

Abstract
This review focuses on how smooth muscle sarcoplasmic reticulum (SR), the major releasable Ca2+ store in these cells, performs its many functions by communicating with the plasma membrane (PM) and other organelles across cytoplasmic nanospaces, defined by membrane–membrane junctions less than 50 nm across. In spite of accumulating evidence in favour of the view that cytoplasmic nanospaces are a prerequisite for effective control of diverse cellular functions, our current understanding of how smooth muscle cells accomplish site- and function-specific Ca2+ signalling remains in its infancy. We first present evidence in support of the view that effective Ca2+ signalling depends on the restricted diffusion of Ca2+ within cytoplasmic nanospaces. We then develop an evidence-based model of the smooth muscle SR – the ‘pan-junctional SR’ model – that incorporates a network of tubules and quilts that are capable of auto-regulating their Ca2+ content and determining junctional [Ca2+]i through loading and unloading at membrane–membrane nanojunctions. Thereby, we provide a novel working hypothesis in order to inform future investigation into the control of a variety of cellular functions by local Ca2+ signals at junctional nanospaces, from contraction and energy metabolism to nuclear transcription. Based on the current literature, we discuss the molecular mechanisms whereby the SR mediates these multiple functions through the interaction of ion channels and pumps embedded in apposing membranes within inter-organellar junctions. We finally highlight the fact that although most current hypotheses are qualitatively supported by experimental data, solid quantitative simulations are seriously lacking. Considering that at physiological concentrations the number of calcium ions in a typical junctional nanospace between the PM and SR is of the order of 1, ion concentration variability plays a major role as the currency of information transfer and stochastic quantitative modelling will be required to both test and develop working hypotheses.
 |
Mark Evans (left) began his investigations into the regulation of cell and organ function by ion channels and calcium with his PhD in 1990 at the University of Edinburgh, under the supervision of Prof. B. L. Ginsborg. Subsequently, he has focused on pulmonary arterial smooth muscle as a Wellcome Trust Research Fellow at the University of Oxford. He was appointed to a Lectureship at the University of St Andrews in 2001, and then to the Chair of Cellular Pharmacology at the University of Edinburgh in 2009. Nicola Fameli (centre) obtained his PhD in 2000 from the Department of Physics and Astronomy of the University of British Columbia (UBC) in Vancouver, Canada, in experimental condensed matter physics. In the Dept of Anaesthesiology, Pharmacology and Therapeutics at UBC, under the supervision of Prof. Cornelis van Breemen, he embarked on quantitative physiology, including the development of realistic stochastic simulations of cation transport in cytoplasmic nanospaces during activation of vascular smooth muscle cells. Cornelis (Casey) van Breemen (right) obtained his DVM from the University of Toronto. He became interested in Ca2+ signalling and obtained his PhD at the University of Alberta under the supervision of Prof. E. E. Daniel. He is currently Professor Emeritus in the Dept of Anesthesiology, Pharmacology and Therapeutics at UBC, and Senior Emeritus at the Child and Family Research Institute also in Vancouver, researching the effects of Marfan Syndrome and ageing on vascular Ca2+ signalling at the Child and Family Research Institute.
Multiple smooth muscle activating mechanisms
Excitation–contraction coupling in all types of muscle (skeletal, cardiac and smooth) is largely controlled by interactions between voltage-gated Ca2+ channels (VGCCs) in the plasma membrane (PM) or its invaginations (T-tubules or caveolae) and Ca2+ release channels in the sarcoplasmic reticulum (SR). However, in smooth muscle cellular membrane interactions appear much more varied and crosstalk between the SR and other organelles, such as lysosomes and mitochondria also appears to contribute significantly to shaping the Ca2+ signal. In addition, smooth muscles display far greater heterogeneity and plasticity than skeletal and cardiac muscles. This should not be surprising considering that smooth muscle supplies the physical force for all homeostatic and reproductive functions in the body from functional hyperaemia in the brain for providing food for thought, to the delivery of babies to preserve our species. Not only does the function of smooth muscle vary from organ to organ, but it can also change over time in the same organ. Notable examples of such plasticity are uterine smooth muscle, which changes from quiescent to highly reactive during consecutive stages of pregnancy and birth, and the changes seen in arterial smooth muscle phenotype, from contractile to proliferative and migratory, during the cycle of injury and wound healing. Since these multiple functions are all controlled by Ca2+ signalling the overarching question is: how can fluctuations in the concentration of one ion, Ca2+, exert such selective and multifaceted control? The generally accepted answer to this question is that both spatial and temporal details of the Ca2+ transients code for selective modulation of
molecular targets and thereby transduce their multiple physiological effects. The underlying mechanisms are complex in nature, requiring strategic spatial positioning of cellular Ca2+ transporters and targets, each of which may be characterized by different kinetics and affinities for Ca2+ (Clark et al. 2010). Detailed discussion of the various channels and transporters that regulate SR-mediated Ca2+ signalling in smooth muscle has recently been provided by others (Wray & Burdyga, 2010). In this short review, therefore, we focus on how junctions of the main Ca2+ regulatory organelle, the sarcoplasmic reticulum (SR), provide cytoplasmic nanospaces, in which highly localized Ca2+ signals can be generated to select for different smooth muscle functions, from contraction and relaxation to gene expression and cell division. The main emphasis will be on vascular smooth muscle, while other types are briefly mentioned for comparison.
We propose that a ‘pan-junctional SR’ forms many different types of nanojunction with the PM, mitochondria, lysosomes and possibly other organelles, each performing separate, but coordinated functions. Figure 1 illustrates that smooth muscle SR may have eleven and possibly more types of nanojunction, hence the title ‘pan-junctional SR’. The most abundant PM–SR junctions selectively regulate luminal calcium concentration ([Ca2+]SR), hyperpolarization and relaxation, depolarization and vasomotion; the mitochondria–SR junctions regulate mitochondrial energy metabolism, apoptosis and SR Ca2+ loading, while the lysosomal–SR junction is essential for nicotinic acid adenine dinucleotide phosphate (NAADP)-initiated calcium-induced calcium release (CICR) from the SR, which may in turn modulate, for example, autophagy and cholesterol metabolism.
Figure 1.
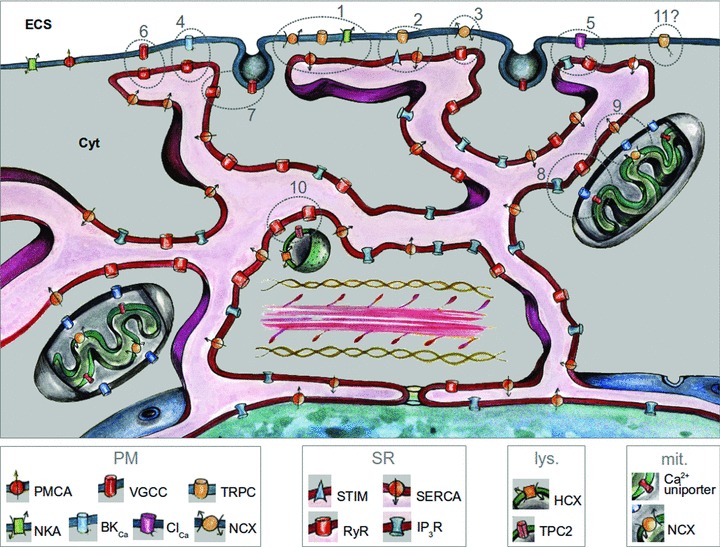
The above graphic illustration of the hypothetical pan-junctional SR features multiple nanojunctions between the SR, on the one hand, and the PM, mitochondria and lysosomes, on the other. Each nanojunction is identified with a number corresponding with its description in the text below. The ion transporter (codes shown below the picture) content of each junction is based on experimental evidence in the literature, which varies from solid to hypothetical. ECS: extra-cellular space; cyt: cytoplasm.
