

Abstract
Studies of regulation of the haematopoietic growth factor erythropoietin led to the unexpected discovery of a widespread system of direct oxygen sensing that regulates gene expression in animals. The oxygen-sensitive signal is generated by a series of non-haem Fe(II)- and 2-oxoglutarate-dependent dioxygenases that catalyse the post-translational hydroxylation of specific residues in the transcription factor hypoxia-inducible factor (HIF). These hydroxylations promote both oxygen-dependent degradation and oxygen-dependent inactivation of HIF, but are suppressed in hypoxia, leading to the accumulation of HIF and assembly of an active transcriptional complex in hypoxic cells. Hypoxia-inducible factor activates an extensive transcriptional cascade that interfaces with other cell signalling pathways, microRNA networks and RNA–protein translational control systems. The relationship of these cellular signalling pathways to the integrated physiology of oxygen homeostasis and the implication of dysregulating these massive physiological pathways in diseases such as cancer are discussed.
 |
Peter Ratcliffe is Nuffield Professor of Clinical Medicine at the University of Oxford. He trained as a Nephrologist, researching the pathophysiology of renal injury in shock, before switching fields to study the regulation of erythropoietin by the kidneys. The work on erythropoietin led to the discovery of a widespread system of gene regulation by oxygen and to the elucidation of the underlying oxygen sensing process. He has directed the hypoxia biology laboratory at Oxford for more than 20 years and has won many awards for this work including the Louis-Jeantet Prize for Medicine, the Canada Gairdner International Award and the Grand Prix Lefoulon-Delalande of the Institut de France.
Introduction
All large organisms face the fundamental physiological challenge of matching oxygen supplies to the needs of respiring tissues. In many animal species, both large size and the capacity for rapid movement enhance the difficulty of this task. In higher animals, specialized organs evolved to facilitate oxygen delivery, and the lungs, heart, blood and vascular system are all devoted to this purpose. The co-ordination of these complex homeostatic systems requires robust mechanisms for detecting and responding to error signals in oxygen homeostasis.
A direct oxygen sensing system that regulates gene expression in animals
In principle, detection systems that ‘sense’ an ‘error signal’ in oxygen homeostasis might operate at many different levels. For instance, inadequate oxygen levels might be detected either indirectly, through the compromise of metabolism, or directly, through changes in the availability of oxygen itself. The central role of oxygen in energy metabolism makes these possibilities difficult to distinguish. However, early observations on one highly dynamic response, the increased production of the haematopoietic growth factor erythropoietin (Epo) by the kidneys, when blood oxygen is reduced, strongly suggested the existence of a process that was responding to the lack of oxygen itself (for review see Jelkmann, 1992; Bunn & Poyton, 1996). Notably, the demonstration that cobalt intoxication produces striking increases in erythropoiesis without obvious metabolic compromise (Berk et al. 1949) and that metabolic inhibitors were unable to mimic this effect (Necas & Thorling, 1972) pointed to a distinct oxygen sensing process. Nevertheless, the prevailing view was that this oxygen sensing system was restricted to Epo and not of general relevance to other cellular and systemic responses that defend oxygen homeostasis, a perspective that was reinforced by the distinctive, high-amplitude dynamics of the Epo response. This initially led to major efforts to identify and isolate the erythropoietin producing cells from kidney. Somewhat contrary to expectations, these cells were shown to be a population of interstitial fibroblasts without obvious specialized features (Bachmann et al. 1993; Maxwell et al. 1993a).
In parallel with this, and again contrary to expectations, early studies of the transcriptional control of the erythropoietin gene revealed that the underlying oxygen sensing system that regulated control DNA sequences at the erythropoietin locus was in fact not restricted to erythropoietin producing cells. A key oxygen-regulated control element, termed the erythropoietin 3′ enhancer, was shown to manifest activity that was inducible by both hypoxia and cobaltous ions (thus mimicking the characteristics of endogenous erythropoietin) when introduced by transfection into a very wide range of cells (Maxwell et al. 1993b). Many of these cells were entirely unrelated to the kidney or to the production of erythropoietin, indicating that the system must be widespread and involved in other responses. In subsequent work, it became clear that the key transcription factor, hypoxia-inducible factor (HIF), originally identified in erythropoietin producing hepatoma cells (Semenza & Wang, 1992), is indeed expressed very widely (probably universally) in mammalian cells (Wang & Semenza, 1993; Firth et al. 1994) and that the HIF system is conserved in primitive animal species that lack erythropoietin, red blood cells or even any specialized oxygen-delivery apparatus (Nagao et al. 1996; Loenarz et al. 2011). In human cells, pan-genomic analyses of HIF binding to DNA have now revealed the existence of in excess of 500 direct transcriptional targets of HIF in a given cell line (Mole et al. 2009; Xia et al. 2009; Schödel et al. 2012b). Recent work has demonstrated that HIF also interacts with microRNA networks (Kulshreshtha et al. 2007), engages in cross-talk with other signal pathways (Koshiji et al. 2004; Gustafsson et al. 2005) and acts as a non-transcriptional regulator of gene expression, for instance by affecting translation of specific genes (Uniacke et al. 2012). These primary activities will initiate secondary cascades of pathway activation, so that the overall complexity of activating the HIF response is enormous.
Hypoxia-inducible factor binds to DNA as an α/β heterodimer; both α and β subunits belonging to the basic helix-loop-helix PAS protein family (Wang et al. 1995). The β subunits are constitutively expressed and form heterodimers with several other members of this family to mediate other transcriptional responses, whilst the α subunits are specifically involved in the response to hypoxia. Early work that aimed at defining the upstream pathways connecting HIF to the signalling of oxygen levels defined discrete domains within the HIF polypeptide that could independently convey oxygen sensitivity on heterologous transcription factors (Jiang et al. 1997; Pugh et al. 1997; Huang et al. 1998). This work suggested a dual process of regulation, because some of these domains conveyed oxygen-dependent instability, whereas some appeared to affect transcriptional activity independent of stability (Fig. 1; Pugh et al. 1997). All domains shared the property of responding both to hypoxia and to cobalt stimulation, suggesting that they interacted independently with the same, or a closely similar, oxygen sensing signal. It was widely believed that, in common with most intracellular signalling pathways, protein phosphorylation would be involved in signal transduction. However, mutation of every phospho-acceptor residue in one of the isolated regulatory domains of HIF failed to ablate its oxygen sensitivity, suggesting that this signal process could operate independently of protein phosphorylation, thus excluding many known transduction mechanisms (Pugh et al. 1997).
Figure 1. Schematic diagram illustrating the dual regulation of hypoxia-inducible factor (HIF) by oxygen-dependent prolyl and asparaginyl hydroxylation.
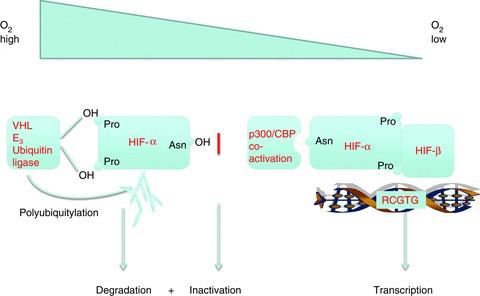
Pro, proline; OH Pro, hydroxyproline; Asn, asparagine; Asn OH, hydroxyasparagine; p300/CBP, E1A binding protein p300/CREB-binding protein.
An important insight into the upstream processes regulating HIF was gained through the recognition that the von Hippel-Lindau tumour suppressor (pVHL) was essential for proteolytic regulation of HIF-α, functioning as a ubiquitin ligase, targeting each of two internal HIF-α domains to mediate oxygen-dependent instability (Maxwell et al. 1999; Cockman et al. 2000; Ohh et al. 2000; Tanimoto et al. 2000; Masson et al. 2001). Detailed biochemical, mutational and mass spectrometric analysis of this interaction revealed the regulatory modification that governs the binding of HIF-α to pVHL to be oxygen-dependent trans-4-hydroxylation at specific prolyl residues within each of two regulatory domains in HIF-α (Pro-402 and -564 in human HIF-1α; Ivan et al. 2001; Jaakkola et al. 2001; Masson et al. 2001). A combination of structurally informed bioinformatic prediction and testing of candidate enzymes then led to the identification of a single non-redundant prolyl hydroxylase in Caenorhabditis elegans and to the recognition that the human genome encodes three closely related HIF prolyl hydroxylases, which we termed PHD (prolyl hydroxylase domain) 1, 2 and 3 (also known as Egln 2, 1 and 3, respectively; Bruick & McKnight, 2001; Epstein et al. 2001). These enzymes are dioxygenases, and their absolute requirement for molecular oxygen as cosubstrate immediately suggested a mechanism for oxygen sensing. Though all three enzymes contribute to the regulation of HIF, in most cells PHD2 (Egln1) is the most abundant enzyme and therefore has a dominant role in the oxygen sensing process (Berra et al. 2003; Appelhoff et al. 2004). Prolyl hydroxylated HIF-α is recognized by the β-domain of pVHL through a hydrogen bonding network to the hydroxyproline residue, which cannot be formed by proline (Hon et al. 2002; Min et al. 2002). Added specificity to this ‘molecular switch’ is achieved by stereoelectronic effects that alter the bias of the pyrrolidine ring of the prolyl residue from the C4-endo to the C4-exo conformation upon hydroxylation. Specific binding of pVHL to the C4-exo conformation of the hydroxyprolyl residue results in a >1000-fold increase in affinity of HIF-α for pVHL upon hydroxylation, thus promoting the destruction of HIF-α in the presence of oxygen (Loenarz et al. 2009).
