- 128.Nevalainen T.J., Märki F., Kortesuo P.T., Grütter M.G., di Marco S., Schmitz A. Synovial type (group II) phospholipase A2 in cartilage. J. Rheumatol. 1993;20:325–330. [PubMed] [Google Scholar]
- 129.Nevalainen T.J. Serum phospholipases A2 in inflammatory diseases. Clin. Chem. 1993;39:2453–2459. [PubMed] [Google Scholar]
- 130.Vignon E., Mathieu P., Louisot P., Vilamitjana J., Harmand M.F., Richard M. Phospholipase A2 activity in human osteoarthritic cartilage. J. Rheumatol. Suppl. 1989;18:35–38. [PubMed] [Google Scholar]
- 131.Pruzanski W., Bogoch E., Katz A., Wloch M., Stefanski E., Grouix B., Sakotic G., Vadas P. Induction of release of secretory nonpancreatic phospholipase A2 from human articular chondrocytes. J. Rheumatol. 1995;22:2114–2119. [PubMed] [Google Scholar]
- 132.Lyons-Giordano B., Davis G.L., Galbraith W., Pratta M.A., Arner E.C. Interleukin-1 beta stimulates phospholipase A2 mRNA synthesis in rabbit articular chondrocytes. Biochem. Biophys. Res. Commun. 1989;164:488–495. doi: 10.1016/0006-291x(89)91746-4. [DOI] [PubMed] [Google Scholar]
- 133.Nakano T., Ohara O., Teraoka H., Arita H. Glucocorticoids suppress group II phospholipase A2 production by blocking mRNA synthesis and post-transcriptional expression. J. Biol. Chem. 1990;265:12745–12748. [PubMed] [Google Scholar]
- 134.Gilman S.C. Activation of rabbit articular chondrocytes by recombinant human cytokines. J. Rheumatol. 1987;14:1002–1007. [PubMed] [Google Scholar]
- 135.Pfeilschifter J., Pignat W., Vosbeck K., Märki F. Interleukin 1 and tumor necrosis factor synergistically stimulate prostaglandin synthesis and phospholipase A2 release from rat renal mesangial cells. Biochem. Biophys. Res. Commun. 1989;159:385–394. doi: 10.1016/0006-291x(89)90003-x. [DOI] [PubMed] [Google Scholar]
- 136.Stevens T.M., Chin J.E., McGowan M., Giannaras J., Kerr J.S. Phospholipase A2 (PLA2) activity in rabbit chondrocytes. Agents Actions. 1989;27:385–387. doi: 10.1007/BF01972829. [DOI] [PubMed] [Google Scholar]
- 137.Suffys P., Van Roy F., Fiers W. Tumor necrosis factor and interleukin 1 activate phospholipase in rat chondrocytes. FEBS Lett. 1988;232:24–28. doi: 10.1016/0014-5793(88)80379-x. [DOI] [PubMed] [Google Scholar]
- 138.Kudo I., Murakami M., Hara S., Inoue K. Mammalian non-pancreatic phospholipases A2. Biochim. Biophys. Acta. 1993;1170:217–231. doi: 10.1016/0005-2760(93)90003-r. [DOI] [PubMed] [Google Scholar]
- 139.Leistad L., Feuerherm A.J., Faxvaag A., Johansen B. Multiple phospholipase A2 enzymes participate in the inflammatory process in osteoarthritic cartilage. Scand. J. Rheumatol. 2011;40:308–316. doi: 10.3109/03009742.2010.547872. [DOI] [PubMed] [Google Scholar]
- 140.Vadas P., Pruzanski W., Stefanski E., Ellies L.G., Aubin J.E., Sos A., Melcher A. Extracellular phospholipase A2 secretion is a common effector pathway of interleukin-1 and tumour necrosis factor action. Immunol. Lett. 1991;28:187–193. doi: 10.1016/0165-2478(91)90002-r. [DOI] [PubMed] [Google Scholar]
- 141.Bonewald L.F., Schwartz Z., Swain L.D., Boyan B.D. Stimulation of matrix vesicle enzyme activity in osteoblast-like cells by 1,25(OH)2D3 and transforming growth factor beta (TGF beta) Bone Miner. 1992;17:139–144. doi: 10.1016/0169-6009(92)90725-s. [DOI] [PubMed] [Google Scholar]
- 142.Schwartz Z., Dennis R., Bonewald L., Swain L., Gomez R., Boyan B.D. Differential regulation of prostaglandin E2 synthesis and phospholipase A2 activity by 1,25-(OH)2D3 in three osteoblast-like cell lines (MC-3T3-E1, ROS 17/2.8, and MG-63) Bone. 1992;13:51–58. doi: 10.1016/8756-3282(92)90361-y. [DOI] [PubMed] [Google Scholar]
- 143.Tokuda H., Oiso Y., Kozawa O. Protein kinase C activation amplifies prostaglandin F2 alpha-induced prostaglandin E2 synthesis in osteoblast-like cells. J. Cell. Biochem. 1992;48:262–268. doi: 10.1002/jcb.240480306. [DOI] [PubMed] [Google Scholar]
- 144.Suzuki N., Matsunaga T., Kanaho Y., Nozawa Y. The mechanism of bradykinin-induced arachidonic acid release in osteoblast-like MC3T3-E1 cells phospholipase A2 activation by bradykinin and its regulation by protein kinase C and calcium. Nihon Seikeigeka Gakkai Zasshi. 1993;67:935–943. [PubMed] [Google Scholar]
- 145.Suzuki A., Kozawa O., Shinoda J., Watanabe-Tomita Y., Saito H., Oiso Y. Mechanism of thrombin-induced arachidonic acid release in osteoblast-like cells. Prostaglandins Leukot Essent Fatty Acids. 1997;56:467–472. doi: 10.1016/s0952-3278(97)90601-3. [DOI] [PubMed] [Google Scholar]
- 146.Miwa M., Kozawa O.O., Tokuda H., Uematsu T. Involvement of arachidonic acid in chemical stress-induced interleukin-6 synthesis in osteoblast-like cells: Comparison with heat shock protein 27 induction. Prostaglandins Leukot Essent Fatty Acids. 2000;62:189–193. doi: 10.1054/plef.2000.0140. [DOI] [PubMed] [Google Scholar]
- 147.Chu S.T., Cheng H.H., Huang C.J., Chang H.C., Chi C.C., Su H.H., Hsu S.S., Wang J.L., Chen I.S., Liu S.I., et al. Phospholipase A2-independent Ca2+ entry and subsequent apoptosis induced by melittin in human MG63 osteosarcoma cells. Life Sci. 2007;80:364–369. doi: 10.1016/j.lfs.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 148.Pruzanski W., Kennedy B.P., van den Bosch H., Stefanski E., Vadas P. Microtubule depolymerization selectively down-regulates the synthesis of proinflammatory secretory nonpancreatic phospholipase A2. Lab. Invest. 1997;76:171–178. [PubMed] [Google Scholar]
- 149.Murakami M., Kuwata H., Amakasu Y., Shimbara S., Nakatani Y., Atsumi G., Kudo I. Prostaglandin E2 amplifies cytosolic phospholipase A2- and cyclooxygenase-2-dependent delayed prostaglandin E2 generation in mouse osteoblastic cells. Enhancement by secretory phospholipase A2. J. Biol. Chem. 1997;272:19891–19897. doi: 10.1074/jbc.272.32.19891. [DOI] [PubMed] [Google Scholar]
- 150.Kudo I., Murakami M. Diverse functional coupling of prostanoid biosynthetic enzymes in various cell types. Adv. Exp. Med. Biol. 1999;469:29–35. doi: 10.1007/978-1-4615-4793-8_5. [DOI] [PubMed] [Google Scholar]
- 151.Higashi S., Ohishi H., Kudo I. Augmented prostaglandin E2 generation resulting from increased activities of cytosolic and secretory phospholipase A2 and induction of cyclooxygenase-2 in interleukin-1 beta-stimulated rat calvarial cells during the mineralizing phase. Inflamm. Res. 2000;49:102–111. doi: 10.1007/s000110050566. [DOI] [PubMed] [Google Scholar]
- 152.Chen Q.R., Miyaura C., Higashi S., Murakami M., Kudo I., Saito S., Hiraide T., Shibasaki Y., Suda T. Activation of cytosolic phospholipase A2 by platelet-derived growth factor is essential for cyclooxygenase-2-dependent prostaglandin E2 synthesis in mouse osteoblasts cultured with interleukin-1. J. Biol. Chem. 1997;272:5952–5958. doi: 10.1074/jbc.272.9.5952. [DOI] [PubMed] [Google Scholar]
- 153.Miyahara T., Katoh T., Watanabe M., Mikami Y., Uchida S., Hosoe M., Sakuma T., Nemoto N., Takayama K., Komurasaki T. Involvement of mitogen-activated protein kinases and protein kinase C in cadmium-induced prostaglandin E2 production in primary mouse osteoblastic cells. Toxicology. 2004;200:159–167. doi: 10.1016/j.tox.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 154.Miyahara T., Tonoyama H., Watanabe M., Okajima A., Miyajima S., Sakuma T., Nemoto N., Takayama K. Stimulative effect of cadmium on prostaglandin E2 production in primary mouse osteoblastic cells. Calcif. Tissue Int. 2001;68:185–191. doi: 10.1007/s002230001216. [DOI] [PubMed] [Google Scholar]
- 155.Leis H.J., Windischhofer W. Inhibition of cyclooxygenases 1 and 2 by the phospholipase-blocker, arachidonyl trifluoromethyl ketone. Br. J. Pharmacol. 2008;155:731–737. doi: 10.1038/bjp.2008.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Miyaura C., Inada M., Matsumoto C., Ohshiba T., Uozumi N., Shimizu T., Ito A. An essential role of cytosolic phospholipase A2alpha in prostaglandin E2-mediated bone resorption associated with inflammation. J. Exp. Med. 2003;197:1303–1310. doi: 10.1084/jem.20030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hackett J.A., Allard-Chamard H., Sarrazin P., de Fatima Lucena M., Gallant M.A., Fortier I., Nader M., Parent J.L., Bkaily G., de Brum-Fernandes A.J. Prostaglandin production by human osteoclasts in culture. J. Rheumatol. 2006;33:1320–1328. [PubMed] [Google Scholar]
- 158.Gregory L.S., Kelly W.L., Reid R.C., Fairlie D.P., Forwood M.R. Inhibitors of cyclooxygenase-2 and secretory phospholipase A2 preserve bone architecture following ovariectomy in adult rats. Bone. 2006;39:134–142. doi: 10.1016/j.bone.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 159.Kurihara H., Nakano T., Takasu N., Arita H. Intracellular localization of group II phospholipase A2 in rat vascular smooth muscle cells and its possible relationship to eicosanoid formation. Biochim. Biophys. Acta. 1991;1082:285–292. doi: 10.1016/0005-2760(91)90204-u. [DOI] [PubMed] [Google Scholar]
- 160.Nevalainen T.J., Grönroos J.M., Kallajoki M. Expression of group II phospholipase A2 in the human gastrointestinal tract. Lab. Invest. 1995;72:201–208. [PubMed] [Google Scholar]
- 161.Romano M., Romano E., Björkerud S., Hurt-Camejo E. Ultrastructural localization of secretory type II phospholipase A2 in atherosclerotic and nonatherosclerotic regions of human arteries. Arterioscler. Thromb. Vasc. Biol. 1998;18:519–525. doi: 10.1161/01.atv.18.4.519. [DOI] [PubMed] [Google Scholar]
- 162.Sartipy P., Johansen B., Gâsvik K., Hurt-Camejo E. Molecular basis for the association of group IIA phospholipase A(2) and decorin in human atherosclerotic lesions. Circ. Res. 2000;86:707–714. doi: 10.1161/01.res.86.6.707. [DOI] [PubMed] [Google Scholar]
- 163.Hurt-Camejo E., Camejo G., Peilot H., Oörni K., Kovanen P. Phospholipase A(2) in vascular disease. Circ. Res. 2001;89:298–304. doi: 10.1161/hh1601.095598. [DOI] [PubMed] [Google Scholar]
- 164.Masuda S., Murakami M., Ishikawa Y., Ishii T., Kudo I. Diverse cellular localizations of secretory phospholipase A2 enzymes in several human tissues. Biochim. Biophys. Acta. 2005;1736:200–210. doi: 10.1016/j.bbalip.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 165.Pruzanski W., Keystone E.C., Sternby B., Bombardier C., Snow K.M., Vadas P. Serum phospholipase A2 correlates with disease activity in rheumatoid arthritis. J. Rheumatol. 1988;15:1351–1355. [PubMed] [Google Scholar]
- 166.Pruzanski W., Vadas P. Phospholipase A2—A mediator between proximal and distal effectors of inflammation. Immunol. Today. 1991;12:143–146. doi: 10.1016/S0167-5699(05)80042-8. [DOI] [PubMed] [Google Scholar]
- 167.Vignon E., Balblanc J.C., Mathieu P., Louisot P., Richard M. Metalloprotease activity, phospholipase A2 activity and cytokine concentration in osteoarthritis synovial fluids. Osteoarthritis Cartilage. 1993;1:115–120. doi: 10.1016/s1063-4584(05)80026-3. [DOI] [PubMed] [Google Scholar]
- 168.Kortekangas P., Aro H.T., Nevalainen T.J. Group II phospholipase A2 in synovial fluid and serum in acute arthritis. Scand. J. Rheumatol. 1994;23:68–72. doi: 10.3109/03009749409103030. [DOI] [PubMed] [Google Scholar]
- 169.Pruzanski W., Albin-Cook K., Laxer R.M., MacMillan J., Stefanski E., Vadas P., Silverman E.D. Phospholipase A2 in juvenile rheumatoid arthritis: Correlation to disease type and activity. J. Rheumatol. 1994;21:1951–1954. [PubMed] [Google Scholar]
- 170.Lin M.K., Farewell V., Vadas P., Bookman A.A., Keystone E.C., Pruzanski W. Secretory phospholipase A2 as an index of disease activity in rheumatoid arthritis. Prospective double blind study of 212 patients. J. Rheumatol. 1996;23:1162–1166. [PubMed] [Google Scholar]
- 171.Michaels R.M., Reading J.C., Beezhold D.H., Ward J.R. Serum phospholipase A2 activity in patients with rheumatoid arthritis before and after treatment with methotrexate, auranofin, or combination of the two. J. Rheumatol. 1996;23:226–229. [PubMed] [Google Scholar]
- 172.Bidgood M.J., Jamal O.S., Cunningham A.M., Brooks P.M., Scott K.F. Type IIA secretory phospholipase A2 up-regulates cyclooxygenase-2 and amplifies cytokine-mediated prostaglandin production in human rheumatoid synoviocytes. J. Immunol. 2000;165:2790–2797. doi: 10.4049/jimmunol.165.5.2790. [DOI] [PubMed] [Google Scholar]
- 173.Ellies L.G., Heersche J.N., Pruzanski W., Vadas P., Aubin J.E. The role of phospholipase A2 in interleukin-1 alpha-mediated inhibition of mineralization of the osteoid formed by fetal rat calvaria cells in vitro. J. Dent. Res. 1993;72:18–24. doi: 10.1177/00220345930720010101. [DOI] [PubMed] [Google Scholar]
- 174.Hegen M., Sun L., Uozumi N., Kume K., Goad M.E., Nickerson-Nutter C.L., Shimizu T., Clark J.D. Cytosolic phospholipase A2alpha-deficient mice are resistant to collagen-induced arthritis. J. Exp. Med. 2003;197:1297–1302. doi: 10.1084/jem.20030016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Murakami M., Taketomi Y., Girard C., Yamamoto K., Lambeau G. Emerging roles of secreted phospholipase A2 enzymes: Lessons from transgenic and knockout mice. Biochimie. 2010;92:561–582. doi: 10.1016/j.biochi.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 176.Bonventre J.V., Huang Z., Taheri M.R., O’Leary E., Li E., Moskowitz M.A., Sapirstein A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 177.Uozumi N., Kume K., Nagase T., Nakatani N., Ishii S., Tashiro F., Komagata Y., Maki K., Ikuta K., Ouchi Y., et al. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997;390:618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 178.Sapirstein A., Bonventre J.V. Specific physiological roles of cytosolic phospholipase A(2) as defined by gene knockouts. Biochim. Biophys. Acta. 2000;1488:139–148. doi: 10.1016/s1388-1981(00)00116-5. [DOI] [PubMed] [Google Scholar]
- 179.Bao S., Miller D.J., Ma Z., Wohltmann M., Eng G., Ramanadham S., Moley K., Turk J. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J. Biol. Chem. 2004;279:38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bao S., Jacobson D.A., Wohltmann M., Bohrer A., Jin W., Philipson L.H., Turk J. Glucose homeostasis, insulin secretion, and islet phospholipids in mice that overexpress iPLA2beta in pancreatic beta-cells and in iPLA2beta-null mice. Am. J. Physiol. Endocrinol. Metab. 2008;294:E217–E229. doi: 10.1152/ajpendo.00474.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mancuso D.J., Kotzbauer P., Wozniak D.F., Sims H.F., Jenkins C.M., Guan S., Han X., Yang K., Sun G., Malik I., et al. Genetic ablation of calcium-independent phospholipase A2 gamma leads to alterations in hippocampal cardiolipin content and molecular species distribution, mitochondrial degeneration, autophagy, and cognitive dysfunction. J. Biol. Chem. 2009;284:35632–35644. doi: 10.1074/jbc.M109.055194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mancuso D.J., Sims H.F., Han X., Jenkins C.M., Guan S.P., Yang K., Moon S.H., Pietka T., Abumrad N.A., Schlesinger P.H., et al. Genetic ablation of calcium-independent phospholipase A2gamma leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J. Biol. Chem. 2007;282:34611–34622. doi: 10.1074/jbc.M707795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mancuso D.J., Sims H.F., Yang K., Kiebish M.A., Su X., Jenkins C.M., Guan S., Moon S.H., Pietka T., Nassir F., et al. Genetic ablation of calcium-independent phospholipase A2gamma prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. J. Biol. Chem. 2010;285:36495–36510. doi: 10.1074/jbc.M110.115766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Boilard E., Lai Y., Larabee K., Balestrieri B., Ghomashchi F., Fujioka D., Gobezie R., Coblyn J.S., Weinblatt M.E., Massarotti E.M., et al. A novel anti-inflammatory role for secretory phospholipase A2 in immune complex-mediated arthritis. EMBO Mol. Med. 2010;2:172–187. doi: 10.1002/emmm.201000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Adler D.H., Cogan J.D., Phillips J.A., Schnetz-Boutaud N., Milne G.L., Iverson T., Stein J.A., Brenner D.A., Morrow J.D., Boutaud O., et al. Inherited human cPLA(2alpha) deficiency is associated with impaired eicosanoid biosynthesis, small intestinal ulceration, and platelet dysfunction. J. Clin. Invest. 2008;118:2121–2131. doi: 10.1172/JCI30473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Uozumi N., Shimizu T. Roles for cytosolic phospholipase A2alpha as revealed by gene-targeted mice. Prostaglandins Other Lipid Mediat. 2002;68–69:59–69. doi: 10.1016/s0090-6980(02)00021-7. [DOI] [PubMed] [Google Scholar]
- 187.Balsinde J., Balboa M.A., Insel P.A., Dennis E.A. Regulation and inhibition of phospholipase A2. Annu. Rev. Pharmacol. Toxicol. 1999;39:175–189. doi: 10.1146/annurev.pharmtox.39.1.175. [DOI] [PubMed] [Google Scholar]
- 188.Pruzanski W., Stefanski E., Vadas P., Ramamurthy N.S. Inhibition of extracellular release of proinflammatory secretory phospholipase A2 (sPLA2) by sulfasalazine: A novel mechanism of anti-inflammatory activity. Biochem. Pharmacol. 1997;53:1901–1907. doi: 10.1016/s0006-2952(97)00137-8. [DOI] [PubMed] [Google Scholar]
- 189.Bradley J.D., Dmitrienko A.A., Kivitz A.J., Gluck O.S., Weaver A.L., Wiesenhutter C., Myers S.L., Sides G.D. A randomized, double-blinded, placebo-controlled clinical trial of LY333013, a selective inhibitor of group II secretory phospholipase A2, in the treatment of rheumatoid arthritis. J. Rheumatol. 2005;32:417–423. [PubMed] [Google Scholar]
- 190.Bryant K.J., Bidgood M.J., Lei P.W., Taberner M., Salom C., Kumar V., Lee L., Church W.B., Courtenay B., Smart B.P., et al. A bifunctional role for group IIA secreted phospholipase A2 in human rheumatoid fibroblast-like synoviocyte arachidonic acid metabolism. J. Biol. Chem. 2011;286:2492–2503. doi: 10.1074/jbc.M110.123927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Granata F., Petraroli A., Boilard E., Bezzine S., Bollinger J., Del Vecchio L., Gelb M.H., Lambeau G., Marone G., Triggiani M. Activation of cytokine production by secreted phospholipase A2 in human lung macrophages expressing the M-type receptor. J. Immunol. 2005;174:464–474. doi: 10.4049/jimmunol.174.1.464. [DOI] [PubMed] [Google Scholar]
- 192.Rosenson R.S., Hislop C., Elliott M., Stasiv Y., Goulder M., Waters D. Effects of varespladib methyl on biomarkers and major cardiovascular events in acute coronary syndrome patients. J. Am. Coll. Cardiol. 2010;56:1079–1088. doi: 10.1016/j.jacc.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 193.Divchev D., Schieffer B. The secretory phospholipase A2 group IIA: A missing link between inflammation, activated renin-angiotensin system, and atherogenesis? Vasc. Health Risk Manag. 2008;4:597–604. doi: 10.2147/vhrm.s2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tai N., Kuwabara K., Kobayashi M., Yamada K., Ono T., Seno K., Gahara Y., Ishizaki J., Hori Y. Cytosolic phospholipase A2 alpha inhibitor, pyrroxyphene, displays anti-arthritic and anti-bone destructive action in a murine arthritis model. Inflamm. Res. 2010;59:53–62. doi: 10.1007/s00011-009-0069-8. [DOI] [PubMed] [Google Scholar]
