

Abstract
Multiple sclerosis (MS) is an inflammatory, demyelinating disorder of the central nervous system that develops in genetically susceptible individuals, probably triggered by common environmental factors. Human leukocyte antigen (HLA) loci were early shown to confer the strongest genetic associations in MS. Now, more than 50 non-HLA MS susceptibility loci are identified, of which the majority are located in immune-regulatory genes. Single nucleotide polymorphisms (SNPs) in the C-type lectin-like domain family 16A (CLEC16A) gene were among the first non-HLA genetic variants that were confirmed to be associated with MS. Fine-mapping has indicated a primary association in MS and also other autoimmune diseases to intronic CLEC16A SNPs. Here, we review the identification of MS susceptibility variants in the CLEC16A gene region, functional studies of the CLEC16A molecule and the recent progress in understanding the implications thereof for MS development. This may serve as an example of the importance for further molecular investigation of the loci identified in genetic studies, with the aim to translate this knowledge into the clinic.
Keywords: autoimmunity, multiple sclerosis, CLEC16A, SOCS1, DEXI, CIITA
1. Introduction
Multiple sclerosis (MS) is an inflammatory, demyelinating disorder of the central nervous system (CNS) that causes relapsing-remitting attacks (RR-MS) or a progressive disease (primary or secondary progressive MS) leading to different degrees of physical and cognitive disability. An increasing number of immune-modulatory medications are used in RR-MS with variable effects on relapse rate and progression of MS lesions, evaluated by magnetic resonance imaging (MRI) [1]. There is a global, latitudinal gradient of MS prevalence, with lower disease prevalence near the equator and an increasing prevalence in the Northern and Southern hemisphere, with 0.5–1.5 cases per 1000 inhabitants, compared to 0.2 cases per 1000 inhabitants in Latin America, Middle East and Africa [2,3]. MS typically appears in young adults and affects females more than twice as often as males [4]. The leading hypothesis is that MS is caused by a complex interaction between multiple genes and environmental factors, which leads to CNS inflammation, causing demyelination and axonal degeneration. To date, the best supported environmental risk factors in MS are a low serum level of vitamin D, Epstein-Barr virus (EBV) infection and smoking [5]. Genetic studies of MS all point to MS as a complex, polygenic disease, where several common genetic variants each exert a relatively small effect [6,7]. Gene identification is the first step in order to identify the biological pathways important for the disease. Characterization of these genes is crucial to facilitate the understanding of the molecular basis for MS pathogenesis.
2. Identification of MS Risk Loci
Already in 1972, the human leukocyte antigen (HLA) gene region was identified as a susceptibility locus for MS [8]. However, due to strong linkage disequilibrium (LD), it was difficult to clearly define the primary association. It is now well established that the HLA class II-DRB1 locus constitutes the primary genetic association, with the HLA-DRB1*15:01 allele as the major genetic risk factor for MS in most populations (odds ratio (OR) = 3.1) [9,10]. Furthermore, it has been convincingly shown that also HLA class I alleles have an effect independent of HLA-DRB1*15:01 [11,12], most strongly the HLA-A*02:01 allele, which is negatively associated with the risk for MS development (OR = 0.73) [10].
Phenotype-genotype studies of associated HLA alleles in MS have shown somewhat conflicting results, but a significant correlation was identified between age at onset of MS and the HLA-DRB1*15:01 allele [10]. Our recent analysis of the MS sub-phenotype, characterized by the presence of oligoclonal bands (OCB) in the cerebrospinal fluid, has shown association of HLA-DRB*04:04 with OCB negative MS and with HLA-DRB*15:01 more strongly in OCB-positive than OCB-negative MS [13].
In spite of intensive genetic research using both linkage studies and candidate gene approaches, it took close to 40 years before non-HLA loci were convincingly identified as associated with MS. In 2007, a moderately powered genome-wide association study (GWAS) identified the first non-HLA MS susceptibility loci, i.e., the interleukin-7 receptor α (IL7Rα) and IL2Rα genes [14]. Simultaneously, the IL7Rα gene was confirmed as an MS susceptibility gene in other cohorts [15,16]. In the following years, a series of GWAS- and meta-analyses were carried out, adding more regions to the list of confirmed MS associated loci, which in the spring of 2011 included 26 non-HLA genetic regions [6,17–26]. In August 2011, the largest GWAS in MS, including 9 772 MS cases and 17 376 controls, was published by the International Multiple Sclerosis Genetics Consortium (IMSGC) and the Wellcome Trust Case Control Consortium 2 (WTCCC2). This GWAS confirmed association to 23 of the previously suggested MS risk loci and identified an additional 29 novel susceptibility loci. Interestingly, the majority of these 52 MS-associated loci were located inside or close to genes of importance for the immune system, especially T-cell immunity [10]. These findings supported the notion that immunological mechanisms are crucial for MS development. When summarizing results from MS GWAS studies, as well as recent meta-analyses, close to 60 MS susceptibility genes have been identified by the end of 2012 [10,23,27,28]. Most of these variants are common and contribute only modestly to MS susceptibility (OR = 1.1–1.3) [7,10]. The list of MS susceptibility genes is expected to grow even further in the near future, when ongoing fine-mapping and replication studies will be completed.
A single nucleotide polymorphism (SNP) in the C-type lectin-like domain family 16A (CLEC16A) gene was among the genetic variants that showed suggestive association (rs6498169, p = 3.83 × 10−6) with MS in the first MS GWAS published in 2007 [14]. CLEC16A has thereafter been convincingly replicated as an MS susceptibility gene in a series of studies [29–37], including the large MS GWAS published in 2011 (rs7200786, p = 6.3 × 10−14, OR = 1.15) [10]. Of note, one-third of the identified MS loci have been reported to be associated with at least one additional autoimmune disease [7,10], and CLEC16A SNPs have shown association with several other autoimmune diseases (see Table 1). CLEC16A is preferentially expressed in immune cells [38], which also indicates that the CLEC16A molecule might play an important role in immune regulation. However, the function of CLEC16A is still not well defined. Interestingly, the CLEC16A gene is located at chromosome 16p13, in a region including several other immune-regulatory genes. Intensive efforts are ongoing, aiming to identify the implication of the identified CLEC16A associations and the functional impact on the encoded protein, as well as on its potential regulatory role on neighboring genes. In this paper, we review the current knowledge about this gene and the encoded molecule, which will serve as an example on how the field is moving from identification of MS susceptibility genes to functional molecular studies.
Table 1.
CLEC16A SNPs associated with autoimmune diseases. The table displays published CLEC16A SNPs that have shown association with indicated autoimmune diseases in different cohorts. Crohn’s = Crohn’s disease; Addisons = Addisons disease; A. Areata = Alopecia Areata; Aus = Australia; Bel = Belgium; Can = Canada; Eur = Europe; Fin = Finland; Nor = Norway; Rom = Romania; Swe = Sweden.