Similar biochemical and mass spectrometric analysis of the third (C-terminal) sequence, which had been shown to mediate oxygen-dependent transcriptional activity independently of any change in protein stability, revealed a third site of HIF-α hydroxylation (Lando et al. 2002b). In this case, the target of hydroxylation is a specific asparaginyl residue (Asn-803 in human HIF-1α). This residue is buried in a hydrophobic region that is formed when C-terminal HIF-α sequences are bound by the CH1 domain of the p300 coactivator (Dames et al. 2002). Hydroxylation at the asparaginyl residue reduces transcriptional activity of HIF-α, at least in part by preventing this interaction with p300 (Fig. 1). Further bioinformatic analyses identified the HIF asparaginyl hydroxylase as a molecule termed FIH (factor inhibiting HIF; Hewitson et al. 2002; Lando et al. 2002a), a protein that had previously been identified in a screen for HIF interacting proteins and shown to inhibit HIF activity (Mahon et al. 2001).
The HIF hydroxylases (three HIF prolyl hydroxylases and the HIF asparaginyl hydroxylase) are all members of the 2-oxoglutarate (2-OG)-dependent dioxygenase superfamily (for review see Loenarz & Schofield, 2008). These enzymes are non-haem iron-dependent enzymes that bind the catalytic iron using a 2-histidine-1-carboxylate ‘facial triad’ that occupies three of the six iron co-ordination sites (Fig. 2). The enzymes use the Krebs cycle intermediate 2-OG (α-ketoglutarate) as cosubstrate; as ‘dioxygenases’, they split molecular oxygen and incorporate both atoms directly into their reaction products (for review see Loenarz & Schofield, 2011). Thus, oxidization of the prime substrate (HIF-α) is coupled to the oxidative decarboxylation of 2-OG to succinate. Based on mechanistic studies of other 2-OG dioxygenases, it is likely that the reaction proceeds via the creation of a highly reactive [e.g. Fe(IV) = O (ferryl iron)] intermediate at the catalytic site (Price et al. 2003). Failure of the coupling process can leave the enzyme in an inactive oxidized state, and reduction of the catalytic iron centre is then required for activity. This process is considered to be the basis of the dependence of many members of this class of enzyme (including the HIF hydroxylases) on the reducing agent ascorbate. Direct utilization of molecular oxygen confers sensitivity to hypoxia, underpinning the role of these enzymes as intracellular oxygen sensors. Interestingly, whilst 2-OG oxygenases are prevalent in both eukaryotic and prokaryotic species (Loenarz & Schofield, 2008), and many primitive organisms exhibit well-defined programmes of gene regulation by oxygen (Bunn & Poyton, 1996), including systems that use 2-OG oxygenases (West et al. 2007; Hughes & Espenshade, 2008), the HIF hydroxylase (PHD/pVHL/HIF) system appears to be specific to (and universal within) the animal kingdom (Loenarz et al. 2011). Most vertebrate organisms express multiple PHD and HIF-α isoforms as a result of gene duplication, PHD2 and HIF-1α being the most universally expressed (Loenarz et al. 2011).
Figure 2. The catalytic iron centre of a typical 2-oxoglutarate (2-OG)-dependent dioxygenase.
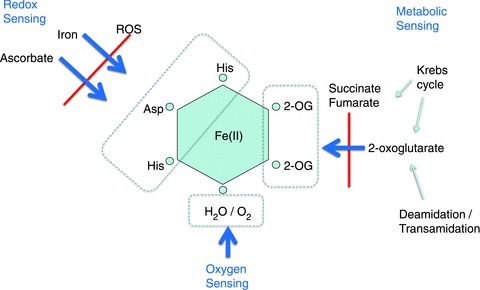
Of six co-ordinate positions, three are used to bind the 2-histidine-1-carboxylate ‘facial triad’ of the apo-enzyme, two are used to bind the cosubstrate 2-oxoglutarate and the sixth is occupied by a water molecule or (upon activation of the enzyme) molecular oxygen. Potentially, these interactions provide signalling interfaces with molecular oxygen, redox stresses and metabolism.
In the following sections, some of the biochemical and physiological characteristics of this system of oxygen sensing are reviewed, the challenges faced in linking molecular mechanisms to integrated physiology are highlighted, and the implications of dysregulation of these massive ‘hard-wired’ HIF hydroxylase pathways in disease are considered, particularly in relation to solid tumours, which commonly manifest tissue hypoxia and upregulation of HIF.
Biochemical aspects of oxygen sensing by HIF hydroxylases
It is estimated that the human genome encodes as many as 60–70 predicted or biochemically assigned 2-OG oxygenases with diverse roles in biology (Loenarz & Schofield, 2008). This has raised an important question as to whether the oxygen sensing function of the HIF hydroxylases is associated with some special biochemical property of these particular enzymes or whether they are biochemically similar to other 2-OG dioxygenases, but derive the oxygen sensing property from the physiological context in which they operate.
Values for KmO2 (the concentration of oxygen required for half-maximal initial catalytic rate) have been reported for the HIF prolyl hydroxylases to be in the range 200–250 μm (Hirsiläet al. 2003). These values are unusually high, but were obtained using recombinant enzymes and short HIF-α peptide substrates. Subsequent studies using longer HIF polypeptides have given lower values, in the region of 100 μm, which are similar to those reported for other 2-OG dioxygenases (Koivunen et al. 2006; Ehrismann et al. 2007). In any case, physiological tissue 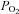 levels are generally found to be substantially below even the lower KmO2 values (Vaupel et al. 1991), and intracellular gradients probably create even lower values in the intracellular microenvironment. Therefore, even enzymes with a much lower KmO2 would have the potential to act as oxygen sensors if their catalytic action was rate limiting for the overall physiological function of the pathway. Thus, it appears unlikely that the unusually high KmO2 values are relevant to the function of the HIF hydroxylases as oxygen sensors.
levels are generally found to be substantially below even the lower KmO2 values (Vaupel et al. 1991), and intracellular gradients probably create even lower values in the intracellular microenvironment. Therefore, even enzymes with a much lower KmO2 would have the potential to act as oxygen sensors if their catalytic action was rate limiting for the overall physiological function of the pathway. Thus, it appears unlikely that the unusually high KmO2 values are relevant to the function of the HIF hydroxylases as oxygen sensors.
A different characteristic that has attracted considerable interest is the possibility that the multiple cofactor requirements that are common to this class of enzyme provide additional means of regulating activity that assist the complex task of maintaining oxygen homeostasis (Schofield & Ratcliffe, 2004; Kaelin & Ratcliffe, 2008). For instance, 2-oxoglutarate is a Krebs cycle intermediate which (via transamination and deamidation reactions) also occupies a pivotal position in amino acid metabolism. Thus, the use of both molecular oxygen and 2-oxoglutarate in the hydroxylation reaction might provide a link between hypoxic and metabolic signals (Isaacs et al. 2005; Hewitson et al. 2007; Koivunen et al. 2007). To date, however, it is unclear whether and in what circumstances 2-oxoglutarate levels are limiting for HIF hydroxylase activity. Perhaps of greater interest to oxygen sensing is the requirement for Fe(II) as cofactor. The binding of Fe(II) by the 2-histidine-1-carboxylate facial triad of the apo-enzyme is relatively labile. For instance, in contrast with haem enzymes, the catalytic Fe(II) can readily be chelated, thus inactivating the enzyme (Hirsiläet al. 2005). This lability (and the need for ascorbate to maintain the iron centre in a reduced and active form) could provide an interface both with cellular iron availability and with cellular redox signals. Redox signals are linked to the availability of oxygen. Thus, the possibility that such processes could provide a second oxygen-sensitive signal interacting with regulation by molecular oxygen availability, or even dominate the regulation of HIF hydroxylase activity, has attracted considerable interest. Substantial evidence indicates that the HIF hydroxylases can indeed be inhibited by oxidant stress.