Nanojunctions
The first nanojunction described in terms of its functional importance was the neuromuscular junction, and there can be no doubt as to how important this was to our understanding of neurotransmitter release and function (Del Castillo & Katz, 1956). However, until recently little attention has been given to the presence, function and plasticity of nanojunctions between intracellular membranes. Perhaps the one exception is in skeletal and cardiac muscles, where the importance to excitation–contraction coupling of the junctional complexes formed between the T-tubules of the sarcolemma and terminal cisternae of the SR is well recognized. The essence of a nanojunction, as suggested from observations in several cell types (Rosenbluth, 1962; Franzini-Armstrong, 1964; Ramesh et al. 1998), is that portions of two biological membranes, typically a few 100 nm in extension and each containing ion transporters and sensors, are separated by a highly structured cytoplasmic space 10–30 nm wide. Both the ultra-structure and electrostatic properties of the nanojunction, together with the composition of transport molecules embedded in their limiting membranes, ensure that cytoplasmic cation concentrations, [Ca2+] in particular, are locally determined. Cations may thus target sites of different affinities and modulate function appropriately.
Pan-junctional SR in vascular smooth muscle
In the early seventies the first convincing electron micrographs of smooth muscle SR revealed that in a number of peripheral areas of the long thin cells only a narrow gap of approximately 20 nm separated the SR from the PM (Gabella, 1971; Devine et al. 1972). A few years later careful 45Ca2+ measurements, employing La3+ quenching of extracellular Ca2+, demonstrated the existence of peripheral cytoplasmic domains between the PM and peripheral SR, which were characterized by restricted diffusion (van Breemen, 1977). Once Ca2+ entered such a nanospace it was either pumped into the SR or diffused into the bulk myoplasm. The same study also showed that both the rates of Ca2+ entry through PM channels and the rate of SR Ca2+ uptake from the restricted peripheral cytoplasmic space determined the fraction of Ca2+ entry that was captured by the SR (see below for recent quantitative modelling). Since then progress has been slow and the concept that site- and function-specific Ca2+ signals may be supported by nanojunctions has received little attention. Perhaps we are now ready for a sea-change, given that the organelle-targeted fluorescent indicators developed in the past two decades (Rizzuto et al. 1993; Miyawaki et al. 1999) have opened the doors to elucidating the highly localized Ca2+ signalling events about the interface of junctional membrane pairs. We must ‘mind the gap’, however, as the dimensions of the nanojunctions are still well below the resolution of light microscopy and most supporting evidence remains indirect in nature. For this reason we will illustrate a stochastic modelling approach that helps formulate and test our hypotheses.
Our ‘pan-junctional SR’ hypothesis is encapsulated in Fig. 1, which illustrates a variety of nanojunctions, and the specific transporters that participate in shaping cation concentration transients within and between them. Below we describe their specific roles in regulating vascular smooth muscle function and the ionic mechanisms by which Ca2+ signalling within each nanospace may be modulated. Throughout the following discussion it is important to realize that the Ca2+ transients within the junctional nanospaces are segregated from those in the bulk myoplasm that determine contractile activity. Several factors participate in restricting [Ca2+] transients to nanojunctions:
The geometry of the junctions, especially the distance between membranes, appears to control the retention of Ca2+ in the nanospace, as suggested by preliminary models (Fameli et al. 2007);
The relatively low diffusivity of (free or buffered) cytosolic Ca2+ (Kushmerick & Podolsky, 1969; Allbritton et al. 1992), in combination with the above-mentioned restricted geometry, favours Ca2+ buffering by nanojunctions;
The kinetics of Ca2+ sinks in the junctions, is another important element, tightly linked to the previous two factors; for example, if, as predicted, sarco/endoplasmic reticulum Ca2+ ATPase 2b (SERCA2b) is resident within PM–SR junctions of pulmonary arterial smooth muscle, its high affinity for Ca2+ (Verboomen et al. 1992) may provide a barrier to Ca2+ flux between the PM and the myofilaments and vice versa (Clark et al. 2010);
There is ample evidence of electron opaque junction-spanning structures that are likely to provide physical obstacles to ion mobility in the junctions by increasing path tortuosity (Devine et al. 1972; Poburko et al. 2008); this is likely to be a more restrictive effect for diffusion than actual chemical buffering, since Ca2+ buffers likely contribute little at these spatial scales (Allbritton et al. 1992; Allbritton & Meyer, 1993).
However, it is important to note that even though the Ca2+ signalling domain for contraction may be of a larger scale, its distribution is far from homogeneous. Separate PM regions have been described for filament attachment and caveolae (Moore et al. 2004) and the density of myosin filaments appears to be less in the cell periphery than central myoplasm (Lee et al. 2002). In addition, it was shown that the functional Ca2+-binding protein calmodulin is tethered to the myofilaments rather than free in solution (Wilson et al. 2002).
SR auto-regulation
For the SR to control and coordinate local cytoplasmic Ca2+ signals it must to some degree be able to regulate its luminal Ca2+ content independently from fluctuations in cytoplasmic [Ca2+]. This is accomplished, in part, by PM–SR junctions, which may load the SR from the extracellular space to replenish it during activating waves of SR Ca2+ release (Lee et al. 2001) or extrude Ca2+ from the SR when it is resting, overloaded with Ca2+ (Nazer & van Breemen, 1998) or signalled to do so by vasodilators (Boittin et al. 2003).
SR loading (Fig. 1: 1 and 2)
Consistent with smooth muscle heterogeneity and plasticity, refilling of the SR from the extracelluar space during stimulated Ca2+ release may be achieved by a variety of different mechanisms. We will focus our attention on one well-documented mechanism, which is illustrated in Fig. 1: 1. In this instance, the process is initiated by the opening of non-selective cation-permeable, receptor-operated channels (ROCs; e.g. transient receptor potential canonical channel TRPC6). These deliver Na+ to the junctional nanospace in a manner coupled to Ca2+ entry mode Na+/Ca2+ exchangers (NCX) in the PM, and thus supply Ca2+ to SERCA on the peripheral SR membrane (Lemos et al. 2007; Poburko et al. 2007). Both depolarization and local Na+ accumulation favour reversal of NCX, and a transient rise in junctional [Na+] may be facilitated by the low-Na+-affinity α2 and α3 isomers of the PM Na+/K+ ATPase (NKA) (Sahin-Erdemli et al. 1994). These isomers are typically localized near NCX and SERCA, probably in PM–SR junctions, and their lower Na+ affinities (Kd∼22–33 mm as reported in Zahler et al. 1997) would favour the generation of a higher junctional [Na+] prior to Na+ extrusion by the NKA (Juhaszova & Blaustein, 1997). The idea that NCX could be associated with regulation of SR Ca2+ content was first presented earlier (Reuter et al. 1973) although supporting data was not available at that time. The extensive present knowledge regarding the dynamics of the transport proteins, molecules and 3-D architecture of the nanojunctions finally permitted the formulation of quantitative hypotheses of this junctional Ca2+ transfer, from Ca2+-entry-mode NCX to SERCA (Fameli et al. 2007). However, subsequent modelling outcomes suggested that greater structural complexity must limit Na+ diffusion. This was adequately provided for by the inclusion of transjunctional protein complexes that had been originally identified in electron-micrographs (Gabella, 1971; Devine et al. 1972; Fameli et al. 2009), although we have yet to consider the possibility that the diffusion path could be shortened by observed tethering of NCX to TRPC channels (Rosker et al. 2004).