| Disease | SNP | Intron | Cases/Controls/Trios | Subject cohort | Reference |
|---|---|---|---|---|---|
| Multiple Sclerosis | rs6498168 | 22 | 2322/5418/1540 | UK, USA | [14] |
| rs6498169 | 22 | 1146/1309 | Australia | [30] | |
| rs725613 | 19 | 1498/1706 | Sardinia | [33] | |
| rs12708716 | 19 | 5737/10296/2369 | Aus., Bel., Nor., Swe., UK, USA | [31] | |
| rs6498169 | 19 | ||||
| rs6498146 | 10 | ||||
| rs741177 | 18 | 1146/1309 | Europe | [34] | |
| rs876476 | 18 | ||||
| rs11863016 | 22 | ||||
| rs9937607 | 22 | ||||
| rs11865121 | 19 | 2624/7220 | UK, USA | [23] | |
| rs6498169 | 22 | 435/550 | Spain | [35] | |
| rs6498169 | 22 | 211/182 (+521 multiplex controls) | UK | [36] | |
| rs6498169 | 22 | 1853/2128 | Holland, Can. | [29] | |
| rs12708716 | 19 | 918/656 | USA (afro American) | [37] | |
| rs6498169 | 22 | ||||
| rs2080272 | 19 | ||||
| rs2041670 | 19 | 603/825 | Europe | [47] | |
| rs998592 | 19 | ||||
| rs12708716 | 19 | 197/197 | India | [48] | |
| rs12708716 | 19 | ||||
| rs7206912 | 22 | 3102/5047/1113 | Nor., UK | [32] | |
| rs6498169 | 22 | ||||
| rs7184083 | 22 | 1343/1379 | UK, US | [49] | |
|
| |||||
| Type 1 Diabetes | rs12708716 | 19 | 2000/3000 | UK | [50] |
| rs725613 | 19 | ||||
| rs2903692 | 22 | 1896/1146/873 | Eur., USA, Aus., Can. | [40] | |
| rs17673553 | 22 | ||||
| rs12708716 | 19 | 4000/5000/2997 | UK, USA, Fin., Nor., Rom. | [26] | |
| rs725613 | 19 | 1037/1706 | Italy (Sardinia) | [33] | |
| rs2903692 | 22 | 735/621 | Japan | [51] | |
| rs725613 | 19 | 205/422 | China (Han) | [52] | |
| rs6498169 | 22 | 316/550 | Spain | [35] | |
| rs12921922 | 4 | 131/121 | China | [53] | |
| rs12931878 | 1 | ||||
| rs12708716 | 19 | 1212/2513 | Germany | [54] | |
| rs2903692 | 22 | 1743/790 | Japan | [55] | |
|
| |||||
| Rheumatoid arthritis | rs6498169 | 22 | 600/550 | Spain | [35] |
| rs6498169 | 22 | 1318/2149 | Norway | [44] | |
| rs6498169 | 22 | 600/550 | Spain | [35] | |
|
| |||||
| Primary biliary cirrhosis | rs58102322 | 18 | 1450/2967 | Europe | [43] |
| rs12924129 | 19 | ||||
| rs12924729 | 19 | 2460/7677 | UK | [56] | |
|
| |||||
| Crohn’s | rs2903692 | 22 | 1264/890 | Spain | [42] |
|
| |||||
| Addisons | rs12917716 | 19 | 542/1220 | Norway, UK | [41] |
|
| |||||
| A. Areata | rs998592 | 19 | 1702/1723 | Europe | [45] |
3. CLEC16A—An Autoimmune Candidate Gene
The CLEC16A gene (previously known as KIAA0350; also designated Gop-1) is 238 kb, consists of 24 exons (http://www.ncbi.nlm.nih.gov/nuccore/NM_015226.2) [39] and is located on chromosome 16p13 (Figure 1A). In addition to MS, CLEC16A is associated with susceptibility to several autoimmune diseases (see Figure 1B and Table 1), including type 1 diabetes (T1D) [40], primary adrenal insufficiency (Addison’s disease) [41], Crohn’s disease [42], primary biliary cirrhosis [43], juvenile idiopathic arthritis [44], rheumatoid arthritis [35,44] and alopecia areata [45]. Many of the reported SNPs are in strong LD (Figure 1C). Therefore, it has been difficult to identify the primary genetic association in this gene region.
Figure 1.

Schematic drawing of (a) the chromosome 16p13 genetic region comprising CIITA, DEXI, CLEC16A and SOCS1 (Genome Reference Consortium Human Build 37 (GRCh37), chromosome 16: 10.971.055–11.349.335) and (b) the 238 kb CLEC16A gene (GRCh37, chromosome 16: 11.038.345–11.276.046), where the autoimmunity-associated single nucleotide polymorphisms (SNPs) and their localization are depicted; (c) Linkage disequilibrium plot (Haploview v. 4.2, CEU-population [46]) for the CLEC16A region (chromosome 16: 11.042.194 (CLEC16A intron 1)–11.249.329 (CLEC16A intron 22), GRCh37) harboring the autoimmune-associated SNPs indicated in (b).
Common for the disease-associated CLEC16A SNPs is that they are located in intronic regions of CLEC16A, the majority in intron 19 and intron 22 of the gene (Figure 1B and Table 1). One rare variant, rs2241100 in exon 23 of the CLEC16A gene, results in a non-synonymous (ns) amino acid change at position 906 (G906E). To examine whether the T1D association could be due to the predisposing effect from this rare ns-CLEC16A SNP, Hakonarson and colleagues sequenced exon 23 in 20 T1D patients that were homozygous for the T1D rs2286973 risk SNP in CLEC16A. However, all individuals were homozygous for the common ns-rs224100 SNP [40]. Thus, no predisposition effect from the rare exon 23 variant was detected.
To validate CLEC16A as an MS susceptibility gene and to fine-map the association, 44 CLEC16A SNPs were genotyped in a collection of Australian samples. This study pointed towards rs6498169 in intron 22 as the most significantly associated CLEC16A SNP [34]. IMSGC reported that the CLEC16A rs12708716 SNP (in intron 19 of CLEC16A and in LD with rs6498169; D′ = 1, R2 = 0.259) was associated with MS at a genome-wide significance level [31]. We conducted a fine-mapping of the CLEC16A region in Norwegian samples, followed by replication of the top hits in two large Norwegian and British sample sets. Among the 57 SNPs tested, rs12708716 appeared to yield the superior association, followed by two other intronic CLEC16A SNPs; rs7206912 and rs6498169 [32]. Importantly, rs12708716 is in LD with the rs7200786 SNP (D′ = 1, R2 = 0.61) (Haploview v. 4.2, CEU-population [46]) in intron 19 of CLEC16A that later was confirmed to be associated with MS at a genome-wide significance level in the large GWAS published in 2011 [10].
4. The CIITA-DEXI-CLEC16A-SOCS1 Gene Complex
The 16p13 chromosomal region where CLEC16A is situated has been the focus of several studies on MS, as well as other autoimmune diseases. Two immune-regulatory genes of potential interest for autoimmunity (see Tables 2 and 3), i.e., the major histocompatibility complex (MHC) class II transactivator (CIITA) gene and the suppressor of cytokine signaling 1 (SOCS1) gene, are located close to CLEC16A (Figure 1A). CIITA encodes an essential transcription factor regulating gene expression of HLA class II molecules [57,58]. Due to the strong association of HLA-DRB*15:01 in MS, CIITA has been suggested as an MS candidate gene. Indeed, some reports have shown association between a CIITA promoter variant (rs3087456) and MS [59,60]. However, a multi-stage investigation did not confirm this association, but reported evidence for interaction between rs4774 (+1614 G/C missense mutation) and HLA-DRB1*15:01 in MS [61].
Table 2.
CIITA SNPs associated with autoimmune diseases. The table displays published CIITA SNPs that have shown association with indicated autoimmune diseases in different cohorts.
| Disease | SNP | SNP localization | Cases/Controls/Trios | Subject cohort | References |
|---|---|---|---|---|---|
| Multiple sclerosis | rs4774 1 | Exon 11 | 1320/1363 | Europe | [61] |
| rs4774 | Exon 11 | 1343/1379 | UK, US | [49] | |
| rs3087456 | Promoter | 548/528 | Scandinavia | [59,60] | |
|
| |||||
| Type 1 Diabetes | rs11074932 | Promoter | 5 cohorts | Sweden | [62] |
| rs3087456 | Promoter | ||||
|
| |||||
| Rheumatoid arthritis | rs3087456 | Promoter | 1288/709 | Scandinavia | [60] |
| rs3087456/ rs4774 2 | Promoter/ exon 11 | 580/454 | Spain | [35] | |
| rs8048002 | Intron 3 | 3551/4827 | Norway, Sweden | [63] | |
| rs3087456 | Promoter | ||||
|
| |||||
| SLE | rs4774 | Exon 11 | 1342/4301 | Europe | [64] |
|
| |||||
| Addisons disease | rs8048002 | Intron 3 | 542/1220 | Norway, UK | [41] |
| rs3087465 | Promoter | 128/406 | Italy | [65] | |
Significant in the presence of HLA-DRB1*15:01.
rs3087456G/rs4774C haplotype is associated with indicated disease.
SLE = systemic lupus erythematosus.
Table 3.
SOCS1 SNPs associated with autoimmune diseases. The table displays published SOCS1 SNPs that have shown association with indicated autoimmune diseases in different cohorts.
SOCS1 is a suppressor of cytokine signaling that is important for immune cell homeostasis and regulation of inflammation [67]. Gene variants in the 5′ untranslated region (UTR) of SOCS1 (rs243324 and rs441349) have been identified in cytokine pathway gene screenings as MS susceptibility variants [66,68]. Furthermore, association to MS has been found for a SNP (rs243315) in the 5′ UTR of the protamine 1 (PRM1) gene [69], which is located downstream of this genomic region. However, this signal might be explained by strong LD between rs243315 and rs441349 within the 5′ UTR of the SOCS1 gene (Haploview 4.2 [46]). Further evidence for involvement of SOCS1 in MS comes from our recent study, showing that SOCS1 expression is significantly lower in thymic tissue samples collected from children undergoing cardiac surgery, carrying at least one CLEC16A risk allele (at rs12708716, rs6498169 or rs7206912) compared to non-carriers of the risk allele [70]. Additionally, the dexamethasone-induced gene (DEXI), located between the CLEC16A and CIITA genes, has recently been suggested to be a novel autoimmune regulatory gene [70,71]. An expression quantitative trait locus for the DEXI gene was identified within intron 19 of CLEC16A in monocytes and in lymphoblastoid cell lines [71,72]. Additionally, DEXI expression was significantly lower in the above mentioned thymic tissue samples from individuals carrying the CLEC16A rs6498169 risk allele (in intron 22) compared to non-carriers of the risk allele [70]. The function of the protein encoded by DEXI is unknown; however, its transcript is induced by dexamethasone [73], which is a glucocorticoid analogue and an immunosuppressive drug. Of note, when SOCS1 and DEXI expression was measured in whole blood samples from healthy controls genotyped for the same CLEC16A SNPs, we were not able to detect any correlation between gene expression and CLEC16A genotypes. This lack of correlation could be due to cell-specific, genetically determined variations that are overshadowed by the variation in the proportions of the various cell types within the blood samples. The discrepancies can also be caused by individual differences, as the whole-blood and thymic samples are harvested from different donor groups of different age and health states [70].