- 195.Kramer R.M., Hession C., Johansen B., Hayes G., McGray P., Chow E.P., Tizard R., Pepinsky R.B. Structure and properties of a human non-pancreatic phospholipase A2. J. Biol. Chem. 1989;264:5768–5775. [PubMed] [Google Scholar]
- 196.Singer A.G., Ghomashchi F., Le Calvez C., Bollinger J., Bezzine S., Rouault M., Sadilek M., Nguyen E., Lazdunski M., Lambeau G., et al. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. J. Biol. Chem. 2002;277:48535–48549. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- 197.Bezzine S., Bollinger J.G., Singer A.G., Veatch S.L., Keller S.L., Gelb M.H. On the binding preference of human groups IIA and X phospholipases A2 for membranes with anionic phospholipids. J. Biol. Chem. 2002;277:48523–48534. doi: 10.1074/jbc.M203137200. [DOI] [PubMed] [Google Scholar]
- 198.Mounier C.M., Ghomashchi F., Lindsay M.R., James S., Singer A.G., Parton R.G., Gelb M.H. Arachidonic acid release from mammalian cells transfected with human groups IIA and X secreted phospholipase A(2) occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A(2)-alpha. J. Biol. Chem. 2004;279:25024–25038. doi: 10.1074/jbc.M313019200. [DOI] [PubMed] [Google Scholar]
- 199.Balsinde J., Balboa M.A., Dennis E.A. Functional coupling between secretory phospholipase A2 and cyclooxygenase-2 and its regulation by cytosolic group IV phospholipase A2. Proc. Natl. Acad. Sci. USA. 1998;95:7951–7956. doi: 10.1073/pnas.95.14.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Han S.K., Kim K.P., Koduri R., Bittova L., Munoz N.M., Leff A.R., Wilton D.C., Gelb M.H., Cho W. Roles of Trp31 in high membrane binding and proinflammatory activity of human group V phospholipase A2. J. Biol. Chem. 1999;274:11881–11888. doi: 10.1074/jbc.274.17.11881. [DOI] [PubMed] [Google Scholar]
- 201.Hanasaki K., Ono T., Saiga A., Morioka Y., Ikeda M., Kawamoto K., Higashino K., Nakano K., Yamada K., Ishizaki J., et al. Purified group X secretory phospholipase A(2) induced prominent release of arachidonic acid from human myeloid leukemia cells. J. Biol. Chem. 1999;274:34203–34211. doi: 10.1074/jbc.274.48.34203. [DOI] [PubMed] [Google Scholar]
- 202.Murakami M., Kambe T., Shimbara S., Higashino K., Hanasaki K., Arita H., Horiguchi M., Arita M., Arai H., Inoue K., et al. Different functional aspects of the group II subfamily (Types IIA and V) and type X secretory phospholipase A(2)s in regulating arachidonic acid release and prostaglandin generation. Implications of cyclooxygenase-2 induction and phospholipid scramblase-mediated cellular membrane perturbation. J. Biol. Chem. 1999;274:31435–31444. doi: 10.1074/jbc.274.44.31435. [DOI] [PubMed] [Google Scholar]
- 203.Bezzine S., Koduri R.S., Valentin E., Murakami M., Kudo I., Ghomashchi F., Sadilek M., Lambeau G., Gelb M.H. Exogenously added human group X secreted phospholipase A(2) but not the group IB, IIA, and V enzymes efficiently release arachidonic acid from adherent mammalian cells. J. Biol. Chem. 2000;275:3179–3191. doi: 10.1074/jbc.275.5.3179. [DOI] [PubMed] [Google Scholar]
- 204.Kim K.P., Rafter J.D., Bittova L., Han S.K., Snitko Y., Munoz N.M., Leff A.R., Cho W. Mechanism of human group V phospholipase A2 (PLA2)-induced leukotriene biosynthesis in human neutrophils. A potential role of heparan sulfate binding in PLA2 internalization and degradation. J. Biol. Chem. 2001;276:11126–11134. doi: 10.1074/jbc.M004604200. [DOI] [PubMed] [Google Scholar]
- 205.Murakami M., Koduri R.S., Enomoto A., Shimbara S., Seki M., Yoshihara K., Singer A., Valentin E., Ghomashchi F., Lambeau G., et al. Distinct arachidonate-releasing functions of mammalian secreted phospholipase A2s in human embryonic kidney 293 and rat mastocytoma RBL-2H3 cells through heparan sulfate shuttling and external plasma membrane mechanisms. J. Biol. Chem. 2001;276:10083–10096. doi: 10.1074/jbc.M007877200. [DOI] [PubMed] [Google Scholar]
- 206.Kim Y.J., Kim K.P., Rhee H.J., Das S., Rafter J.D., Oh Y.S., Cho W. Internalized group V secretory phospholipase A2 acts on the perinuclear membranes. J. Biol. Chem. 2002;277:9358–9365. doi: 10.1074/jbc.M110987200. [DOI] [PubMed] [Google Scholar]
- 207.Muñoz N.M., Kim Y.J., Meliton A.Y., Kim K.P., Han S.K., Boetticher E., O’Leary E., Myou S., Zhu X., Bonventre J.V., et al. Human group V phospholipase A2 induces group IVA phospholipase A2-independent cysteinyl leukotriene synthesis in human eosinophils. J. Biol. Chem. 2003;278:38813–38820. doi: 10.1074/jbc.M302476200. [DOI] [PubMed] [Google Scholar]
- 208.Murakami M., Shimbara S., Kambe T., Kuwata H., Winstead M.V., Tischfield J.A., Kudo I. The functions of five distinct mammalian phospholipase A2S in regulating arachidonic acid release. Type IIa and type V secretory phospholipase A2S are functionally redundant and act in concert with cytosolic phospholipase A2. J. Biol. Chem. 1998;273:14411–14423. doi: 10.1074/jbc.273.23.14411. [DOI] [PubMed] [Google Scholar]
- 209.Murakami M., Kambe T., Shimbara S., Yamamoto S., Kuwata H., Kudo I. Functional association of type IIA secretory phospholipase A(2) with the glycosylphosphatidylinositol-anchored heparan sulfate proteoglycan in the cyclooxygenase-2-mediated delayed prostanoid-biosynthetic pathway. J. Biol. Chem. 1999;274:29927–29936. doi: 10.1074/jbc.274.42.29927. [DOI] [PubMed] [Google Scholar]
- 210.Murakami M., Kambe T., Shimbara S., Kudo I. Functional coupling between various phospholipase A2s and cyclooxygenases in immediate and delayed prostanoid biosynthetic pathways. J. Biol. Chem. 1999;274:3103–3115. doi: 10.1074/jbc.274.5.3103. [DOI] [PubMed] [Google Scholar]
- 211.Balboa M.A., Shirai Y., Gaietta G., Ellisman M.H., Balsinde J., Dennis E.A. Localization of group V phospholipase A2 in caveolin-enriched granules in activated P388D1 macrophage-like cells. J. Biol. Chem. 2003;278:48059–48065. doi: 10.1074/jbc.M305904200. [DOI] [PubMed] [Google Scholar]
- 212.Lambeau G., Lazdunski M. Receptors for a growing family of secreted phospholipases A2. Trends Pharmacol. Sci. 1999;20:162–170. doi: 10.1016/s0165-6147(99)01300-0. [DOI] [PubMed] [Google Scholar]
- 213.Reynolds L.J., Hughes L.L., Louis A.I., Kramer R.M., Dennis E.A. Metal ion and salt effects on the phospholipase A2, lysophospholipase, and transacylase activities of human cytosolic phospholipase A2. Biochim. Biophys. Acta. 1993;1167:272–280. doi: 10.1016/0005-2760(93)90229-3. [DOI] [PubMed] [Google Scholar]
- 214.Huber L.C., Distler O., Tarner I., Gay R.E., Gay S., Pap T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatology. 2006;45:669–675. doi: 10.1093/rheumatology/kel065. [DOI] [PubMed] [Google Scholar]
- 215.Myers L.K., Kang A.H., Postlethwaite A.E., Rosloniec E.F., Morham S.G., Shlopov B.V., Goorha S., Ballou L.R. The genetic ablation of cyclooxygenase 2 prevents the development of autoimmune arthritis. Arthritis Rheum. 2000;43:2687–2693. doi: 10.1002/1529-0131(200012)43:12<2687::AID-ANR8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 216.Ochi T., Ohkubo Y., Mutoh S. Role of cyclooxygenase-2, but not cyclooxygenase-1, on type II collagen-induced arthritis in DBA/1J mice. Biochem. Pharmacol. 2003;66:1055–1060. doi: 10.1016/s0006-2952(03)00420-9. [DOI] [PubMed] [Google Scholar]
- 217.Trebino C.E., Stock J.L., Gibbons C.P., Naiman B.M., Wachtmann T.S., Umland J.P., Pandher K., Lapointe J.M., Saha S., Roach M.L., et al. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc. Natl. Acad. Sci. USA. 2003;100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kamei D., Yamakawa K., Takegoshi Y., Mikami-Nakanishi M., Nakatani Y., Oh-Ishi S., Yasui H., Azuma Y., Hirasawa N., Ohuchi K., et al. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J. Biol. Chem. 2004;279:33684–33695. doi: 10.1074/jbc.M400199200. [DOI] [PubMed] [Google Scholar]
- 219.Vane J.R., Botting R.M. New insights into the mode of action of anti-inflammatory drugs. Inflamm. Res. 1995;44:1–10. doi: 10.1007/BF01630479. [DOI] [PubMed] [Google Scholar]
- 220.Altman R.D., Latta L.L., Keer R., Renfree K., Hornicek F.J., Banovac K. Effect of nonsteroidal antiinflammatory drugs on fracture healing: A laboratory study in rats. J. Orthop. Trauma. 1995;9:392–400. doi: 10.1097/00005131-199505000-00006. [DOI] [PubMed] [Google Scholar]
- 221.Törnkvist H., Lindholm T.S., Netz P., Strömberg L., Lindholm T.C. Effect of ibuprofen and indomethacin on bone metabolism reflected in bone strength. Clin. Orthop. Relat. Res. 1984;187:255–259. [PubMed] [Google Scholar]
- 222.Ro J., Langeland N., Sander J. Effect of indomethacin on collagen metabolism of rat fracture callus in vitro. Acta Orthop. Scand. 1978;49:323–328. doi: 10.3109/17453677809050082. [DOI] [PubMed] [Google Scholar]
- 223.Keller J., Bünger C., Andreassen T.T., Bak B., Lucht U. Bone repair inhibited by indomethacin. Effects on bone metabolism and strength of rabbit osteotomies. Acta Orthop. Scand. 1987;58:379–383. doi: 10.3109/17453678709146360. [DOI] [PubMed] [Google Scholar]
- 224.Ritter M.A., Gioe T.J. The effect of indomethacin on para-articular ectopic ossification following total hip arthroplasty. Clin. Orthop. Relat. Res. 1982;167:113–117. [PubMed] [Google Scholar]
- 225.Sodemann B., Persson P.E., Nilsson O.S. Prevention of periarticular heterotopic ossification following total hip arthroplasty: Clinical experience with indomethacin and ibuprofen. Arch. Orthop. Trauma Surg. 1988;107:329–333. doi: 10.1007/BF00381057. [DOI] [PubMed] [Google Scholar]
- 226.Kjaersgaard-Andersen P., Ritter M.A. Short-term treatment with nonsteroidal antiinflammatory medications to prevent heterotopic bone formation after total hip arthroplasty. A preliminary report. Clin. Orthop. Relat. Res. 1992;279:157–162. [PubMed] [Google Scholar]
- 227.Dimar J.R., Ante W.A., Zhang Y.P., Glassman S.D. The effects of nonsteroidal anti-inflammatory drugs on posterior spinal fusions in the rat. Spine (Phila Pa 1976) 1996;21:1870–1876. doi: 10.1097/00007632-199608150-00006. [DOI] [PubMed] [Google Scholar]
- 228.Glassman S.D., Rose S.M., Dimar J.R., Puno R.M., Campbell M.J., Johnson J.R. The effect of postoperative nonsteroidal anti-inflammatory drug administration on spinal fusion. Spine (Phila Pa 1976) 1998;23:834–838. doi: 10.1097/00007632-199804010-00020. [DOI] [PubMed] [Google Scholar]
- 229.Chang J.K., Li C.J., Wu S.C., Yeh C.H., Chen C.H., Fu Y.C., Wang G.J., Ho M.L. Effects of anti-inflammatory drugs on proliferation, cytotoxicity and osteogenesis in bone marrow mesenchymal stem cells. Biochem. Pharmacol. 2007;74:1371–13782. doi: 10.1016/j.bcp.2007.06.047. [DOI] [PubMed] [Google Scholar]
- 230.Stafford J.B., Marnett L.J. Prostaglandin E2 inhibits tumor necrosis factor-alpha RNA through PKA type I. Biochem. Biophys. Res. Commun. 2008;366:104–109. doi: 10.1016/j.bbrc.2007.11.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Crofford L.J., Lipsky P.E., Brooks P., Abramson S.B., Simon L.S., van de Putte L.B. Basic biology and clinical application of specific cyclooxygenase-2 inhibitors. Arthritis Rheum. 2000;43:4–13. doi: 10.1002/1529-0131(200001)43:1<4::AID-ANR2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 232.Martel-Pelletier J., Pelletier J.P., Fahmi H. Cyclooxygenase-2 and prostaglandins in articular tissues. Semin. Arthritis Rheum. 2003;33:155–167. doi: 10.1016/s0049-0172(03)00134-3. [DOI] [PubMed] [Google Scholar]
- 233.Blackwell K.A., Raisz L.G., Pilbeam C.C. Prostaglandins in bone: Bad cop, good cop? Trends Endocrinol. Metab. 2010;21:294–301. doi: 10.1016/j.tem.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Miller S.B. Prostaglandins in health and disease: An overview. Semin. Arthritis Rheum. 2006;36:37–49. doi: 10.1016/j.semarthrit.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 235.Martel-Pelletier J., Lajeunesse D., Fahmi H., Tardif G., Pelletier J.P. New thoughts on the pathophysiology of osteoarthritis: One more step toward new therapeutic targets. Curr. Rheumatol. Rep. 2006;8:30–36. doi: 10.1007/s11926-006-0022-6. [DOI] [PubMed] [Google Scholar]
- 236.Zhang X., Schwarz E.M., Young D.A., Puzas J.E., Rosier R.N., O’Keefe R.J. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J. Clin. Invest. 2002;109:1405–1415. doi: 10.1172/JCI15681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Atik O.S., Uslu M.M., Eksioglu F., Satana T. Etiology of senile osteoporosis: A hypothesis. Clin. Orthop. Relat. Res. 2006;443:25–27. doi: 10.1097/01.blo.0000200235.76565.c8. [DOI] [PubMed] [Google Scholar]
- 238.Hikiji H., Takato T., Shimizu T., Ishii S. The roles of prostanoids, leukotrienes, and platelet-activating factor in bone metabolism and disease. Prog. Lipid Res. 2008;47:107–126. doi: 10.1016/j.plipres.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 239.Lettesjö H., Nordström E., Ström H., Nilsson B., Glinghammar B., Dahlstedt L., Möller E. Synovial fluid cytokines in patients with rheumatoid arthritis or other arthritic lesions. Scand. J. Immunol. 1998;48:286–292. doi: 10.1046/j.1365-3083.1998.00399.x. [DOI] [PubMed] [Google Scholar]
- 240.Hidaka T., Suzuki K., Kawakami M., Okada M., Kataharada K., Shinohara T., Takamizawa-Matsumoto M., Ohsuzu F. Dynamic changes in cytokine levels in serum and synovial fluid following filtration leukocytapheresis therapy in patients with rheumatoid arthritis. J. Clin. Apher. 2001;16:74–81. doi: 10.1002/jca.1016. [DOI] [PubMed] [Google Scholar]
- 241.Hochberg M.C. COX-2 selective inhibitors in the treatment of arthritis: A rheumatologist perspective. Curr. Top. Med. Chem. 2005;5:443–448. doi: 10.2174/1568026054201695. [DOI] [PubMed] [Google Scholar]
- 242.Boyan B.D., Sylvia V.L., Liu Y., Sagun R., Cochran D.L., Lohmann C.H., Dean D.D., Schwartz Z. Surface roughness mediates its effects on osteoblasts via protein kinase A and phospholipase A2. Biomaterials. 1999;20:2305–2310. doi: 10.1016/s0142-9612(99)00159-3. [DOI] [PubMed] [Google Scholar]
- 243.Kawaguchi H., Pilbeam C.C., Harrison J.R., Raisz L.G. The role of prostaglandins in the regulation of bone metabolism. Clin. Orthop. Relat. Res. 1995:36–46. [PubMed] [Google Scholar]
- 244.Watkins B.A., Lippman H.E., Le Bouteiller L., Li Y., Seifert M.F. Bioactive fatty acids: Role in bone biology and bone cell function. Prog. Lipid Res. 2001;40:125–148. doi: 10.1016/s0163-7827(00)00016-3. [DOI] [PubMed] [Google Scholar]
- 245.Knippenberg M., Helder M.N., de Blieck-Hogervorst J.M., Wuisman P.I., Klein-Nulend J. Prostaglandins differentially affect osteogenic differentiation of human adipose tissue-derived mesenchymal stem cells. Tissue Eng. 2007;13:2495–2503. doi: 10.1089/ten.2006.0420. [DOI] [PubMed] [Google Scholar]
- 246.Sato I., Suzuki A., Kakita A., Ono Y., Miura Y., Itoh M., Oiso Y. Stimulatory effect of prostaglandin F(2alpha) on Na-dependent phosphate transport in osteoblast-like cells. Prostaglandins Leukotrienes Essent. Fatty Acids. 2003;68:311–315. doi: 10.1016/s0952-3278(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 247.Montessuit C., Caverzasio J., Bonjour J.P. Characterization of a Pi transport system in cartilage matrix vesicles. Potential role in the calcification process. J. Biol. Chem. 1991;266:17791–17797. [PubMed] [Google Scholar]
- 248.Khan E., Abu-Amer Y. Activation of peroxisome proliferator-activated receptor-gamma inhibits differentiation of preosteoblasts. J. Lab. Clin. Med. 2003;142:29–34. doi: 10.1016/S0022-2143(03)00058-1. [DOI] [PubMed] [Google Scholar]
- 249.Koshihara Y., Kawamura M. Prostaglandin D2 stimulates calcification of human osteoblastic cells. Biochem. Biophys. Res. Commun. 1989;159:1206–1212. doi: 10.1016/0006-291x(89)92238-9. [DOI] [PubMed] [Google Scholar]
- 250.Baudry A., Bitard J., Mouillet-Richard S., Locker M., Poliard A., Launay J.M., Kellermann O. Serotonergic 5-HT(2B) receptor controls tissue-nonspecific alkaline phosphatase activity in osteoblasts via eicosanoids and phosphatidylinositol-specific phospholipase C. J. Biol. Chem. 2010;285:26066–26073. doi: 10.1074/jbc.M109.073791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Tsushita K., Kozawa O., Tokuda H., Oiso Y., Saito H. Proliferative effect of PGD2 on osteoblast-like cells; independent activation of pertussis toxin-sensitive GTP-binding protein from PGE2 or PGF2 alpha. Prostaglandins Leukotrienes Essent. Fatty Acids. 1992;45:267–274. doi: 10.1016/0952-3278(92)90082-t. [DOI] [PubMed] [Google Scholar]
- 252.Makide K., Kitamura H., Sato Y., Okutani M., Aoki J. Emerging lysophospholipid mediators, lysophosphatidylserine, lysophosphatidylthreonine, lysophosphatidylethanolamine and lysophosphatidylglycerol. Prostaglandins Other Lipid Mediat. 2009;89:135–139. doi: 10.1016/j.prostaglandins.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 253.Oka S., Nakajima K., Yamashita A., Kishimoto S., Sugiura T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem. Biophys. Res. Commun. 2007;362:928–934. doi: 10.1016/j.bbrc.2007.08.078. [DOI] [PubMed] [Google Scholar]
- 254.Henstridge C.M., Balenga N.A., Ford L.A., Ross R.A., Waldhoer M., Irving A.J. The GPR55 ligand l-alpha-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB J. 2009;23:183–193. doi: 10.1096/fj.08-108670. [DOI] [PubMed] [Google Scholar]
- 255.Henstridge C.M. Off-target cannabinoid effects mediated by GPR55. Pharmacology. 2012;89:179–187. doi: 10.1159/000336872. [DOI] [PubMed] [Google Scholar]
- 256.Sharir H., Abood M.E. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol. Ther. 2010;126:301–313. doi: 10.1016/j.pharmthera.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Brown A.J., Daniels D.A., Kassim M., Brown S., Haslam C.P., Terrell V.R., Brown J., Nichols P.L., Staton P.C., Wise A., et al. Pharmacology of GPR55 in yeast and identification of GSK494581A as a mixed-activity glycine transporter subtype 1 inhibitor and GPR55 agonist. J. Pharmacol. Exp. Ther. 2011;337:236–246. doi: 10.1124/jpet.110.172650. [DOI] [PubMed] [Google Scholar]
- 258.Oka S., Toshida T., Maruyama K., Nakajima K., Yamashita A., Sugiura T. 2-Arachidonoyl-sn-glycero-3-phosphoinositol: A possible natural ligand for GPR55. J. Biochem. 2009;145:13–20. doi: 10.1093/jb/mvn136. [DOI] [PubMed] [Google Scholar]