Smooth muscle SR reloading via SERCA may also be facilitated by Ca2+ influx through VGCCs (Takeda et al. 2011), ROCs (Albert et al. 2009; Shi et al. 2012), and the stromal interaction molecule (STIM)/Orai system (Fig. 1:2; Takahashi et al. 2007a,b; Berra-Romani et al. 2008), respectively, all of which have been shown to support homogeneous increases in SR calcium content. However, even when both VGCCs and ROCs are blocked the SR can be slowly refilled, presumably through an elusive Ca2+ leak in the PM (Deth & van Breemen, 1974). The extent to which these different SR-loading mechanisms may overlap in a single smooth muscle type or its proliferative and migratory phenotypes remains to be determined, but it seems likely that they will contribute to the heterogeneity in Ca2+ signalling mechanisms between different smooth muscles.
SR unloading (Fig. 1: 3)
During rest, when the SR is not actively releasing Ca2+, NCX operates in the Ca2+ exit mode favouring unloading of the SR via a PM–SR junction, and functions together with Ca2+ extrusion via the plasma membrane Ca2+ ATPase (PMCA), located outside the PM–SR junctions, to maintain cellular Ca2+ homeostasis (Lee et al. 2002). Ca2+ exit mode NCX is clearly favoured by inactivation of ROCs and repolarization of the membrane. That aside, the mechanism proposed is that under these conditions Ca2+ released into PM–SR junctions from the peripheral SR would raise the local [Ca2+] to stimulate Ca2+ extrusion (van Breemen et al. 1995; Nazer & van Breemen, 1998). Ryanodine receptors (RyRs) and inositol 1,4,5-trisphosphate receptors (IP3Rs) are present at PM–SR junctions and may well serve this function, but at this time there is no concrete evidence for which release channel supplies Ca2+ for Ca2+ exit mode NCX in smooth muscle, although in endothelial cells it is clear that the RyR fulfils this function (Liang et al. 2004). Since the NCX is rheogenic, the hyperpolarization induced via RyRs linked to Ca2+-activated large conductance potassium (BKCa) channels, described below, will promote SR Ca2+ unloading. For the sake of clarity, Fig. 1 shows separate nanojunctions for loading and unloading the SR (Fig. 1: 1, 2 and 3), but it is also possible that a single more complex PM–SR junction would allow NCX to oscillate between the Ca2+ exit and entry modes depending on the SR Ca2+ content, PM potential and the junctional Na+ concentration.
STOCs, sparks and hyperpolarization (Fig. 1: 4)
It was originally proposed that α-adrenergic stimulation of the guinea pig taenia coli released Ca2+ from an internal store to activate BKCa channels and cause large transient hyperpolarization (Bülbring & Tomita, 1977, 1987). Since this hyperpolarization was associated with relaxation, they proposed that the Ca2+ release was localized to a plasmalemmal domain proximal to the target channels. In 1986, spontaneous transient outward currents (STOCs) in visceral smooth muscle were observed (Benham & Bolton, 1986), which resulted from the activation of 75–100 BKCa channels localized in less than 3% of the cell PM and that this occurred in response to local RyR-mediated SR Ca2+ release. The picture was completed with the demonstration of Ca2+ sparks in vascular smooth muscle and their role in determining basal tension and evoked vasodilatation of cerebral arteries (Nelson et al. 1995). In these vessels, sparks play a critical role in the regulation of myogenic tone, and thus autoregulation of cerebral blood flow, by providing variable feedback regulation on the opening of VGCCs (Ledoux et al. 2006). Analysis of the relationship between spark intensity and STOC size suggests a distance between RyR in the SR and BKCa in the PM comparable with the width of diadic junctions in cardiac muscle (Pérez et al. 1999). Moreover, there is little doubt that SERCA maintains a releasable pool of Ca2+ within the superficial SR, which is linked to relaxation (Boittin et al. 2002, 2003). Evidence suggests that the type of SERCA (SERCA2b) located in the superficial SR may differ (by kinetics and mechanisms of regulation) from the SERCA type(s) that recycle Ca2+ into the deep SR (Clark et al. 2010).
Ca2+-induced Ca2+ release (Fig. 1: 6 and 7)
Following initial descriptions in skeletal muscle (Endo et al. 1970), CICR was first demonstrated in intestinal smooth muscle (Saida, 1982) and vascular smooth muscle (Saida & van Breemen, 1983) by showing that, in saponin-skinned fibres, a sudden increase in [Ca2+]i released additional Ca2+ from a caffeine-sensitive store. Since the ultra-structural relationship between the VGCC and RyR varies from one smooth muscle to another (compare, for example, Moore et al. 2004 with Gordienko et al. 2008), it might be expected that the nature of CICR will vary as well. The observation of a structural linkage between dihydropyridine receptors and RyRs in the bladder detrusor muscle of the guinea pig led to the hypothesis of a tight coupling between the two channels similar to that seen in cardiac muscle (10–20 nm; Moore et al. 2004). In contrast, it was proposed that CICR in smooth muscle of the rabbit urinary bladder is generally loosely coupled, meaning that the distance between VGCC and RyR is greater than 100 nm (Kotlikoff, 2003; Ji et al. 2006). That different smooth muscles display a range of coupling between Ca2+ entry and RyR activation, from tight to none at all, seems all the more likely when one considers the fact that constriction induced by membrane depolarization in pulmonary arteries appears insensitive to block of RyRs (Dipp & Evans, 2001; Dipp et al. 2001). The significance of this may lie in the fact that different smooth muscle types may utilise CICR in different ways, and may not necessarily require coupling between VGCCs and RyRs or IP3Rs.