In cultured cells, supplementation with both iron and ascorbate reduces the levels of HIF-α that are observed in standard (oxygenated) conditions (Knowles et al. 2003, 2006; Kuiper et al. 2010), implying that the availability of these cofactors can be limiting for full enzyme activity even in the presence of oxygen. Exogenous sources of reactive oxygen species inhibit HIF hydroxylases both in vitro and in cells (Gerald et al. 2004; Pan et al. 2007). Taken together, these findings strongly suggest that ‘oxygen sensing’ hydroxylase activity is modulated by redox signals, at least in pathophysiological conditions. What is less clear is how this relates to the physiological signalling of hypoxia. While excess oxygen (hyperoxia) can cause cell injury through the excess production of reactive oxygen species, and some investigators have reported that hypoxia reduces the production of reactive oxygen species (Hoffman et al. 2007), other investigators have assembled considerable evidence that inhibition of electron transport in hypoxia leads to increased production of reactive oxygen species and have proposed that this, rather than lack of molecular oxygen itself, is responsible for the reduction in HIF hydroxylase activity that signals hypoxia (Chandel et al. 1998; Bell et al. 2007). Of relevance to this, researchers in my laboratory recently performed comparative analyses of HIF prolyl and asparaginyl hydroxylation in cells. These studies revealed that whilst HIF prolyl hydroxylation is more sensitive to hypoxia than HIF asparaginyl hydroxylation, the reverse was true when cells were exposed to hydrogen peroxide to mimic oxidant stress (Tian, 2011; Masson et al. 2012). Hypoxia-inducible factor asparaginyl hydroxylation was found to be strongly inhibited at very low concentrations of hydrogen peroxide that had little, if any, effect on HIF prolyl hydroxylation (Masson et al. 2012). Furthermore, reoxygenation of hypoxic cells promotes immediate hydroxylation and degradation of HIF, whereas recovery from oxidant inactivation, at least for the HIF asparaginyl hydroxylase FIH, was found to be delayed (Tian, 2011; Masson et al. 2012). These findings suggest that hypoxia and oxidant stress represent separate regulatory inputs to the HIF hydroxylase pathways.
What is not currently clear is whether differential sensitivity of HIF prolyl and asparaginyl hydroxylation to peroxide represents unusual sensitivity of FIH or unusual resistance of PHD2 (among the 2-oxoglutarate dioxygenase enzyme family). Interestingly, biophysical analyses have revealed two unusual properties of PHD2 (the major HIF prolyl hydroxylase) that could be relevant; firstly, the enzyme binds to Fe(II) unusually tightly (McNeill et al. 2005); and secondly, in the absence of HIF substrate, the enzyme complex is unusually stable, even in the presence of oxygen (Flashman et al. 2010; Loenarz et al. 2011). Furthermore, structural studies of the HIF prolyl hydroxylase PHD2 and the HIF asparaginyl hydroxylase FIH complexed to their respective HIF peptide substrates show major differences that might relate to the functional findings. Whilst FIH binds its substrate in an open cleft (Elkins et al. 2003), the binding of target HIF-α peptides to PHD2 is more closed and results in a conformational change in which a loop of PHD2 shifts to form a clamp-like structure over the substrate (Chowdhury et al. 2009). Both the tight binding of Fe(II) and the stability of the PHD2 enzyme complex in the absence of substrate (reduced susceptibility to uncoupled turnover) may be predicted to reduce sensitivity to inactivation by oxidant stress. It is thus tempting to speculate that these properties are important for the function of PHD2 as a sensor that responds most specifically to molecular oxygen.
Nevertheless, it is clear that all HIF hydroxylases are, to different degrees, sensitive to oxidant injury (Pan et al. 2007; Diebold et al. 2010). Moreover, given that hypoxia does not suppress the sensitivity to peroxide, it is likely that the two stimuli interact (Masson et al. 2012). This raises the intriguing possibility that oxidant stress, by inactivating a proportion of the expressed HIF hydroxylase enzyme, acts as a ‘range finding’ mechanism that alters the effective oxygen sensing ‘set point’. In this model, the rate of hydroxylation at any particular oxygen tension is reduced by oxidant stress, making the system more sensitive to hypoxia and switching on HIF pathways that may defend against the stress.
Range finding mechanisms; linking biochemistry to physiology
Such ‘range finding’ effects are likely to be of fundamental importance in linking the biochemistry of HIF hydroxylases to integrated physiology. Thus, a key principle for operation as ‘oxygen sensors’ is that oxygen-dependent rates of hydroxylation are matched to the rates of other processes involved in the regulation of HIF (Fig. 3). For instance, if rates of HIF-α translation and/or rates of HIF-α degradation downstream of the hydroxylation step were low, then hydroxylation of HIF-α might readily run to completion even in very severe hypoxia. The system would then not be oxygen regulated. Therefore the biochemical process of oxygen-dependent hydroxylation must be ‘on range’ to convey oxygen sensitivity on the physiological pathway.
Figure 3. The principle of ‘range finding’ processes that match HIF prolyl hydroxylation to the rate of synthesis of HIF and to the capacity of HIF degradation pathways downstream of hydroxylation.

NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; mTOR, mammalian target of rapamycin.
The need for ‘range finding’ mechanisms is emphasized by the diversity of the integrated physiology that is connected to HIF. This includes not only well-characterized aspects of oxygen homeostasis, such as cardiopulmonary and ventilatory control (Smith et al. 2008; Prabhakar & Semenza, 2012), but also diverse aspects of cell physiology, such as lymphocyte, neutrophil and macrophage functions that are not classically associated with hypoxia signalling but are dysregulated in animals bearing inactivating alleles of HIF-α isoforms (Cramer et al. 2003; Walmsley et al. 2005; Takeda et al. 2010; Dang et al. 2011). Although the exact role of tissue hypoxia in many of this processes is not known, it is clear that in the intact organism oxygen-dependent regulation of HIF can be observed in diverse tissues operating at strikingly different ‘set points’. This is well illustrated by studies in the kidney, which harbours particularly large oxygen gradients (Leichtweiss et al. 1969). Immunohistochemical studies of the induction of HIF-α protein in rodents subjected to anaemic/hypoxic stimulation reveal similar regulatory characteristics for HIF in tubular segments that are operating at tissue  levels that differ by an order of magnitude (Leichtweiss et al. 1969; Rosenberger et al. 2002). Although the integrative mechanisms that define tissue/cell-specific ‘range finding’ are not yet understood, many analyses have defined properties (both of the hydroxylases and connected pathways) that could contribute to such ‘range finding’ processes.
levels that differ by an order of magnitude (Leichtweiss et al. 1969; Rosenberger et al. 2002). Although the integrative mechanisms that define tissue/cell-specific ‘range finding’ are not yet understood, many analyses have defined properties (both of the hydroxylases and connected pathways) that could contribute to such ‘range finding’ processes.
Abundance and location of the HIF hydroxylases
Any process that alters the effective activity of the hydroxylase enzyme has the potential to change the oxygen sensing ‘set point’. Thus, it is of interest that transcriptional induction of the HIF prolyl hydroxylases by HIF itself is a highly conserved feature of the pathway, observed for at least one PHD enzyme in all species examined to date, from the ‘basal’ animal Trichoplax adherens to man (Epstein et al. 2001; Stiehl et al. 2006; Loenarz et al. 2011). Although, experimentally, these responses are usually analysed with respect to time (hence, they are described as a dynamic feedback response, which limits HIF activation over a period of time), they would be equally well fitted for a physiological role, perhaps in ‘range finding’ across cells operating at different oxygen tensions; i.e. in tissues operating at low oxygen levels, induction of PHD expression would lower the operating range for regulation by hydroxylation and match this range more closely to tissue oxygen tension.
Other controls over effective hydroxylase activity are likely to be exerted by intracellular processes that affect the subcellular location of the enzyme and/or its access to HIF substrate. For instance, binding of the principal HIF prolyl hydroxylase enzyme PHD2 to the peptidyl-prolyl cis-trans isomerase FKBP38 is proposed to limit PHD2 activity by a process that involves membrane anchoring and proteasomal degradation but is independent of the isomerase activity (Barth et al. 2009). It has also been proposed that PHD enzymes operate in a spatially restricted context through association with scaffold proteins (Foxler et al. 2012). Cellular compartmentalization may provide another control. For instance, it has been shown that PHD2 undergoes nuclear–cytoplasmic shuttling and that the deletion of PHD2 sequences required for nuclear entry is associated with a greatly impaired capacity to downregulate HIF (Pientka et al. 2012). Whether and how such processes are used to tune the PHD/pVHL/HIF system physiologically is unclear, but biological control at this level is supported by the observation of associations between nuclear localization of PHD2 and aggressive cancer phenotypes (Jokilehto et al. 2010).