- 259.Whyte L.S., Ryberg E., Sims N.A., Ridge S.A., Mackie K., Greasley P.J., Ross R.A., Rogers M.J. The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo. Proc. Natl. Acad. Sci. USA. 2009;106:16511–16516. doi: 10.1073/pnas.0902743106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Staton P.C., Hatcher J.P., Walker D.J., Morrison A.D., Shapland E.M., Hughes J.P., Chong E., Mander P.K., Green P.J., Billinton A., et al. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain. 2008;139:225–236. doi: 10.1016/j.pain.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 261.Blackburn J., Mansell J.P. The emerging role of lysophosphatidic acid (LPA) in skeletal biology. Bone. 2012;50:756–762. doi: 10.1016/j.bone.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 262.Hurst-Kennedy J., Boyan B.D., Schwartz Z. Lysophosphatidic acid signaling promotes proliferation, differentiation, and cell survival in rat growth plate chondrocytes. Biochim. Biophys. Acta. 2009;1793:836–846. doi: 10.1016/j.bbamcr.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 263.Tigyi G., Miledi R. Lysophosphatidates bound to serum albumin activate membrane currents in Xenopus oocytes and neurite retraction in PC12 pheochromocytoma cells. J. Biol. Chem. 1992;267:21360–21367. [PubMed] [Google Scholar]
- 264.Baker D.L., Umstot E.S., Desiderio D.M., Tigyi G.J. Quantitative analysis of lysophosphatidic acid in human blood fractions. Ann. N. Y. Acad. Sci. 2000;905:267–269. doi: 10.1111/j.1749-6632.2000.tb06557.x. [DOI] [PubMed] [Google Scholar]
- 265.Sano T., Baker D., Virag T., Wada A., Yatomi Y., Kobayashi T., Igarashi Y., Tigyi G. Multiple mechanisms linked to platelet activation result in lysophosphatidic acid and sphingosine 1-phosphate generation in blood. J. Biol. Chem. 2002;277:21197–21206. doi: 10.1074/jbc.M201289200. [DOI] [PubMed] [Google Scholar]
- 266.Aoki J., Taira A., Takanezawa Y., Kishi Y., Hama K., Kishimoto T., Mizuno K., Saku K., Taguchi R., Arai H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J. Biol. Chem. 2002;277:48737–48744. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- 267.Hosogaya S., Yatomi Y., Nakamura K., Ohkawa R., Okubo S., Yokota H., Ohta M., Yamazaki H., Koike T., Ozaki Y. Measurement of plasma lysophosphatidic acid concentration in healthy subjects: Strong correlation with lysophospholipase D activity. Ann. Clin. Biochem. 2008;45:364–368. doi: 10.1258/acb.2008.007242. [DOI] [PubMed] [Google Scholar]
- 268.Ishii I., Fukushima N., Ye X., Chun J. Lysophospholipid receptors: Signaling and biology. Annu. Rev. Biochem. 2004;73:321–354. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 269.Gerrard J.M., Robinson P. Identification of the molecular species of lysophosphatidic acid produced when platelets are stimulated by thrombin. Biochim. Biophys. Acta. 1989;1001:282–285. doi: 10.1016/0005-2760(89)90112-4. [DOI] [PubMed] [Google Scholar]
- 270.Xiao Y., Chen Y., Kennedy A.W., Belinson J., Xu Y. Evaluation of plasma lysophospholipids for diagnostic significance using electrospray ionization mass spectrometry (ESI-MS) analyses. Ann. N. Y. Acad. Sci. 2000;905:242–259. doi: 10.1111/j.1749-6632.2000.tb06554.x. [DOI] [PubMed] [Google Scholar]
- 271.Baker D.L., Desiderio D.M., Miller D.D., Tolley B., Tigyi G.J. Direct quantitative analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray ionization liquid chromatography-mass spectrometry. Anal. Biochem. 2001;292:287–295. doi: 10.1006/abio.2001.5063. [DOI] [PubMed] [Google Scholar]
- 272.Aoki J. Mechanisms of lysophosphatidic acid production. Semin. Cell Dev. Biol. 2004;15:477–489. doi: 10.1016/j.semcdb.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 273.Aoki J., Inoue A., Okudaira S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta. 2008;1781:513–518. doi: 10.1016/j.bbalip.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 274.Nakanaga K., Hama K., Aoki J. Autotaxin—An LPA producing enzyme with diverse functions. J. Biochem. 2010;148:13–24. doi: 10.1093/jb/mvq052. [DOI] [PubMed] [Google Scholar]
- 275.Tokumura A., Fujimoto H., Yoshimoto O., Nishioka Y., Miyake M., Fukuzawa K. Production of lysophosphatidic acid by lysophospholipase D in incubated plasma of spontaneously hypertensive rats and Wistar Kyoto rats. Life Sci. 1999;65:245–253. doi: 10.1016/s0024-3205(99)00243-x. [DOI] [PubMed] [Google Scholar]
- 276.Tokumura A., Majima E., Kariya Y., Tominaga K., Kogure K., Yasuda K., Fukuzawa K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002;277:39436–39442. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- 277.Umezu-Goto M., Kishi Y., Taira A., Hama K., Dohmae N., Takio K., Yamori T., Mills G.B., Inoue K., Aoki J., et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell. Biol. 2002;158:227–233. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Jonas A. Lecithin cholesterol acyltransferase. Biochim. Biophys. Acta. 2000;1529:245–256. doi: 10.1016/s1388-1981(00)00153-0. [DOI] [PubMed] [Google Scholar]
- 279.Sato T., Aoki J., Nagai Y., Dohmae N., Takio K., Doi T., Arai H., Inoue K. Serine phospholipid-specific phospholipase A that is secreted from activated platelets. A new member of the lipase family. J. Biol. Chem. 1997;272:2192–2198. doi: 10.1074/jbc.272.4.2192. [DOI] [PubMed] [Google Scholar]
- 280.Fourcade O., Simon M.F., Viodé C., Rugani N., Leballe F., Ragab A., Fournié B., Sarda L., Chap H. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell. 1995;80:919–927. doi: 10.1016/0092-8674(95)90295-3. [DOI] [PubMed] [Google Scholar]
- 281.Mauco G., Dajeans P., Chap H., Douste-Blazy L. Subcellular localization of inositol lipids in blood platelets as deduced from the use of labelled precursors. Biochem. J. 1987;244:757–761. doi: 10.1042/bj2440757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Pages C., Simon M.F., Valet P., Saulnier-Blache J.S. Lysophosphatidic acid synthesis and release. Prostaglandins Other Lipid Mediat. 2001;64:1–10. doi: 10.1016/s0090-6980(01)00110-1. [DOI] [PubMed] [Google Scholar]
- 283.Pasternack S.M., von Kügelgen I., Al Aboud K., Lee Y.A., Rüschendorf F., Voss K., Hillmer A.M., Molderings G.J., Franz T., Ramirez A., et al. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat. Genet. 2008;40:329–334. doi: 10.1038/ng.84. [DOI] [PubMed] [Google Scholar]
- 284.Thumser A.E., Voysey J.E., Wilton D.C. The binding of lysophospholipids to rat liver fatty acid-binding protein and albumin. Biochem. J. 1994;301:801–806. doi: 10.1042/bj3010801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Nochi H., Tomura H., Tobo M., Tanaka N., Sato K., Shinozaki T., Kobayashi T., Takagishi K., Ohta H., Okajima F., et al. Stimulatory role of lysophosphatidic acid in cyclooxygenase-2 induction by synovial fluid of patients with rheumatoid arthritis in fibroblast-like synovial cells. J. Immunol. 2008;181:5111–5119. doi: 10.4049/jimmunol.181.7.5111. [DOI] [PubMed] [Google Scholar]
- 286.Ferry G., Tellier E., Try A., Grés S., Naime I., Simon M.F., Rodriguez M., Boucher J., Tack I., Gesta S., et al. Autotaxin is released from adipocytes, catalyzes lysophosphatidic acid synthesis, and activates preadipocyte proliferation. Up-regulated expression with adipocyte differentiation and obesity. J. Biol. Chem. 2003;278:18162–18169. doi: 10.1074/jbc.M301158200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Boucharaba A., Serre C.M., Grès S., Saulnier-Blache J.S., Bordet J.C., Guglielmi J., Clézardin P., Peyruchaud O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Invest. 2004;114:1714–1725. doi: 10.1172/JCI22123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Panupinthu N., Zhao L., Possmayer F., Ke H.Z., Sims S.M., Dixon S.J. P2X7 nucleotide receptors mediate blebbing in osteoblasts through a pathway involving lysophosphatidic acid. J. Biol. Chem. 2007;282:3403–3412. doi: 10.1074/jbc.M605620200. [DOI] [PubMed] [Google Scholar]
- 289.Panupinthu N., Rogers J.T., Zhao L., Solano-Flores L.P., Possmayer F., Sims S.M., Dixon S.J. P2X7 receptors on osteoblasts couple to production of lysophosphatidic acid: A signaling axis promoting osteogenesis. J. Cell. Biol. 2008;181:859–871. doi: 10.1083/jcb.200708037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Gaits F., Fourcade O., Le Balle F., Gueguen G., Gaigé B., Gassama-Diagne A., Fauvel J., Salles J.P., Mauco G., Simon M.F., et al. Lysophosphatidic acid as a phospholipid mediator: Pathways of synthesis. FEBS Lett. 1997;410:54–58. doi: 10.1016/s0014-5793(97)00411-0. [DOI] [PubMed] [Google Scholar]
- 291.Chun J., Hla T., Lynch K.R., Spiegel S., Moolenaar W.H. International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 2010;62:579–587. doi: 10.1124/pr.110.003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Grey A., Banovic T., Naot D., Hill B., Callon K., Reid I., Cornish J. Lysophosphatidic acid is an osteoblast mitogen whose proliferative actions involve G(i) proteins and protein kinase C, but not P42/44 mitogen-activated protein kinases. Endocrinology. 2001;142:1098–1106. doi: 10.1210/endo.142.3.8011. [DOI] [PubMed] [Google Scholar]
- 293.Caverzasio J., Palmer G., Suzuki A., Bonjour J.P. Evidence for the involvement of two pathways in activation of extracellular signal-regulated kinase (Erk) and cell proliferation by Gi and Gq protein-coupled receptors in osteoblast-like cells. J. Bone Miner. Res. 2000;15:1697–1706. doi: 10.1359/jbmr.2000.15.9.1697. [DOI] [PubMed] [Google Scholar]
- 294.Dziak R., Yang B.M., Leung B.W., Li S., Marzec N., Margarone J., Bobek L. Effects of sphingosine-1-phosphate and lysophosphatidic acid on human osteoblastic cells. Prostaglandins Leukotrienes Essent. Fatty Acids. 2003;68:239–249. doi: 10.1016/s0952-3278(02)00277-6. [DOI] [PubMed] [Google Scholar]
- 295.Gidley J., Openshaw S., Pring E.T., Sale S., Mansell J.P. Lysophosphatidic acid cooperates with 1alpha,25(OH)2D3 in stimulating human MG63 osteoblast maturation. Prostaglandins Other Lipid Mediat. 2006;80:46–61. doi: 10.1016/j.prostaglandins.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 296.Kim M.K., Lee H.Y., Park K.S., Shin E.H., Jo S.H., Yun J., Lee S.W., Yoo Y.H., Lee Y.S., Baek S.H., et al. Lysophosphatidic acid stimulates cell proliferation in rat chondrocytes. Biochem. Pharmacol. 2005;70:1764–1771. doi: 10.1016/j.bcp.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 297.Grey A., Chen Q., Callon K., Xu X., Reid I.R., Cornish J. The phospholipids sphingosine-1-phosphate and lysophosphatidic acid prevent apoptosis in osteoblastic cells via a signaling pathway involving G(i) proteins and phosphatidylinositol-3 kinase. Endocrinology. 2002;143:4755–4763. doi: 10.1210/en.2002-220347. [DOI] [PubMed] [Google Scholar]
- 298.Lyons J.M., Karin N.J. A role for G protein-coupled lysophospholipid receptors in sphingolipid-induced Ca2+ signaling in MC3T3-E1 osteoblastic cells. J. Bone Miner. Res. 2001;16:2035–2042. doi: 10.1359/jbmr.2001.16.11.2035. [DOI] [PubMed] [Google Scholar]
- 299.Masiello L.M., Fotos J.S., Galileo D.S., Karin N.J. Lysophosphatidic acid induces chemotaxis in MC3T3-E1 osteoblastic cells. Bone. 2006;39:72–82. doi: 10.1016/j.bone.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 300.Checovich W.J., Mosher D.F. Lysophosphatidic acid enhances fibronectin binding to adherent cells. Arterioscler. Thromb. 1993;13:1662–1667. doi: 10.1161/01.atv.13.11.1662. [DOI] [PubMed] [Google Scholar]
- 301.Zhang Q., Checovich W.J., Peters D.M., Albrecht R.M., Mosher D.F. Modulation of cell surface fibronectin assembly sites by lysophosphatidic acid. J. Cell. Biol. 1994;127:1447–1459. doi: 10.1083/jcb.127.5.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Zhang Q., Magnusson M.K., Mosher D.F. Lysophosphatidic acid and microtubule-destabilizing agents stimulate fibronectin matrix assembly through Rho-dependent actin stress fiber formation and cell contraction. Mol. Biol. Cell. 1997;8:1415–1425. doi: 10.1091/mbc.8.8.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Karagiosis S.A., Karin N.J. Lysophosphatidic acid induces osteocyte dendrite outgrowth. Biochem. Biophys. Res. Commun. 2007;357:194–199. doi: 10.1016/j.bbrc.2007.03.121. [DOI] [PubMed] [Google Scholar]
- 304.Liu Y.B., Kharode Y., Bodine P.V., Yaworsky P.J., Robinson J.A., Billiard J. LPA induces osteoblast differentiation through interplay of two receptors: LPA1 and LPA4. J. Cell. Biochem. 2010;109:794–800. doi: 10.1002/jcb.22471. [DOI] [PubMed] [Google Scholar]
- 305.Contos J.J., Fukushima N., Weiner J.A., Kaushal D., Chun J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc. Natl. Acad. Sci. USA. 2000;97:13384–13389. doi: 10.1073/pnas.97.24.13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Gennero I., Laurencin-Dalicieux S., Conte-Auriol F., Briand-Mésange F., Laurencin D., Rue J., Beton N., Malet N., Mus M., Tokumura A., et al. Absence of the lysophosphatidic acid receptor LPA1 results in abnormal bone development and decreased bone mass. Bone. 2011;49:395–403. doi: 10.1016/j.bone.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Lapierre D.M., Tanabe N., Pereverzev A., Spencer M., Shugg R.P., Dixon S.J., Sims S.M. Lysophosphatidic acid signals through multiple receptors in osteoclasts to elevate cytosolic calcium concentration, evoke retraction, and promote cell survival. J. Biol. Chem. 2010;285:25792–25801. doi: 10.1074/jbc.M110.109322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Itoh R., Miura S., Takimoto A., Kondo S., Sano H., Hiraki Y. Stimulatory actions of lysophosphatidic acid on mouse ATDC5 chondroprogenitor cells. J. Bone Miner. Metab. 2010;28:659–671. doi: 10.1007/s00774-010-0184-1. [DOI] [PubMed] [Google Scholar]
- 309.Hale J.E., Wuthier R.E. The mechanism of matrix vesicle formation. Studies on the composition of chondrocyte microvilli and on the effects of microfilament-perturbing agents on cellular vesiculation. J. Biol. Chem. 1987;262:1916–1925. [PubMed] [Google Scholar]
- 310.Peress N.S., Anderson H.C., Sajdera S.W. The lipids of matrix vesicles from bovine fetal epiphyseal cartilage. Calcif. Tissue Res. 1974;14:275–281. doi: 10.1007/BF02060301. [DOI] [PubMed] [Google Scholar]
- 311.Wuthier R.E. Lipids of matrix vesicles. Fed. Proc. 1976;35:117–121. [PubMed] [Google Scholar]
- 312.Genge B.R., Wu L.N., Wuthier R.E. Separation and quantification of chicken and bovine growth plate cartilage matrix vesicle lipids by high-performance liquid chromatography using evaporative light scattering detection. Anal. Biochem. 2003;322:104–115. doi: 10.1016/S0003-2697(03)00462-7. [DOI] [PubMed] [Google Scholar]
- 313.Say J.C., Ciuffi K., Furriel R.P., Ciancaglini P., Leone F.A. Alkaline phosphatase from rat osseous plates: Purification and biochemical characterization of a soluble form. Biochim. Biophys. Acta. 1991;1074:256–262. doi: 10.1016/0304-4165(91)90161-9. [DOI] [PubMed] [Google Scholar]
- 314.Pizauro J.M., Ciancaglini P., Leone F.A. Phosphotransferase activity associated with rat osseous plate alkaline phosphatase: A possible role in biomineralization. Int. J. Biochem. 1992;24:1391–1396. doi: 10.1016/0020-711x(92)90064-8. [DOI] [PubMed] [Google Scholar]
- 315.Zhang L., Balcerzak M., Radisson J., Thouverey C., Pikula S., Azzar G., Buchet R. Phosphodiesterase activity of alkaline phosphatase in ATP-initiated Ca(2+) and phosphate deposition in isolated chicken matrix vesicles. J. Biol. Chem. 2005;280:37289–37296. doi: 10.1074/jbc.M504260200. [DOI] [PubMed] [Google Scholar]
- 316.Schwartz Z., Boyan B. The effects of vitamin D metabolites on phospholipase A2 activity of growth zone and resting zone cartilage cells in vitro. Endocrinology. 1988;122:2191–2198. doi: 10.1210/endo-122-5-2191. [DOI] [PubMed] [Google Scholar]
- 317.Schwartz Z., Schlader D.L., Swain L.D., Boyan B.D. Direct effects of 1,25-dihydroxyvitamin D3 and 24,25-dihydroxyvitamin D3 on growth zone and resting zone chondrocyte membrane alkaline phosphatase and phospholipase-A2 specific activities. Endocrinology. 1988;123:2878–2884. doi: 10.1210/endo-123-6-2878. [DOI] [PubMed] [Google Scholar]
- 318.Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature. 1984;308:693–698. doi: 10.1038/308693a0. [DOI] [PubMed] [Google Scholar]
- 319.Vines C.M. Phospholipase C. Adv. Exp. Med. Biol. 2012;740:235–254. doi: 10.1007/978-94-007-2888-2_10. [DOI] [PubMed] [Google Scholar]
- 320.Li H., Zhang L., Yin D., Zhang Y., Miao J. Targeting phosphatidylcholine-specific phospholipase C for atherogenesis therapy. Trends Cardiovasc. Med. 2010;20:172–176. doi: 10.1016/j.tcm.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 321.Spadaro F., Ramoni C., Mezzanzanica D., Miotti S., Alberti P., Cecchetti S., Iorio E., Dolo V., Canevari S., Podo F. Phosphatidylcholine-specific phospholipase C activation in epithelial ovarian cancer cells. Cancer Res. 2008;68:6541–6549. doi: 10.1158/0008-5472.CAN-07-6763. [DOI] [PubMed] [Google Scholar]
- 322.Tsai C.C., Kai J.I., Huang W.C., Wang C.Y., Wang Y., Chen C.L., Fang Y.T., Lin Y.S., Anderson R., Chen S.H., et al. Glycogen synthase kinase-3beta facilitates IFN-gamma-induced STAT1 activation by regulating Src homology-2 domain-containing phosphatase 2. J. Immunol. 2009;183:856–864. doi: 10.4049/jimmunol.0804033. [DOI] [PubMed] [Google Scholar]
- 323.Horstmeyer A., Licht C., Scherr G., Eckes B., Krieg T. Signalling and regulation of collagen I synthesis by ET-1 and TGF-beta1. FEBS J. 2005;272:6297–6309. doi: 10.1111/j.1742-4658.2005.05016.x. [DOI] [PubMed] [Google Scholar]
- 324.Weber C., Erl W., Pietsch A., Danesch U., Weber P.C. Docosahexaenoic acid selectively attenuates induction of vascular cell adhesion molecule-1 and subsequent monocytic cell adhesion to human endothelial cells stimulated by tumor necrosis factor-alpha. Arterioscler. Thromb. Vasc. Biol. 1995;15:622–628. doi: 10.1161/01.atv.15.5.622. [DOI] [PubMed] [Google Scholar]
- 325.Zhang L., Zhao J., Su L., Huang B., Wang L., Su H., Zhang Y., Zhang S., Miao J. D609 inhibits progression of preexisting atheroma and promotes lesion stability in apolipoprotein e−/− mice: A role of phosphatidylcholine-specific phospholipase in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010;30:411–418. doi: 10.1161/ATVBAHA.109.195768. [DOI] [PubMed] [Google Scholar]
- 326.Rapuano B.E., Bockman R.S. Protein kinase C-independent activation of a novel nonspecific phospholipase C pathway by phorbol myristate acetate releases arachidonic acid for prostaglandin synthesis in MC3T3-E1 osteoblasts. Prostaglandins. 1997;53:163–186. doi: 10.1016/s0090-6980(97)00011-7. [DOI] [PubMed] [Google Scholar]
- 327.Ishimi Y., Miyaura C., Jin C.H., Akatsu T., Abe E., Nakamura Y., Yamaguchi A., Yoshiki S., Matsuda T., Hirano T. IL-6 is produced by osteoblasts and induces bone resorption. J. Immunol. 1990;145:3297–3303. [PubMed] [Google Scholar]
- 328.Nijweide P.J., Burger E.H., Feyen J.H. Cells of bone: Proliferation, differentiation, and hormonal regulation. Physiol. Rev. 1986;66:855–886. doi: 10.1152/physrev.1986.66.4.855. [DOI] [PubMed] [Google Scholar]
- 329.Kozawa O., Suzuki A., Tokuda H., Kaida T., Uematsu T. Protein kinase C activation by interleukin (IL)-1 limits IL-1-induced IL-6 synthesis in osteoblast-like cells: Involvement of phosphatidylcholine-specific phospholipase C. J. Cell. Biochem. 1997;67:103–111. [PubMed] [Google Scholar]