Electrical synchronization and vasomotion (Fig. 1: 5)
All smooth muscles exhibit Ca2+ wave activity. In large blood vessels, be they conduit arteries or capacitative veins, agonist-mediated activation elicits asynchronous Ca2+ waves generated by CICR at IP3-sensitized IP3R (Iino et al. 1994; Ruehlmann et al. 2000; McCarron et al. 2010). As mentioned above, PM–SR junctions are required to maintain this type of Ca2+ oscillation by refilling the SR. When small resistance arteries are stimulated with agonists they initially also exhibit asynchronous Ca2+ waves, which quickly convert into synchronous non-wave-like Ca2+ oscillations in adjoining smooth muscle cells. The resulting synchronized contractions are the basis of vasomotion and are achieved by rapid spread of Ca2+-activated chloride channel (ClCa)-induced depolarization (Boedtkjer et al. 2008). According to this view the original periodic IP3R/Ca2+-mediated Ca2+ release near the PM is responsible for the initiation of each [Ca2+]i oscillation in mesenteric resistance arteries (Peng et al. 2001). By contrast, auto-regulation of cerebral arteries presents an alternative example of an SR Ca2+ release-activated PM channel, the cation-permeable channel transient receptor potential melastatin-4 (TRPM4) (Gonzales & Earley, 2012). TRPM4 is essential for pressure-induced depolarization and contraction in cerebral arteries, which also exhibit vasomotion. TRPM4 is activated by SR Ca2+ release via IP3R; however, prolonged exposure to Ca2+ inactivates the channel due to activation of phospholipase C and decreases in levels of phosphatidylinositol 4,5-bisphosphate (PIP2). This is kept in check by local endogenous buffering, which ensures that Ca2+ release events are short and localized. This led to the conclusion that TRPM4 channels on the PM are less than 50 nm from the SR membrane, but not physically coupled to IP3R. In short, the all-important electrical synchronization and vasomotion processes also appear to revolve around nanojunctions between SR and PM, and may vary between different types of smooth muscles. The two examples provided above illustrate that different junctional compositions may regulate the same function of synchronized smooth muscle activity and that Fig. 1 is only meant to provide a limited array of plausible examples. On the other hand it is equally clear that certain combinations of channels, for example, BKCa plus ClCa, within the same junction would obviate any useful function.
Vasomotion and Ca2+ waves and oscillations are strongly influenced by mitochondria, not only because they require a regulated energy supply, but also due to complex Ca2+ exchange between the SR and mitochondria.
SR-mitochondrial Ca2+ exchange (Fig. 1: 8, 9)
Mitochondria–SR junctions regulate mitochondrial energy supply, apoptosis and SR Ca2+ loading. There is now a vast literature on this subject, which has seen exciting recent progress with the identification of the main Ca2+ transporters: the mitochondrial uniporter (MCU) and mitochondrial Na+/Ca2+ exchanger (NLCX), and may be accessed via an excellent recent review (Pizzo et al. 2012). Although mitochondria are capable of slow Ca2+ uptake from the bulk cytoplasm, which is in steady state with extrusion mainly via NLCX and partly H+/Ca2+ exchangers (HCX), rapid mitochondrial Ca2+ transients depend on close approximations of the SR release channels, IP3R and possibly RyR and the MCU. This process has been elegantly visualized using Ca2+ indicators targeted to the outer mitochondrial membrane, which record hot spots of 20–30 μm Ca2+ during activation of IP3R (Giacomello et al. 2010). Ca2+ accesses the MCU via the large voltage-dependent anion channels in the outer membrane, which may be rate limiting. In smooth muscle the large transient mitochondrial Ca2+ gain is subsequently extruded via the NLCX which fuels SERCA-mediated re-uptake into the SR of smooth muscle cells (Szado et al. 2003; Poburko et al. 2009). In addition, evidence suggests that during the refilling process described above, peripheral mitochondria ‘funnel’ Ca2+ lost from the PM–SR junction back into the SR (Poburko et al. 2009). Evidence has also been presented for a more central population of mitochondria that are insensitive to activity of the PM NCX (Szado et al. 2003). Besides the direct involvement of MCU and NLCX in cellular Ca2+ movements, mitochondrial Ca2+ transport also regulates Ca2+ channels in the SR and PM. Recent work showed that strategically localized, immobile mitochondria remove Ca2+ from active clusters of IP3R to prevent their inhibition, thus allowing propagation of CICR-mediated Ca2+ release waves (Olson et al. 2010). An interesting variation on such mitochondrial control over excitatory Ca2+ waves in vascular smooth muscle was suggested in an article showing that the return supply of Ca2+ delivered by NLCX towards the IP3R cluster served as an activating pacemaker mechanism (Ishii et al. 2006). Both studies cited above provide a mechanistic explanation for the earlier observation that rotenone-induced mitochondrial depolarization drastically interfered with smooth muscle Ca2+ oscillations (Swärd et al. 2002). Mitochondrial modulation of VGCCs and store-operated channels in the PM has also been documented, but this has not as yet been shown in smooth muscle. The limited data presented above indicate that SR–mitochondrial nanojunctions are important in smooth muscle Ca2+ signalling; however, caution is warranted in relation to any results obtained with pharmacological agents, as they modify many other mitochondrial functions. Rapid advances are now anticipated due to genetic manipulation of MCU and NLCX (Pizzo et al. 2012).
The lysosome–SR junction (Fig. 1: 10)
In pulmonary arterial smooth muscle cells NAADP appears to activate a unique, but converging, Ca2+ signalling pathway via lysosome–SR (L–SR) junctions, and one which may mediate Ca2+ signalling by endothelin-1. NAADP releases Ca2+ from lysosome-related stores in a manner that requires two pore segment channel subtype 2 (TPC2; Calcraft et al. 2009; Agbani et al. 2011). Subsequently this lysosomal Ca2+ release is amplified, in an all-or-none manner (Boittin et al. 2002), by CICR from the SR via RyR3, which is resident on the SR membrane participating in L-SR junctions (Kinnear et al. 2004, 2008). Thereafter, RyR2 may be recruited to carry, by CICR, a propagating Ca2+ wave across the wider cell in order to facilitate contraction (Kinnear et al. 2004; Clark et al. 2010). This is supported by observations that SERCA2a, RyR2 and RyR3 are located on the deep SR, which may comprise a segregated contractile domain.
What could be the advantage of RyR3 in the lysosome–SR junction? With respect to CICR, RyR3 exhibits the highest EC50, and would therefore provide for a greater ‘margin of safety’ with respect to the all-or-none initiation of CICR by Ca2+ bursts from lysosomal Ca2+ stores at the L–SR junction. Furthermore, RyR3 exhibits a higher gain in open probability (Po). Thus, once the threshold for activation is breached RyR3 would offer greater amplification of Ca2+ bursts from lysosomal Ca2+ stores. On the other hand RyR2 having a more diffuse cellular distribution (Kinnear et al. 2004; Clark et al. 2010) and lower EC50 for CICR would ensure that once initiated a propagating Ca2+ wave would be less prone to failure.
Although such dynamic considerations point intriguingly toward specific mechanisms, they remain in the realm of conjecture due to the near-inaccessibility of the lysosome–SR junctions by current calcium imaging techniques. However, as we briefly consider next, precise hypotheses can further our understanding by means of quantitative simulations.