To clarify the role of the different MS-associated SNPs within the 16p13 chromosomal region harboring the CIITA-DEXI-CLEC16A-SOCS1 gene complex, Zuvich and colleagues genotyped 149 SNPs in a combined American and British sample set and performed a detailed LD pattern and logistic regression analysis. Their data indicate that this region likely contains three independent MS disease loci; however, the CLEC16A rs7184083, which is in LD with rs12708716 (D′ = 1.00, r2 = 0.31), displayed the most significant p-value in this region [49]. Altogether, these data highlight the importance for further studies of this genetic region in relation to MS pathogenesis. Such efforts are already ongoing; for instance, in the “MS Immunochip project”, where IMSGC is genotyping samples from a large number of MS cases and controls using the “Immunochip”, a chip that was designed to replicate and fine-map disease risk loci associated with several autoimmune diseases [74–76]. Risk loci, among them the CIITA-DEXI-CLEC16A-SOCS1 region (see Tables 1–3), are shared among autoimmune diseases [6]. With the established roles of SOCS1 and CIITA in inflammation and autoimmunity [77,78] and the likely role of CLEC16A in immune regulation (see below), the refinement of the genetic association, as well as the molecular function, holds promises to elucidate the mechanisms behind autoimmune diseases.
5. CLEC16A Expression
Microarray expression data indicate that CLEC16A is extensively expressed in different immune cells, in certain parts of the brain, i.e., cerebellum, spinal cord and pineal gland, and in testis [38,79]. Reverse-transcriptase polymerase chain reaction analyses have also detected low levels of CLEC16A expression in ovary and small intestine [80]. Laser scanning microscopy of the rat brain showed CLEC16A protein expression in astrocytes and neurons, but not in microglia. CLEC16A expression was further increased in rat astrocytes upon intraspinal injection of lipopolysaccharide (LPS) [81].
The human CLEC16A gene has been suggested to encode at least three different splice variants [79], two long isoforms expressed from all 24 exons or from 21 exons and a short transcript encoded from the last four exons. The two long CLEC16A isoforms are expressed in whole blood and in thymic tissue samples [32]. Unpublished data from our laboratory further show expression of these two long isoforms in a wide range of primary immune cells, as well as in immune cell lines and in the HEK293T cell line (a human embryonic kidney cell line). The third isoform was not tested in these analyses. Normal genetic variation may contribute to disease susceptibility and severity by affecting splicing of mRNA [82]. Interestingly, the relative expression of the two long CLEC16A isoforms did correlate with the MS associated, intronic rs12708716 variant in human thymic tissue samples. However, when whole blood samples, which are heterogeneous in their cellular composition, were analyzed, no such correlation was found [32]. This indicates that the relative expression of CLEC16A isoforms could be cell type specific.
The fact that non-coding, disease associated CLEC16A SNPs might affect CLEC16A expression in a cell-type specific manner has also been suggested for the non-coding rs2903692 (in strong LD (r2 = 0.88) with rs12708716), which shows genome-wide significant association with T1D. Investigation of a synonymous exon 19 SNP (rs2286973, a marker for the intronic T1D rs2903692 risk allele) did not reveal genotype dependent differences in CLEC16A expression in ten lymphoblastoid cell lines. However, when CLEC16A expression was analyzed in four different natural killer cell lines, a trend towards higher CLEC16A expression was observed in the cell line that was homozygous for the A-allele in rs2903692 [40]. When we analyzed the expression of total CLEC16A (including the two long isoforms) in relation with CLEC16A genotype (rs12708716, rs6498169 and rs7206912), we did not find any significant correlation, neither in whole blood, nor in thymic tissue samples [32,70]. However, we did observe a trend towards lower CLEC16A expression in the presence of an MS associated risk SNP at either rs6498169 or rs7206912 (Figure 2) [70]. Lack of power could explain the absence of a significant correlation; therefore, CLEC16A expression analyses in increased numbers of samples are needed.
Figure 2.
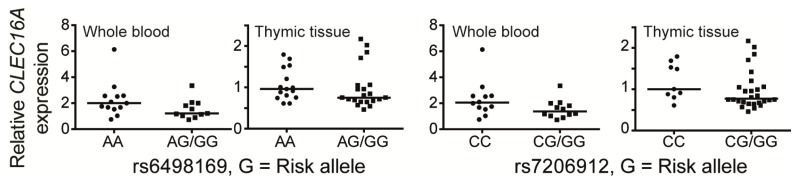
The graphs show relative expression of total CLEC16A (including the two long isoforms) normalized to the TATA-binding protein (TBP) gene in thymic tissue samples from 37 Norwegian children under the age of 13 undergoing corrective cardiac surgery and whole blood samples from 24 healthy, normal controls genotyped for the indicated SNPs in CLEC16A intron 22, as described [70]. Individuals carrying the risk allele were compared with individuals homozygous for the protective allele. Left graphs: rs6498169 (risk allele G): GG/AG: n = 21 (thymus), n = 11 (whole blood), AA: n = 15 (thymus), n = 13 (whole blood); right graphs: rs7206912 (risk allele G): GG/CG: n = 28 (thymus), n = 12 (whole blood), CC: n = 9 (thymus), n = 12 (whole blood). Correlation between gene expression levels and genotypes were assessed by Mann-Whitney U-test (GraphPad Prism 5, GraphPad Software, Inc., San Diego, CA, USA) and were found to be non-significant. Horizontal lines indicate the median values within the groups.
Of note, when sorting the samples according to the rs12708716 genotype (in CLEC16A intron 19), we did not reveal any sign of genotype-dependent CLEC16A expression in thymic samples [70]. However, no such comparison could be analyzed in whole blood, as only one whole blood sample was homozygous for the protective rs12708716 G-allele. In conclusion, CLEC16A expression analysis in purified immune-cell subsets will be important to evaluate a potential cell specific regulation of CLEC16A.
6. The Structural Domains of the CLEC16A Molecule
The full-length CLEC16A gene encodes a protein of 1053 amino acids (Q2KHT3-1, see Figure 3A), whose name was designated C-type lectin-like domain family 16A (CLEC16A) due to the presence of a C-type lectin-like domain (CTLD). Interestingly, other members of the C-type lectin family, such as DCIR (for dendritic cell immunoreceptor [83]) have been linked to autoimmunity in rodent models [84]. The most common CTLD function in vertebrates, and the ancestral mode of action, is Ca2+-dependent carbohydrate binding. Consequently, these proteins are involved in diverse processes, such as cell-cell adhesion, plasma glycoprotein turnover, as well as innate pathogen recognition [85]. The short nature of the CLEC16A CTLD (only 23 amino acids [86]), makes it unlikely that the CLEC16A protein region harboring this CTLD can fold properly into the compact 110–130 amino-acid domain of a carbohydrate recognition element [85,87,88]. However, many CTLDs have evolved to specifically recognize other ligands, such as proteins, lipids, as well as inorganic ligands [87]. Whether CLEC16A can interact with other potential ligands through its CTLD has not yet been studied.
Figure 3.

(a) Schematic representation of proposed CLEC16A protein variants (Q2KHT3-1, -2 and -3) highlighting from the N-terminal: a highly conserved uncharacterized protein domain, the FPL motif (amino acid (aa) 51–199 in Q2KHT3-1 and Q2KHT3-2,), a predicted transmembrane (TM) domain (aa 308–330 in Q2KHT3-1), an immunoreceptor-tyrosine based activation motif (ITAM, aa 483–499 in Q2KHT3-1) and the C-type lectin-like domain (CTLD, aa 566–588 in Q2KHT3-1). Black boxes indicate sequences where the amino acid identity differs from the canonical sequence; (b) Protein sequence alignment of the transmembrane region (TM, left), the immunoreceptor tyrosine-based activation motif (ITAM) sequence (middle) and the CTLD (right) of CLEC16A from Homo sapiens, Rattus norwegicus and Drosophila melanogaster. Boxed grey areas highlight fully conserved amino acids between the indicated species. “*” indicates identical amino acids in all three sequences; “:” indicates conserved substitution of amino acids; “.” indicates semi-conserved substitution of amino acids; and open space indicates no conservation of amino acid between the three species.