- 330.Kozawa O., Suzuki A., Kaida T., Tokuda H., Uematsu T. Tumor necrosis factor-alpha autoregulates interleukin-6 synthesis via activation of protein kinase C. Function of sphingosine 1-phosphate and phosphatidylcholine-specific phospholipase C. J. Biol. Chem. 1997;272:25099–25104. doi: 10.1074/jbc.272.40.25099. [DOI] [PubMed] [Google Scholar]
- 331.Sakai T., Sugiyama T., Banno Y., Kato Y., Nozawa Y. Involvement of phosphatidylcholine hydrolysis by phospholipase C in prostaglandin F2alpha-induced 1,2-diacylglycerol formation in osteoblast-like MC3T3-E1 cells. J. Bone Miner. Metab. 2004;22:198–206. doi: 10.1007/s00774-003-0470-2. [DOI] [PubMed] [Google Scholar]
- 332.Ma Y., Fu D., Liu Z. Effect of lead on apoptosis in cultured rat primary osteoblasts. Toxicol. Ind. Health. 2012;28:136–146. doi: 10.1177/0748233711407956. [DOI] [PubMed] [Google Scholar]
- 333.Xia L., Zhang D., Wang C., Wei F., Hu Y. PC-PLC is involved in osteoclastogenesis induced by TNF-α through upregulating IP3R1 expression. FEBS Lett. 2012;586:3341–3348. doi: 10.1016/j.febslet.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 334.Hofbauer L.C., Schrader J., Niebergall U., Viereck V., Burchert A., Hörsch D., Preissner K.T., Schoppet M. Interleukin-4 differentially regulates osteoprotegerin expression and induces calcification in vascular smooth muscle cells. Thromb. Haemost. 2006;95:708–714. [PubMed] [Google Scholar]
- 335.Whyte M.P., Landt M., Ryan L.M., Mulivor R.A., Henthorn P.S., Fedde K.N., Mahuren J.D., Coburn S.P. Alkaline phosphatase: Placental and tissue-nonspecific isoenzymes hydrolyze phosphoethanolamine, inorganic pyrophosphate, and pyridoxal 5′-phosphate. Substrate accumulation in carriers of hypophosphatasia corrects during pregnancy. J. Clin. Invest. 1995;95:1440–1445. doi: 10.1172/JCI117814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Fedde K.N., Lane C.C., Whyte M.P. Alkaline phosphatase is an ectoenzyme that acts on micromolar concentrations of natural substrates at physiologic pH in human osteosarcoma (SAOS-2) cells. Arch. Biochem. Biophys. 1988;264:400–409. doi: 10.1016/0003-9861(88)90305-0. [DOI] [PubMed] [Google Scholar]
- 337.Berridge M.J. Inositol trisphosphate and calcium signaling. Ann. N. Y. Acad. Sci. 1995;766:31–43. doi: 10.1111/j.1749-6632.1995.tb26646.x. [DOI] [PubMed] [Google Scholar]
- 338.Yin H.L., Janmey P.A. Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol. 2003;65:761–789. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 339.Di Paolo G., de Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 340.Suh B.C., Hille B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr. Opin. Neurobiol. 2005;15:370–378. doi: 10.1016/j.conb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 341.Wong K.K., Engelman J.A., Cantley L.C. Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Kashiwada M., Lu P., Rothman P.B. PIP3 pathway in regulatory T cells and autoimmunity. Immunol. Res. 2007;39:194–224. doi: 10.1007/s12026-007-0075-2. [DOI] [PubMed] [Google Scholar]
- 343.Suh P.G., Ryu S.H., Moon K.H., Suh H.W., Rhee S.G. Cloning and sequence of multiple forms of phospholipase C. Cell. 1988;54:161–169. doi: 10.1016/0092-8674(88)90548-x. [DOI] [PubMed] [Google Scholar]
- 344.Kim J.K., Lim S., Kim J., Kim S., Kim J.H., Ryu S.H., Suh P.G. Subtype-specific roles of phospholipase C-β via differential interactions with PDZ domain proteins. Adv. Enzyme Regul. 2011;51:138–151. doi: 10.1016/j.advenzreg.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 345.Cocco L., Follo M.Y., Faenza I., Fiume R., Ramazzotti G., Weber G., Martelli A.M., Manzoli F.A. Physiology and pathology of nuclear phospholipase C β1. Adv. Enzyme Regul. 2011;51:2–12. doi: 10.1016/j.advenzreg.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 346.Faccio R., Cremasco V. PLCgamma2: Where bone and immune cells find their common ground. Ann. N. Y. Acad. Sci. 2010;1192:124–130. doi: 10.1111/j.1749-6632.2009.05217.x. [DOI] [PubMed] [Google Scholar]
- 347.Choi J.H., Ryu S.H., Suh P.G. On/off-regulation of phospholipase C-gamma 1-mediated signal transduction. Adv. Enzyme Regul. 2007;47:104–116. doi: 10.1016/j.advenzreg.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 348.Kelley G.G., Reks S.E., Ondrako J.M., Smrcka A.V. Phospholipase C(epsilon): A novel Ras effector. EMBO J. 2001;20:743–754. doi: 10.1093/emboj/20.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Lopez I., Mak E.C., Ding J., Hamm H.E., Lomasney J.W. A novel bifunctional phospholipase c that is regulated by Galpha 12 and stimulates the Ras/mitogen-activated protein kinase pathway. J. Biol. Chem. 2001;276:2758–2765. doi: 10.1074/jbc.M008119200. [DOI] [PubMed] [Google Scholar]
- 350.Smrcka A.V., Brown J.H., Holz G.G. Role of phospholipase Cɛ in physiological phosphoinositide signaling networks. Cell. Signal. 2012;24:1333–1343. doi: 10.1016/j.cellsig.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Song C., Hu C.D., Masago M., Kariyai K., Yamawaki-Kataoka Y., Shibatohge M., Wu D., Satoh T., Kataoka T. Regulation of a novel human phospholipase C, PLCepsilon, through membrane targeting by Ras. J. Biol. Chem. 2001;276:2752–2757. doi: 10.1074/jbc.M008324200. [DOI] [PubMed] [Google Scholar]
- 352.Saunders C.M., Larman M.G., Parrington J., Cox L.J., Royse J., Blayney L.M., Swann K., Lai F.A. PLC zeta: A sperm-specific trigger of Ca(2+) oscillations in eggs and embryo development. Development. 2002;129:3533–3544. doi: 10.1242/dev.129.15.3533. [DOI] [PubMed] [Google Scholar]
- 353.Hwang J.I., Oh Y.S., Shin K.J., Kim H., Ryu S.H., Suh P.G. Molecular cloning and characterization of a novel phospholipase C, PLC-eta. Biochem. J. 2005;389:181–186. doi: 10.1042/BJ20041677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Nakahara M., Shimozawa M., Nakamura Y., Irino Y., Morita M., Kudo Y., Fukami K. A novel phospholipase C, PLC(eta)2, is a neuron-specific isozyme. J. Biol. Chem. 2005;280:29128–29134. doi: 10.1074/jbc.M503817200. [DOI] [PubMed] [Google Scholar]
- 355.Zhou Y., Wing M.R., Sondek J., Harden T.K. Molecular cloning and characterization of PLC-eta2. Biochem. J. 2005;391:667–676. doi: 10.1042/BJ20050839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Suh P.G., Park J.I., Manzoli L., Cocco L., Peak J.C., Katan M., Fukami K., Kataoka T., Yun S., Ryu S.H. Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep. 2008;41:415–434. doi: 10.5483/bmbrep.2008.41.6.415. [DOI] [PubMed] [Google Scholar]
- 357.Harden T.K., Sondek J. Regulation of phospholipase C isozymes by ras superfamily GTPases. Annu. Rev. Pharmacol. Toxicol. 2006;46:355–379. doi: 10.1146/annurev.pharmtox.46.120604.141223. [DOI] [PubMed] [Google Scholar]
- 358.Rhee S.G., Suh P.G., Ryu S.H., Lee S.Y. Studies of inositol phospholipid-specific phospholipase C. Science. 1989;244:546–550. doi: 10.1126/science.2541501. [DOI] [PubMed] [Google Scholar]
- 359.Katan M. Families of phosphoinositide-specific phospholipase C: Structure and function. Biochim. Biophys. Acta. 1998;1436:5–17. doi: 10.1016/s0005-2760(98)00125-8. [DOI] [PubMed] [Google Scholar]
- 360.Sorli S.C., Bunney T.D., Sugden P.H., Paterson H.F., Katan M. Signaling properties and expression in normal and tumor tissues of two phospholipase C epsilon splice variants. Oncogene. 2005;24:90–100. doi: 10.1038/sj.onc.1208168. [DOI] [PubMed] [Google Scholar]
- 361.Kim C.G., Park D., Rhee S.G. The role of carboxyl-terminal basic amino acids in Gqalpha-dependent activation, particulate association, and nuclear localization of phospholipase C-beta1. J. Biol. Chem. 1996;271:21187–21192. doi: 10.1074/jbc.271.35.21187. [DOI] [PubMed] [Google Scholar]
- 362.Manzoli L., Martelli A.M., Billi A.M., Faenza I., Fiume R., Cocco L. Nuclear phospholipase C: Involvement in signal transduction. Prog. Lipid Res. 2005;44:185–206. doi: 10.1016/j.plipres.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 363.Martelli A.M., Fiume R., Faenza I., Tabellini G., Evangelista C., Bortul R., Follo M.Y., Falà F., Cocco L. Nuclear phosphoinositide specific phospholipase C (PI-PLC)-beta 1: A central intermediary in nuclear lipid-dependent signal transduction. Histol. Histopathol. 2005;20:1251–1260. doi: 10.14670/HH-20.1251. [DOI] [PubMed] [Google Scholar]
- 364.Bae Y.S., Cantley L.G., Chen C.S., Kim S.R., Kwon K.S., Rhee S.G. Activation of phospholipase C-gamma by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998;273:4465–4469. doi: 10.1074/jbc.273.8.4465. [DOI] [PubMed] [Google Scholar]
- 365.Huang P.S., Davis L., Huber H., Goodhart P.J., Wegrzyn R.E., Oliff A., Heimbrook D.C. An SH3 domain is required for the mitogenic activity of microinjected phospholipase C-gamma 1. FEBS Lett. 1995;358:287–292. doi: 10.1016/0014-5793(94)01453-8. [DOI] [PubMed] [Google Scholar]
- 366.Smith M.R., Ryu S.H., Suh P.G., Rhee S.G., Kung H.F. S-phase induction and transformation of quiescent NIH 3T3 cells by microinjection of phospholipase C. Proc. Natl. Acad. Sci. USA. 1989;86:3659–3663. doi: 10.1073/pnas.86.10.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Wang Z., Glück S., Zhang L., Moran M.F. Requirement for phospholipase C-gamma1 enzymatic activity in growth factor-induced mitogenesis. Mol. Cell. Biol. 1998;18:590–597. doi: 10.1128/mcb.18.1.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Cuvillier O. Sphingosine in apoptosis signaling. Biochim. Biophys. Acta. 2002;1585:153–162. doi: 10.1016/s1388-1981(02)00336-0. [DOI] [PubMed] [Google Scholar]
- 369.Park D., Jhon D.Y., Kriz R., Knopf J., Rhee S.G. Cloning, sequencing, expression, and Gq-independent activation of phospholipase C-beta 2. J. Biol. Chem. 1992;267:16048–16055. [PubMed] [Google Scholar]
- 370.Homma Y., Takenawa T., Emori Y., Sorimachi H., Suzuki K. Tissue- and cell type-specific expression of mRNAs for four types of inositol phospholipid-specific phospholipase C. Biochem. Biophys. Res. Commun. 1989;164:406–412. doi: 10.1016/0006-291x(89)91734-8. [DOI] [PubMed] [Google Scholar]
- 371.Sun L., Mao G., Kunapuli S.P., Dhanasekaran D.N., Rao A.K. Alternative splice variants of phospholipase C-beta2 are expressed in platelets: Effect on Galphaq-dependent activation and localization. Platelets. 2007;18:217–223. doi: 10.1080/09537100601016133. [DOI] [PubMed] [Google Scholar]
- 372.Jhon D.Y., Lee H.H., Park D., Lee C.W., Lee K.H., Yoo O.J., Rhee S.G. Cloning, sequencing, purification, and Gq-dependent activation of phospholipase C-beta 3. J. Biol. Chem. 1993;268:6654–6661. [PubMed] [Google Scholar]
- 373.Adamski F.M., Timms K.M., Shieh B.H. A unique isoform of phospholipase Cbeta4 highly expressed in the cerebellum and eye. Biochim. Biophys. Acta. 1999;1444:55–60. doi: 10.1016/s0167-4781(98)00260-7. [DOI] [PubMed] [Google Scholar]
- 374.Mizuguchi M., Yamada M., Kim S.U., Rhee S.G. Phospholipase C isozymes in neurons and glial cells in culture: An immunocytochemical and immunochemical study. Brain Res. 1991;548:35–40. doi: 10.1016/0006-8993(91)91103-8. [DOI] [PubMed] [Google Scholar]
- 375.Lin F.G., Cheng H.F., Lee I.F., Kao H.J., Loh S.H., Lee W.H. Downregulation of phospholipase C delta3 by cAMP and calcium. Biochem. Biophys. Res. Commun. 2001;286:274–280. doi: 10.1006/bbrc.2001.5371. [DOI] [PubMed] [Google Scholar]
- 376.Lee S.B., Rhee S.G. Molecular cloning, splice variants, expression, and purification of phospholipase C-delta 4. J. Biol. Chem. 1996;271:25–31. doi: 10.1074/jbc.271.1.25. [DOI] [PubMed] [Google Scholar]
- 377.Nameroff M., Trotter J.A., Keller J.M., Munar E. Inhibition of cellular differentiation by phospholipase C.I. Effects of the enzyme on myogenesis and chondrogenesis in vitro. J. Cell. Biol. 1973;58:107–118. doi: 10.1083/jcb.58.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Das P., Schurman D.J., Smith R.L. Nitric oxide and G proteins mediate the response of bovine articular chondrocytes to fluid-induced shear. J. Orthop. Res. 1997;15:87–93. doi: 10.1002/jor.1100150113. [DOI] [PubMed] [Google Scholar]
- 379.D’Andrea P., Calabrese A., Capozzi I., Grandolfo M., Tonon R., Vittur F. Intercellular Ca2+ waves in mechanically stimulated articular chondrocytes. Biorheology. 2000;37:75–83. [PubMed] [Google Scholar]
- 380.Nong L., Yin G., Ren K., Tang J., Fan W. Periodic mechanical stress enhances rat chondrocyte area expansion and migration through Src-PLCgamma1-ERK1/2 signaling. Eur. J. Cell. Biol. 2010;89:705–711. doi: 10.1016/j.ejcb.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 381.Rhee S.G. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Bleasdale J.E., Bundy G.L., Bunting S., Fitzpatrick F.A., Huff R.M., Sun F.F., Pike J.E. Inhibition of phospholipase C dependent processes by U-73, 122. Adv. Prostaglandin Thromboxane Leukotriene Res. 1989;19:590–593. [PubMed] [Google Scholar]
- 383.Poiraudeau S., Lieberherr M., Kergosie N., Corvol M.T. Different mechanisms are involved in intracellular calcium increase by insulin-like growth factors 1 and 2 in articular chondrocytes: Voltage-gated calcium channels, and/or phospholipase C coupled to a pertussis-sensitive G-protein. J. Cell. Biochem. 1997;64:414–422. [PubMed] [Google Scholar]
- 384.Grandolfo M., Calabrese A., D’Andrea P. Mechanism of mechanically induced intercellular calcium waves in rabbit articular chondrocytes and in HIG-82 synovial cells. J. Bone Miner. Res. 1998;13:443–453. doi: 10.1359/jbmr.1998.13.3.443. [DOI] [PubMed] [Google Scholar]
- 385.Capozzi I., Tonon R., D’andrea P. Ca2+-sensitive phosphoinositide hydrolysis is activated in synovial cells but not in articular chondrocytes. Biochem. J. 1999;344:545–553. [PMC free article] [PubMed] [Google Scholar]
- 386.Boyan B.D., Sylvia V.L., Dean D.D., Schwartz Z. Membrane mediated signaling mechanisms are used differentially by metabolites of vitamin D(3) in musculoskeletal cells. Steroids. 2002;67:421–427. doi: 10.1016/s0039-128x(01)00178-7. [DOI] [PubMed] [Google Scholar]
- 387.Boyan B.D., Sylvia V.L., Dean D.D., Del Toro F., Schwartz Z. Differential regulation of growth plate chondrocytes by 1alpha,25-(OH)2D3 and 24R,25-(OH)2D3 involves cell-maturation-specific membrane-receptor-activated phospholipid metabolism. Crit. Rev. Oral Biol. Med. 2002;13:143–154. doi: 10.1177/154411130201300205. [DOI] [PubMed] [Google Scholar]
- 388.Schwartz Z., Graham E.J., Wang L., Lossdörfer S., Gay I., Johnson-Pais T.L., Carnes D.L., Sylvia V.L., Boyan B.D. Phospholipase A2 activating protein (PLAA) is required for 1alpha,25(OH)2D3 signaling in growth plate chondrocytes. J. Cell. Physiol. 2005;203:54–70. doi: 10.1002/jcp.20212. [DOI] [PubMed] [Google Scholar]
- 389.Boyan B.D., Wang L., Wong K.L., Jo H., Schwartz Z. Plasma membrane requirements for 1alpha,25(OH)2D3 dependent PKC signaling in chondrocytes and osteoblasts. Steroids. 2006;71:286–290. doi: 10.1016/j.steroids.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 390.Sylvia V.L., Schwartz Z., Curry D.B., Chang Z., Dean D.D., Boyan B.D. 1,25(OH)2D3 regulates protein kinase C activity through two phospholipid-dependent pathways involving phospholipase A2 and phospholipase C in growth zone chondrocytes. J. Bone Miner. Res. 1998;13:559–569. doi: 10.1359/jbmr.1998.13.4.559. [DOI] [PubMed] [Google Scholar]
- 391.Schwartz Z., Sylvia V.L., Luna M.H., DeVeau P., Whetstone R., Dean D.D., Boyan B.D. The effect of 24R,25-(OH)(2)D(3) on protein kinase C activity in chondrocytes is mediated by phospholipase D whereas the effect of 1alpha,25-(OH)(2)D(3) is mediated by phospholipase C. Steroids. 2001;66:683–694. doi: 10.1016/s0039-128x(01)00100-3. [DOI] [PubMed] [Google Scholar]
- 392.Schwartz Z., Ehland H., Sylvia V.L., Larsson D., Hardin R.R., Bingham V., Lopez D., Dean D.D., Boyan B.D. 1alpha,25-dihydroxyvitamin D(3) and 24R,25-dihydroxyvitamin D(3) modulate growth plate chondrocyte physiology via protein kinase C-dependent phosphorylation of extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase. Endocrinology. 2002;143:2775–2786. doi: 10.1210/endo.143.7.8889. [DOI] [PubMed] [Google Scholar]
- 393.Schwartz Z., Shaked D., Hardin R.R., Gruwell S., Dean D.D., Sylvia V.L., Boyan B.D. 1alpha,25(OH)2D3 causes a rapid increase in phosphatidylinositol-specific PLC-beta activity via phospholipase A2-dependent production of lysophospholipid. Steroids. 2003;68:423–437. doi: 10.1016/s0039-128x(03)00044-8. [DOI] [PubMed] [Google Scholar]
- 394.Ekstein J., Nasatzky E., Boyan B.D., Ornoy A., Schwartz Z. Growth-plate chondrocytes respond to 17beta-estradiol with sex-specific increases in IP3 and intracellular calcium ion signalling via a capacitative entry mechanism. Steroids. 2005;70:775–786. doi: 10.1016/j.steroids.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 395.Sylvia V.L., Boyan B.D., Dean D.D., Schwartz Z. The membrane effects of 17beta-estradiol on chondrocyte phenotypic expression are mediated by activation of protein kinase C through phospholipase C and G-proteins. J. Steroid Biochem. Mol. Biol. 2000;73:211–224. doi: 10.1016/s0960-0760(00)00078-9. [DOI] [PubMed] [Google Scholar]
- 396.Sylvia V.L., Walton J., Lopez D., Dean D.D., Boyan B.D., Schwartz Z. 17 beta-estradiol-BSA conjugates and 17 beta-estradiol regulate growth plate chondrocytes by common membrane associated mechanisms involving PKC dependent and independent signal transduction. J. Cell. Biochem. 2001;81:413–429. doi: 10.1002/1097-4644(20010601)81:3<413::aid-jcb1055>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 397.Hurst-Kennedy J., Zhong M., Gupta V., Boyan B.D., Schwartz Z. 24R,25-Dihydroxyvitamin D3, lysophosphatidic acid, and p53: A signaling axis in the inhibition of phosphate-induced chondrocyte apoptosis. J. Steroid Biochem. Mol. Biol. 2010;122:264–271. doi: 10.1016/j.jsbmb.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 398.Evans J.F., Shen C.L., Pollack S., Aloia J.F., Yeh J.K. Adrenocorticotropin evokes transient elevations in intracellular free calcium ([Ca2+]i) and increases basal [Ca2+]i in resting chondrocytes through a phospholipase C-dependent mechanism. Endocrinology. 2005;146:3123–3132. doi: 10.1210/en.2004-1612. [DOI] [PubMed] [Google Scholar]
- 399.Riggs B.L., Khosla S., Melton L.J. Sex steroids and the construction and conservation of the adult skeleton. Endocr. Rev. 2002;23:279–302. doi: 10.1210/edrv.23.3.0465. [DOI] [PubMed] [Google Scholar]
- 400.ElBaradie K., Wang Y., Boyan B.D., Schwartz Z. Rapid membrane responses to dihydrotestosterone are sex dependent in growth plate chondrocytes. J. Steroid Biochem. Mol. Biol. 2012;132:15–23. doi: 10.1016/j.jsbmb.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 401.Chao P.Z., Hsieh M.S., Cheng C.W., Lin Y.F., Chen C.H. Regulation of MMP-3 expression and secretion by the chemokine eotaxin-1 in human chondrocytes. J. Biomed. Sci. 2011;18:86. doi: 10.1186/1423-0127-18-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Harada D., Yamanaka Y., Ueda K., Nishimura R., Morishima T., Seino Y., Tanaka H. Sustained phosphorylation of mutated FGFR3 is a crucial feature of genetic dwarfism and induces apoptosis in the ATDC5 chondrogenic cell line via PLCgamma-activated STAT1. Bone. 2007;41:273–281. doi: 10.1016/j.bone.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 403.Suzuki Y., Hruska K.A., Reid I., Alvarez U.M., Avioli L.V. Characterization of phospholipase C activity of the plasma membrane and cytosol of an osteoblast-like cell line. Am. J. Med. Sci. 1989;297:135–144. doi: 10.1097/00000441-198903000-00001. [DOI] [PubMed] [Google Scholar]