3-D quantitative modelling: an invaluable tool to study transport processes in nanojunctions
All documented instances of the nanojunctions outlined in Fig. 1 require an experimental resolution of <5 nm to visualize the important Ca2+ (and Na+, and possibly other species) transients generated therein. This cannot be satisfactorily accomplished by currently available instrumentation. Nevertheless, by quantitative 3-D modelling we can study nanojunction-based hypotheses on mechanisms of Ca2+ signalling, develop them further and thereby generate new questions that may drive further experimentation. This cycle of renewal leads naturally to the development of yet more accurate quantitative models by incorporating all new biophysical features as they become available from experimental study. A very small population of ions is predicted to be present in the volumes of cytoplasm that reside within a single junction, and increases of one single Ca2+ ion may increase the local concentration from nanomolar to micromolar. A stochastic approach is thus required to model these signalling events. The quantitative stochastic models we develop are typically informed by confocal and electron microscopy, including immunofluorescence and immunogold labelling, transporter densities and kinetics.
Initial simplified models focused on Ca2+ flux across the PM–SR junction. While they only accounted for the stochastic element of diffusion, encouraging results were obtained. However, they highlighted the need to incorporate in the models more realistic intracellular architecture reconstructions, transporter kinetics and density, and reaction implementations. That aside, it appeared that the functional integrity of PM–SR junctions relies heavily on the close apposition of the two membranes, since in the models a separation of less than 50 nm adequately provided for compartmentalized Ca2+ signalling, which was lost when the separation of PM and junctional SR was greater than approximately 50 nm (Fameli et al. 2007).
Employing a more sophisticated set of modelling tools, we have explored the L–SR nanojunction described in the previous section (Fameli, 2011).
Figure 2 illustrates some of the steps in the process of building a 3-D stochastic Monte Carlo model of Ca2+ transport in L–SR nanojunctions and some preliminary results. From several electron micrographs like the one in Fig. 2A, we recreated a 3-D representation of the relevant junctional geometry including one lysosome (grey object in Fig. 2B), partially wrapped by a portion of the SR (blue object in Fig. 2B), using open source ‘3-D content creation’ software (blender.org). We then simulated the junctional [Ca2+] ([Ca2+]NJ) transient formed by the release of lysosomal Ca2+ (small white dots in Fig. 2B) via TPC2 (red objects in Fig. 2B) as stimulated by NAADP, in the presence of SERCA pumps on the nanojunctional SR (yellow objects in Fig. 2B) and mobile Ca2+ buffers (larger green dots in Fig. 2B) in the junctional and neighbouring cytoplasm. To study the influence of the junctional geometry on the transient, we repeated the simulations with different L–SR junctional widths. A plot of the [Ca2+]NJ transient peak values vs. the junction width (Fig. 2C) suggests that Ca2+ accumulation in the lysosome–SR junction is extremely sensitive to applied changes in junctional dimensions and in particular on junctional width. Based on the information that RyR3 requires about 8–10 μm Ca2+ to release about 60–90% of SR Ca2+ (green band in Fig. 2C; Takeshima et al. 1995), our simulation results so far suggest that sufficiently high [Ca2+]NJ transients to activate the L–SR-resident RyR3s can occur in L–SR junctions if these are less than about 50 nm wide. In other words, it is evident from this model that ‘merely’ altering the nanojunctional geometry causes significant disruption of a key element in the NAADP-dependent Ca2+ signalling pathway that leads to RyR-based CICR and eventual vascular smooth muscle contraction. Preliminary outcomes support the view that, as found for the PM–SR junctions, appropriate regulation and control of Ca2+ signalling is compromised when junctional membranes of the lysosome and SR are separated by distances greater than 50 nm. This also implies that ‘functional uncoupling’ of lysosome–SR junctions could be readily achieved and may contribute to plasticity of function.
Figure 2.
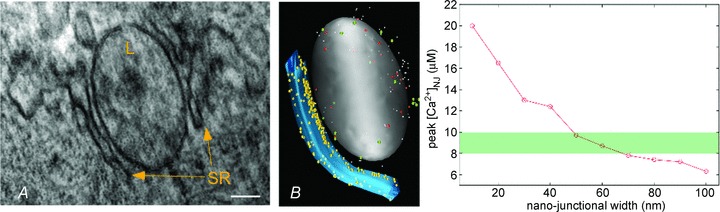
A, electron micrograph of a partially SR-surrounded lysosome (L). Tissue: rat pulmonary artery smooth muscle. Scale bar: 100 nm. B, 3-D software reproduction of a lysosome (grey)–SR (blue) nanojunction inspired by a series of observations from micrographs as in A; red transporters represent TPC2 Ca2+ release channels, yellow transporters stand for SERCA pumps; white molecules are Ca2+ and larger green ones are mobile buffers. C, simulation output as peak [Ca2+]NJ vs. width of junction. Initial [Ca2+]lys was set to 500 μm as reported in mast cells (Lloyd-Evans et al. 2008). Ca2+ Brownian motion trajectories are reproduced by means of accurate and extensively validated random walk algorithms implemented in the stochastic particle-simulator MCell (mcell.org) (Stiles et al. 1996; Stiles & Bartol, 2001; Kerr et al. 2008), which also allows the placement of relevant transporters and their appropriate reaction kinetics on the recreated object geometries. The green bar indicates the approximate threshold value for CICR at RyR3.
It is clear, therefore, that quantitative simulations are essential for testing hypotheses related to transport processes occurring in specific nanojunctions. However, we also contemplate the need for a similar approach to the integration of activities within the entire population of nanojunctions, including the mechanisms of regenerative waves of SR Ca2+ release. As discussed above, the SR performs a multitude of functions at different sites within the smooth muscle cell even though its entire lumen, including that of the nuclear envelope, appears contiguous (McCarron & Olson, 2008). This paradox has been kept alive for more than 50 years as a running controversy related to the pros and cons of smooth muscle SR compartmentalization, eloquently summarized in the recent review (Wray & Burdyga, 2010). We envision future development of a dynamic model based on the SR as a continuous quilted network of membranes containing clusters of Ca2+ release channels as well as SERCA pumps, which are probably also inhomogeneously distributed over the entire surface. For example, SERCA may be concentrated in PM–SR junctions functioning in refilling during excitatory Ca2+ waves. If, as can be clearly observed in both electron microscopy and confocal imaging, the lumen width varies considerably, then there would be regions within the continuous lumen that impede rapid diffusion between adjacent SR domains. The clustering of release channels (SR Ca2+ sinks) and SERCA (luminal Ca2+ sources) combined with SR sections offering resistance to diffusion would be ideal to create dynamic apparent Ca2+ compartments without the need to postulate the existence of membranous barriers between them. Such a system would be ideally suited for the propagation of Ca2+ waves as well as for replenishment of SR luminal Ca2+ for maintenance of the Ca2+ oscillations.
Does appropriate control of transcription require nanojunctions of the SR (Fig. 1: 11)?