Yet another possible means of regulating enzyme activity has been raised by recent studies of the HIF asparaginyl hydroxylase, FIH. This enzyme catalyses the hydroxylation of many other substrates, in particular asparaginyl residues that form part of the consensus ‘ankyrin repeat’ in the family of ‘ankyrin repeat domain’ containing proteins (Cockman et al. 2006). These proteins show a high affinity for FIH and are abundant in cells, so that they might be predicted to compete with HIF for hydroxylation (Coleman et al. 2007; Wilkins et al. 2009). Given that competition from ankyrin substrates will be dependent on the hydroxylation status of those substrates (itself determined by oxygen availability), this type of ‘endogenous competition’ could increase the complexity of the interface with oxygen levels and contribute to ‘range finding’.
Components of the VHL/HIF system
In addition to control of the ‘oxygen sensing’ hydroxylase enzymes, the integrated function of the PHD/pVHL/HIF pathway could potentially be tuned by multiple feedback controls operating at other steps in the pathway, which effectively downregulate the pathway after a period of HIF activation. Although these processes have generally been viewed as temporal feedback responses, they also have the potential to act as range finding mechanisms. They include the upregulation of an antisense HIF-1α transcript, which downregulates the HIF-1α coding mRNA (Rossignol et al. 2002; Uchida et al. 2004), induction of HIF-3α, an isoform that can antagonize HIF transcription by forming transcriptionally inactive complexes with other HIF-α proteins (Makino et al. 2001, 2007), and the induction of Rbx-2 (Tan et al. 2008), a component of the pVHL ubiquitin E3 ligase that promotes more rapid destruction of HIF-α proteins. In all of these processes, the upregulated ‘antagonist’ is a direct transcriptional target of HIF itself. Interestingly, given that they impinge differently on different HIF-α isoforms (HIF-1α and HIF-2α), which have different spectra of transcriptional targets, these mechanisms also have the potential to alter the character of the transcriptional output.
Finally, one crucial property of the PHD/pVHL/HIF system is that bidirectional signalling is achieved by the rapid resynthesis of HIF protein. Although the signalling pathways that regulate HIF protein synthesis have not been studied as intensively as pathways of degradation, it is clear that steady-state levels will reflect a balance between degradation and synthesis. Thus, at any given oxygen concentration (and rate of hydroxylation) the steady-state level of HIF (hence, the ‘oxygen sensing poise’ of the system) will be affected by the synthetic rate (Fig. 3). In this context, many growth promoting stimuli enhance HIF translation, presumably reflecting the need to entrain mechanisms for modulating basal oxygen homeostasis as demand is increased (Zundel et al. 2000; Laughner et al. 2001; Brugarolas & Kaelin, 2004).
Overall, whilst many mechanisms have been defined that could potentially provided ‘range finding’ controls for the oxygen sensing HIF hydroxylases, understanding whether and precisely how these operate to maintain oxygen homeostasis in large animals remains far from being completely understood. Nevertheless, genetic data clearly indicate that genes and physiological processes operating with different dynamics and at different oxygen tensions are regulated by the same HIF hydroxylase oxygen sensing system. This has some general implications for understanding interfaces between molecular and integrative physiology. In particular, it implies that inferring the existence of distinct molecular mechanisms from the quantitative properties of individual integrated physiological responses may be misleading (as witnessed by the erroneous view that erythropoietin was regulated by a ‘private’ oxygen sensing system in kidney cells). As a corollary, it suggests that precise predictions of effects on integrated physiology from a simple understanding of the molecular process of signal generation will also be difficult. This is now of fundamental importance in assessing the therapeutic potential of HIF hydroxylase inhibitors (Hewitson & Schofield, 2004; Fraisl et al. 2009); careful empirical analysis through experimental medicine and integrated physiological approaches will probably be required for this to succeed.
Hypoxia and cancer
Molecular analysis of the HIF hydroxylase system and hypoxia signalling pathways has revealed enormous complexity. In addition to the challenge of understanding the operation of these pathways in integrated physiology, this has major implications for the understanding of disease. In this final section, the importance of these physiological considerations is illustrated by reference to hypoxia and cancer.
It has long been recognized that solid tumours frequently contain regions of profound hypoxia and that tumour hypoxia is associated with adverse prognosis. This association has been reported to be independent of treatment modality (surgery, radiotherapy or chemotherapy) and to be observed in many different types of tumours (Höckel et al. 1993; Brizel et al. 1996). In keeping with these early observations, numerous studies have reported that activation of HIF is associated with aggressive tumour behaviour and adverse prognosis (Chi et al. 2006; Winter et al. 2007; for review see Semenza, 2003, 2012). These associations are extremely robust and, indeed, enhanced glucose uptake (in part, a consequence of transcriptional upregulation of specific glucose transporters and glycolytic enzymes by HIF; Semenza, 2010) underpins 18-fluorodeoxyglucose positron emission tomographic scanning in the diagnosis of cancer. Therefore, a central question has emerged concerning the role played by the activation of HIF signalling pathways in cancer and whether this drives the malignant phenotype.
Two classes of observation have lent support to this possibility. Firstly, as successive HIF targets have emerged from investigation of the molecular physiology of the system, it has become clear that many classical ‘cancer-associated’ properties (such as invasion and angiogenesis) involve the activation of HIF target genes (for review see Semenza, 2003). Secondly, many oncogenic pathways have been found to be mechanistically linked to the activation of HIF (Maxwell et al. 1999; Zundel et al. 2000; Laughner et al. 2001; Brugarolas & Kaelin, 2004; Bernardi et al. 2006). Most strikingly, mutation of the von Hippel-Lindau tumour suppressor (pVHL) disables the key ubiquitin E3 ligase complex that is necessary for degradation of HIF, leading to constitutive stabilization of HIF-α proteins and activation of the pathway (Maxwell et al. 1999). Other tumour suppressor and oncogenic pathways are linked to HIF activation through different mechanisms. For instance, classical tumour suppressor mutations in PTEN, promyelocytic leukaemia gene (PML) and TSC activate HIF by promoting translation of HIF-α proteins (Zundel et al. 2000; Brugarolas & Kaelin, 2004; Bernardi et al. 2006). Even unusual and poorly understood oncogenic pathways, such as those activated by mutations in the Krebs cycle enzymes fumarate hydratase and succinate dehydrogenase activate HIF (Isaacs et al. 2005; Pollard et al. 2005; Selak et al. 2005). In these conditions, HIF activation is a consequence of inhibition of the HIF prolyl hydroxylases by accumulated fumarate and/or succinate, which act as 2-oxoglutarate analogues (Hewitson et al. 2007; O’Flaherty et al. 2010). If oncogenic mutations activate HIF, HIF activation is associated with aggressive cancer, and HIF target genes include many with established oncogenic associations, it is therefore tempting to infer the existence of a simple connection whereby general activation of HIF causes cancer or at least causes cancer progression.
Oddly, however, despite the mechanistic links between oncogenic mutation and upregulation of HIF, activating mutations of HIF itself (for instance deletions or mutations in the degradation domain) have not been described in cancer genome sequencing programmes. Moreover, although in experimental systems genetic inactivation of HIF usually has a negative effect on tumour growth (reviewed by Semenza, 2003), this is not always the case. For instance, HIF activation is clearly observed in fumarate hydratase-associated human kidney cysts and cancer (Isaacs et al. 2005; Pollard et al. 2005). However, in a mouse model of the disease caused by kidney-specific inactivation of fumarate hydratase and characterized by hyperplastic renal cyst development, combined inactivation of HIF-1α in association with inactivation of fumarate hydratase exacerbates rather than suppresses disease (Adam et al.). The effect of inactivating a particular component of the HIF system also appears to be highly context specific. For instance, genetic inactivation of HIF-1α in transformed murine astrocytes has positive or negative effects on tumour growth depending on the microenvironment in which the cells are grown (Blouw et al. 2003).