- 404.Hoberg M., Gratz H.H., Noll M., Jones D.B. Mechanosensitivity of human osteosarcoma cells and phospholipase C beta2 expression. Biochem. Biophys. Res. Commun. 2005;333:142–149. doi: 10.1016/j.bbrc.2005.05.088. [DOI] [PubMed] [Google Scholar]
- 405.Bai X.C., Lu D., Bai J., Zheng H., Ke Z.Y., Li X.M., Luo S.Q. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem. Biophys. Res. Commun. 2004;314:197–207. doi: 10.1016/j.bbrc.2003.12.073. [DOI] [PubMed] [Google Scholar]
- 406.Godwin S.L., Soltoff S.P. Calcium-sensing receptor-mediated activation of phospholipase C-gamma1 is downstream of phospholipase C-beta and protein kinase C in MC3T3-E1 osteoblasts. Bone. 2002;30:559–566. doi: 10.1016/s8756-3282(01)00700-1. [DOI] [PubMed] [Google Scholar]
- 407.Takuwa Y., Ohue Y., Takuwa N., Yamashita K. Endothelin-1 activates phospholipase C and mobilizes Ca2+ from extra- and intracellular pools in osteoblastic cells. Am. J. Physiol. 1989;257:E797–E803. doi: 10.1152/ajpendo.1989.257.6.E797. [DOI] [PubMed] [Google Scholar]
- 408.Takuwa Y., Masaki T., Yamashita K. The effects of the endothelin family peptides on cultured osteoblastic cells from rat calvariae. Biochem. Biophys. Res. Commun. 1990;170:998–1005. doi: 10.1016/0006-291x(90)90491-5. [DOI] [PubMed] [Google Scholar]
- 409.Lam H.C., Lee J.K., Lai K.H. Detection and characterization of endothelin in transformed human osteoblast cell culture medium. Endocrine. 2000;12:77–80. doi: 10.1385/ENDO:12:1:77. [DOI] [PubMed] [Google Scholar]
- 410.Masukawa H., Miura Y., Sato I., Oiso Y., Suzuki A. Stimulatory effect of endothelin-1 on Na-dependent phosphate transport and its signaling mechanism in osteoblast-like cells. J. Cell. Biochem. 2001;83:47–55. doi: 10.1002/jcb.1206. [DOI] [PubMed] [Google Scholar]
- 411.Zach D., Windischhofer W., Leis H.J. Endothelin- and sarafotoxin-induced receptor-mediated calcium mobilization in a clonal murine osteoblast-like cell line, MC3T3-E1/B. Bone. 2001;28:595–602. doi: 10.1016/s8756-3282(01)00461-6. [DOI] [PubMed] [Google Scholar]
- 412.Suzuki A., Palmer G., Bonjour J.P., Caverzasio J. Stimulation of sodium-dependent phosphate transport and signaling mechanisms induced by basic fibroblast growth factor in MC3T3-E1 osteoblast-like cells. J. Bone Miner. Res. 2000;15:95–102. doi: 10.1359/jbmr.2000.15.1.95. [DOI] [PubMed] [Google Scholar]
- 413.Tang C.H., Yang R.S., Chen Y.F., Fu W.M. Basic fibroblast growth factor stimulates fibronectin expression through phospholipase C gamma, protein kinase C alpha, c-Src, NF-kappaB, and p300 pathway in osteoblasts. J. Cell. Physiol. 2007;211:45–55. doi: 10.1002/jcp.20896. [DOI] [PubMed] [Google Scholar]
- 414.Zhen X., Bonjour J.P., Caverzasio J. Platelet-derived growth factor stimulates sodium-dependent Pi transport in osteoblastic cells via phospholipase Cgamma and phosphatidylinositol 3′-kinase. J. Bone Miner. Res. 1997;12:36–44. doi: 10.1359/jbmr.1997.12.1.36. [DOI] [PubMed] [Google Scholar]
- 415.Godwin S.L., Soltoff S.P. Extracellular calcium and platelet-derived growth factor promote receptor-mediated chemotaxis in osteoblasts through different signaling pathways. J. Biol. Chem. 1997;272:11307–11312. doi: 10.1074/jbc.272.17.11307. [DOI] [PubMed] [Google Scholar]
- 416.Moenning A., Jäger R., Egert A., Kress W., Wardelmann E., Schorle H. Sustained platelet-derived growth factor receptor alpha signaling in osteoblasts results in craniosynostosis by overactivating the phospholipase C-gamma pathway. Mol. Cell. Biol. 2009;29:881–891. doi: 10.1128/MCB.00885-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Swarthout J.T., Doggett T.A., Lemker J.L., Partridge N.C. Stimulation of extracellular signal-regulated kinases and proliferation in rat osteoblastic cells by parathyroid hormone is protein kinase C-dependent. J. Biol. Chem. 2001;276:7586–7592. doi: 10.1074/jbc.M007400200. [DOI] [PubMed] [Google Scholar]
- 418.Hömme M., Schmitt C.P., Himmele R., Hoffmann G.F., Mehls O., Schaefer F. Vitamin D and dexamethasone inversely regulate parathyroid hormone-induced regulator of G protein signaling-2 expression in osteoblast-like cells. Endocrinology. 2003;144:2496–2504. doi: 10.1210/en.2002-0160. [DOI] [PubMed] [Google Scholar]
- 419.Hilal G., Massicotte F., Martel-Pelletier J., Fernandes J.C., Pelletier J.P., Lajeunesse D. Endogenous prostaglandin E2 and insulin-like growth factor 1 can modulate the levels of parathyroid hormone receptor in human osteoarthritic osteoblasts. J. Bone Miner. Res. 2001;16:713–721. doi: 10.1359/jbmr.2001.16.4.713. [DOI] [PubMed] [Google Scholar]
- 420.Reeve J., Zanelli J.M. Parathyroid hormone and bone. Clin. Sci. 1986;71:231–238. doi: 10.1042/cs0710231. [DOI] [PubMed] [Google Scholar]
- 421.Wu Y., Kumar R. Parathyroid hormone regulates transforming growth factor beta1 and beta2 synthesis in osteoblasts via divergent signaling pathways. J. Bone Miner. Res. 2000;15:879–884. doi: 10.1359/jbmr.2000.15.5.879. [DOI] [PubMed] [Google Scholar]
- 422.Erclik M.S., Mitchell J. The role of protein kinase C-delta in PTH stimulation of IGF-binding protein-5 mRNA in UMR-106–01 cells. Am. J. Physiol. Endocrinol. Metab. 2002;282:E534–E541. doi: 10.1152/ajpendo.00417.2001. [DOI] [PubMed] [Google Scholar]
- 423.Cheung R., Erclik M.S., Mitchell J. Increased expression of G11alpha in osteoblastic cells enhances parathyroid hormone activation of phospholipase C and AP-1 regulation of matrix metalloproteinase-13 mRNA. J. Cell. Physiol. 2005;204:336–343. doi: 10.1002/jcp.20299. [DOI] [PubMed] [Google Scholar]
- 424.Imamura Y., Kozawa O., Suzuki A., Watanabe Y., Saito H., Oiso Y. Mechanism of phospholipase D activation induced by prostaglandin D2 in osteoblast-like cells: Function of Ca2+/calmodulin. Cell. Signal. 1995;7:45–51. doi: 10.1016/0898-6568(94)00059-k. [DOI] [PubMed] [Google Scholar]
- 425.Babich M., King K.L., Nissenson R.A. Thrombin stimulates inositol phosphate production and intracellular free calcium by a pertussis toxin-insensitive mechanism in osteosarcoma cells. Endocrinology. 1990;126:948–954. doi: 10.1210/endo-126-2-948. [DOI] [PubMed] [Google Scholar]
- 426.Kawase T., Suzuki A. Initial responses of a clonal osteoblast-like cell line, MOB 3–4, to phosphatidic acid in vitro. Bone Miner. 1990;10:61–70. doi: 10.1016/0169-6009(90)90049-l. [DOI] [PubMed] [Google Scholar]
- 427.Tokuda H., Kozawa O., Niwa M., Matsuno H., Kato K., Uematsu T. Mechanism of prostaglandin E2-stimulated heat shock protein 27 induction in osteoblast-like MC3T3-E1 cells. J. Endocrinol. 2002;172:271–281. doi: 10.1677/joe.0.1720271. [DOI] [PubMed] [Google Scholar]
- 428.Kondo A., Togari A. Activation of osteoblastic functions by a mediator of pain, bradykinin. Biochem. Pharmacol. 2004;68:1423–1431. doi: 10.1016/j.bcp.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 429.Toriyama K., Morita I., Seyama Y., Yamashita S., Murota S. Prostaglandin E2 evokes intracellular calcium rise in mouse osteoblastic cell line, MC3T3E-1. Eicosanoids. 1990;3:157–160. [PubMed] [Google Scholar]
- 430.Tomura H., Wang J.Q., Liu J.P., Komachi M., Damirin A., Mogi C., Tobo M., Nochi H., Tamoto K., Im D.S., et al. Cyclooxygenase-2 expression and prostaglandin E2 production in response to acidic pH through OGR1 in a human osteoblastic cell line. J. Bone Miner. Res. 2008;23:1129–1139. doi: 10.1359/jbmr.080236. [DOI] [PubMed] [Google Scholar]
- 431.Hakeda Y., Shiokawa M., Mano H., Kameda T., Raisz L.G., Kumegawa M. Prostaglandin F2alpha stimulates tyrosine phosphorylation and mitogen-activated protein kinase in osteoblastic MC3T3-E1 cells via protein kinase C activation. Endocrinology. 1997;138:1821–1828. doi: 10.1210/endo.138.5.5107. [DOI] [PubMed] [Google Scholar]
- 432.Quarles L.D., Haupt D.M., Davidai G., Middleton J.P. Prostaglandin F2 alpha-induced mitogenesis in MC3T3-E1 osteoblasts: Role of protein kinase-C-mediated tyrosine phosphorylation. Endocrinology. 1993;132:1505–1513. doi: 10.1210/endo.132.4.8462449. [DOI] [PubMed] [Google Scholar]
- 433.Hatakeyama D., Kozawa O., Otsuka T., Shibata T., Uematsu T. Zinc suppresses IL-6 synthesis by prostaglandin F2alpha in osteoblasts: Inhibition of phospholipase C and phospholipase D. J. Cell. Biochem. 2002;85:621–628. doi: 10.1002/jcb.10166. [DOI] [PubMed] [Google Scholar]
- 434.Kozawa O., Tokuda H., Suzuki A., Kotoyori J., Ito Y., Oiso Y. Effect of glucocorticoid on prostaglandin F2 alpha-induced prostaglandin E2 synthesis in osteoblast-like cells: Inhibition of phosphoinositide hydrolysis by phospholipase C as well as phospholipase A2. Eur. J. Endocrinol. 1994;131:510–515. doi: 10.1530/eje.0.1310510. [DOI] [PubMed] [Google Scholar]
- 435.Kozawa O., Tokuda H., Matsuno H., Uematsu T. Activation of mitogen-activated protein kinase is involved in sphingosine 1-phosphate-stimulated interleukin-6 synthesis in osteoblasts. FEBS Lett. 1997;418:149–151. doi: 10.1016/s0014-5793(97)01366-5. [DOI] [PubMed] [Google Scholar]
- 436.Kozawa O., Kawamura H., Matsuno H., Uematsu T. p38 MAP kinase is involved in the signalling of sphingosine in osteoblasts: Sphingosine inhibits prostaglandin F(2alpha)-induced phosphoinositide hydrolysis. Cell. Signal. 2000;12:447–450. doi: 10.1016/s0898-6568(00)00086-3. [DOI] [PubMed] [Google Scholar]
- 437.Le Mellay V., Grosse B., Lieberherr M. Phospholipase C beta and membrane action of calcitriol and estradiol. J. Biol. Chem. 1997;272:11902–11907. doi: 10.1074/jbc.272.18.11902. [DOI] [PubMed] [Google Scholar]
- 438.Le Mellay V., Lasmoles F., Lieberherr M. Galpha(q/11) and gbetagamma proteins and membrane signaling of calcitriol and estradiol. J. Cell. Biochem. 1999;75:138–146. doi: 10.1002/(sici)1097-4644(19991001)75:1<138::aid-jcb14>3.3.co;2-9. [DOI] [PubMed] [Google Scholar]
- 439.Schwartz Z., Lohmann C.H., Vocke A.K., Sylvia V.L., Cochran D.L., Dean D.D., Boyan B.D. Osteoblast response to titanium surface roughness and 1alpha,25-(OH)(2)D(3) is mediated through the mitogen-activated protein kinase (MAPK) pathway. J. Biomed. Mater. Res. 2001;56:417–426. doi: 10.1002/1097-4636(20010905)56:3<417::aid-jbm1111>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 440.Marmiroli S., Ognibene A., Bavelloni A., Cinti C., Cocco L., Maraldi N.M. Interleukin 1 alpha stimulates nuclear phospholipase C in human osteosarcoma SaOS-2 cells. J. Biol. Chem. 1994;269:13–16. [PubMed] [Google Scholar]
- 441.Zini N., Sabatelli P., Faenza I., Ognibene A., Maraldi N.M. Interleukin-1 alpha induces variations of the intranuclear amount of phosphatidylinositol 4,5-bisphosphate and phospholipase C beta 1 in human osteosarcoma Saos-2 cells. Histochem. J. 1996;28:495–504. doi: 10.1007/BF02331409. [DOI] [PubMed] [Google Scholar]
- 442.Rodan S.B., Wesolowski G., Chin J., Limjuco G.A., Schmidt J.A., Rodan G.A. IL-1 binds to high affinity receptors on human osteosarcoma cells and potentiates prostaglandin E2 stimulation of cAMP production. J. Immunol. 1990;145:1231–1237. [PubMed] [Google Scholar]
- 443.Martelli A.M., Billi A.M., Manzoli L., Faenza I., Aluigi M., Falconi M., De Pol A., Gilmour R.S., Cocco L. Insulin selectively stimulates nuclear phosphoinositide-specific phospholipase C (PI-PLC) beta1 activity through a mitogen-activated protein (MAP) kinase-dependent serine phosphorylation. FEBS Lett. 2000;486:230–236. doi: 10.1016/s0014-5793(00)02313-9. [DOI] [PubMed] [Google Scholar]
- 444.Piecyk M., Anderson P. Signal transduction in rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2001;15:789–803. doi: 10.1053/berh.2001.0194. [DOI] [PubMed] [Google Scholar]
- 445.Lax A.J., Grigoriadis A.E. Pasteurella multocida toxin: The mitogenic toxin that stimulates signalling cascades to regulate growth and differentiation. Int. J. Med. Microbiol. 2001;291:261–268. doi: 10.1078/1438-4221-00129. [DOI] [PubMed] [Google Scholar]
- 446.Chang H.T., Hsu S.S., Chou C.T., Cheng J.S., Wang J.L., Lin K.L., Fang Y.C., Chen W.C., Chien J.M., Lu T., et al. Effect of thymol on Ca2+ homeostasis and viability in MG63 human osteosarcoma cells. Pharmacology. 2011;88:201–212. doi: 10.1159/000331864. [DOI] [PubMed] [Google Scholar]
- 447.Shin M.K., Jang Y.H., Yoo H.J., Kang D.W., Park M.H., Kim M.K., Song J.H., Kim S.D., Min G., You H.K., et al. N-formyl-methionyl-leucyl-phenylalanine (fMLP) promotes osteoblast differentiation via the N-formyl peptide receptor 1-mediated signaling pathway in human mesenchymal stem cells from bone marrow. J. Biol. Chem. 2011;286:17133–17143. doi: 10.1074/jbc.M110.197772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Aki Y., Kondo A., Nakamura H., Togari A. Lysophosphatidic acid-stimulated interleukin-6 and -8 synthesis through LPA1 receptors on human osteoblasts. Arch. Oral Biol. 2008;53:207–213. doi: 10.1016/j.archoralbio.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 449.Katz S., Boland R., Santillán G. Modulation of ERK 1/2 and p38 MAPK signaling pathways by ATP in osteoblasts: Involvement of mechanical stress-activated calcium influx, PKC and Src activation. Int. J. Biochem. Cell. Biol. 2006;38:2082–2091. doi: 10.1016/j.biocel.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 450.Nishii N., Nejime N., Yamauchi C., Yanai N., Shinozuka K., Nakabayashi T. Effects of ATP on the intracellular calcium level in the osteoblastic TBR31–2 cell line. Biol. Pharm. Bull. 2009;32:18–23. doi: 10.1248/bpb.32.18. [DOI] [PubMed] [Google Scholar]
- 451.Nakamura I., Lipfert L., Rodan G.A., Duong L.T. Convergence of alpha(v)beta(3) integrin- and macrophage colony stimulating factor-mediated signals on phospholipase Cgamma in prefusion osteoclasts. J. Cell. Biol. 2001;152:361–373. doi: 10.1083/jcb.152.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Epple H., Cremasco V., Zhang K., Mao D., Longmore G.D., Faccio R. Phospholipase Cgamma2 modulates integrin signaling in the osteoclast by affecting the localization and activation of Src kinase. Mol. Cell. Biol. 2008;28:3610–3622. doi: 10.1128/MCB.00259-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Lucht U. Effects of calcitonin on osteoclasts in vivo. An ultrastructural and histochemical study. Zeitschrift Zellforsch. Mikrosk. Anat. 1973;145:75–87. doi: 10.1007/BF00307190. [DOI] [PubMed] [Google Scholar]
- 454.Baron R., Neff L., Brown W., Louvard D., Courtoy P.J. Selective internalization of the apical plasma membrane and rapid redistribution of lysosomal enzymes and mannose 6-phosphate receptors during osteoclast inactivation by calcitonin. J. Cell. Sci. 1990;97:439–447. doi: 10.1242/jcs.97.3.439. [DOI] [PubMed] [Google Scholar]
- 455.Henriksen K., Bay-Jensen A.C., Christiansen C., Karsdal M.A. Oral salmon calcitonin— Pharmacology in osteoporosis. Expert Opin. Biol. Ther. 2010;10:1617–1629. doi: 10.1517/14712598.2010.526104. [DOI] [PubMed] [Google Scholar]
- 456.Stenbeck G., Lawrence K.M., Albert A.P. Hormone-stimulated modulation of endocytic trafficking in osteoclasts. Front. Endocrinol. 2012;3:103. doi: 10.3389/fendo.2012.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Silver I.A., Murrills R.J., Etherington D.J. Microelectrode studies on the acid microenvironment beneath adherent macrophages and osteoclasts. Exp. Cell. Res. 1988;175:266–276. doi: 10.1016/0014-4827(88)90191-7. [DOI] [PubMed] [Google Scholar]
- 458.Malgaroli A., Meldolesi J., Zallone A.Z., Teti A. Control of cytosolic free calcium in rat and chicken osteoclasts. The role of extracellular calcium and calcitonin. J. Biol. Chem. 1989;264:14342–14347. [PubMed] [Google Scholar]
- 459.Miyauchi A., Hruska K.A., Greenfield E.M., Duncan R., Alvarez J., Barattolo R., Colucci S., Zambonin-Zallone A., Teitelbaum S.L., Teti A. Osteoclast cytosolic calcium, regulated by voltage-gated calcium channels and extracellular calcium, controls podosome assembly and bone resorption. J. Cell. Biol. 1990;111:2543–2552. doi: 10.1083/jcb.111.6.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Zaidi M., Moonga B.S., Adebanjo O.A. Novel mechanisms of calcium handling by the osteoclast: A review-hypothesis. Proc. Assoc. Am. Physicians. 1999;111:319–327. doi: 10.1046/j.1525-1381.1999.99233.x. [DOI] [PubMed] [Google Scholar]
- 461.Lorget F., Kamel S., Mentaverri R., Wattel A., Naassila M., Maamer M., Brazier M. High extracellular calcium concentrations directly stimulate osteoclast apoptosis. Biochem. Biophys. Res. Commun. 2000;268:899–903. doi: 10.1006/bbrc.2000.2229. [DOI] [PubMed] [Google Scholar]
- 462.Chamoux E., Bisson M., Payet M.D., Roux S. TRPV-5 mediates a receptor activator of NF-kappaB (RANK) ligand-induced increase in cytosolic Ca2+ in human osteoclasts and down-regulates bone resorption. J. Biol. Chem. 2010;285:25354–25362. doi: 10.1074/jbc.M109.075234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Kajiya H., Okamoto F., Nemoto T., Kimachi K., Toh-Goto K., Nakayana S., Okabe K. RANKL-induced TRPV2 expression regulates osteoclastogenesis via calcium oscillations. Cell Calcium. 2010;48:260–269. doi: 10.1016/j.ceca.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 464.Mentaverri R., Yano S., Chattopadhyay N., Petit L., Kifor O., Kamel S., Terwilliger E.F., Brazier M., Brown E.M. The calcium sensing receptor is directly involved in both osteoclast differentiation and apoptosis. FASEB J. 2006;20:2562–2564. doi: 10.1096/fj.06-6304fje. [DOI] [PubMed] [Google Scholar]
- 465.Hurtel-Lemaire A.S., Mentaverri R., Caudrillier A., Cournarie F., Wattel A., Kamel S., Terwilliger E.F., Brown E.M., Brazier M. The calcium-sensing receptor is involved in strontium ranelate-induced osteoclast apoptosis. New insights into the associated signaling pathways. J. Biol. Chem. 2009;284:575–584. doi: 10.1074/jbc.M801668200. [DOI] [PubMed] [Google Scholar]
- 466.Seuwen K., Boddeke H.G., Migliaccio S., Perez M., Taranta A., Teti A. A novel calcium sensor stimulating inositol phosphate formation and [Ca2+]i signaling expressed by GCT23 osteoclast-like cells. Proc. Assoc. Am. Physicians. 1999;111:70–81. doi: 10.1046/j.1525-1381.1999.09866.x. [DOI] [PubMed] [Google Scholar]