The link between Ca2+ signalling and long-term changes in terms of remodelling (proliferation and migration) and cell death is not yet elucidated. In this respect, nuclear factor of activated T-cells (NFAT) translocation to the nucleus is significant. We know that released Ca2+ activates calcineurin, which is bound to the scaffolding protein AKAP79 on the PM. The Ca2+–calcineurin complex dephosphorylates NFAT and thus induces translocation from the cytoplasm to the nucleus (Nilsson et al. 2008). Yet this apparently simple process is controlled by a complex, but poorly understood Ca2+ signalling mechanism. This is clear from the fact that Ca2+ entry through TRPC1, Ca2+ entry through STIM/Orai complexes, Ca2+ release through IP3Rs and mitochondrial Ca2+ release have all been shown to be required for NFAT translocation, which also appears to require the presence of both PM–SR and SR–mitochondria nanojunctions. This is perhaps not surprising given that protection of transcription-activating sites from physiological Ca2+ transients activating myofilaments is clearly essential in order to prevent inappropriate vascular remodelling in response to normal cell and tissue function. Consistent with this idea, Ca2+ uptake by SERCA2 is required not only to preserve smooth muscle Ca2+ waves, but to afford protection against NFAT signalling (Bobe et al. 2011). However, while the case for different roles for SERCA, TRPC1 and STIM/Orai complexes in calcineurin/NFAT activation is evident, future studies must address the fact that this site on the PM is segregated from the nuclear membrane by SR, mitochondria and, not least, by the contractile domain itself. In this respect it is interesting that recent experiments have shown that smooth muscle Ca2+ waves evoked by endothelin-1 are deflected by the nuclear envelope (Esfandiarei et al. 2013), consistent with previous proposals that the nuclear membrane provides a buffer barrier (al-Mohanna et al. 1994; Wamhoff et al. 2002) that may determine in some respect Ca2+ signalling to the nucleus. It seems likely, therefore, that additional nuclear nanospaces may be conferred by the pan-junctional SR.
Conclusion
We hypothesize that nanojunctions between the SR, on the one hand, and PM, mitochondria, lysosomes and other organelles, on the other, are the basis for segregating localized calcium signals for the independent regulation of contraction, relaxation, energy metabolism, apoptosis, proliferation and migration. Variations in the prevalence, ultra-structure and molecular makeup of the nanojunctions could explain both smooth muscle heterogeneity and plasticity. Therefore, elucidating the mechanisms of ion transport within nanospaces is essential to our further understanding of calcium signalling not just in smooth muscle, but all cell types. A combination of high-resolution dynamic imaging of localized ion concentrations, ultrastructural 3-D reconstruction and (stochastic) quantitative modelling could lead to specific testable hypotheses, with the caveat that the signalling architecture of each cell type be considered unique and studied separately.
Acknowledgments
We are grateful to Garnet Martens and the University of British Columbia Bioimaging Facility for their input and assistance on electron microscopy imaging and to Elke van Breemen for the artwork. This work was supported by Grant No. CIHR MOP-84309 from the Canadian Institutes of Health Research and by the British Heart Foundation Project Grants PG/03/065 and PG/10/95/28657, and Programme Grant RG/12/14/29885.
Glossary
- BKCa
Ca2+-activated large conductance potassium (channel)
- CICR
calcium-induced calcium release
- ClCa
Ca2+-activated chloride channel
- HCX
H+/Ca2+ exchanger
- IP3R
inositol 1,4,5-trisphosphate receptor; lysosome–SR (junction)
- MCU
mitochondrial uniporter
- NAADP
nicotinic acid adenine dinucleotide phosphate
- NCX
Na+/Ca2+ exchanger
- NFAT
nuclear factor of activated T-cells
- NLCX
mitochondrial Na+/Ca2+ exchanger
- NKA
Na+/K+ ATPase
- PM
plasma membrane
- PMCA
plasma membrane Ca2+ ATPase
- ROC
receptor-operated channel
- RyR
ryanodine receptor
- SERCA
sarco/endoplasmic reticulum Ca2+ ATPase
- SR
sarcoplasmic reticulum
- STIM
stromal interaction molecule
- STOC
spontaneous transient outward current
- TRPM4
transient receptor potential melastatin-4
- TPC
two pore segment channel
- TRPC
transient receptor potential canonical
- VGCC
voltage-gated Ca2+ channel
References
- Agbani EO, Ogunbayo OA, Parrington J, Galione A, Ma J, Zhu MX, Evans AM. Nicotinic acid adenine dinucleotide phosphate evokes global calcium signals in mouse pulmonary arterial smooth muscle cells by activating Two Pore Segment Channel 2. Proc Physiol Soc. 2011;25 http://www.physoc.org/proceedings/abstract/Proc%20Physiol%20Soc%2025PC39. [Google Scholar]
- Albert AP, Saleh SN, Large WA. Identification of canonical transient receptor potential (TRPC) channel proteins in native vascular smooth muscle cells. Curr Med Chem. 2009;16:1158–1165. doi: 10.2174/092986709787581815. [DOI] [PubMed] [Google Scholar]
- Allbritton NL, Meyer T. Localized calcium spikes and propagating calcium waves. Cell Calcium. 1993;14:691–697. doi: 10.1016/0143-4160(93)90095-n. [DOI] [PubMed] [Google Scholar]
- Allbritton NL, Meyer T, Stryer L. Range of messenger action of calcium ion and inositol 1,4,5-trisphosphate. Science. 1992;258:1812–1815. doi: 10.1126/science.1465619. [DOI] [PubMed] [Google Scholar]
- al-Mohanna FA, Caddy KW, Bolsover SR. The nucleus is insulated from large cytosolic calcium ion changes. Nature. 1994;367:745–750. doi: 10.1038/367745a0. [DOI] [PubMed] [Google Scholar]
- Benham CD, Bolton TB. Spontaneous transient outward currents in single visceral and vascular smooth muscle cells of the rabbit. J Physiol. 1986;381:385–406. doi: 10.1113/jphysiol.1986.sp016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berra-Romani R, Mazzocco-Spezzia A, Pulina MV, Golovina VA. Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am J Physiol Cell Physiol. 2008;295:C779–C790. doi: 10.1152/ajpcell.00173.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobe R, Hadri L, Lopez JJ, Sassi Y, Atassi F, Karakikes I, Liang L, Limon I, Lompré A-M, Hatem SN, Hajjar RJ, Lipskaia L. SERCA2a controls the mode of agonist-induced intracellular Ca2+ signal, transcription factor NFAT and proliferation in human vascular smooth muscle cells. J Mol Cell Cardiol. 2011;50:621–633. doi: 10.1016/j.yjmcc.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boedtkjer DMB, Matchkov VV, Boedtkjer E, Nilsson H, Aalkjaer C. Vasomotion has chloride-dependency in rat mesenteric small arteries. Pflugers Arch. 2008;457:389–404. doi: 10.1007/s00424-008-0532-3. [DOI] [PubMed] [Google Scholar]