In other situations, inactivation of different components of the HIF pathway has different or even opposing effects on tumour growth. This has been most clearly established in pVHL-defective clear cell renal cancer (CCRC). Inactivation of the pVHL ubiquitin E3 ligase leads to constitutive activation of the entire HIF pathway (Maxwell et al. 1999). However, genetic studies on CCRC cell lines grown as xenografts have clearly indicated that oncogenic drive is restricted to specific components of the HIF pathway. Thus, whilst HIF-2α overexpression promotes tumour growth, HIF-1α overexpression has the reverse effect (Maranchie et al. 2002; Kondo et al. 2003; Raval et al. 2005; Shen et al. 2011). Interestingly, in support of an antitumourigenic action of HIF-1α in this context, human VHL-defective CCRC is associated with a high prevalence of copy number reduction for a region of chromosome 14q containing HIF-1α, amongst other genes (Shen et al. 2011). Furthermore, cancer DNA sequencing has revealed a small but significant number of inactivating mutations in HIF-1α in VHL-defective CCRC (Morris et al. 2009; Dalgliesh et al. 2010). Large-scale studies of human CCRC predisposition have revealed polymorphisms at the HIF-2α locus and a HIF-2α-dependent transcriptional enhancer of cyclin D1 as CCRC susceptibility determinants (Purdue et al. 2010; Schödel et al.). Although it has been argued that HIF-2α is generally more protumourigenic than HIF-1α (Keith et al. 2012), and this is strongly supported by data in CCRC, protumourigenic effects of HIF-2α are again inconsistent, with inactivation of HIF-2α promoting tumour growth in other contexts (Acker et al. 2005; Majmundar et al. 2010).
Thus, despite extraordinarily strong and consistent associations between hypoxia, general activation of the HIF system and aggressive cancer, a simple causal association is not consistently supported by genetic evidence. Rather, these studies suggest that specific components (as opposed to general activation of the HIF system) are responsible for protumourigenic actions, that some components of the HIF system are antitumourigenic, and that pro- versus antitumourigenic actions of HIF are highly context specific. This review concludes with an outline of how consideration of the massive complexity of physiological hypoxic signalling may explain this paradox and consideration of the implications for the understanding of cancer and its treatment.
Given that increases in tissue mass create a demand for oxygen, growth pathways (which are dysregulated in cancer) are physiologically linked to the activation of HIF pathways in order to maintain oxygen homeostasis. However, physiological homeostatic responses to hypoxia may achieve their purpose by diverse mechanisms, encompassing both enhancement of oxygen delivery (e.g. angiogenesis, erythropoiesis) and restriction of oxygen demand (e.g. cytostasis, altered energy metabolism). As outlined above, recent analyses of HIF hydroxylase pathways have revealed that they are extraordinarily complex. When activated by oncogenic mutations or even by microenvironmental hypoxia in cancer, these massive pathways, which evolved to be fit for a physiological (not oncogenic) purpose, will be activated in their entirety. Whether they are promoting oncogenesis, neutral or even restricting the oncogenic process, all components of the pathway will be activated in the cancer as a consequence of the physiological ‘hard wiring’ of the network (Fig. 4). What are the implications of this?
Figure 4. Coselection of extensive hard-wired physiological pathways in cancer.
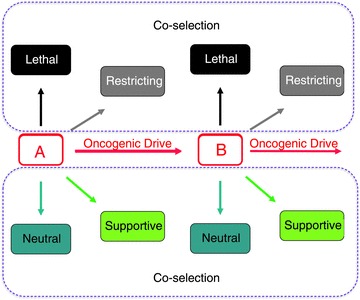
Oncogenic mutations (A) and (B) provide a cell with autonomous advantage, hence oncogenic drive, but are physiologically linked to pathways whose activation is supportive, neutral, restrictive or even lethal.
The first implication is simple; that tight associations may arise simply because of physiological ‘hard wiring’ and do not necessarily imply causation, explaining the discrepancies between the robust association of HIF activation with cancer and the variable findings in genetic intervention studies. Other implications are less obvious. For instance, coselection provides a potential explanation for the appearance in cancer of properties that are difficult to rationalize on the basis of the standard genetic model of cancer, which is classically considered to be a multistep process involving clonal selection of cells on the basis of cell autonomous advantage (Vogelstein & Kinzler, 1993). For instance, it is difficult to understand how such selection could operate to generate an ‘angiogenic switch’ (Hanahan & Folkman, 1996) to improve the blood supply to the tumour, because the advantage would be equally available to neighbouring cells in clonal competition. Coselection of physiologically hard-wired pathways connecting growth to oxygen supply (angiogenesis) would provide a more credible explanation (Fig. 5).
Figure 5.
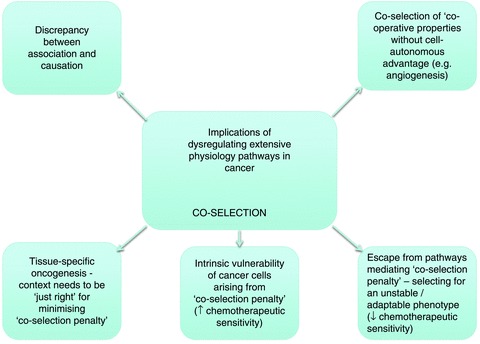
Implications of the physiological ‘coselection penalty’ in cancer
However, the most profound implications arise from consideration of the penalty that is entrained by the switching of massive physiological pathways, such as the hypoxia response, where not all components of the pathway are protumourigenic (Fig. 5). The balance between penalty and oncogenic advantage of dysregulating a physiological pathway is likely to be specific to the cellular context; in the case of hypoxia signalling pathways, reflecting cell-specific strategies for restoration of oxygen homeostasis. Such mechanisms would explain the contextual specificity of genetic interventions on the HIF system and (more generally) the striking tissue specificity of cancer predisposition that is often manifested by ubiquitously expressed tumour suppressors. For instance, the PHD/pVHL/HIF system is ubiquitous in animal cells, but VHL-associated cancer is highly tissue specific, being largely confined to renal cancer, phaeochromocytoma and ocular/CNS haemangioblastoma (reviewed by Kaelin, 2008). Moreover, across the mutational spectrum of VHL disease there is a strong correlation between the quantitative level of HIF dysregulation and the type of tumour predisposition (Clifford et al. 2001; Hoffman et al. 2001; Li et al. 2007; Shen et al. 2011). High-level HIF dysregulation appears to be inconsistent with the development of phaeochromocytoma but consistent (or even required) for renal clear cell carcinoma development. Thus, it appears that whether the HIF pathway is driving cancer or simply needing to be compatible with cancer development, the extent of activation needs to be ‘just right’, reflecting the need to balance the penalty incurred by coselection of ‘hard-wired’ physiological pathways against the oncogenic drive. Assuming this, one might expect alterations in the physiological pathway to become apparent during the course of cancer development. This is clearly illustrated in VHL-associated CCRC. Following VHL inactivation in kidney tubules, the HIF system shifts from being dominantly represented by HIF-1α (the normal renal tubular epithelial isoform) to being dominantly represented by HIF-2α (Mandriota et al. 2002; Rosenberger et al. 2002). The mechanisms behind the switch are largely unclear, but the shift appears to be associated with an alteration in the balance of activated HIF targets towards a more oncogenic profile (Raval et al. 2005; Keith & Simon, 2007).
I would like to conclude with two further speculations on the implications of these arguments for cancer therapy. Firstly, the need to avoid or limit the coselection penalty entrained by the activation of physiological pathways provides strong selective pressures for plasticity and heterogeneity. Given that multiple microenvironmental stresses (not limited to hypoxia) will be entrained as a solid tumour grows, it seems likely that the need to evade the penalty of the dysregulated activation of massive physiological pathways will select for the plasticity and heterogeneity that are characteristic of solid tumours and are considered to underlie escape from targeted chemotherapeutics. Secondly, the penalty of coselecting massive physiological pathways, such as the hypoxia response, makes cancer cells intrinsically vulnerable. For this reason alone, they may therefore manifest enhanced susceptibility to further stress, such as chemotherapeutic agents; i.e. the chemotherapeutic selectivity of many agents may be due (at least in part) to general cellular sensitivity of cancer cells, as much as to intrinsic selectivity of drug action. Thus, the penalty (and means of escape) from the antitumourigenic components of activating massive physiological pathways in cancer might account for much of the recent positive and negative experience in targeted cancer therapeutics; useful chemotherapeutic selectivity from agents that might otherwise be predicted to affect both neoplastic and non-neoplastic cells, and rapid escape from control in tumours that initially appear very sensitive to such agents.
Acknowledgments
Work in the author's laboratory was supported by the Wellcome Trust, the Biotechnology and Biological Sciences Research Council, Cancer Research UK and the Ludwig Institute for Cancer Research.