Upon ligand binding, C-type lectin receptors within the innate immunity commonly induce multiple signal transduction cascades through their own immunoreceptor tyrosine-based activation motifs (ITAMs) or through ITAMs of interacting proteins [89]. Interestingly, CLEC16A contains a potential ITAM ([90,91]; amino acids (aa) 483–499, see Figure 3A), thereby indicating a role for CLEC16A in immune-cell signaling. In addition to the CTLD and the ITAM, CLEC16A contains an N-terminally, highly conserved and uncharacterized domain, designated FPL (pfam09758) [92] (aa 51–199) and it is predicted to contain a trans-membrane region [93] (aa 308–330). As can be seen from the schematic drawing of CLEC16A in Figure 3A, these four domains are intact in the truncated CLEC16A isoform of 906 amino acids (Q2KHT3-2). The sequence of this isoform differs from the canonical sequence in that the following regions are missing; aa 201–202 (encoded by exon 6), aa 419–434 (encoded by parts of exon 11–12), as well as aa 925–1053 (encoded by the last two exons). Additionally, amino acids corresponding to aa 882–924 (exon 23) in Q2KHT3-1 differ in identity between the two isoforms [94]. Furthermore, a third transcript encoding a protein of only 138 amino acids, designated Q2KHT3-3, is listed at http://genome.ucsc.edu [79]. This splice variant is encoded from the four last exons of the CLE16A gene and lacks amino acids 1–913. In addition, the amino acids 914–935 differ from the canonical sequence [94,95], and this short isoform lacks the motifs that are identified in Q2KHT3-1 and -2.
7. The Function of CLEC16A
Up to this date, there is to our knowledge no published work on the functional aspects of human CLEC16A. However, two recent papers report studies of the CLEC16A orthologue, Ema, in Drosophila melanogaster[96,97]. Kim and colleagues have demonstrated that Ema is an endosomal protein with an important role in endosomal maturation and trafficking. They demonstrated that absence of Ema in the Drosophila garland cells induced accumulation of large endosomal intermediates, disrupted membrane trafficking and failure of proper lysosomal degradation [96]. In addition, Ema has been suggested to promote membrane traffic from the Golgi apparatus to the autophagosomes, since it is required for autophagosomal growth and efficient autophagy [97]. As there is 41% amino acid identity between human CLEC16A and Ema (clustalW [98]), these studies could pinpoint relevant functions also for the human counterpart. Interestingly, human CLEC16A can rescue the membrane-trafficking defects in Ema-mutant fly [96], and it restores normal autophagosomal growth when expressed in Ema-mutant fat body cells [97]. This suggests that Ema and CLEC16A are functionally conserved. The CLEC16A CTLD is not conserved from human to Drosophila, and Ema does not contain an ITAM consensus sequence (YXXI/LX6–8YXXI/L) (see Figure 3B) [90,91]. Of note, the FPL motif is well-conserved between human and Drosophila CLEC16A (clustalW [98]) and the putative transmembrane domain of Ema (Tmpred at http://www.ch.embnet.org) [93] lies in a highly conserved region of the protein (Figure 3B and data not shown), indicating that these two domains may be of importance for the conserved functions reported by Kim and colleagues [96,97].
The endolysosomal pathway regulates the levels and distribution of membrane receptors [99]. The study of Kim and colleagues [96] suggests that CLEC16A might confer susceptibility to autoimmune disorders through its role in endosomal regulation of immune receptor signaling pathways, leading to aberrant immune responses of autoreactive T-cells. Furthermore, its endosomal localization within the cell suggests that it might affect antigen presentation by the HLA-complex, which could affect T-cell immune responses in the periphery by autoreactive T-cells, as well as establishing self-tolerance during thymic T-cell development. Interestingly, CLEC16A shares localization to endosomal structures with molecules encoded by other MS susceptibility genes, such as signal transducer and activator of transcription 3 (STAT3) [100], tyrosine kinase 2 (Tyk2) [101] and ectopic viral integration site 5 (Evi5) [102,103].
Autophagy is a lysosomal catalytic process for bulk degradation of unwanted cytoplasmic substances and for recycling of nutrients. Autophagy is also involved in innate and adaptive immune responses, playing an important role for combating microbes, in antigen processing for MHC presentation and in lymphocyte development, survival and proliferation. Furthermore, interfering autophagy-related processes have been implicated in several diseases, including autoimmunity [104]. A number of GWAS analyses have linked SNPs in autophagy-related genes to susceptibility for autoimmune diseases, e.g., autophagy-specific gene 1 (ATG5) in systemic lupus erythematosus [105] and unc-51-like kinase 1 (ULK1) in Crohn’s disease [106]. A potential role for CLEC16A in autophagy would add this gene to the list of autophagy-related autoimmune associated genes. Furthermore, it has been reported that a couple of MS risk loci might play a role in autophagy; CYP27B1 expression is required for TLR2/1 mediated antibacterial autophagy in monocytes [107], whereas CD40 triggers killing of Toxoplasma gondii in an autophagy-dependent manner [108].
Inflammation leading to demyelination and axonal damage in the CNS is the main pathological feature of MS. During CNS inflammation, glial cells are activated and secrete cytokines. Astrocytes are the major constituents of the glial cell population in the CNS and do not only have the ability to enhance immune responses and inhibit myelin repair, but they can also limit CNS inflammation by supporting oligodendrocyte and axonal regeneration [109]. Interestingly, Wu and colleagues were able to show that CLEC16A is important for astrocyte activation in rats, as LPS-induced production of the pro-inflammatory cytokine, tumor necrosis factor alpha (TNFα), was reduced in astrocytes when CLEC16A expression was knocked down after transfection with CLEC16A-specific small-interfering RNA [81]. Taken together, this suggests that CLEC16A might be of importance during CNS inflammation, at least in a rat model system.
8. The CLEC16 Gene Region and Disease Susceptibility
In summary, fine-mapping analyses of MS associated SNPs in the 16p13 region point to CLEC16A SNPs as the most strongly associated MS genetic variants in this gene-rich region [49]. However, as these SNPs are intronic, we do not know whether CLEC16A per se is important for development of MS or whether these SNPs are expression quantitative trait loci for CLEC16A or other genes, if they tag a causal rare SNP or if they are in LD with other regulatory SNPs in the same genetic region. Non-coding SNPs may confer susceptibility to disease development by affecting expression of nearby genes in cis[110,111]. Indeed, MS associated SNPs in the CLEC16A region have been shown to correlate with expression of the neighboring SOCS1 and DEXI genes [70,71]. Interestingly, chromosome conformation capture assays performed by Davison et al.[71] proposed that a loop can be formed between the DEXI promoter and a 20 kb region of CLEC16A intron 19 (containing the MS associated rs12708716) shown in three tested cell lines (THP-1 (a monocytic cell line), A549 (a lung epithelial cell line) and a human EBV-transformed B-cell line). This supports that intronic, disease-associated SNPs in CLEC16A may be in physical proximity with a distant gene, such as DEXI, thereby potentially affecting the binding of transcription factors or other regulatory proteins important for gene expression. The same close proximity was not observed between the SOCS1 gene and the intron 19 of CLEC16A in these cell lines [71].
CLEC16A, SOCS1 and DEXI are co-expressed in thymic tissue samples [70] and in human lymphoblastoid cell lines [49], but not in whole blood [70] nor in monocytes [71]. This cell-specific co-regulation could be due to a common, regulatory element controlling cell-specific expression of all three genes. In support of this notion, the co-expressed DEXI, CLEC16A and SOCS1 genes constitute an “active” chromatin domain, at least in lymphoblastoid cells, as analyzed by methylation of lysine 27 on histone H3 and CCCTC-binding factor (CTCF) occupancy [49,79]. If one or more autoimmune-associated SNPs are in LD with a SNP in a common regulatory region, it could affect gene expression of several genes in this region, including DEXI, CLEC16A and SOCS1, meaning that all three genes might have an impact on disease susceptibility or phenotype.
Gene identification is an important first step to pinpoint a relevant gene region; however, GWASs do not tell whether the disease-associated variant is causal. By combining GWAS data with data from the Encyclopedia DNA Elements (ENCODE) project [112] for a number of traits and diseases, Schaub and colleagues were able to show that, for the majority of associations, the SNP most likely to play a functional role is usually a different SNP in strong LD with the reported association [113]. In fact, in MS, it was recently published that a majority of MS-associated SNPs identified in GWASs were located in functionally active DNA sites, so-called DNase hypersensitive sites. Interestingly, this co-localization was more pronounced in immune cells that are relevant for MS pathogenesis compared to other cell types [114]. After finalizing the fine-mapping of this genetic region, it will be possible to combine these results with the ENCODE data to define functional DNA elements that will be interesting for functional studies of this gene and neighboring genes in the time to come.