- 467.Yoon S.H., Lee Y., Kim H.J., Lee Z.H., Hyung S.W., Lee S.W., Kim H.H. Lyn inhibits osteoclast differentiation by interfering with PLCgamma1-mediated Ca2+ signaling. FEBS Lett. 2009;583:1164–1170. doi: 10.1016/j.febslet.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 468.Sakai H., Moriura Y., Notomi T., Kawawaki J., Ohnishi K., Kuno M. Phospholipase C-dependent Ca2+-sensing pathways leading to endocytosis and inhibition of the plasma membrane vacuolar H+-ATPase in osteoclasts. Am. J. Physiol. Cell. Physiol. 2010;299:C570–C578. doi: 10.1152/ajpcell.00486.2009. [DOI] [PubMed] [Google Scholar]
- 469.Kim T., Kim K., Lee S.H., So H.S., Lee J., Kim N., Choi Y. Identification of LRRc17 as a negative regulator of receptor activator of NF-kappaB ligand (RANKL)-induced osteoclast differentiation. J. Biol. Chem. 2009;284:15308–15316. doi: 10.1074/jbc.M807722200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Bennett B.D., Alvarez U., Hruska K.A. Receptor-operated osteoclast calcium sensing. Endocrinology. 2001;142:1968–1974. doi: 10.1210/endo.142.5.8125. [DOI] [PubMed] [Google Scholar]
- 471.Weidema A.F., Dixon S.J., Sims S.M. Activation of P2Y but not P2X(4) nucleotide receptors causes elevation of [Ca2+]i in mammalian osteoclasts. Am. J. Physiol. Cell. Physiol. 2001;280:C1531–C1539. doi: 10.1152/ajpcell.2001.280.6.C1531. [DOI] [PubMed] [Google Scholar]
- 472.Komarova S.V., Pilkington M.F., Weidema A.F., Dixon S.J., Sims S.M. RANK ligand-induced elevation of cytosolic Ca2+ accelerates nuclear translocation of nuclear factor kappa B in osteoclasts. J. Biol. Chem. 2003;278:8286–8293. doi: 10.1074/jbc.M206421200. [DOI] [PubMed] [Google Scholar]
- 473.Komarova S.V., Pereverzev A., Shum J.W., Sims S.M., Dixon S.J. Convergent signaling by acidosis and receptor activator of NF-kappaB ligand (RANKL) on the calcium/calcineurin/NFAT pathway in osteoclasts. Proc. Natl. Acad. Sci. USA. 2005;102:2643–2648. doi: 10.1073/pnas.0406874102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Yang S., Li Y.P. RGS12 is essential for RANKL-evoked signaling for terminal differentiation of osteoclasts in vitro. J. Bone Miner. Res. 2007;22:45–54. doi: 10.1359/jbmr.061007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Mao D., Epple H., Uthgenannt B., Novack D.V., Faccio R. PLCgamma2 regulates osteoclastogenesis via its interaction with ITAM proteins and GAB2. J. Clin. Invest. 2006;116:2869–2879. doi: 10.1172/JCI28775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Stein N.C., Kreutzmann C., Zimmermann S.P., Niebergall U., Hellmeyer L., Goettsch C., Schoppet M., Hofbauer L.C. Interleukin-4 and interleukin-13 stimulate the osteoclast inhibitor osteoprotegerin by human endothelial cells through the STAT6 pathway. J. Bone Miner. Res. 2008;23:750–758. doi: 10.1359/jbmr.080203. [DOI] [PubMed] [Google Scholar]
- 477.Shinohara M., Koga T., Okamoto K., Sakaguchi S., Arai K., Yasuda H., Takai T., Kodama T., Morio T., Geha R.S., et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132:794–806. doi: 10.1016/j.cell.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 478.Taguchi Y., Gohda J., Koga T., Takayanagi H., Inoue J. A unique domain in RANK is required for Gab2 and PLCgamma2 binding to establish osteoclastogenic signals. Genes Cells. 2009;14:1331–1345. doi: 10.1111/j.1365-2443.2009.01351.x. [DOI] [PubMed] [Google Scholar]
- 479.Kim K., Kim J.H., Moon J.B., Lee J., Kwak H.B., Park Y.W., Kim N. The transmembrane adaptor protein, linker for activation of T cells (LAT), regulates RANKL-induced osteoclast differentiation. Mol. Cells. 2012;33:401–406. doi: 10.1007/s10059-012-0009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Kim M.S., Yang Y.M., Son A., Tian Y.S., Lee S.I., Kang S.W., Muallem S., Shin D.M. RANKL-mediated reactive oxygen species pathway that induces long lasting Ca2+ oscillations essential for osteoclastogenesis. J. Biol. Chem. 2010;285:6913–6921. doi: 10.1074/jbc.M109.051557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Lietman S.A., Yin L., Levine M.A. SH3BP2 mutations potentiate osteoclastogenesis via PLCγ. J. Orthop. Res. 2010;28:1425–1430. doi: 10.1002/jor.21164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Kawamoto T., Fan C., Gaivin R.J., Levine M.A., Lietman S.A. Decreased SH3BP2 inhibits osteoclast differentiation and function. J. Orthop. Res. 2011;29:1521–1527. doi: 10.1002/jor.21408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Guo J., Liu M., Yang D., Bouxsein M.L., Thomas C.C., Schipani E., Bringhurst F.R., Kronenberg H.M. Phospholipase C signaling via the parathyroid hormone (PTH)/PTH-related peptide receptor is essential for normal bone responses to PTH. Endocrinology. 2010;151:3502–3513. doi: 10.1210/en.2009-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Liu B.Y., Wu P.W., Bringhurst F.R., Wang J.T. Estrogen inhibition of PTH-stimulated osteoclast formation and attachment in vitro: Involvement of both PKA and PKC. Endocrinology. 2002;143:627–635. doi: 10.1210/endo.143.2.8614. [DOI] [PubMed] [Google Scholar]
- 485.Baines R.J., Brown C., Ng L.L., Boarder M.R. Angiotensin II-stimulated phospholipase C responses of two vascular smooth muscle-derived cell lines. Role of cyclic GMP. Hypertension. 1996;28:772–778. doi: 10.1161/01.hyp.28.5.772. [DOI] [PubMed] [Google Scholar]
- 486.Boss V., Abbott K.L., Wang X.F., Pavlath G.K., Murphy T.J. The cyclosporin A-sensitive nuclear factor of activated T cells (NFAT) proteins are expressed in vascular smooth muscle cells. Differential localization of NFAT isoforms and induction of NFAT-mediated transcription by phospholipase C-coupled cell surface receptors. J. Biol. Chem. 1998;273:19664–19671. doi: 10.1074/jbc.273.31.19664. [DOI] [PubMed] [Google Scholar]
- 487.Paulhe F., Bogyo A., Chap H., Perret B., Racaud-Sultan C. Vascular smooth muscle cell spreading onto fibrinogen is regulated by calpains and phospholipase C. Biochem. Biophys. Res. Commun. 2001;288:875–881. doi: 10.1006/bbrc.2001.5859. [DOI] [PubMed] [Google Scholar]
- 488.Kelly J., Brazil D., Clyne C., McHale N.G., Gierschik P., Keenan A.K. Evidence for the presence of G-proteins, adenylyl cyclase and phospholipase C activities in lymphatic smooth muscle cell membranes. Cell. Signal. 1996;8:425–432. doi: 10.1016/s0898-6568(96)00098-8. [DOI] [PubMed] [Google Scholar]
- 489.Grillone L.R., Clark M.A., Godfrey R.W., Stassen F., Crooke S.T. Vasopressin induces V1 receptors to activate phosphatidylinositol- and phosphatidylcholine-specific phospholipase C and stimulates the release of arachidonic acid by at least two pathways in the smooth muscle cell line, A-10. J. Biol. Chem. 1988;263:2658–2663. [PubMed] [Google Scholar]
- 490.Liu B., Itoh H., Louie O., Kubota K., Kent K.C. The role of phospholipase C and phosphatidylinositol 3-kinase in vascular smooth muscle cell migration and proliferation. J. Surg. Res. 2004;120:256–265. doi: 10.1016/j.jss.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 491.Roztocil E., Nicholl S.M., Davies M.G. Sphingosine-1-phosphate-induced oxygen free radical generation in smooth muscle cell migration requires Galpha12/13 protein-mediated phospholipase C activation. J. Vasc. Surg. 2007;46:1253–1259. doi: 10.1016/j.jvs.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Shibukawa Y., Suzuki T. Ca2+ signaling mediated by IP3-dependent Ca2+ releasing and store-operated Ca2+ channels in rat odontoblasts. J. Bone Miner. Res. 2003;18:30–38. doi: 10.1359/jbmr.2003.18.1.30. [DOI] [PubMed] [Google Scholar]
- 493.Sooampon S., Manokawinchoke J., Pavasant P. Transient receptor potential vanilloid-1 regulates osteoprotegerin/RANKL homeostasis in human periodontal ligament cells. J. Periodontal Res. 2012 doi: 10.1111/j.1600-0765.2012.01493.x. [DOI] [PubMed] [Google Scholar]
- 494.Osathanon T., Nowwarote N., Pavasant P. Basic fibroblast growth factor inhibits mineralization but induces neuronal differentiation by human dental pulp stem cells through a FGFR and PLCγ signaling pathway. J. Cell. Biochem. 2011;112:1807–1816. doi: 10.1002/jcb.23097. [DOI] [PubMed] [Google Scholar]
- 495.Nakamura Y., Fukami K. Roles of phospholipase C isozymes in organogenesis and embryonic development. Physiology. 2009;24:332–341. doi: 10.1152/physiol.00031.2009. [DOI] [PubMed] [Google Scholar]
- 496.Liao H.J., Kume T., McKay C., Xu M.J., Ihle J.N., Carpenter G. Absence of erythrogenesis and vasculogenesis in Plcg1-deficient mice. J. Biol. Chem. 2002;277:9335–9341. doi: 10.1074/jbc.M109955200. [DOI] [PubMed] [Google Scholar]
- 497.Nakamura Y., Hamada Y., Fujiwara T., Enomoto H., Hiroe T., Tanaka S., Nose M., Nakahara M., Yoshida N., Takenawa T., et al. Phospholipase C-delta1 and -delta3 are essential in the trophoblast for placental development. Mol. Cell. Biol. 2005;25:10979–10988. doi: 10.1128/MCB.25.24.10979-10988.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Bhattacharya R., Kwon J., Li X., Wang E., Patra S., Bida J.P., Bajzer Z., Claesson-Welsh L., Mukhopadhyay D. Distinct role of PLCbeta3 in VEGF-mediated directional migration and vascular sprouting. J. Cell. Sci. 2009;122:1025–1034. doi: 10.1242/jcs.041913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Shirane M., Sawa H., Kobayashi Y., Nakano T., Kitajima K., Shinkai Y., Nagashima K., Negishi I. Deficiency of phospholipase C-gamma1 impairs renal development and hematopoiesis. Development. 2001;128:5173–5180. doi: 10.1242/dev.128.24.5173. [DOI] [PubMed] [Google Scholar]
- 500.Hashimoto A., Takeda K., Inaba M., Sekimata M., Kaisho T., Ikehara S., Homma Y., Akira S., Kurosaki T. Cutting edge: Essential role of phospholipase C-gamma 2 in B cell development and function. J. Immunol. 2000;165:1738–1742. doi: 10.4049/jimmunol.165.4.1738. [DOI] [PubMed] [Google Scholar]
- 501.Tassi I., Presti R., Kim S., Yokoyama W.M., Gilfillan S., Colonna M. Phospholipase C-gamma 2 is a critical signaling mediator for murine NK cell activating receptors. J. Immunol. 2005;175:749–754. doi: 10.4049/jimmunol.175.2.749. [DOI] [PubMed] [Google Scholar]
- 502.Tassi I., Cella M., Castro I., Gilfillan S., Khan W.N., Colonna M. Requirement of phospholipase C-gamma2 (PLCgamma2) for Dectin-1-induced antigen presentation and induction of TH1/TH17 polarization. Eur. J. Immunol. 2009;39:1369–1378. doi: 10.1002/eji.200839313. [DOI] [PubMed] [Google Scholar]
- 503.Cremasco V., Graham D.B., Novack D.V., Swat W., Faccio R. Vav/Phospholipase Cgamma2-mediated control of a neutrophil-dependent murine model of rheumatoid arthritis. Arthritis. Rheum. 2008;58:2712–2722. doi: 10.1002/art.23757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Cremasco V., Benasciutti E., Cella M., Kisseleva M., Croke M., Faccio R. Phospholipase C gamma 2 is critical for development of a murine model of inflammatory arthritis by affecting actin dynamics in dendritic cells. PLoS One. 2010;5:e8909. doi: 10.1371/journal.pone.0008909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Graham D.B., Robertson C.M., Bautista J., Mascarenhas F., Diacovo M.J., Montgrain V., Lam S.K., Cremasco V., Dunne W.M., Faccio R., et al. Neutrophil-mediated oxidative burst and host defense are controlled by a Vav-PLCgamma2 signaling axis in mice. J. Clin. Invest. 2007;117:3445–3452. doi: 10.1172/JCI32729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Zhang K., Kim S., Cremasco V., Hirbe A.C., Collins L., Piwnica-Worms D., Novack D.V., Weilbaecher K., Faccio R. CD8+ T cells regulate bone tumor burden independent of osteoclast resorption. Cancer Res. 2011;71:4799–4808. doi: 10.1158/0008-5472.CAN-10-3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Matsuo K., Galson D.L., Zhao C., Peng L., Laplace C., Wang K.Z., Bachler M.A., Amano H., Aburatani H., Ishikawa H., et al. Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J. Biol. Chem. 2004;279:26475–26480. doi: 10.1074/jbc.M313973200. [DOI] [PubMed] [Google Scholar]
- 508.Chen Y., Wang X., Di L., Fu G., Bai L., Liu J., Feng X., McDonald J.M., Michalek S., He Y., et al. Phospholipase Cgamma2 mediates RANKL-stimulated lymph node organogenesis and osteoclastogenesis. J. Biol. Chem. 2008;283:29593–29601. doi: 10.1074/jbc.M802493200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Kertész Z., Gyori D., Körmendi S., Fekete T., Kis-Tóth K., Jakus Z., Schett G., Rajnavölgyi E., Dobó-Nagy C., Mócsai A. Phospholipase Cγ2 is required for basal but not oestrogen deficiency-induced bone resorption. Eur. J. Clin. Invest. 2012;42:49–60. doi: 10.1111/j.1365-2362.2011.02556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Kanematsu T., Takeya H., Watanabe Y., Ozaki S., Yoshida M., Koga T., Iwanaga S., Hirata M. Putative inositol 1,4,5-trisphosphate binding proteins in rat brain cytosol. J. Biol. Chem. 1992;267:6518–6525. [PubMed] [Google Scholar]
- 511.Kanematsu T., Yoshimura K., Hidaka K., Takeuchi H., Katan M., Hirata M. Domain organization of p130, PLC-related catalytically inactive protein, and structural basis for the lack of enzyme activity. Eur. J. Biochem. 2000;267:2731–2737. doi: 10.1046/j.1432-1327.2000.01291.x. [DOI] [PubMed] [Google Scholar]
- 512.Yoshimura K., Takeuchi H., Sato O., Hidaka K., Doira N., Terunuma M., Harada K., Ogawa Y., Ito Y., Kanematsu T., et al. Interaction of p130 with, and consequent inhibition of, the catalytic subunit of protein phosphatase 1alpha. J. Biol. Chem. 2001;276:17908–17913. doi: 10.1074/jbc.M009677200. [DOI] [PubMed] [Google Scholar]
- 513.Kanematsu T., Yasunaga A., Mizoguchi Y., Kuratani A., Kittler J.T., Jovanovic J.N., Takenaka K., Nakayama K.I., Fukami K., Takenawa T., et al. Modulation of GABA(A) receptor phosphorylation and membrane trafficking by phospholipase C-related inactive protein/protein phosphatase 1 and 2A signaling complex underlying brain-derived neurotrophic factor-dependent regulation of GABAergic inhibition. J. Biol. Chem. 2006;281:22180–22189. doi: 10.1074/jbc.M603118200. [DOI] [PubMed] [Google Scholar]
- 514.Fujii M., Kanematsu T., Ishibashi H., Fukami K., Takenawa T., Nakayama K.I., Moss S.J., Nabekura J., Hirata M. Phospholipase C-related but catalytically inactive protein is required for insulin-induced cell surface expression of gamma-aminobutyric acid type A receptors. J. Biol. Chem. 2010;285:4837–4846. doi: 10.1074/jbc.M109.070045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Takeuchi H., Kanematsu T., Misumi Y., Yaakob H.B., Yagisawa H., Ikehara Y., Watanabe Y., Tan Z., Shears S.B., Hirata M. Localization of a high-affinity inositol 1,4,5-trisphosphate/inositol 1,4,5,6-tetrakisphosphate binding domain to the pleckstrin homology module of a new 130 kDa protein: Characterization of the determinants of structural specificity. Biochem. J. 1996;318:561–568. doi: 10.1042/bj3180561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Mizokami A., Kanematsu T., Ishibashi H., Yamaguchi T., Tanida I., Takenaka K., Nakayama K.I., Fukami K., Takenawa T., Kominami E., et al. Phospholipase C-related inactive protein is involved in trafficking of gamma2 subunit-containing GABA(A) receptors to the cell surface. J. Neurosci. 2007;27:1692–1701. doi: 10.1523/JNEUROSCI.3155-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Matsuda M., Tsutsumi K., Kanematsu T., Fukami K., Terada Y., Takenawa T., Nakayama K.I., Hirata M. Involvement of phospholipase C-related inactive protein in the mouse reproductive system through the regulation of gonadotropin levels. Biol. Reprod. 2009;81:681–689. doi: 10.1095/biolreprod.109.076760. [DOI] [PubMed] [Google Scholar]
- 518.Tsutsumi K., Matsuda M., Kotani M., Mizokami A., Murakami A., Takahashi I., Terada Y., Kanematsu T., Fukami K., Takenawa T., et al. Involvement of PRIP, phospholipase C-related, but catalytically inactive protein, in bone formation. J. Biol. Chem. 2011;286:31032–31042. doi: 10.1074/jbc.M111.235903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Gao J., Takeuchi H., Zhang Z., Fukuda M., Hirata M. Phospholipase C-related but catalytically inactive protein (PRIP) modulates synaptosomal-associated protein 25 (SNAP-25) phosphorylation and exocytosis. J. Biol. Chem. 2012;287:10565–10578. doi: 10.1074/jbc.M111.294645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Spiegel S., Milstien S. Sphingosine-1-phosphate: Signaling inside and out. FEBS Lett. 2000;476:55–57. doi: 10.1016/s0014-5793(00)01670-7. [DOI] [PubMed] [Google Scholar]
- 521.Maceyka M., Payne S.G., Milstien S., Spiegel S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim. Biophys. Acta. 2002;1585:193–201. doi: 10.1016/s1388-1981(02)00341-4. [DOI] [PubMed] [Google Scholar]
- 522.Khavandgar Z., Poirier C., Clarke C.J., Li J., Wang N., McKee M.D., Hannun Y.A., Murshed M. A cell-autonomous requirement for neutral sphingomyelinase 2 in bone mineralization. J. Cell. Biol. 2011;194:277–289. doi: 10.1083/jcb.201102051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Gilbert S.J., Blain E.J., Jones P., Duance V.C., Mason D.J. Exogenous sphingomyelinase increases collagen and sulphated glycosaminoglycan production by primary articular chondrocytes: An in vitro study. Arthritis. Res. Ther. 2006;8:89. doi: 10.1186/ar1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Sabatini M., Rolland G., Léonce S., Thomas M., Lesur C., Pérez V., de Nanteuil G., Bonnet J. Effects of ceramide on apoptosis, proteoglycan degradation, and matrix metalloproteinase expression in rabbit articular cartilage. Biochem. Biophys. Res. Commun. 2000;267:438–444. doi: 10.1006/bbrc.1999.1983. [DOI] [PubMed] [Google Scholar]
- 525.Ehlert K., Frosch M., Fehse N., Zander A., Roth J., Vormoor J. Farber disease: Clinical presentation, pathogenesis and a new approach to treatment. Pediatr. Rheumatol. Online J. 2007;5:15:1–15:7. doi: 10.1186/1546-0096-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Gilbert S.J., Blain E.J., Duance V.C., Mason D.J. Sphingomyelinase decreases type II collagen expression in bovine articular cartilage chondrocytes via the ERK signaling pathway. Arthritis. Rheum. 2008;58:209–220. doi: 10.1002/art.23172. [DOI] [PubMed] [Google Scholar]
- 527.Kitajima I., Soejima Y., Takasaki I., Beppu H., Tokioka T., Maruyama I. Ceramide-induced nuclear translocation of NF-kappa B is a potential mediator of the apoptotic response to TNF-alpha in murine clonal osteoblasts. Bone. 1996;19:263–270. doi: 10.1016/8756-3282(96)00181-0. [DOI] [PubMed] [Google Scholar]
- 528.Takeshita A., Watanabe A., Takada Y., Hanazawa S. Selective stimulation by ceramide of the expression of the alpha isoform of retinoic acid and retinoid X receptors in osteoblastic cells. A role of sphingosine 1-phosphate-mediated AP-1 in the ligand-dependent transcriptional activity of these receptors. J. Biol. Chem. 2000;275:32220–32226. doi: 10.1074/jbc.M002569200. [DOI] [PubMed] [Google Scholar]
- 529.Kozawa O., Hatakeyama D., Tokuda H., Oiso Y., Matsuno H., Uematsu T. Sphingomyelinase amplifies BMP-4-induced osteocalcin synthesis in osteoblasts: Role of ceramide. Cell. Signal. 2002;14:999–1004. doi: 10.1016/s0898-6568(02)00033-5. [DOI] [PubMed] [Google Scholar]