- Boittin F-X, Dipp M, Kinnear NP, Galione A, Evans AM. Vasodilation by the calcium-mobilizing messenger cyclic ADP-ribose. J Biol Chem. 2003;278:9602–9608. doi: 10.1074/jbc.M204891200. [DOI] [PubMed] [Google Scholar]
- Boittin F-X, Galione A, Evans AM. Nicotinic acid adenine dinucleotide phosphate mediates Ca2+ signals and contraction in arterial smooth muscle via a two-pool mechanism. Circ Res. 2002;91:1168–1175. doi: 10.1161/01.res.0000047507.22487.85. [DOI] [PubMed] [Google Scholar]
- Bülbring E, Tomita T. Calcium requirement for the alpha-action of catecholamines on guinea-pig taenia coli. Proc R Soc Lond B Biol Sci. 1977;197:271–284. doi: 10.1098/rspb.1977.0070. [DOI] [PubMed] [Google Scholar]
- Bülbring E, Tomita T. Catecholamine action on smooth muscle. Pharmacol Rev. 1987;39:49–96. [PubMed] [Google Scholar]
- Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, Zhu MX. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459:596–600. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JH, Kinnear NP, Kalujnaia S, Cramb G, Fleischer S, Jeyakumar LH, Wuytack F, Evans AM. Identification of functionally segregated sarcoplasmic reticulum calcium stores in pulmonary arterial smooth muscle. J Biol Chem. 2010;285:13542–13549. doi: 10.1074/jbc.M110.101485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo J, Katz B. Biophysical aspects of neuro-muscular transmission. Prog Biophys Biophys Chem. 1956;6:121–170. [PubMed] [Google Scholar]
- Deth R, van Breemen C. Relative contributions of Ca2+ influx and cellular Ca2+ release during drug induced activation of the rabbit aorta. Pflugers Arch. 1974;348:13–22. doi: 10.1007/BF00587735. [DOI] [PubMed] [Google Scholar]
- Devine CE, Somlyo AV, Somlyo AP. Sarcoplasmic reticulum and excitation-contraction coupling in mammalian smooth muscles. J Cell Biol. 1972;52:690–718. doi: 10.1083/jcb.52.3.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dipp M, Evans AM. Cyclic ADP-ribose is the primary trigger for hypoxic pulmonary vasoconstriction in the rat lung in situ. Circ Res. 2001;89:77–83. doi: 10.1161/hh1301.093616. [DOI] [PubMed] [Google Scholar]
- Dipp M, Nye PC, Evans AM. Hypoxic release of calcium from the sarcoplasmic reticulum of pulmonary artery smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2001;281:L318–L325. doi: 10.1152/ajplung.2001.281.2.L318. [DOI] [PubMed] [Google Scholar]
- Endo M, Tanaka M, Ogawa Y. Calcium induced release of calcium from the sarcoplasmic reticulum of skinned skeletal muscle fibres. Nature. 1970;228:34–36. doi: 10.1038/228034a0. [DOI] [PubMed] [Google Scholar]
- Esfandiarei M, Fameli N, Choi YY, Tehrani AY, Hoskins JG, van Breemen C. Waves of calcium depletion in the sarcoplasmic reticulum of vascular smooth muscle cells: an inside view of spatiotemporal ca(2+) regulation. PLoS One. 2013;8:e55333. doi: 10.1371/journal.pone.0055333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fameli N, Kuo K-H, van Breemen C. A model for the generation of localized transient [Na+] elevations in vascular smooth muscle. Biochem Biophys Res Commun. 2009;389:461–465. doi: 10.1016/j.bbrc.2009.08.166. [DOI] [PubMed] [Google Scholar]
- Fameli N, van Breeman C. Stochastic three dimensional modelling of ionic transport in cytoplasmic nanospaces. Proc Physiol Soc. 2011;25:SA06. [Google Scholar]
- Fameli N, van Breemen C, Kuo K-H. A quantitative model for linking Na+/Ca2+ exchanger to SERCA during refilling of the sarcoplasmic reticulum to sustain [Ca2+] oscillations in vascular smooth muscle. Cell Calcium. 2007;42:565–575. doi: 10.1016/j.ceca.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C. Fine structure of sarcoplasmic reticulum and tranverse tubular system in muscle fibers. Fed Proc. 1964;23:887–895. [PubMed] [Google Scholar]
- Gabella G. Caveolae intracellulares and sarcoplasmic reticulum in smooth muscle. J Cell Sci. 1971;8:601–609. doi: 10.1242/jcs.8.3.601. [DOI] [PubMed] [Google Scholar]
- Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell. 2010;38:280–290. doi: 10.1016/j.molcel.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Gonzales AL, Earley S. Endogenous cytosolic Ca2+ buffering is necessary for TRPM4 activity in cerebral artery smooth muscle cells. Cell Calcium. 2012;51:82–93. doi: 10.1016/j.ceca.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordienko DV, Harhun MI, Kustov MV, Pucovský V, Bolton TB. Sub-plasmalemmal [Ca2+]i upstroke in myocytes of the guinea-pig small intestine evoked by muscarinic stimulation: IP3R-mediated Ca2+ release induced by voltage-gated Ca2+ entry. Cell Calcium. 2008;43:122–141. doi: 10.1016/j.ceca.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M, Kasai H, Yamazawa T. Visualization of neural control of intracellular Ca2+ concentration in single vascular smooth muscle cells in situ. EMBO J. 1994;13:5026–5031. doi: 10.1002/j.1460-2075.1994.tb06831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K, Hirose K, Iino M. Ca2+ shuttling between endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO Rep. 2006;7:390–396. doi: 10.1038/sj.embor.7400620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Feldman M, Doran R, Zipfel W, Kotlikoff MI. Ca2+-induced Ca2+ release through localized Ca2+ uncaging in smooth muscle. J Gen Physiol. 2006;127:225–235. doi: 10.1085/jgp.200509422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhaszova M, Blaustein MP. Na+ pump low and high ouabain affinity alpha subunit isoforms are differently distributed in cells. Proc Natl Acad Sci U S A. 1997;94:1800–1805. doi: 10.1073/pnas.94.5.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr RA, Bartol TM, Kaminsky B, Dittrich M, Chang J-CJ, Baden SB, Sejnowski TJ, Stiles JR. Fast Monte Carlo simulation methods for biological reaction-diffusion systems in solution and on surfaces. SIAM J Sci Comput. 2008;30:3126. doi: 10.1137/070692017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnear NP, Boittin F-X, Thomas JM, Galione A, Evans AM. Lysosome-sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J Biol Chem. 2004;279:54319–54326. doi: 10.1074/jbc.M406132200. [DOI] [PubMed] [Google Scholar]
- Kinnear NP, Wyatt CN, Clark JH, Calcraft PJ, Fleischer S, Jeyakumar LH, Nixon GF, Evans AM. Lysosomes co-localize with ryanodine receptor subtype 3 to form a trigger zone for calcium signalling by NAADP in rat pulmonary arterial smooth muscle. Cell Calcium. 2008;44:190–201. doi: 10.1016/j.ceca.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlikoff MI. Calcium-induced calcium release in smooth muscle: the case for loose coupling. Prog Biophys Mol Biol. 2003;83:171–191. doi: 10.1016/s0079-6107(03)00056-7. [DOI] [PubMed] [Google Scholar]