Glossary
- CCRC
clear cell renal cancer
- Epo
erythropoietin
- FIH
factor inhibiting HIF
- HIF
hypoxia-inducible factor
- 2-OG
2-oxoglutarate
- PHD
prolyl hydroxylase domain
- pVHL
von Hippel-Lindau tumour suppressor
References
- Acker T, Diez-Juan A, Aragones J, Tjwa M, Brusselmans K, Moons L, Fukumura D, Moreno-Murciano MP, Herbert JM, Burger A, Riedel J, Elvert G, Flamme I, Maxwell PH, Collen D, Dewerchin M, Jain RK, Plate KH, Carmeliet P. Genetic evidence for a tumor suppressor role of HIF-2α. Cancer Cell. 2005;8:131–141. doi: 10.1016/j.ccr.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, Stevens M, Fischer R, Carmeliet P, Maxwell PH, Pugh CW, Frizzell N, Soga T, Kessler BM, El-Bahrawy M, Ratcliffe PJ, Pollard PJ. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2 and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- Bachmann S, Le Hir M, Eckardt K-U. Co-localization of erythropoietin messenger RNA and ecto-5′-nucleotidase immunoreactivity in peritubular cells of rat renal cortex indicates that fibroblasts produce erythropoietin. J Histochem Cytochem. 1993;41:335–341. doi: 10.1177/41.3.8429197. [DOI] [PubMed] [Google Scholar]
- Barth S, Edlich F, Berchner-Pfannschmidt U, Gneuss S, Jahreis G, Hasgall PA, Fandrey J, Wenger RH, Camenisch G. Hypoxia-inducible factor prolyl-4-hydroxylase PHD2 protein abundance depends on integral membrane anchoring of FKBP38. J Biol Chem. 2009;284:23046–23058. doi: 10.1074/jbc.M109.032631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, Chandel NS. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol. 2007;177:1029–1036. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk L, Burchenal JH, Castle WB. Erythropoietic effect of cobalt in patients with or without anemia. N Engl J Med. 1949;240:754–761. doi: 10.1056/NEJM194905122401903. [DOI] [PubMed] [Google Scholar]
- Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, Cordon-Cardo C, Simon MC, Rafii S, Pandolfi PP. PML inhibits HIF-1α translation and neoangiogenesis through repression of mTOR. Nature. 2006;442:779–785. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouw B, Song H, Tihan T, Bosze J, Ferrara N, Gerber HP, Johnson RS, Bergers G. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell. 2003;4:133–146. doi: 10.1016/s1535-6108(03)00194-6. [DOI] [PubMed] [Google Scholar]
- Brizel DM, Scully SP, Harrelson JM, Layfield LJ, Bean JM, Prosnitz LR, Dewhirst MW. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996;56:941–943. [PubMed] [Google Scholar]
- Brugarolas J, Kaelin WG., Jr Dysregulation of HIF and VEGF is a unifying feature of the familial hamartoma syndromes. Cancer Cell. 2004;6:7–10. doi: 10.1016/j.ccr.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev. 1996;76:839–885. doi: 10.1152/physrev.1996.76.3.839. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi JT, Wang Z, Nuyten DS, Rodriguez EH, Schaner ME, Salim A, Wang Y, Kristensen GB, Helland A, Borresen-Dale AL, Giaccia A, Longaker MT, Hastie T, Yang GP, van de Vijver MJ, Brown PO. Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med. 2006;3:e47. doi: 10.1371/journal.pmed.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R, McDonough MA, Mecinović J, Loenarz C, Flashman E, Hewitson KS, Domene C, Schofield CJ. Structural basis for binding of hypoxia-inducible factor to the oxygen-sensing prolyl hydroxylases. Structure. 2009;17:981–989. doi: 10.1016/j.str.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH, Ratcliffe PJ, Maher ER. Contrasting effects on HIF-1α regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet. 2001;10:1029–1038. doi: 10.1093/hmg/10.10.1029. [DOI] [PubMed] [Google Scholar]
- Cockman ME, Lancaster DE, Stolze IP, Hewitson KS, McDonough MA, Coleman ML, Coles CH, Yu X, Hay RT, Ley SC, Pugh CW, Oldham NJ, Masson N, Schofield CJ, Ratcliffe PJ. Posttranslational hydroxylation of ankyrin repeats in IκB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH) Proc Natl Acad Sci U S A. 2006;103:14767–14772. doi: 10.1073/pnas.0606877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275:25733–25741. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- Coleman ML, McDonough MA, Hewitson KS, Coles C, Mecinovic J, Edelmann M, Cook KM, Cockman ME, Lancaster DE, Kessler BM, Oldham NJ, Ratcliffe PJ, Schofield CJ. Asparaginyl hydroxylation of the Notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. J Biol Chem. 2007;282:24027–24038. doi: 10.1074/jbc.M704102200. [DOI] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber H-P, Ferrara N, Johnson RS. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, Teague J, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Forbes S, Jia M, Jones D, Knott H, Kok CY, Lau KW, Leroy C, Lin ML, McBride DJ, Maddison M, Maguire S, McLay K, Menzies A, Mironenko T, Mulderrig L, Mudie L, O’Meara S, Pleasance E, Rajasingham A, Shepherd R, Smith R, Stebbings L, Stephens P, Tang G, Tarpey PS, Turrell K, Dykema KJ, Khoo SK, Petillo D, Wondergem B, Anema J, Kahnoski RJ, Teh BT, Stratton MR, Futreal PA. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dames SA, Martinez-Yamout M, Guzman RND, Dyson HJ, Wright PE. Structural basis for HIF-1α/CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci U S A. 2002;99:5271–5276. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, Luo W, Zeller K, Shimoda L, Topalian SL, Semenza GL, Dang CV, Pardoll DM, Pan F. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold I, Flügel D, Becht S, Belaiba RS, Bonello S, Hess J, Kietzmann T, Görlach A. The hypoxia-inducible factor-2α is stabilized by oxidative stress involving NOX4. Antioxid Redox Signal. 2010;13:425–436. doi: 10.1089/ars.2009.3014. [DOI] [PubMed] [Google Scholar]
- Ehrismann D, Flashman E, Genn DN, Mathioudakis N, Hewitson KS, Ratcliffe PJ, Schofield CJ. Studies on the activity of the hypoxia-inducible factor hydroxylases using an oxygen consumption assay. Biochem J. 2007;401:227–234. doi: 10.1042/BJ20061151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ. Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1α. J Biol Chem. 2003;278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian Y-M, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologues define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Firth JD, Ebert BL, Pugh CW, Ratcliffe PJ. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: similarities with the erythropoeitin 3′ enhancer. Proc Natl Acad Sci U S A. 1994;91:6496–6500. doi: 10.1073/pnas.91.14.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flashman E, Hoffart LM, Hamed RB, Bollinger JM, Jr, Krebs C, Schofield CJ. Evidence for the slow reaction of hypoxia-inducible factor prolyl hydroxylase 2 with oxygen. FEBS J. 2010;277:4089–4099. doi: 10.1111/j.1742-4658.2010.07804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foxler DE, Bridge KS, James V, Webb TM, Mee M, Wong SC, Feng Y, Constantin-Teodosiu D, Petursdottir TE, Bjornsson J, Ingvarsson S, Ratcliffe PJ, Longmore GD, Sharp TV. The LIMD1 protein bridges an association between the prolyl hydroxylases and VHL to repress HIF-1 activity. Nat Cell Biol. 2012;14:201–208. doi: 10.1038/ncb2424. [DOI] [PubMed] [Google Scholar]
- Fraisl P, Aragones J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov. 2009;8:139–152. doi: 10.1038/nrd2761. [DOI] [PubMed] [Google Scholar]
- Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. Hypoxia requires Notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Hewitson KS, Lienard BM, McDonough MA, Clifton IJ, Butler D, Soares AS, Oldham NJ, McNeill LA, Schofield CJ. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J Biol Chem. 2007;282:3293–3301. doi: 10.1074/jbc.M608337200. [DOI] [PubMed] [Google Scholar]
- Hewitson KS, McNeill LA, Riordan MV, Tian Y-M, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ. Hypoxia inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277:26351–26355. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- Hewitson KS, Schofield CJ. The HIF pathway as a therapeutic target. Drug Discov Today. 2004;9:704–711. doi: 10.1016/S1359-6446(04)03202-7. [DOI] [PubMed] [Google Scholar]
- Hirsilä M, Koivunen P, Günzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor HIF. J Biol Chem. 2003;278:30772–30780. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- Hirsilä M, Koivunen P, Xu L, Seeley T, Kivirikko KI, Myllyharju J. Effect of desferrioxamine and metals on the hydroxylases in the oxygen sensing pathway. FASEB J. 2005;19:1308–1310. doi: 10.1096/fj.04-3399fje. [DOI] [PubMed] [Google Scholar]