9. Conclusions
CLEC16A has been convincingly replicated as an MS susceptibility gene in a series of studies (see Table 1). In the gene-rich region of chromosome 16p13, several autoimmune disease-associated SNPs have also been identified in the neighboring SOCS1 and CIITA genes, as well as in intergenic regions (see Tables 2 and 3). However, SNPs within intronic regions of CLEC16A appear to display the strongest association, at least in MS [49]. Together with its selective expression in immune cells [38] and due to its association with a wide variety of autoimmune diseases, the CLEC16A gene and the encoded molecule is indeed of interest for further functional studies. In addition, as intronic CLEC16A sequences, harboring disease-associated SNPs, are correlated with expression of neighboring genes, i.e., SOCS1 and DEXI[70,71], the interaction between the CLEC16A gene and these genes needs to be further analyzed to understand new pathological mechanisms for development of autoimmunity.
Acknowledgements
The authors are funded by grants from The South-Eastern Norway Regional Health Authority, the Research Council of Norway, University of Oslo, Ullevål University Hospital Scientific Advisory Council (VIRUUS) and the Odd Fellow society. Our research group has received an unrestricted grant for CLEC16A gene studies from Biogen Idec, Norway.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Giovannoni G., Southam E., Waubant E. Systematic review of disease-modifying therapies to assess unmet needs in multiple sclerosis: Tolerability and adherence. Mult. Scler. 2012;18:932–946. doi: 10.1177/1352458511433302. [DOI] [PubMed] [Google Scholar]
- 2.Taylor B.V., Pearson J.F., Clarke G., Mason D.F., Abernethy D.A., Willoughby E., Sabel C. MS prevalence in New Zealand, an ethnically and latitudinally diverse country. Mult. Scler. 2010;16:1422–1431. doi: 10.1177/1352458510379614. [DOI] [PubMed] [Google Scholar]
- 3.Simpson S., Jr, Blizzard L., Otahal P., van der Mei I., Taylor B. Latitude is significantly associated with the prevalence of multiple sclerosis: A meta-analysis. J. Neurol. Neurosurg. Psychiatry. 2011;82:1132–1141. doi: 10.1136/jnnp.2011.240432. [DOI] [PubMed] [Google Scholar]
- 4.Alonso A., Hernan M.A. Temporal trends in the incidence of multiple sclerosis: A systematic review. Neurology. 2008;71:129–135. doi: 10.1212/01.wnl.0000316802.35974.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Gorman C., Lucas R., Taylor B. Environmental risk factors for multiple sclerosis: A review with a focus on molecular mechanisms. Int. J. Mol. Sci. 2012;13:11718–11752. doi: 10.3390/ijms130911718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kemppinen A., Sawcer S., Compston A. Genome-wide association studies in multiple sclerosis: Lessons and future prospects. Brief. Funct. Genomics. 2011;10:61–70. doi: 10.1093/bfgp/elr004. [DOI] [PubMed] [Google Scholar]
- 7.Gourraud P.A., Harbo H.F., Hauser S.L., Baranzini S.E. The genetics of multiple sclerosis: An up-to-date review. Immunol. Rev. 2012;248:87–103. doi: 10.1111/j.1600-065X.2012.01134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jersild C., Svejgaard A., Fog T. HL-A antigens and multiple sclerosis. Lancet. 1972;1:1240–1241. doi: 10.1016/s0140-6736(72)90962-2. [DOI] [PubMed] [Google Scholar]
- 9.Olerup O., Zetterquist H. HLA-DRB1*01 subtyping by allele-specific PCR amplification: A sensitive, specific and rapid technique. Tissue Antigens. 1991;37:197–204. doi: 10.1111/j.1399-0039.1991.tb01872.x. [DOI] [PubMed] [Google Scholar]
- 10.International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2. Sawcer S., Hellenthal G., Pirinen M., Spencer C.C., Patsopoulos N.A., Moutsianas L., Dilthey A., Su Z., et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brynedal B., Duvefelt K., Jonasdottir G., Roos I.M., Akesson E., Palmgren J., Hillert J. HLA-A confers an HLA-DRB1 independent influence on the risk of multiple sclerosis. PLoS One. 2007;2:e664. doi: 10.1371/journal.pone.0000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fogdell-Hahn A., Ligers A., Gronning M., Hillert J., Olerup O. Multiple sclerosis: A modifying influence of HLA class I genes in an HLA class II associated autoimmune disease. Tissue Antigens. 2000;55:140–148. doi: 10.1034/j.1399-0039.2000.550205.x. [DOI] [PubMed] [Google Scholar]
- 13.Mero I.-L., Gustavsen M.W., Sæther H.S., Flåm S.T., Berg-Hansen P., Søndergaard H.B., Jensen P.E.H., Berge T., Bjølgerud A., Muggerud A., et al. Oligoclonal band status in Scandinavian multiple sclerosis patients is associated with specific genetic risk alleles. PLoS One. 2012 doi: 10.1371/journal.pone.0058352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.International Multiple Sclerosis Genetics Consortium. Hafler D.A., Compston A., Sawcer S., Lander E.S., Daly M.J., de Jager P.L., de Bakker P.I., Gabriel S.B., Mirel D.B., et al. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 15.Gregory S.G., Schmidt S., Seth P., Oksenberg J.R., Hart J., Prokop A., Caillier S.J., Ban M., Goris A., Barcellos L.F., et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet. 2007;39:1083–1091. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- 16.Lundmark F., Duvefelt K., Iacobaeus E., Kockum I., Wallstrom E., Khademi M., Oturai A., Ryder L.P., Saarela J., Harbo H.F., et al. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat. Genet. 2007;39:1108–1113. doi: 10.1038/ng2106. [DOI] [PubMed] [Google Scholar]
- 17.Australia and New Zealand Multiple Sclerosis Genetics Consortium. Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat. Genet. 2009;41:824–828. doi: 10.1038/ng.396. [DOI] [PubMed] [Google Scholar]
- 18.Jakkula E., Leppa V., Sulonen A.M., Varilo T., Kallio S., Kemppinen A., Purcell S., Koivisto K., Tienari P., Sumelahti M.L., et al. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am. J. Hum. Genet. 2010;86:285–291. doi: 10.1016/j.ajhg.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanna S., Pitzalis M., Zoledziewska M., Zara I., Sidore C., Murru R., Whalen M.B., Busonero F., Maschio A., Costa G., et al. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat. Genet. 2010;42:495–497. doi: 10.1038/ng.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nischwitz S., Cepok S., Kroner A., Wolf C., Knop M., Muller-Sarnowski F., Pfister H., Roeske D., Rieckmann P., Hemmer B., et al. Evidence for VAV2 and ZNF433 as susceptibility genes for multiple sclerosis. J. Neuroimmunol. 2010;227:162–166. doi: 10.1016/j.jneuroim.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 21.Briggs F.B., Bartlett S.E., Goldstein B.A., Wang J., McCauley J.L., Zuvich R.L., de Jager P.L., Rioux J.D., Ivinson A.J., Compston A., et al. Evidence for CRHR1 in multiple sclerosis using supervised machine learning and meta-analysis in 12,566 individuals. Hum. Mol. Genet. 2010;19:4286–4295. doi: 10.1093/hmg/ddq328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Jager P.L., Baecher-Allan C., Maier L.M., Arthur A.T., Ottoboni L., Barcellos L., McCauley J.L., Sawcer S., Goris A., Saarela J., et al. The role of the CD58 locus in multiple sclerosis. Proc. Natl. Acad. Sci. USA. 2009;106:5264–5269. doi: 10.1073/pnas.0813310106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Jager P.L., Jia X., Wang J., de Bakker P.I., Ottoboni L., Aggarwal N.T., Piccio L., Raychaudhuri S., Tran D., Aubin C., et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 2009;41:776–782. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baranzini S.E., Wang J., Gibson R.A., Galwey N., Naegelin Y., Barkhof F., Radue E.W., Lindberg R.L., Uitdehaag B.M., Johnson M.R., et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum. Mol. Genet. 2009;18:767–778. doi: 10.1093/hmg/ddn388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Comabella M., Craig D.W., Camina-Tato M., Morcillo C., Lopez C., Navarro A., Rio J., Biomarker M.S.S.G., Montalban X., Martin R. Identification of a novel risk locus for multiple sclerosis at 13q31.3 by a pooled genome-wide scan of 500,000 single nucleotide polymorphisms. PLoS One. 2008;3:e3490. doi: 10.1371/journal.pone.0003490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wellcome Trust Case Control Consortium, Australo-Anglo-American Spondylitis Consortium. Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., McCarthy M.I., et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patsopoulos N.A. Bayer Pharma MS Genetics Working Group, Steering Committees of Studies Evaluating IFNβ-1b and a CCR1-Antagonist; ANZgene Consortium, GeneMSA; International Multiple Sclerosis Genetics Consortium. Esposito F., Reischl J., Lehr S., Bauer D., et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann. Neurol. 2011;70:897–912. doi: 10.1002/ana.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu Z.X., Zhang K., Qiu X.S., Zhou M., Li W.M. CD226 Gly307Ser association with multiple autoimmune diseases: A meta-analysis. Hum. Immunol. 2012;74:249–255. doi: 10.1016/j.humimm.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 29.Hoppenbrouwers I.A., Aulchenko Y.S., Janssens A.C., Ramagopalan S.V., Broer L., Kayser M., Ebers G.C., Oostra B.A., van Duijn C.M., Hintzen R.Q. Replication of CD58 and CLEC16A as genome-wide significant risk genes for multiple sclerosis. J. Hum. Genet. 2009;54:676–680. doi: 10.1038/jhg.2009.96. [DOI] [PubMed] [Google Scholar]
- 30.Rubio J.P., Stankovich J., Field J., Tubridy N., Marriott M., Chapman C., Bahlo M., Perera D., Johnson L.J., Tait B.D., et al. Replication of KIAA0350, IL2RA, RPL5 and CD58 as multiple sclerosis susceptibility genes in Australians. Genes Immun. 2008;9:624–630. doi: 10.1038/gene.2008.59. [DOI] [PubMed] [Google Scholar]
- 31.International Multiple Sclerosis Genetics Consortium. The expanding genetic overlap between multiple sclerosis and type I diabetes. Genes Immun. 2009;10:11–14. doi: 10.1038/gene.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mero I.L., Ban M., Lorentzen A.R., Smestad C., Celius E.G., Saether H., Saeedi H., Viken M.K., Skinningsrud B., Undlien D.E., et al. Exploring the CLEC16A gene reveals a MS-associated variant with correlation to the relative expression of CLEC16A isoforms in thymus. Genes Immun. 2011;12:191–198. doi: 10.1038/gene.2010.59. [DOI] [PubMed] [Google Scholar]
- 33.Zoledziewska M., Costa G., Pitzalis M., Cocco E., Melis C., Moi L., Zavattari P., Murru R., Lampis R., Morelli L., et al. Variation within the CLEC16A gene shows consistent disease association with both multiple sclerosis and type 1 diabetes in Sardinia. Genes Immun. 2009;10:15–17. doi: 10.1038/gene.2008.84. [DOI] [PubMed] [Google Scholar]
- 34.Perera D., Stankovich J., Butzkueven H., Taylor B.V., Foote S.J., Kilpatrick T.J., Rubio J.P. Fine mapping of multiple sclerosis susceptibility genes provides evidence of allelic heterogeneity at the IL2RA locus. J. Neuroimmunol. 2009;211:105–109. doi: 10.1016/j.jneuroim.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 35.Martinez A., Perdigones N., Cenit M.C., Espino L., Varade J., Lamas J.R., Santiago J.L., Fernandez-Arquero M., de la Calle H., Arroyo R., et al. Chromosomal region 16p13: Further evidence of increased predisposition to immune diseases. Ann. Rheum. Dis. 2010;69:309–311. doi: 10.1136/ard.2008.098376. [DOI] [PubMed] [Google Scholar]
- 36.D’Netto M.J., Ward H., Morrison K.M., Ramagopalan S.V., Dyment D.A., DeLuca G.C., Handunnetthi L., Sadovnick A.D., Ebers G.C. Risk alleles for multiple sclerosis in multiplex families. Neurology. 2009;72:1984–1988. doi: 10.1212/WNL.0b013e3181a92c25. [DOI] [PubMed] [Google Scholar]
- 37.Johnson B.A., Wang J., Taylor E.M., Caillier S.J., Herbert J., Khan O.A., Cross A.H., de Jager P.L., Gourraud P.A., Cree B.C., et al. Multiple sclerosis susceptibility alleles in African Americans. Genes Immun. 2010;11:343–350. doi: 10.1038/gene.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.BioGPS Gene Annotation Portal. [accessed on 15 December 2012]. Available online: http://biogps.org.
- 39.National Center for Biotechnology Information, reference sequence NM_015226.2 (Homo sapiens C-type lectin domain family 16, member A (CLEC16A)) [accessed on 15 December 2012]. Available online: http://www.ncbi.nlm.nih.gov/nuccore/NM_015226.2.
- 40.Hakonarson H., Grant S.F., Bradfield J.P., Marchand L., Kim C.E., Glessner J.T., Grabs R., Casalunovo T., Taback S.P., Frackelton E.C., et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007;448:591–594. doi: 10.1038/nature06010. [DOI] [PubMed] [Google Scholar]
- 41.Skinningsrud B., Husebye E.S., Pearce S.H., McDonald D.O., Brandal K., Wolff A.B., Lovas K., Egeland T., Undlien D.E. Polymorphisms in CLEC16A and CIITA at 16p13 are associated with primary adrenal insufficiency. J. Clin. Endocrinol. Metab. 2008;93:3310–3317. doi: 10.1210/jc.2008-0821. [DOI] [PubMed] [Google Scholar]
- 42.Marquez A., Varade J., Robledo G., Martinez A., Mendoza J.L., Taxonera C., Fernandez-Arquero M., Diaz-Rubio M., Gomez-Garcia M., Lopez-Nevot M.A., et al. Specific association of a CLEC16A/KIAA0350 polymorphism with NOD2/CARD15(−) Crohn’s disease patients. Eur. J. Hum. Genet. 2009;17:1304–1308. doi: 10.1038/ejhg.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirschfield G.M., Xie G., Lu E., Sun Y., Juran B.D., Chellappa V., Coltescu C., Mason A.L., Milkiewicz P., Myers R.P., et al. Association of primary biliary cirrhosis with variants in the CLEC16A, SOCS1, SPIB and SIAE immunomodulatory genes. Genes Immun. 2012;13:328–335. doi: 10.1038/gene.2011.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skinningsrud B., Lie B.A., Husebye E.S., Kvien T.K., Forre O., Flato B., Stormyr A., Joner G., Njolstad P.R., Egeland T., et al. A CLEC16A variant confers risk for juvenile idiopathic arthritis and anti-cyclic citrullinated peptide antibody negative rheumatoid arthritis. Ann. Rheum. Dis. 2010;69:1471–1474. doi: 10.1136/ard.2009.114934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jagielska D., Redler S., Brockschmidt F.F., Herold C., Pasternack S.M., Garcia Bartels N., Hanneken S., Eigelshoven S., Refke M., Barth S., et al. Follow-up study of the first genome-wide association scan in alopecia areata: IL13 and KIAA0350 as susceptibility loci supported with genome-wide significance. J. Invest. Dermatol. 2012;132:2192–2197. doi: 10.1038/jid.2012.129. [DOI] [PubMed] [Google Scholar]
- 46.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 47.Nischwitz S., Cepok S., Kroner A., Wolf C., Knop M., Muller-Sarnowski F., Pfister H., Rieckmann P., Hemmer B., Ising M., et al. More CLEC16A gene variants associated with multiple sclerosis. Acta Neurol. Scand. 2011;123:400–406. doi: 10.1111/j.1600-0404.2010.01421.x. [DOI] [PubMed] [Google Scholar]
- 48.Pandit L., Ban M., Sawcer S., Singhal B., Nair S., Radhakrishnan K., Shetty R., Misri Z., Hegde S., Bhat I.G. Evaluation of the established non-MHC multiple sclerosis loci in an Indian population. Mult. Scler. 2011;17:139–143. doi: 10.1177/1352458510384011. [DOI] [PubMed] [Google Scholar]
- 49.Zuvich R.L., Bush W.S., McCauley J.L., Beecham A.H., De Jager P.L., Ivinson A.J., Compston A., Hafler D.A., Hauser S.L., Sawcer S.J., et al. Interrogating the complex role of chromosome 16p13.13 in multiple sclerosis susceptibility: Independent genetic signals in the CIITA-CLEC16A-SOCS1 gene complex. Hum. Mol. Genet. 2011;20:3517–3524. doi: 10.1093/hmg/ddr250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Todd J.A., Walker N.M., Cooper J.D., Smyth D.J., Downes K., Plagnol V., Bailey R., Nejentsev S., Field S.F., Payne F., et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Awata T., Kawasaki E., Tanaka S., Ikegami H., Maruyama T., Shimada A., Nakanishi K., Kobayashi T., Iizuka H., Uga M., et al. Association of type 1 diabetes with two Loci on 12q13 and 16p13 and the influence coexisting thyroid autoimmunity in Japanese. J. Clin. Endocrinol. Metab. 2009;94:231–235. doi: 10.1210/jc.2008-0718. [DOI] [PubMed] [Google Scholar]