- 530.Klein B.Y., Kerem Z., Rojansky N. LDL induces Saos2 osteoblasts death via Akt pathways responsive to a neutral sphingomyelinase inhibitor. J. Cell. Biochem. 2006;98:661–671. doi: 10.1002/jcb.20807. [DOI] [PubMed] [Google Scholar]
- 531.Hill P.A., Tumber A. Ceramide-induced cell death/survival in murine osteoblasts. J. Endocrinol. 2010;206:225–233. doi: 10.1677/JOE-10-0068. [DOI] [PubMed] [Google Scholar]
- 532.Tokuda H., Kozawa O., Harada A., Uematsu T. Extracellular sphingomyelinase induces interleukin-6 synthesis in osteoblasts. J. Cell. Biochem. 1999;72:262–268. doi: 10.1002/(sici)1097-4644(19990201)72:2<262::aid-jcb10>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 533.Takeshita A., Shinoda H., Nakabayashi Y., Takano A., Matsumoto K., Suetsugu M., Miyazawa K., Tanaka S., Endo H., Ueyama Y., et al. Sphingosine 1-phosphate acts as a signal molecule in ceramide signal transduction of TNF-alpha-induced activator protein-1 in osteoblastic cell line MC3T3-E1 cells. J. Oral Sci. 2005;47:43–51. doi: 10.2334/josnusd.47.43. [DOI] [PubMed] [Google Scholar]
- 534.Spiegel S., Merrill A.H. Sphingolipid metabolism and cell growth regulation. FASEB J. 1996;10:1388–1397. doi: 10.1096/fasebj.10.12.8903509. [DOI] [PubMed] [Google Scholar]
- 535.Bao X., Ogawa T., Se S., Akiyama M., Bahtiar A., Takeya T., Ishida-Kitagawa N. Acid sphingomyelinase regulates osteoclastogenesis by modulating sphingosine kinases downstream of RANKL signaling. Biochem. Biophys. Res. Commun. 2011;405:533–537. doi: 10.1016/j.bbrc.2011.01.061. [DOI] [PubMed] [Google Scholar]
- 536.Takeda H., Ozaki K., Yasuda H., Ishida M., Kitano S., Hanazawa S. Sphingomyelinase and ceramide inhibit formation of F-actin ring in and bone resorption by rabbit mature osteoclasts. FEBS Lett. 1998;422:255–258. doi: 10.1016/s0014-5793(98)00005-2. [DOI] [PubMed] [Google Scholar]
- 537.Aubin I., Adams C.P., Opsahl S., Septier D., Bishop C.E., Auge N., Salvayre R., Negre-Salvayre A., Goldberg M., Guénet J.L., et al. A deletion in the gene encoding sphingomyelin phosphodiesterase 3 (Smpd3) results in osteogenesis and dentinogenesis imperfecta in the mouse. Nat. Genet. 2005;37:803–805. doi: 10.1038/ng1603. [DOI] [PubMed] [Google Scholar]
- 538.Stoffel W., Jenke B., Blöck B., Zumbansen M., Koebke J. Neutral sphingomyelinase 2 (smpd3) in the control of postnatal growth and development. Proc. Natl. Acad. Sci. USA. 2005;102:4554–4559. doi: 10.1073/pnas.0406380102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Stoffel W., Jenke B., Holz B., Binczek E., Günter R.H., Knifka J., Koebke J., Niehoff A. Neutral sphingomyelinase (SMPD3) deficiency causes a novel form of chondrodysplasia and dwarfism that is rescued by Col2A1-driven smpd3 transgene expression. Am. J. Pathol. 2007;171:153–161. doi: 10.2353/ajpath.2007.061285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Thouverey C., Malinowska A., Balcerzak M., Strzelecka-Kiliszek A., Buchet R., Dadlez M., Pikula S. Proteomic characterization of biogenesis and functions of matrix vesicles released from mineralizing human osteoblast-like cells. J. Proteomics. 2011;74:1123–1134. doi: 10.1016/j.jprot.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 541.Balcerzak M., Malinowska A., Thouverey C., Sekrecka A., Dadlez M., Buchet R., Pikula S. Proteome analysis of matrix vesicles isolated from femurs of chicken embryo. Proteomics. 2008;8:192–205. doi: 10.1002/pmic.200700612. [DOI] [PubMed] [Google Scholar]
- 542.Ponting C.P., Kerr I.D. A novel family of phospholipase D homologues that includes phospholipid synthases and putative endonucleases: Identification of duplicated repeats and potential active site residues. Protein Sci. 1996;5:914–922. doi: 10.1002/pro.5560050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Lavieri R., Scott S.A., Lewis J.A., Selvy P.E., Armstrong M.D., Alex Brown H., Lindsley C.W. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part II. Identification of the 1,3,8-triazaspiro[4,5]decan-4-one privileged structure that engenders PLD2 selectivity. Bioorg. Med. Chem. Lett. 2009;19:2240–2243. doi: 10.1016/j.bmcl.2009.02.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Stace C.L., Ktistakis N.T. Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim. Biophys. Acta. 2006;1761:913–926. doi: 10.1016/j.bbalip.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 545.Exton J.H. Phosphatidylcholine breakdown and signal transduction. Biochim. Biophys. Acta. 1994;1212:26–42. doi: 10.1016/0005-2760(94)90186-4. [DOI] [PubMed] [Google Scholar]
- 546.Exton J.H. Phospholipase D: Enzymology, mechanisms of regulation, and function. Physiol. Rev. 1997;77:303–320. doi: 10.1152/physrev.1997.77.2.303. [DOI] [PubMed] [Google Scholar]
- 547.Kanoh H., Sakane F., Imai S., Wada I. Diacylglycerol kinase and phosphatidic acid phosphatase—Enzymes metabolizing lipid second messengers. Cell. Signal. 1993;5:495–503. doi: 10.1016/0898-6568(93)90045-n. [DOI] [PubMed] [Google Scholar]
- 548.Martin T.W. Formation of diacylglycerol by a phospholipase d-phosphatidate phosphatase pathway specific for phosphatidylcholine in endothelial cells. Biochim. Biophys. Acta. 1988;962:282–296. doi: 10.1016/0005-2760(88)90258-5. [DOI] [PubMed] [Google Scholar]
- 549.Mackay H.J., Twelves C.J. Targeting the protein kinase C family: Are we there yet? Nat. Rev. Cancer. 2007;7:554–562. doi: 10.1038/nrc2168. [DOI] [PubMed] [Google Scholar]
- 550.Khan W.A., Blobe G.C., Hannun Y.A. Arachidonic acid and free fatty acids as second messengers and the role of protein kinase C. Cell. Signal. 1995;7:171–184. doi: 10.1016/0898-6568(94)00089-t. [DOI] [PubMed] [Google Scholar]
- 551.Ghosh S., Strum J.C., Sciorra V.A., Daniel L., Bell R.M. Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-o-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J. Biol. Chem. 1996;271:8472–8480. doi: 10.1074/jbc.271.14.8472. [DOI] [PubMed] [Google Scholar]
- 552.Rizzo M.A., Shome K., Watkins S.C., Romero G. The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J. Biol. Chem. 2000;275:23911–23918. doi: 10.1074/jbc.M001553200. [DOI] [PubMed] [Google Scholar]
- 553.Fang Y., Vilella-Bach M., Bachmann R., Flanigan A., Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 554.Exton J.H. Phospholipase d-structure, regulation and function. Rev. Physiol. Biochem. Pharmacol. 2002;144:1–94. doi: 10.1007/BFb0116585. [DOI] [PubMed] [Google Scholar]
- 555.Steed P.M., Clark K.L., Boyar W.C., Lasala D.J. Characterization of human PLD2 and the analysis of PLD isoform splice variants. FASEB J. 1998;12:1309–1317. doi: 10.1096/fasebj.12.13.1309. [DOI] [PubMed] [Google Scholar]
- 556.Cao J.X., Koop B.F., Upton C. A human homolog of the vaccinia virus HindIII K4L gene is a member of the phospholipase D superfamily. Virus Res. 1997;48:11–18. doi: 10.1016/s0168-1702(96)01422-0. [DOI] [PubMed] [Google Scholar]
- 557.Choi S.Y., Huang P., Jenkins G.M., Chan D.C., Schiller J., Frohman M.A. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell. Biol. 2006;8:1255–1262. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- 558.McDermott M., Wakelam M.J., Morris A.J. Phospholipase D. Biochem. Cell. Biol. 2004;82:225–253. doi: 10.1139/o03-079. [DOI] [PubMed] [Google Scholar]
- 559.Watanabe-Tomita Y., Suzuki A., Shinoda J., Oiso Y., Kozawa O. Arachidonic acid release induced by extracellular ATP in osteoblasts: Role of phospholipase D. Prostaglandins Leukotrienes Essent. Fatty Acids. 1997;57:335–339. doi: 10.1016/s0952-3278(97)90553-6. [DOI] [PubMed] [Google Scholar]
- 560.Hughes W.E., Parker P.J. Endosomal localization of phospholipase D 1a and 1b is defined by the C-termini of the proteins, and is independent of activity. Biochem. J. 2001;356:727–736. doi: 10.1042/0264-6021:3560727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Lee S., Park J.B., Kim J.H., Kim Y., Shin K.J., Lee J.S., Ha S.H., Suh P.G., Ryu S.H. Actin directly interacts with phospholipase D, inhibiting its activity. J. Biol. Chem. 2001;276:28252–28260. doi: 10.1074/jbc.M008521200. [DOI] [PubMed] [Google Scholar]
- 562.Sugars J.M., Cellek S., Manifava M., Coadwell J., Ktistakis N.T. Fatty acylation of phospholipase D1 on cysteine residues 240 and 241 determines localization on intracellular membranes. J. Biol. Chem. 1999;274:30023–30027. doi: 10.1074/jbc.274.42.30023. [DOI] [PubMed] [Google Scholar]
- 563.Xie Z., Ho W.T., Exton J.H. Functional implications of post-translational modifications of phospholipases D1 and D2. Biochim. Biophys. Acta. 2002;1580:9–21. doi: 10.1016/s1388-1981(01)00168-8. [DOI] [PubMed] [Google Scholar]
- 564.Jang J.H., Lee C.S., Hwang D., Ryu S.H. Understanding of the roles of phospholipase D and phosphatidic acid through their binding partners. Prog. Lipid Res. 2012;51:71–81. doi: 10.1016/j.plipres.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 565.Rizzo M., Romero G. Pharmacological importance of phospholipase D and phosphatidic acid in the regulation of the mitogen-activated protein kinase cascade. Pharmacol. Ther. 2002;94:35–50. doi: 10.1016/s0163-7258(02)00170-5. [DOI] [PubMed] [Google Scholar]
- 566.Szumiło M., Rahden-Staroń I. Phospholipase D in mammalian cells: Structure, properties, physiological and pathological role. Postepy Hig. Med. Dosw. 2006;60:421–430. [PubMed] [Google Scholar]
- 567.Park M.K., Her Y.M., Cho M.L., Oh H.J., Park E.M., Kwok S.K., Ju J.H., Park K.S., Min D.S., Kim H.Y., et al. IL-15 promotes osteoclastogenesis via the PLD pathway in rheumatoid arthritis. Immunol. Lett. 2011;139:42–51. doi: 10.1016/j.imlet.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 568.Jenkins G.M., Frohman M.A. Phospholipase D: A lipid centric review. Cell. Mol. Life Sci. 2005;62:2305–2316. doi: 10.1007/s00018-005-5195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Conquer J.A., Jones S.A., Cruz T.F. Effect of interleukin 1, lipopolysaccharide and phorbol esters on phospholipase D activity in chondrocytes. Osteoarthritis Cartilage. 1994;2:269–273. [PubMed] [Google Scholar]
- 570.Sylvia V.L., Schwartz Z., Del Toro F., DeVeau P., Whetstone R., Hardin R.R., Dean D.D., Boyan B.D. Regulation of phospholipase D (PLD) in growth plate chondrocytes by 24R,25-(OH)2D3 is dependent on cell maturation state (resting zone cells) and is specific to the PLD2 isoform. Biochim. Biophys. Acta. 2001;1499:209–221. doi: 10.1016/s0167-4889(00)00120-8. [DOI] [PubMed] [Google Scholar]
- 571.Dean D.D., Muniz O.E., Berman I., Pita J.C., Carreno M.R., Woessner J.F., Howell D.S. Localization of collagenase in the growth plate of rachitic rats. J. Clin. Invest. 1985;76:716–722. doi: 10.1172/JCI112026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Dean D.D., Muniz O.E., Howell D.S. Association of collagenase and tissue inhibitor of metalloproteinases (TIMP) with hypertrophic cell enlargement in the growth plate. Matrix. 1989;9:366–375. doi: 10.1016/s0934-8832(89)80041-1. [DOI] [PubMed] [Google Scholar]
- 573.Boyan B.D., Dean D.D., Sylvia V.L., Schwartz Z. Steroid hormone action in musculoskeletal cells involves membrane receptor and nuclear receptor mechanisms. Connect. Tissue Res. 2003;44:130–135. [PubMed] [Google Scholar]
- 574.Langston G.G., Swain L.D., Schwartz Z., Del Toro F., Gomez R., Boyan B.D. Effect of 1,25(OH)2D3 and 24,25(OH)2D3 on calcium ion fluxes in costochondral chondrocyte cultures. Calcif. Tissue Int. 1990;47:230–236. doi: 10.1007/BF02555924. [DOI] [PubMed] [Google Scholar]
- 575.Schwartz Z., Schlader D.L., Ramirez V., Kennedy M.B., Boyan B.D. Effects of vitamin D metabolites on collagen production and cell proliferation of growth zone and resting zone cartilage cells in vitro. J. Bone Miner. Res. 1989;4:199–207. doi: 10.1002/jbmr.5650040211. [DOI] [PubMed] [Google Scholar]
- 576.Boyan B.D., Schwartz Z., Swain L.D., Carnes D.L., Zislis T. Differential expression of phenotype by resting zone and growth region costochondral chondrocytes in vitro. Bone. 1988;9:185–194. doi: 10.1016/8756-3282(88)90008-7. [DOI] [PubMed] [Google Scholar]
- 577.Helm S., Sylvia V.L., Harmon T., Dean D.D., Boyan B.D., Schwartz Z. 24,25-(OH)2D3 regulates protein kinase C through two distinct phospholipid-dependent mechanisms. J. Cell. Physiol. 1996;169:509–521. doi: 10.1002/(SICI)1097-4652(199612)169:3<509::AID-JCP11>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 578.Boyan B.D., Sylvia V.L., McKinney N., Schwartz Z. Membrane actions of vitamin D metabolites 1alpha,25(OH)2D3 and 24R,25(OH)2D3 are retained in growth plate cartilage cells from vitamin D receptor knockout mice. J. Cell. Biochem. 2003;90:1207–1223. doi: 10.1002/jcb.10716. [DOI] [PubMed] [Google Scholar]
- 579.Boyan B.D., Jennings E.G., Wang L., Schwartz Z. Mechanisms regulating differential activation of membrane-mediated signaling by 1alpha,25(OH)2D3 and 24R,25(OH)2D3. J. Steroid Biochem. Mol. Biol. 2004;89–90:309–315. doi: 10.1016/j.jsbmb.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 580.Boyan B.D., Sylvia V.L., Dean D.D., Pedrozo H., Del Toro F., Nemere I., Posner G.H., Schwartz Z. 1,25-(OH)2D3 modulates growth plate chondrocytes via membrane receptor-mediated protein kinase C by a mechanism that involves changes in phospholipid metabolism and the action of arachidonic acid and PGE2. Steroids. 1999;64:129–136. doi: 10.1016/s0039-128x(98)00099-3. [DOI] [PubMed] [Google Scholar]
- 581.Schwartz Z., Sylvia V.L., Del Toro F., Hardin R.R., Dean D.D., Boyan B.D. 24R,25-(OH)(2)D(3) mediates its membrane receptor-dependent effects on protein kinase C and alkaline phosphatase via phospholipase A(2) and cyclooxygenase-1 but not cyclooxygenase-2 in growth plate chondrocytes. J. Cell. Physiol. 2000;182:390–401. doi: 10.1002/(SICI)1097-4652(200003)182:3<390::AID-JCP10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 582.Sylvia V.L., Schwartz Z., Holmes S.C., Dean D.D., Boyan B.D. 24,25-(OH)2D3 regulation of matrix vesicle protein kinase C occurs both during biosynthesis and in the extracellular matrix. Calcif. Tissue Int. 1997;61:313–321. doi: 10.1007/s002239900341. [DOI] [PubMed] [Google Scholar]
- 583.Dean D.D., Boyan B.D., Schwart Z., Muniz O.E., Carreno M.R., Maeda S., Howell D.S. Effect of 1alpha,25-dihydroxyvitamin D3 and 24R,25-dihydroxyvitamin D3 on metalloproteinase activity and cell maturation in growth plate cartilage in vivo. Endocrine. 2001;14:311–323. doi: 10.1385/endo:14:3:311. [DOI] [PubMed] [Google Scholar]
- 584.Kusner D.J., Barton J.A., Qin C., Wang X., Iyer S.S. Evolutionary conservation of physical and functional interactions between phospholipase D and actin. Arch. Biochem. Biophys. 2003;412:231–241. doi: 10.1016/s0003-9861(03)00052-3. [DOI] [PubMed] [Google Scholar]
- 585.Blackwood R.A., Smolen J.E., Transue A., Hessler R.J., Harsh D.M., Brower R.C., French S. Phospholipase D activity facilitates Ca2+-induced aggregation and fusion of complex liposomes. Am. J. Physiol. 1997;272:C1279–C1285. doi: 10.1152/ajpcell.1997.272.4.C1279. [DOI] [PubMed] [Google Scholar]
- 586.Burger K.N. Greasing membrane fusion and fission machineries. Traffic. 2000;1:605–613. doi: 10.1034/j.1600-0854.2000.010804.x. [DOI] [PubMed] [Google Scholar]
- 587.Burger K.N., Demel R.A., Schmid S.L., de Kruijff B. Dynamin is membrane-active: Lipid insertion is induced by phosphoinositides and phosphatidic acid. Biochemistry. 2000;39:12485–12493. doi: 10.1021/bi000971r. [DOI] [PubMed] [Google Scholar]
- 588.Bader M.F., Vitale N. Phospholipase D in calcium-regulated exocytosis: Lessons from chromaffin cells. Biochim. Biophys. Acta. 2009;1791:936–941. doi: 10.1016/j.bbalip.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 589.Laulagnier K., Grand D., Dujardin A., Hamdi S., Vincent-Schneider H., Lankar D., Salles J.P., Bonnerot C., Perret B., Record M. PLD2 is enriched on exosomes and its activity is correlated to the release of exosomes. FEBS Lett. 2004;572:11–14. doi: 10.1016/j.febslet.2004.06.082. [DOI] [PubMed] [Google Scholar]
- 590.Kozawa O., Suzuki A., Watanabe Y., Shinoda J., Oiso Y. Effect of platelet-derived growth factor on phosphatidylcholine-hydrolyzing phospholipase D in osteoblast-like cells. Endocrinology. 1995;136:4473–4478. doi: 10.1210/endo.136.10.7664667. [DOI] [PubMed] [Google Scholar]
- 591.Shinoda J., Suzuki A., Oiso Y., Kozawa O. Thromboxane A2-stimulated phospholipase D in osteoblast-like cells: Possible involvement of PKC. Am. J. Physiol. 1995;269:E524–E529. doi: 10.1152/ajpendo.1995.269.3.E524. [DOI] [PubMed] [Google Scholar]
- 592.Tokuda H., Suzuki A., Watanabe-Tomita Y., Shinoda J., Imamura Y., Oiso Y., Igata A., Kozawa O. Function of Ca2+ in phosphatidylcholine-hydrolyzing phospholipase D activation in osteoblast-like cells. Bone. 1996;19:347–352. doi: 10.1016/s8756-3282(96)00185-8. [DOI] [PubMed] [Google Scholar]
- 593.Oiso Y., Suzuki A., Kozawa O. Effect of prostaglandin E2 on phospholipase D activity in osteoblast-like MC3T3-E1 cells. J. Bone Miner. Res. 1995;10:1185–1190. doi: 10.1002/jbmr.5650100807. [DOI] [PubMed] [Google Scholar]
- 594.Suzuki A., Kozawa O., Shinoda J., Watanabe Y., Saito H., Oiso Y. Thrombin induces proliferation of osteoblast-like cells through phosphatidylcholine hydrolysis. J. Cell. Physiol. 1996;168:209–216. doi: 10.1002/(SICI)1097-4652(199607)168:1<209::AID-JCP25>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 595.Sugiyama T., Sakai T., Nozawa Y., Oka N. Prostaglandin F2 alpha-stimulated phospholipase D activation in osteoblast-like MC3T3-E1 cells: Involvement in sustained 1,2-diacylglycerol production. Biochem. J. 1994;298( Pt. 2):479–484. doi: 10.1042/bj2980479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Kozawa O., Suzuki A., Kotoyori J., Tokuda H., Watanabe Y., Ito Y., Oiso Y. Prostaglandin F2 alpha activates phospholipase D independently from activation of protein kinase C in osteoblast-like cells. J. Cell. Biochem. 1994;55:373–379. doi: 10.1002/jcb.240550315. [DOI] [PubMed] [Google Scholar]
- 597.Kozawa O., Suzuki A., Shinoda J., Oiso Y. Effect of retinoic acid on prostaglandin F2 alpha-induced phospholipase D activity in osteoblast-like cells. Prostaglandins Leukotrienes Essent. Fatty Acids. 1996;55:151–154. doi: 10.1016/s0952-3278(96)90090-3. [DOI] [PubMed] [Google Scholar]
- 598.Kozawa O., Suzuki A., Watanabe Y., Shinoda J., Oiso Y. Function of Ca2+ influx in phospholipase D activation induced by prostaglandin F2 alpha in osteoblast-like cells: Involvement of tyrosine kinase. Prostaglandins Leukotrienes Essent. Fatty Acids. 1995;52:319–323. doi: 10.1016/0952-3278(95)90033-0. [DOI] [PubMed] [Google Scholar]
- 599.Kozawa O., Suzuki A., Oiso Y. Tyrosine kinase regulates phospholipase D activation at a point downstream from protein kinase C in osteoblast-like cells. J. Cell. Biochem. 1995;57:251–255. doi: 10.1002/jcb.240570208. [DOI] [PubMed] [Google Scholar]