- Höckel M, Knoop C, Schlenger K, Vorndran B, Baussmann E, Mitze M, Knapstein PG, Vaupel P. Intratumoral pO2 predicts survival in advanced cancer of the uterine cervix. Radiother Oncol. 1993;26:45–50. doi: 10.1016/0167-8140(93)90025-4. [DOI] [PubMed] [Google Scholar]
- Hoffman DL, Salter JD, Brookes PS. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: implications for hypoxic cell signaling. Am J Physiol Heart Circ Physiol. 2007;292:H101–H108. doi: 10.1152/ajpheart.00699.2006. [DOI] [PubMed] [Google Scholar]
- Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M, Kaelin WG., Jr von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet. 2001;10:1019–1027. doi: 10.1093/hmg/10.10.1019. [DOI] [PubMed] [Google Scholar]
- Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY. Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL. Nature. 2002;417:975–978. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependent domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes BT, Espenshade PJ. Oxygen-regulated degradation of fission yeast SREBP by Ofd1, a prolyl hydroxylase family member. EMBO J. 2008;27:1491–1501. doi: 10.1038/emboj.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe PJ, Lineham M, Neckers L. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian Y-M, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Jelkmann W. Erythropoietin: structure, control of production, and function. Physiol Rev. 1992;72:449–489. doi: 10.1152/physrev.1992.72.2.449. [DOI] [PubMed] [Google Scholar]
- Jiang B-H, Zheng JZ, Leung SW, Roe R, Semenza GL. Transactivation and inhibitory domains of hypoxia-inducible factor 1α. Modulation of transcriptional activity by oxygen tension. J Biol Chem. 1997;272:19253–19260. doi: 10.1074/jbc.272.31.19253. [DOI] [PubMed] [Google Scholar]
- Jokilehto T, Högel H, Heikkinen P, Rantanen K, Elenius K, Sundström J, Jaakkola PM. Retention of prolyl hydroxylase PHD2 in the cytoplasm prevents PHD2-induced anchorage-independent carcinoma cell growth. Exp Cell Res. 2010;316:1169–1178. doi: 10.1016/j.yexcr.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–472. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles HJ, Mole DR, Ratcliffe PJ, Harris AL. Normoxic stabilization of hypoxia-inducible factor-1α by modulation of the labile iron pool in differentiating U937 macrophages: effect of natural resistance-associated macrophage protein 1. Cancer Res. 2006;66:2600–2607. doi: 10.1158/0008-5472.CAN-05-2351. [DOI] [PubMed] [Google Scholar]
- Knowles HJ, Raval RR, Harris AL, Ratcliffe PJ. Effect of ascorbate on the activity of hypoxia inducible factor (HIF) in cancer cells. Cancer Res. 2003;63:1764–1768. [PubMed] [Google Scholar]
- Koivunen P, Hirsilä M, Kivirikko KI, Myllyharju J. The length of peptide substrates has a marked effect on hydroxylation by the hypoxia-inducible factor prolyl 4-hydroxylases. J Biol Chem. 2006;281:28712–28720. doi: 10.1074/jbc.M604628200. [DOI] [PubMed] [Google Scholar]
- Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem. 2007;282:4524–4532. doi: 10.1074/jbc.M610415200. [DOI] [PubMed] [Google Scholar]
- Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr Inhibition of HIF2α is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1:439–444. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1α induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23:1949–1956. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper C, Molenaar IG, Dachs GU, Currie MJ, Sykes PH, Vissers MC. Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res. 2010;70:5749–5758. doi: 10.1158/0008-5472.CAN-10-0263. [DOI] [PubMed] [Google Scholar]
- Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, Agosto-Perez FJ, Davuluri R, Liu CG, Croce CM, Negrini M, Calin GA, Ivan M. A microRNA signature of hypoxia. Mol Cell Biol. 2007;27:1859–1867. doi: 10.1128/MCB.01395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002a;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain: a hypoxic switch. Science. 2002b;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1α (HIF-1α) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leichtweiss H-P, Lübbers DW, Weiss C, Baumgärtl N, Reschke W. The oxygen supply of the rat kidney: measurements of intrarenal pO2. Pflugers Arch. 1969;309:328–349. doi: 10.1007/BF00587756. [DOI] [PubMed] [Google Scholar]
- Li L, Zhang L, Zhang X, Yan Q, Minamishima YA, Olumi AF, Mao M, Bartz S, Kaelin WG., Jr Hypoxia-inducible factor linked to differential kidney cancer risk seen with type 2A and type 2B VHL mutations. Mol Cell Biol. 2007;27:5381–5392. doi: 10.1128/MCB.00282-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loenarz C, Coleman ML, Boleininger A, Schierwater B, Holland PWH, Ratcliffe PJ, Schofield CJ. The hypoxia-inducible transcription factor pathway regulates oxygen sensing in the simplest animal, Trichoplax adhaerens. EMBO Rep. 2011;12:63–70. doi: 10.1038/embor.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loenarz C, Mecinović J, Chowdhury R, McNeill LA, Flashman E, Schofield CJ. Evidence for a stereoelectronic effect in human oxygen sensing. Angew Chem Int Ed Engl. 2009;48:1784–1787. doi: 10.1002/anie.200805427. [DOI] [PubMed] [Google Scholar]
- Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol. 2008;4:152–156. doi: 10.1038/nchembio0308-152. [DOI] [PubMed] [Google Scholar]
- Loenarz C, Schofield CJ. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem Sci. 2011;36:7–18. doi: 10.1016/j.tibs.2010.07.002. [DOI] [PubMed] [Google Scholar]
- McNeill LA, Flashman E, Buck MR, Hewitson KS, Clifton IJ, Jeschke G, Claridge TD, Ehrismann D, Oldham NJ, Schofield CJ. Hypoxia-inducible factor prolyl hydroxylase 2 has a high affinity for ferrous iron and 2-oxoglutarate. Mol Biosyst. 2005;1:321–324. doi: 10.1039/b511249b. [DOI] [PubMed] [Google Scholar]
- Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, Cao Y, Berkenstam A, Poellinger L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001;414:550–554. doi: 10.1038/35107085. [DOI] [PubMed] [Google Scholar]
- Makino Y, Uenishi R, Okamoto K, Isoe T, Hosono O, Tanaka H, Kanopka A, Poellinger L, Haneda M, Morimoto C. Transcriptional up-regulation of inhibitory PAS domain protein gene expression by hypoxia-inducible factor 1 (HIF-1): a negative feedback regulatory circuit in HIF-1-mediated signaling in hypoxic cells. J Biol Chem. 2007;282:14073–14082. doi: 10.1074/jbc.M700732200. [DOI] [PubMed] [Google Scholar]
- Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, Wykoff CC, Maher ER, Harris AL, Ratcliffe PJ, Maxwell PH. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor funtion in the nephron. Cancer Cell. 2002;1:459–468. doi: 10.1016/s1535-6108(02)00071-5. [DOI] [PubMed] [Google Scholar]
- Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD. The contribution of VHL substrate binding and HIF1-α to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002;1:247–255. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]
- Masson N, Singleton RS, Sekirnik R, Trudgian DC, Ambrose LJ, Miranda MX, Tian YM, Kessler BM, Schofield CJ, Ratcliffe PJ. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep. 2012;12:251–257. doi: 10.1038/embor.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197–5206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Osmond MK, Pugh CW, Heryet A, Nicholls LG, Tan CC, Doe BG, Ferguson DJP, Johnson MH, Ratcliffe PJ. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int. 1993a;44:1149–1162. doi: 10.1038/ki.1993.362. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Pugh CW, Ratcliffe PJ. Inducible operation of the erythropoietin 3′ enhancer in multiple cell lines: evidence for a widespread oxygen sensing mechanism. Proc Natl Acad Sci U S A. 1993b;90:2423–2427. doi: 10.1073/pnas.90.6.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Min J-H, Yang H, Ivan M, Gertler F, Kaelin WG, Jr, Pavletich NP. Structure of an HIF-1α-pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, Ratcliffe PJ. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284:16767–16775. doi: 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MR, Hughes DJ, Tian YM, Ricketts CJ, Lau KW, Gentle D, Shuib S, Serrano-Fernandez P, Lubinski J, Wiesener MS, Pugh CW, Latif F, Ratcliffe PJ, Maher ER. Mutation analysis of hypoxia-inducible factors HIF1A and HIF2A in renal cell carcinoma. Anticancer Res. 2009;29:4337–4343. [PubMed] [Google Scholar]