- 52.Wu X., Zhu X., Wang X., Ma J., Zhu S., Li J., Liu Y. Intron polymorphism in the KIAA0350 gene is reproducibly associated with susceptibility to type 1 diabetes (T1D) in the Han Chinese population. Clin. Endocrinol. 2009;71:46–49. doi: 10.1111/j.1365-2265.2008.03437.x. [DOI] [PubMed] [Google Scholar]
- 53.Sang Y., Zong W., Yan J., Liu M. The correlation between the CLEC16A gene and genetic susceptibility to type 1 diabetes in Chinese children. Int. J. Endocrinol. 2012;2012:245384. doi: 10.1155/2012/245384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Howson J.M., Rosinger S., Smyth D.J., Boehm B.O., Group A.-E.S., Todd J.A. Genetic analysis of adult-onset autoimmune diabetes. Diabetes. 2011;60:2645–2653. doi: 10.2337/db11-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamashita H., Awata T., Kawasaki E., Ikegami H., Tanaka S., Maruyama T., Shimada A., Nakanishi K., Takahashi K., Kobayashi T., et al. Analysis of the HLA and non-HLA susceptibility loci in Japanese type 1 diabetes. Diabetes/Metab. Res. Rev. 2011;27:844–848. doi: 10.1002/dmrr.1234. [DOI] [PubMed] [Google Scholar]
- 56.Mells G.F., Floyd J.A., Morley K.I., Cordell H.J., Franklin C.S., Shin S.Y., Heneghan M.A., Neuberger J.M., Donaldson P.T., Day D.B., et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat. Genet. 2011;43:329–332. doi: 10.1038/ng.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang C.H., Flavell R.A. Class II transactivator regulates the expression of multiple genes involved in antigen presentation. J. Exp. Med. 1995;181:765–767. doi: 10.1084/jem.181.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang C.H., Guerder S., Hong S.C., van Ewijk W., Flavell R.A. Mice lacking the MHC class II transactivator (CIITA) show tissue-specific impairment of MHC class II expression. Immunity. 1996;4:167–178. doi: 10.1016/s1074-7613(00)80681-0. [DOI] [PubMed] [Google Scholar]
- 59.Rasmussen H.B., Kelly M.A., Clausen J. Genetic susceptibility to multiple sclerosis: Detection of polymorphic nucleotides and an intron in the 3′ untranslated region of the major histocompatibility complex class II transactivator gene. Hum. Immunol. 2001;62:371–377. doi: 10.1016/s0198-8859(01)00215-4. [DOI] [PubMed] [Google Scholar]
- 60.Swanberg M., Lidman O., Padyukov L., Eriksson P., Akesson E., Jagodic M., Lobell A., Khademi M., Borjesson O., Lindgren C.M., et al. MHC2TA is associated with differential MHC molecule expression and susceptibility to rheumatoid arthritis, multiple sclerosis and myocardial infarction. Nat. Genet. 2005;37:486–494. doi: 10.1038/ng1544. [DOI] [PubMed] [Google Scholar]
- 61.Bronson P.G., Caillier S., Ramsay P.P., McCauley J.L., Zuvich R.L., De Jager P.L., Rioux J.D., Ivinson A.J., Compston A., Hafler D.A., et al. CIITA variation in the presence of HLA-DRB1*1501 increases risk for multiple sclerosis. Hum. Mol. Genet. 2010;19:2331–2340. doi: 10.1093/hmg/ddq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gyllenberg A., Asad S., Piehl F., Swanberg M., Padyukov L., Van Yserloo B., Rutledge E.A., McNeney B., Graham J., Orho-Melander M., et al. Age-dependent variation of genotypes in MHC II transactivator gene (CIITA) in controls and association to type 1 diabetes. Genes Immun. 2012;13:632–640. doi: 10.1038/gene.2012.44. [DOI] [PubMed] [Google Scholar]
- 63.Eike M.C., Skinningsrud B., Ronninger M., Stormyr A., Kvien T.K., Joner G., Njolstad P.R., Forre O., Flato B., Alfredsson L., et al. CIITA gene variants are associated with rheumatoid arthritis in Scandinavian populations. Genes Immun. 2012;13:431–436. doi: 10.1038/gene.2012.11. [DOI] [PubMed] [Google Scholar]
- 64.Bronson P.G., Goldstein B.A., Ramsay P.P., Beckman K.B., Noble J.A., Lane J.A., Seldin M.F., Kelly J.A., Harley J.B., Moser K.L., et al. The rs4774 CIITA missense variant is associated with risk of systemic lupus erythematosus. Genes Immun. 2011;12:667–671. doi: 10.1038/gene.2011.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ghaderi M., Gambelunghe G., Tortoioli C., Brozzetti A., Jatta K., Gharizadeh B., de Bellis A., Pecori Giraldi F., Terzolo M., Betterle C., et al. MHC2TA single nucleotide polymorphism and genetic risk for autoimmune adrenal insufficiency. J. Clin. Endocrinol. Metab. 2006;91:4107–4111. doi: 10.1210/jc.2006-0855. [DOI] [PubMed] [Google Scholar]
- 66.Vandenbroeck K., Alvarez J., Swaminathan B., Alloza I., Matesanz F., Urcelay E., Comabella M., Alcina A., Fedetz M., Ortiz M.A., et al. A cytokine gene screen uncovers SOCS1 as genetic risk factor for multiple sclerosis. Genes Immun. 2012;13:21–28. doi: 10.1038/gene.2011.44. [DOI] [PubMed] [Google Scholar]
- 67.Fenner J.E., Starr R., Cornish A.L., Zhang J.G., Metcalf D., Schreiber R.D., Sheehan K., Hilton D.J., Alexander W.S., Hertzog P.J. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat. Immunol. 2006;7:33–39. doi: 10.1038/ni1287. [DOI] [PubMed] [Google Scholar]
- 68.Zuvich R.L., McCauley J.L., Oksenberg J.R., Sawcer S.J., De Jager P.L. International Multiple Sclerosis Genetics, C. Aubin C., Cross A.H., Piccio L., Aggarwal N.T., et al. Genetic variation in the IL7RA/IL7 pathway increases multiple sclerosis susceptibility. Hum. Genet. 2010;127:525–535. doi: 10.1007/s00439-010-0789-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.International Multiple Sclerosis Genetics Consortium. Comprehensive follow-up of the first genome-wide association study of multiple sclerosis identifies KIF21B and TMEM39A as susceptibility loci. Hum. Mol. Genet. 2010;19:953–962. doi: 10.1093/hmg/ddp542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leikfoss I.S., Mero I.L., Dahle M.K., Lie B.A., Harbo H.F., Spurkland A., Berge T. Multiple sclerosis-associated single-nucleotide polymorphisms in CLEC16A correlate with reduced SOCS1 and DEXI expression in the thymus. Genes Immun. 2013;14:62–66. doi: 10.1038/gene.2012.52. [DOI] [PubMed] [Google Scholar]
- 71.Davison L.J., Wallace C., Cooper J.D., Cope N.F., Wilson N.K., Smyth D.J., Howson J.M., Saleh N., Al-Jeffery A., Angus K.L., et al. Long-range DNA looping and gene expression analyses identify DEXI as an autoimmune disease candidate gene. Hum. Mol. Genet. 2012;21:322–333. doi: 10.1093/hmg/ddr468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nica A.C., Montgomery S.B., Dimas A.S., Stranger B.E., Beazley C., Barroso I., Dermitzakis E.T. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet. 2010;6:e1000895. doi: 10.1371/journal.pgen.1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edgar A.J., Birks E.J., Yacoub M.H., Polak J.M. Cloning of dexamethasone-induced transcript: A novel glucocorticoid-induced gene that is upregulated in emphysema. Am. J. Respir. Cell Mol. Biol. 2001;25:119–124. doi: 10.1165/ajrcmb.25.1.4417. [DOI] [PubMed] [Google Scholar]
- 74.Cortes A., Brown M.A. Promise and pitfalls of the Immunochip. Arthritis Res. Ther. 2011;13:101. doi: 10.1186/ar3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eyre S., Bowes J., Diogo D., Lee A., Barton A., Martin P., Zhernakova A., Stahl E., Viatte S., McAllister K., et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 2012;44:1336–1340. doi: 10.1038/ng.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cooper J.D., Simmonds M.J., Walker N.M., Burren O., Brand O.J., Guo H., Wallace C., Stevens H., Coleman G. Wellcome Trust Case Control Consortium, et al. Seven newly identified loci for autoimmune thyroid disease. Hum. Mol. Genet. 2012;21:5202–5208. doi: 10.1093/hmg/dds357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoshimura A., Suzuki M., Sakaguchi R., Hanada T., Yasukawa H. SOCS, Inflammation, and autoimmunity. Front. Immunol. 2012;3:20. doi: 10.3389/fimmu.2012.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Friese M.A., Jones E.Y., Fugger L. MHC II molecules in inflammatory diseases: Interplay of qualities and quantities. Trends Immunol. 2005;26:559–561. doi: 10.1016/j.it.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 79.UCSC Genome Bioinformatics. [accessed on 15 December 2012]. Available online: http://genome.ucsc.edu.