- 600.Suzuki A., Oiso Y., Kozawa O. Effect of endothelin-1 on phospholipase D activity in osteoblast-like cells. Mol. Cell. Endocrinol. 1994;105:193–196. doi: 10.1016/0303-7207(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 601.Kozawa O., Suzuki A., Shinoda J., Ozaki N., Oiso Y., Uematsu T. Involvement of phospholipase D activation in endothelin-1-induced release of arachidonic acid in osteoblast-like cells. J. Cell. Biochem. 1997;64:376–381. [PubMed] [Google Scholar]
- 602.Suzuki A., Shinoda J., Kanda S., Oiso Y., Kozawa O. Basic fibroblast growth factor stimulates phosphatidylcholine-hydrolyzing phospholipase D in osteoblast-like cells. J. Cell. Biochem. 1996;63:491–499. doi: 10.1002/(sici)1097-4644(19961215)63:4<491::aid-jcb10>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 603.Bourgoin S.G., Harbour D., Poubelle P.E. Role of protein kinase C alpha, Arf, and cytoplasmic calcium transients in phospholipase D activation by sodium fluoride in osteoblast-like cells. J. Bone Miner. Res. 1996;11:1655–1665. doi: 10.1002/jbmr.5650111109. [DOI] [PubMed] [Google Scholar]
- 604.Kaneki H., Yokozawa J., Fujieda M., Mizuochi S., Ishikawa C., Ide H. Phorbol ester-induced production of prostaglandin E2 from phosphatidylcholine through the activation of phospholipase D in UMR-106 cells. Bone. 1998;23:213–222. doi: 10.1016/s8756-3282(98)00100-8. [DOI] [PubMed] [Google Scholar]
- 605.Singh A.T., Gilchrist A., Voyno-Yasenetskaya T., Radeff-Huang J.M., Stern P.H. G alpha12/G alpha13 subunits of heterotrimeric G proteins mediate parathyroid hormone activation of phospholipase D in UMR-106 osteoblastic cells. Endocrinology. 2005;146:2171–2175. doi: 10.1210/en.2004-1283. [DOI] [PubMed] [Google Scholar]
- 606.Radeff J.M., Singh A.T., Stern P.H. Role of protein kinase A, phospholipase C and phospholipase D in parathyroid hormone receptor regulation of protein kinase Calpha and interleukin-6 in UMR-106 osteoblastic cells. Cell. Signal. 2004;16:105–114. doi: 10.1016/s0898-6568(03)00131-1. [DOI] [PubMed] [Google Scholar]
- 607.Singh A.T., Frohman M.A., Stern P.H. Parathyroid hormone stimulates phosphatidylethanolamine hydrolysis by phospholipase D in osteoblastic cells. Lipids. 2005;40:1135–1140. doi: 10.1007/s11745-005-1477-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 608.Igarashi K., Hirafuji M., Adachi H., Shinoda H., Mitani H. Role of endogenous PGE2 in osteoblastic functions of a clonal osteoblast-like cell, MC3T3-E1. Prostaglandins Leukotrienes Essent. Fatty Acids. 1994;50:169–172. doi: 10.1016/0952-3278(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 609.Jilka R.L., Weinstein R.S., Bellido T., Roberson P., Parfitt A.M., Manolagas S.C. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Invest. 1999;104:439–446. doi: 10.1172/JCI6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Tashjian A.H., Levine L. Epidermal growth factor stimulates prostaglandin production and bone resorption in cultured mouse calvaria. Biochem. Biophys. Res. Commun. 1978;85:966–975. doi: 10.1016/0006-291x(78)90638-1. [DOI] [PubMed] [Google Scholar]
- 611.Carpio L.C., Dziak R. Activation of phospholipase D signaling pathway by epidermal growth factor in osteoblastic cells. J. Bone Miner. Res. 1998;13:1707–1713. doi: 10.1359/jbmr.1998.13.11.1707. [DOI] [PubMed] [Google Scholar]
- 612.Kim M.J., Choi M.U., Kim C.W. Activation of phospholipase D1 by surface roughness of titanium in MG63 osteoblast-like cell. Biomaterials. 2006;27:5502–5511. doi: 10.1016/j.biomaterials.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 613.Fang M., Olivares-Navarrete R., Wieland M., Cochran D.L., Boyan B.D., Schwartz Z. The role of phospholipase D in osteoblast response to titanium surface microstructure. J. Biomed. Mater. Res. A. 2010;93:897–909. doi: 10.1002/jbm.a.32596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614.Sumikawa K., Okochi T., Adachi K. Differences in phosphatidate hydrolytic activity of human alkaline phosphatase isozymes. Biochim. Biophys. Acta. 1990;1046:27–31. doi: 10.1016/0005-2760(90)90090-k. [DOI] [PubMed] [Google Scholar]
- 615.Al Husaini H., Wheatley-Price P., Clemons M., Shepherd F.A. Prevention and management of bone metastases in lung cancer: A review. J. Thorac. Oncol. 2009;4:251–259. doi: 10.1097/JTO.0b013e31819518fc. [DOI] [PubMed] [Google Scholar]
- 616.Hirsh V., Major P.P., Lipton A., Cook R.J., Langer C.J., Smith M.R., Brown J.E., Coleman R.E. Zoledronic acid and survival in patients with metastatic bone disease from lung cancer and elevated markers of osteoclast activity. J. Thorac. Oncol. 2008;3:228–236. doi: 10.1097/JTO.0b013e3181651c0e. [DOI] [PubMed] [Google Scholar]
- 617.Bendre M.S., Margulies A.G., Walser B., Akel N.S., Bhattacharrya S., Skinner R.A., Swain F., Ramani V., Mohammad K.S., Wessner L.L., et al. Tumor-derived interleukin-8 stimulates osteolysis independent of the receptor activator of nuclear factor-kappaB ligand pathway. Cancer Res. 2005;65:11001–11009. doi: 10.1158/0008-5472.CAN-05-2630. [DOI] [PubMed] [Google Scholar]
- 618.Martin D., Galisteo R., Gutkind J.S. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J. Biol. Chem. 2009;284:6038–6042. doi: 10.1074/jbc.C800207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Waugh D.J., Wilson C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 620.MacManus C.F., Pettigrew J., Seaton A., Wilson C., Maxwell P.J., Berlingeri S., Purcell C., McGurk M., Johnston P.G., Waugh D.J. Interleukin-8 signaling promotes translational regulation of cyclin D in androgen-independent prostate cancer cells. Mol. Cancer Res. 2007;5:737–748. doi: 10.1158/1541-7786.MCR-07-0032. [DOI] [PubMed] [Google Scholar]
- 621.Hsu Y.L., Hung J.Y., Ko Y.C., Hung C.H., Huang M.S., Kuo P.L. Phospholipase D signaling pathway is involved in lung cancer-derived IL-8 increased osteoclastogenesis. Carcinogenesis. 2010;31:587–596. doi: 10.1093/carcin/bgq030. [DOI] [PubMed] [Google Scholar]
- 622.Gravallese E.M., Manning C., Tsay A., Naito A., Pan C., Amento E., Goldring S.R. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis. Rheum. 2000;43:250–258. doi: 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 623.Kong Y.Y., Feige U., Sarosi I., Bolon B., Tafuri A., Morony S., Capparelli C., Li J., Elliott R., McCabe S., et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- 624.Ogata Y., Kukita A., Kukita T., Komine M., Miyahara A., Miyazaki S., Kohashi O. A novel role of IL-15 in the development of osteoclasts: Inability to replace its activity with IL-2. J. Immunol. 1999;162:2754–2760. [PubMed] [Google Scholar]
- 625.Miranda-Carús M.E., Benito-Miguel M., Balsa A., Cobo-Ibáñez T., Pérez de Ayala C., Pascual-Salcedo D., Martín-Mola E. Peripheral blood T lymphocytes from patients with early rheumatoid arthritis express RANKL and interleukin-15 on the cell surface and promote osteoclastogenesis in autologous monocytes. Arthritis. Rheum. 2006;54:1151–1164. doi: 10.1002/art.21731. [DOI] [PubMed] [Google Scholar]
- 626.Elvers M., Stegner D., Hagedorn I., Kleinschnitz C., Braun A., Kuijpers M.E., Boesl M., Chen Q., Heemskerk J.W., Stoll G., et al. Impaired alpha(IIb)beta(3) integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Sci. Signal. 2010;3:ra1. doi: 10.1126/scisignal.2000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Norton L.J., Zhang Q., Saqib K.M., Schrewe H., Macura K., Anderson K.E., Lindsley C.W., Brown H.A., Rudge S.A., Wakelam M.J. PLD1 rather than PLD2 regulates phorbol-ester-, adhesion-dependent and Fc gamma-receptor-stimulated ROS production in neutrophils. J. Cell. Sci. 2011;124:1973–1983. doi: 10.1242/jcs.082008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Balcerzak M., Pikula S., Buchet R. Phosphorylation-dependent phospholipase D activity of matrix vesicles. FEBS Lett. 2006;580:5676–5680. doi: 10.1016/j.febslet.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 629.Tsujioka H., Takami N., Misumi Y., Ikehara Y. Intracellular cleavage of glycosylphosphatidylinositol by phospholipase D induces activation of protein kinase Calpha. Biochem. J. 1999;342:449–455. [PMC free article] [PubMed] [Google Scholar]
- 630.LeBoeuf R.C., Caldwell M., Guo Y., Metz C., Davitz M.A., Olson L.K., Deeg M.A. Mouse glycosylphosphatidylinositol-specific phospholipase D (Gpld1) characterization. Mamm. Genome. 1998;9:710–714. doi: 10.1007/s003359900851. [DOI] [PubMed] [Google Scholar]
- 631.Scallon B.J., Fung W.J., Tsang T.C., Li S., Kado-Fong H., Huang K.S., Kochan J.P. Primary structure and functional activity of a phosphatidylinositol-glycan-specific phospholipase D. Science. 1991;252:446–448. doi: 10.1126/science.2017684. [DOI] [PubMed] [Google Scholar]
- 632.Tang J.H., Gu S.L., Zhang X.J. Preliminary study of the gene structure of human glycosylphosphatidylinositol specific phospholipase D. Hunan Yi Ke Da Xue Xue Bao. 2001;26:95–97. [PubMed] [Google Scholar]
- 633.Brunner G., Metz C.N., Nguyen H., Gabrilove J., Patel S.R., Davitz M.A., Rifkin D.B., Wilson E.L. An endogenous glycosylphosphatidylinositol-specific phospholipase D releases basic fibroblast growth factor-heparan sulfate proteoglycan complexes from human bone marrow cultures. Blood. 1994;83:2115–2125. [PubMed] [Google Scholar]
- 634.Gregory P., Kraemer E., Zürcher G., Gentinetta R., Rohrbach V., Brodbeck U., Andres A.C., Ziemiecki A., Bütikofer P. GPI-specific phospholipase D (GPI-PLD) is expressed during mouse development and is localized to the extracellular matrix of the developing mouse skeleton. Bone. 2005;37:139–147. doi: 10.1016/j.bone.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 635.Larson D.M., Kennedy M.A., Bowen R.F., Verchere C.B., Deeg M.A. Glycosylphosphatidylinositol-specific phospholipase D immunoreactivity is present in islet amyloid in type 2 diabetes. J. Pathol. 2004;203:961–967. doi: 10.1002/path.1600. [DOI] [PubMed] [Google Scholar]
- 636.Rhode H., Lopatta E., Schulze M., Pascual C., Schulze H.P., Schubert K., Schubert H., Reinhart K., Horn A. Glycosylphosphatidylinositol-specific phospholipase D in blood serum: Is the liver the only source of the enzyme? Clin. Chim. Acta. 1999;281:127–145. doi: 10.1016/s0009-8981(98)00218-6. [DOI] [PubMed] [Google Scholar]
- 637.Xiaotong H., Hannocks M.J., Hampson I., Brunner G. GPI-specific phospholipase D mRNA expression in tumor cells of different malignancy. Clin. Exp. Metastasis. 2002;19:291–299. doi: 10.1023/a:1015545407700. [DOI] [PubMed] [Google Scholar]
- 638.Knudson C.B., Knudson W. Cartilage proteoglycans. Semin. Cell Dev. Biol. 2001;12:69–78. doi: 10.1006/scdb.2000.0243. [DOI] [PubMed] [Google Scholar]
- 639.Paine-Saunders S., Viviano B.L., Zupicich J., Skarnes W.C., Saunders S. Glypican-3 controls cellular responses to Bmp4 in limb patterning and skeletal development. Dev. Biol. 2000;225:179–187. doi: 10.1006/dbio.2000.9831. [DOI] [PubMed] [Google Scholar]
- 640.Pilia G., Hughes-Benzie R.M., MacKenzie A., Baybayan P., Chen E.Y., Huber R., Neri G., Cao A., Forabosco A., Schlessinger D. Mutations in GPC3, a glypican gene, cause the Simpson-Golabi-Behmel overgrowth syndrome. Nat. Genet. 1996;12:241–247. doi: 10.1038/ng0396-241. [DOI] [PubMed] [Google Scholar]
- 641.Schwab W., Gavlik J.M., Beichler T., Funk R.H., Albrecht S., Magdolen V., Luther T., Kasper M., Shakibaei M. Expression of the urokinase-type plasminogen activator receptor in human articular chondrocytes: Association with caveolin and beta 1-integrin. Histochem. Cell. Biol. 2001;115:317–323. doi: 10.1007/s004180100255. [DOI] [PubMed] [Google Scholar]
- 642.Daci E., Everts V., Torrekens S., Van Herck E., Tigchelaar-Gutterr W., Bouillon R., Carmeliet G. Increased bone formation in mice lacking plasminogen activators. J. Bone Miner. Res. 2003;18:1167–1176. doi: 10.1359/jbmr.2003.18.7.1167. [DOI] [PubMed] [Google Scholar]
- 643.Xing Y., Gu Y., Xu L.C., Siedlecki C.A., Donahue H.J., You J. Effects of membrane cholesterol depletion and GPI-anchored protein reduction on osteoblastic mechanotransduction. J. Cell. Physiol. 2011;226:2350–2359. doi: 10.1002/jcp.22579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Goding J.W., Terkeltaub R., Maurice M., Deterre P., Sali A., Belli S.I. Ecto-phosphodiesterase/pyrophosphatase of lymphocytes and non-lymphoid cells: Structure and function of the PC-1 family. Immunol. Rev. 1998;161:11–26. doi: 10.1111/j.1600-065x.1998.tb01568.x. [DOI] [PubMed] [Google Scholar]
- 645.Van Meeteren L.A., Ruurs P., Christodoulou E., Goding J.W., Takakusa H., Kikuchi K., Perrakis A., Nagano T., Moolenaar W.H. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J. Biol. Chem. 2005;280:21155–21161. doi: 10.1074/jbc.M413183200. [DOI] [PubMed] [Google Scholar]
- 646.Van Meeteren L.A., Moolenaar W.H. Regulation and biological activities of the autotaxin-LPA axis. Prog. Lipid Res. 2007;46:145–160. doi: 10.1016/j.plipres.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 647.Tania M., Khan M.A., Zhang H., Li J., Song Y. Autotaxin: A protein with two faces. Biochem. Biophys. Res. Commun. 2010;401:493–497. doi: 10.1016/j.bbrc.2010.09.114. [DOI] [PubMed] [Google Scholar]
- 648.Giganti A., Rodriguez M., Fould B., Moulharat N., Cogé F., Chomarat P., Galizzi J.P., Valet P., Saulnier-Blache J.S., Boutin J.A., et al. Murine and human autotaxin alpha, beta, and gamma isoforms: Gene organization, tissue distribution, and biochemical characterization. J. Biol. Chem. 2008;283:7776–7789. doi: 10.1074/jbc.M708705200. [DOI] [PubMed] [Google Scholar]
- 649.Fukushima N. LPA in neural cell development. J. Cell. Biochem. 2004;92:993–1003. doi: 10.1002/jcb.20093. [DOI] [PubMed] [Google Scholar]
- 650.Hu P.F., Chen Y., Cai P.F., Jiang L.F., Wu L.D. Sphingosine-1-phosphate: A potential therapeutic target for rheumatoid arthritis. Mol. Biol. Rep. 2011;38:4225–4230. doi: 10.1007/s11033-010-0545-9. [DOI] [PubMed] [Google Scholar]
- 651.Clair T., Aoki J., Koh E., Bandle R.W., Nam S.W., Ptaszynska M.M., Mills G.B., Schiffmann E., Liotta L.A., Stracke M.L. Autotaxin hydrolyzes sphingosylphosphorylcholine to produce the regulator of migration, sphingosine-1-phosphate. Cancer Res. 2003;63:5446–5453. [PubMed] [Google Scholar]
- 652.Bourgoin S.G., Zhao C. Autotaxin and lysophospholipids in rheumatoid arthritis. Curr. Opin. Investig. Drugs. 2010;11:515–526. [PubMed] [Google Scholar]
- 653.Lai W.Q., Irwan A.W., Goh H.H., Howe H.S., Yu D.T., Valle-Oñate R., McInnes I.B., Melendez A.J., Leung B.P. Anti-inflammatory effects of sphingosine kinase modulation in inflammatory arthritis. J. Immunol. 2008;181:8010–8017. doi: 10.4049/jimmunol.181.11.8010. [DOI] [PubMed] [Google Scholar]
- 654.Kitano M., Hla T., Sekiguchi M., Kawahito Y., Yoshimura R., Miyazawa K., Iwasaki T., Sano H., Saba J.D., Tam Y.Y. Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium: Regulation of synovial proliferation and inflammatory gene expression. Arthritis. Rheum. 2006;54:742–753. doi: 10.1002/art.21668. [DOI] [PubMed] [Google Scholar]
- 655.Deutschman D.H., Carstens J.S., Klepper R.L., Smith W.S., Page M.T., Young T.R., Gleason L.A., Nakajima N., Sabbadini R.A. Predicting obstructive coronary artery disease with serum sphingosine-1-phosphate. Am. Heart J. 2003;146:62–68. doi: 10.1016/S0002-8703(03)00118-2. [DOI] [PubMed] [Google Scholar]
- 656.Pyne S., Pyne N. Sphingosine 1-phosphate signalling via the endothelial differentiation gene family of G-protein-coupled receptors. Pharmacol. Ther. 2000;88:115–131. doi: 10.1016/s0163-7258(00)00084-x. [DOI] [PubMed] [Google Scholar]
- 657.Takeshita H., Kitano M., Iwasaki T., Kitano S., Tsunemi S., Sato C., Sekiguchi M., Azuma N., Miyazawa K., Hla T., et al. Sphingosine 1-phosphate (S1P)/S1P receptor 1 signaling regulates receptor activator of NF-κB ligand (RANKL) expression in rheumatoid arthritis. Biochem. Biophys. Res. Commun. 2012;419:154–159. doi: 10.1016/j.bbrc.2012.01.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 658.Bächner D., Ahrens M., Schröder D., Hoffmann A., Lauber J., Betat N., Steinert P., Flohé L., Gross G. Bmp-2 downstream targets in mesenchymal development identified by subtractive cloning from recombinant mesenchymal progenitors (C3H10T1/2) Dev. Dyn. 1998;213:398–411. doi: 10.1002/(SICI)1097-0177(199812)213:4<398::AID-AJA5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 659.Bächner D., Ahrens M., Betat N., Schröder D., Gross G. Developmental expression analysis of murine autotaxin (ATX) Mech. Dev. 1999;84:121–125. doi: 10.1016/s0925-4773(99)00048-9. [DOI] [PubMed] [Google Scholar]
- 660.Garciadiego-Cázares D., Rosales C., Katoh M., Chimal-Monroy J. Coordination of chondrocyte differentiation and joint formation by alpha5beta1 integrin in the developing appendicular skeleton. Development. 2004;131:4735–4742. doi: 10.1242/dev.01345. [DOI] [PubMed] [Google Scholar]
- 661.Weilbaecher K.N., Guise T.A., McCauley L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer. 2011;11:411–425. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 662.Peyruchaud O., Leblanc R., David M. Pleiotropic activity of lysophosphatidic acid in bone metastasis. Biochim. Biophys. Acta. 2013;1831:99–104. doi: 10.1016/j.bbalip.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 663.Karagiosis S.A., Chrisler W.B., Bollinger N., Karin N.J. Lysophosphatidic acid-induced ERK activation and chemotaxis in MC3T3-E1 preosteoblasts are independent of EGF receptor transactivation. J. Cell. Physiol. 2009;219:716–723. doi: 10.1002/jcp.21720. [DOI] [PubMed] [Google Scholar]
- 664.David M., Wannecq E., Descotes F., Jansen S., Deux B., Ribeiro J., Serre C.M., Grès S., Bendriss-Vermare N., Bollen M., et al. Cancer cell expression of autotaxin controls bone metastasis formation in mouse through lysophosphatidic acid-dependent activation of osteoclasts. PLoS One. 2010;5:e9741. doi: 10.1371/journal.pone.0009741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665.Murakami-Murofushi K., Uchiyama A., Fujiwara Y., Kobayashi T., Kobayashi S., Mukai M., Murofushi H., Tigyi G. Biological functions of a novel lipid mediator, cyclic phosphatidic acid. Biochim. Biophys. Acta. 2002;1582:1–7. doi: 10.1016/s1388-1981(02)00131-2. [DOI] [PubMed] [Google Scholar]
- 666.Mansell J.P., Nowghani M., Pabbruwe M., Paterson I.C., Smith A.J., Blom A.W. Lysophosphatidic acid and calcitriol co-operate to promote human osteoblastogenesis: Requirement of albumin-bound LPA. Prostaglandins Other Lipid Mediat. 2011;95:45–52. doi: 10.1016/j.prostaglandins.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 667.Panchatcharam M., Miriyala S., Yang F., Rojas M., End C., Vallant C., Dong A., Lynch K., Chun J., Morris A.J., et al. Lysophosphatidic acid receptors 1 and 2 play roles in regulation of vascular injury responses but not blood pressure. Circ. Res. 2008;103:662–670. doi: 10.1161/CIRCRESAHA.108.180778. [DOI] [PMC free article] [PubMed] [Google Scholar]