- Nagao M, Ebert BL, Ratcliffe PJ, Pugh CW. Drosophila melanogaster SL2 cells contain a hypoxically inducible DNA binding complex which recognises mammalian HIF-1 binding sites. FEBS Lett. 1996;387:161–166. doi: 10.1016/0014-5793(96)00484-x. [DOI] [PubMed] [Google Scholar]
- Necas E, Thorling EB. Unresponsiveness of erythropoietin-producing cells to cyanide. Am J Physiol. 1972;222:1187–1190. doi: 10.1152/ajplegacy.1972.222.5.1187. [DOI] [PubMed] [Google Scholar]
- O’Flaherty L, Adam J, Heather LC, Zhdanov AV, Chung YL, Miranda MX, Croft J, Olpin S, Clarke K, Pugh CW, Griffiths J, Papkovsky D, Ashrafian H, Ratcliffe PJ, Pollard PJ. Dysregulation of hypoxia pathways in fumarate hydratase-deficient cells is independent of defective mitochondrial metabolism. Hum Mol Genet. 2010;19:3844–3851. doi: 10.1093/hmg/ddq305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel-Lindau protein. Nature Cell Biology. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ, Simon MC. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol. 2007;27:912–925. doi: 10.1128/MCB.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pientka FK, Hu J, Schindler SG, Brix B, Thiel A, Joehren O, Fandrey J, Berchner-Pfannschmidt U, Depping R. Oxygen sensing by prolyl-4-hydroxylase PHD2 within the nuclear compartment and the influence of compartimentalisation on HIF-1 signalling. J Cell Sci. 2012;125:5168–5176. doi: 10.1242/jcs.109041. [DOI] [PubMed] [Google Scholar]
- Pollard PJ, Brière JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IP. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev. 2012;92:967–1003. doi: 10.1152/physrev.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- Pugh CW, O’Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ. Activation of hypoxia inducible factor-1; definition of regulatory domains within the a subunit. J Biol Chem. 1997;272:11205–11214. doi: 10.1074/jbc.272.17.11205. [DOI] [PubMed] [Google Scholar]
- Purdue MP, Johansson M, Zelenika D, Toro JR, Scelo G, Moore LE, Prokhortchouk E, Wu X, Kiemeney LA, Gaborieau V, Jacobs KB, Chow WH, Zaridze D, Matveev V, Lubinski J, Trubicka J, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Bucur A, Bencko V, Foretova L, Janout V, Boffetta P, Colt JS, Davis FG, Schwartz KL, Banks RE, Selby PJ, Harnden P, Berg CD, Hsing AW, Grubb RL, 3rd, Boeing H, Vineis P, Clavel-Chapelon F, Palli D, Tumino R, Krogh V, Panico S, Duell EJ, Quirós JR, Sanchez MJ, Navarro C, Ardanaz E, Dorronsoro M, Khaw KT, Allen NE, Bueno-de-Mesquita HB, Peeters PH, Trichopoulos D, Linseisen J, Ljungberg B, Overvad K, Tjønneland A, Romieu I, Riboli E, Mukeria A, Shangina O, Stevens VL, Thun MJ, Diver WR, Gapstur SM, Pharoah PD, Easton DF, Albanes D, Weinstein SJ, Virtamo J, Vatten L, Hveem K, Njølstad I, Tell GS, Stoltenberg C, Kumar R, Koppova K, Cussenot O, Benhamou S, Oosterwijk E, Vermeulen SH, Aben KK, van der Marel SL, Ye Y, Wood CG, Pu X, Mazur AM, Boulygina ES, Chekanov NN, Foglio M, Lechner D, Gut I, Heath S, Blanche H, Hutchinson A, Thomas G, Wang Z, Yeager M, Fraumeni JF, Jr, Skryabin KG, McKay JD, Rothman N, Chanock SJ, Lathrop M, Brennan P. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat Genet. 2010;43:60–65. doi: 10.1038/ng.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger C, Mandriota SJ, Jürgensen JS, Wiesener MS, Horstrup JH, Frei U, Ratcliffe PJ, Maxwell PH, Bachmann S, Eckardt KU. Expression of hypoxia-inducible factor-1α and -2α in hypoxic and ischemic rat kidneys. J Am Soc Nephrol. 2002;13:1721–1732. doi: 10.1097/01.asn.0000017223.49823.2a. [DOI] [PubMed] [Google Scholar]
- Rossignol F, Vaché C, Clottes E. Natural antisense transcripts of hypoxia-inducible factor 1α are detected in different normal and tumour human tissues. Gene. 2002;299:135–140. doi: 10.1016/s0378-1119(02)01049-1. [DOI] [PubMed] [Google Scholar]
- Schödel J, Bardella C, Sciesielski LK, Brown JM, Pugh CW, Buckle V, Tomlinson IP, Ratcliffe PJ, Mole DR. Common genetic variants at the 11q13.3 renal cancer susceptibility locus influence binding of HIF to an enhancer of cyclin D1 expression. Nat Genet. 2012a;44:420–425. doi: 10.1038/ng.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2012b;117:e207–e217. doi: 10.1182/blood-2010-10-314427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207–214. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cellular Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Beroukhim R, Schumacher SE, Zhou J, Chang M, Signoretti S, Kaelin WG., Jr Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer Discov. 2011;1:222–235. doi: 10.1158/2159-8290.CD-11-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TG, Robbins PA, Ratcliffe PJ. The human side of hypoxia-inducible factor. Br J Haematol. 2008;141:325–334. doi: 10.1111/j.1365-2141.2008.07029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiehl DP, Wirthner R, Köditz J, Spielmann P, Camenisch G, Wenger RH. Increased proyl 4-hydroxylase domain proteins compensate for decreased oxygen levels. J Biol Chem. 2006;281:23482–23491. doi: 10.1074/jbc.M601719200. [DOI] [PubMed] [Google Scholar]
- Takeda N, O’Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, Asagiri M, Simon MC, Hoffmann A, Johnson RS. Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24:491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Gu Q, He H, Pamarthy D, Semenza GL, Sun Y. SAG/ROC2/RBX2 is a HIF-1 target gene that promotes HIF-1α ubiquitination and degradation. Oncogene. 2008;27:1404–1411. doi: 10.1038/sj.onc.1210780. [DOI] [PubMed] [Google Scholar]
- Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1α by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000;19:4298–4309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Yeoh KK, Lee MK, Ericsson T, Kessler BM, Kramer HB, Edelmann MJ, William C, Pugh CW, Schofield CJ, Ratcliffe PJ. Differential sensitivity of HIF hydroxylation sites to hypoxia and hydroxylase inhibitors. J Biol Chem. 2011;286:13041–13051. doi: 10.1074/jbc.M110.211110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida T, Rossignol F, Matthay MA, Mounier R, Couette S, Clottes E, Clerici C. Prolonged hypoxia differentially regulates HIF-1α and HIF-2α expression in lung epithelial cells: implication of HIF-1α natural antisense. J Biol Chem. 2004;279:14871–14878. doi: 10.1074/jbc.M400461200. [DOI] [PubMed] [Google Scholar]
- Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, Payette J, Holcik M, Pause A, Lee S. An oxygen-regulated switch in the protein synthesis machinery. Nature. 2012;486:126–129. doi: 10.1038/nature11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaupel P, Schlenger K, Knoop C, Höckel M. Oxygenation of human tumors: evaluation of tissue oxygen distribution in breast cancers by computerized O2 tension measurements. Cancer Res. 1991;51:3316–3322. [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9:138–141. doi: 10.1016/0168-9525(93)90209-z. [DOI] [PubMed] [Google Scholar]
- Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER. Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-κB activity. J Exp Med. 2005;201:105–115. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Jiang B-H, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic–helix–loop–helix–PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993;90:4304–4308. doi: 10.1073/pnas.90.9.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West CM, van der Wel H, Wang ZA. Prolyl 4-hydroxylase-1 mediates O2 signaling during development of Dictyostelium. Development. 2007;134:3349–3358. doi: 10.1242/dev.000893. [DOI] [PubMed] [Google Scholar]
- Wilkins SE, Hyvärinen J, Chicher J, Gorman JJ, Peet DJ, Bilton RL, Koivunen P. Differences in hydroxylation and binding of Notch and HIF-1α demonstrate substrate selectivity for factor inhibiting HIF-1 (FIH-1) Int J Biochem Cell Biol. 2009;41:1563–1571. doi: 10.1016/j.biocel.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Winter SC, Buffa FM, Silva P, Miller C, Valentine HR, Turley H, Shah KA, Cox GJ, Corbridge RJ, Homer JJ, Musgrove B, Slevin N, Sloan P, Price P, West CM, Harris AL. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res. 2007;67:3441–3449. doi: 10.1158/0008-5472.CAN-06-3322. [DOI] [PubMed] [Google Scholar]
- Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, Kung AL. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci U S A. 2009;106:4260–4265. doi: 10.1073/pnas.0810067106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, Stokoe D, Giaccia AJ. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000;14:391–396. [PMC free article] [PubMed] [Google Scholar]