- 80.Nagase T., Ishikawa K., Nakajima D., Ohira M., Seki N., Miyajima N., Tanaka A., Kotani H., Nomura N., Ohara O. Prediction of the coding sequences of unidentified human genes. VII. The complete sequences of 100 new cDNA clones from brain which can code for large proteins in vitro. DNA Res. 1997;4:141–150. doi: 10.1093/dnares/4.2.141. [DOI] [PubMed] [Google Scholar]
- 81.Wu X., Li J., Chen C., Yan Y., Jiang S., Wu X., Shao B., Xu J., Kang L., Huang Y., et al. Involvement of CLEC16A in activation of astrocytes after LPS treated. Neurochem. Res. 2012;37:5–14. doi: 10.1007/s11064-011-0581-4. [DOI] [PubMed] [Google Scholar]
- 82.Wang G.S., Cooper T.A. Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 83.Bates E.E., Fournier N., Garcia E., Valladeau J., Durand I., Pin J.J., Zurawski S.M., Patel S., Abrams J.S., Lebecque S., et al. APCs express DCIR, a novel C-type lectin surface receptor containing an immunoreceptor tyrosine-based inhibitory motif. J. Immunol. 1999;163:1973–1983. [PubMed] [Google Scholar]
- 84.Fujikado N., Saijo S., Yonezawa T., Shimamori K., Ishii A., Sugai S., Kotaki H., Sudo K., Nose M., Iwakura Y. Dcir deficiency causes development of autoimmune diseases in mice due to excess expansion of dendritic cells. Nat. Med. 2008;14:176–180. doi: 10.1038/nm1697. [DOI] [PubMed] [Google Scholar]
- 85.McGreal E.P., Martinez-Pomares L., Gordon S. Divergent roles for C-type lectins expressed by cells of the innate immune system. Mol. Immunol. 2004;41:1109–1121. doi: 10.1016/j.molimm.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 86.Prosite—Database of Protein Domains, Families and Functional Sites. [accessed on 15 December 2012]. Available online: http://prosite.expasy.org.
- 87.Zelensky A.N., Gready J.E. The C-type lectin-like domain superfamily. FEBS J. 2005;272:6179–6217. doi: 10.1111/j.1742-4658.2005.05031.x. [DOI] [PubMed] [Google Scholar]
- 88.Cummings R.D., McEver R.P. C-Type Lectins. In: Varki A., Cummings R.D., Esko J.D., Freeze H.H., Stanley P., Bertozzi C.R., Hart G.W., Etzler M.E., editors. Essentials of Glycobiology. 2nd ed. Cold Spring Harbor; New York, NY, USA: 2009. [PubMed] [Google Scholar]
- 89.Kingeter L.M., Lin X. C-type lectin receptor-induced NF-kappaB activation in innate immune and inflammatory responses. Cell. Mol. Immunol. 2012;9:105–112. doi: 10.1038/cmi.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dinkel H., Chica C., Via A., Gould C.M., Jensen L.J., Gibson T.J., Diella F. Phospho.ELM: A database of phosphorylation sites—Update 2011. Nucleic Acids Res. 2011;39:D261–D267. doi: 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.The Eukaryotic Linear Motif Resource for Functional Sites in Proteins. [accessed on 15 December 2012]. Available online: http://elm.eu.org.
- 92.Marchler-Bauer A., Anderson J.B., Chitsaz F., Derbyshire M.K., DeWeese-Scott C., Fong J.H., Geer L.Y., Geer R.C., Gonzales N.R., Gwadz M., et al. CDD: Specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009;37:D205–D210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Swiss EMBnet Node Server. [accessed on 15 December 2012]. Available online http://www.ch.embnet.org.
- 94.National Center for Biotechnology Information. [accessed on 15 December 2012]. Available online: http://www.ncbi.nlm.nih.gov.
- 95.UniProt—A resource of Protein Sequence and Functional Information. [accessed on 15 December 2012]. Available online: www.uniprot.org.
- 96.Kim S., Wairkar Y.P., Daniels R.W., DiAntonio A. The novel endosomal membrane protein Ema interacts with the class C Vps-HOPS complex to promote endosomal maturation. J. Cell Biol. 2010;188:717–734. doi: 10.1083/jcb.200911126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim S., Diantonio A. A role for the membrane Golgi protein Ema in autophagy. Autophagy. 2012;8:1269–1270. doi: 10.4161/auto.20871. [DOI] [PubMed] [Google Scholar]
- 98.Thompson J.D., Gibson T.J., Higgins D.G. Curr. Prot. Bioinform. 2002. Multiple sequence alignment using ClustalW and ClustalX; pp. 2.3.1–2.3.22. [DOI] [PubMed] [Google Scholar]
- 99.Seto E.S., Bellen H.J., Lloyd T.E. When cell biology meets development: Endocytic regulation of signaling pathways. Genes Dev. 2002;16:1314–1336. doi: 10.1101/gad.989602. [DOI] [PubMed] [Google Scholar]
- 100.German C.L., Sauer B.M., Howe C.L. The STAT3 beacon: IL-6 recurrently activates STAT 3 from endosomal structures. Exp. Cell Res. 2011;317:1955–1969. doi: 10.1016/j.yexcr.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Payelle-Brogard B., Pellegrini S. Biochemical monitoring of the early endocytic traffic of the type I interferon receptor. J. Interferon Cytokine Res. 2010;30:89–98. doi: 10.1089/jir.2009.0044. [DOI] [PubMed] [Google Scholar]
- 102.Westlake C.J., Junutula J.R., Simon G.C., Pilli M., Prekeris R., Scheller R.H., Jackson P.K., Eldridge A.G. Identification of Rab11 as a small GTPase binding protein for the Evi5 oncogene. Proc. Natl. Acad. Sci. USA. 2007;104:1236–1241. doi: 10.1073/pnas.0610500104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hehnly H., Chen C.T., Powers C.M., Liu H.L., Doxsey S. The centrosome regulates the rab11-dependent recycling endosome pathway at appendages of the mother centriole. Curr. Biol. 2012;22:1944–1950. doi: 10.1016/j.cub.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pierdominici M., Vomero M., Barbati C., Colasanti T., Maselli A., Vacirca D., Giovannetti A., Malorni W., Ortona E. Role of autophagy in immunity and autoimmunity, with a special focus on systemic lupus erythematosus. FASEB J. 2012;26:1400–1412. doi: 10.1096/fj.11-194175. [DOI] [PubMed] [Google Scholar]
- 105.Zhou X.J., Lu X.L., Lv J.C., Yang H.Z., Qin L.X., Zhao M.H., Su Y., Li Z.G., Zhang H. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann. Rheumatic Dis. 2011;70:1330–1337. doi: 10.1136/ard.2010.140111. [DOI] [PubMed] [Google Scholar]
- 106.Henckaerts L., Cleynen I., Brinar M., John J.M., van Steen K., Rutgeerts P., Vermeire S. Genetic variation in the autophagy gene ULK1 and risk of Crohn’s disease. Inflamm. Bowel Dis. 2011;17:1392–1397. doi: 10.1002/ibd.21486. [DOI] [PubMed] [Google Scholar]
- 107.Shin D.M., Yuk J.M., Lee H.M., Lee S.H., Son J.W., Harding C.V., Kim J.M., Modlin R.L., Jo E.K. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell. Microbiol. 2010;12:1648–1665. doi: 10.1111/j.1462-5822.2010.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Portillo J.A., Okenka G., Reed E., Subauste A., van Grol J., Gentil K., Komatsu M., Tanaka K., Landreth G., Levine B., et al. The CD40-autophagy pathway is needed for host protection despite IFN-Gamma-dependent immunity and CD40 induces autophagy via control of P21 levels. PLoS One. 2010;5:e14472. doi: 10.1371/journal.pone.0014472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nair A., Frederick T.J., Miller S.D. Astrocytes in multiple sclerosis: A product of their environment. Cell. Mol. Life Sci. 2008;65:2702–2720. doi: 10.1007/s00018-008-8059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cookson W., Liang L., Abecasis G., Moffatt M., Lathrop M. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 2009;10:184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. Trait-associated SNPs are more likely to be eQTLs: Annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Consortium E.P., Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schaub M.A., Boyle A.P., Kundaje A., Batzoglou S., Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22:1748–1759. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]