- Kushmerick MJ, Podolsky RJ. Ionic mobility in muscle cells. Science. 1969;166:1297–1298. doi: 10.1126/science.166.3910.1297. [DOI] [PubMed] [Google Scholar]
- Ledoux J, Werner ME, Brayden JE, Nelson MT. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 2006;21:69–78. doi: 10.1152/physiol.00040.2005. [DOI] [PubMed] [Google Scholar]
- Lee C-H, Poburko D, Kuo K-H, Seow CY, van Breemen C. Ca2+ oscillations, gradients, and homeostasis in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2002;282:H1571–H1583. doi: 10.1152/ajpheart.01035.2001. [DOI] [PubMed] [Google Scholar]
- Lee CH, Poburko D, Sahota P, Sandhu J, Ruehlmann DO, van Breemen C. The mechanism of phenylephrine-mediated [Ca2+]i oscillations underlying tonic contraction in the rabbit inferior vena cava. J Physiol. 2001;534:641–650. doi: 10.1111/j.1469-7793.2001.t01-1-00641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos VS, Poburko D, Liao C-H, Cole WC, van Breemen C. Na+ entry via TRPC6 causes Ca2+ entry via NCX reversal in ATP stimulated smooth muscle cells. Biochem Biophys Res Commun. 2007;352:130–134. doi: 10.1016/j.bbrc.2006.10.160. [DOI] [PubMed] [Google Scholar]
- Liang W, Buluc M, van Breemen C, Wang X. Vectorial Ca2+ release via ryanodine receptors contributes to Ca2+ extrusion from freshly isolated rabbit aortic endothelial cells. Cell Calcium. 2004;36:431–443. doi: 10.1016/j.ceca.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- McCarron JG, Chalmers S, MacMillan D, Olson ML. Agonist-evoked Ca2+ wave progression requires Ca2+ and IP3. J Cell Physiol. 2010;224:334–344. doi: 10.1002/jcp.22103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarron JG, Olson ML. A single luminally continuous sarcoplasmic reticulum with apparently separate Ca2+ stores in smooth muscle. J Biol Chem. 2008;283:7206–7218. doi: 10.1074/jbc.M708923200. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Griesbeck O, Heim R, Tsien RY. Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc Natl Acad Sci U S A. 1999;96:2135–2140. doi: 10.1073/pnas.96.5.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore ED, Voigt T, Kobayashi YM, Isenberg G, Fay FS, Gallitelli MF, Franzini-Armstrong C. Organization of Ca2+ release units in excitable smooth muscle of the guinea-pig urinary bladder. Biophys J. 2004;87:1836–1847. doi: 10.1529/biophysj.104.044123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazer MA, van Breemen C. Functional linkage of Na+-Ca2+ exchange and sarcoplasmic reticulum Ca2+ release mediates Ca2+ cycling in vascular smooth muscle. Cell Calcium. 1998;24:275–283. doi: 10.1016/s0143-4160(98)90051-3. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- Nilsson LM, Nilsson-Ohman J, Zetterqvist AV, Gomez MF. Nuclear factor of activated T-cells transcription factors in the vasculature: the good guys or the bad guys. Curr Opin Lipidol. 2008;19:483–490. doi: 10.1097/MOL.0b013e32830dd545. [DOI] [PubMed] [Google Scholar]
- Olson ML, Chalmers S, McCarron JG. Mitochondrial Ca2+ uptake increases Ca2+ release from inositol 1,4,5-trisphosphate receptor clusters in smooth muscle cells. J Biol Chem. 2010;285:2040–2050. doi: 10.1074/jbc.M109.027094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Matchkov V, Ivarsen A, Aalkjaer C, Nilsson H. Hypothesis for the initiation of vasomotion. Circ Res. 2001;88:810–815. doi: 10.1161/hh0801.089603. [DOI] [PubMed] [Google Scholar]
- Pérez GJ, Bonev AD, Patlak JB, Nelson MT. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J Gen Physiol. 1999;113:229–238. doi: 10.1085/jgp.113.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzo P, Drago I, Filadi R, Pozzan T. Mitochondrial Ca2+ homeostasis: mechanism, role, and tissue specificities. Pflugers Arch. 2012;464:3–17. doi: 10.1007/s00424-012-1122-y. [DOI] [PubMed] [Google Scholar]
- Poburko D, Fameli N, Kuo K-H, van Breemen C. Ca2+ signaling in smooth muscle: TRPC6, NCX and LNats in nanodomains. Channels (Austin) 2008;2:10–12. doi: 10.4161/chan.2.1.6053. [DOI] [PubMed] [Google Scholar]
- Poburko D, Liao C-H, Lemos VS, Lin E, Maruyama Y, Cole WC, van Breemen C. Transient receptor potential channel 6-mediated, localized cytosolic [Na+] transients drive Na+/Ca2+ exchanger-mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ Res. 2007;101:1030–1038. doi: 10.1161/CIRCRESAHA.107.155531. [DOI] [PubMed] [Google Scholar]
- Poburko D, Liao C-H, van Breemen C, Demaurex N. Mitochondrial regulation of sarcoplasmic reticulum Ca2+ content in vascular smooth muscle cells. Circ Res. 2009;104:104–112. doi: 10.1161/CIRCRESAHA.108.180612. [DOI] [PubMed] [Google Scholar]
- Ramesh V, Sharma VK, Sheu SS, Franzini-Armstrong C. Structural proximity of mitochondria to calcium release units in rat ventricular myocardium may suggest a role in Ca2+ sequestration. Ann N Y Acad Sci. 1998;853:341–344. doi: 10.1111/j.1749-6632.1998.tb08295.x. [DOI] [PubMed] [Google Scholar]
- Reuter H, Blaustein MP, Haeusler G. Na-Ca exchange and tension development in arterial smooth muscle. Philos Trans R Soc Lond B Biol Sci. 1973;265:87–94. doi: 10.1098/rstb.1973.0011. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Pozzan T. Intracellular targeting of the photoprotein aequorin: a new approach for measuring, in living cells, Ca2+ concentrations in defined cellular compartments. Cytotechnology. 1993;11(Suppl. 1):S44–46. [PubMed] [Google Scholar]
- Rosenbluth J. Subsurface cisterns and their relationship to the neuronal plasma membrane. J Cell Biol. 1962;13:405–421. doi: 10.1083/jcb.13.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosker C, Graziani A, Lukas M, Eder P, Zhu MX, Romanin C, Groschner K. Ca2+ signaling by TRPC3 involves Na+ entry and local coupling to the Na+/Ca2+ exchanger. J Biol Chem. 2004;279:13696–13704. doi: 10.1074/jbc.M308108200. [DOI] [PubMed] [Google Scholar]
- Ruehlmann DO, Lee CH, Poburko D, van Breemen C. Asynchronous Ca2+ waves in intact venous smooth muscle. Circ Res. 2000;86:E72–79. doi: 10.1161/01.res.86.4.e72. [DOI] [PubMed] [Google Scholar]
- Sahin-Erdemli I, Rashed SM, Songu-Mize E. Rat vascular tissues express all three α-isoforms of Na+-K+-ATPase. Am J Physiol Heart Circ Physiol. 1994;266:H350–H353. doi: 10.1152/ajpheart.1994.266.1.H350. [DOI] [PubMed] [Google Scholar]
- Saida K. Intracellular Ca release in skinned smooth muscle. J Gen Physiol. 1982;80:191–202. doi: 10.1085/jgp.80.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saida K, van Breemen C. Mechanism of Ca++ antagonist-induced vasodilation. Intracellular actions. Circ Res. 1983;52:137–142. doi: 10.1161/01.res.52.2.137. [DOI] [PubMed] [Google Scholar]
- Shi J, Ju M, Abramowitz J, Large WA, Birnbaumer L, Albert AP. TRPC1 proteins confer PKC and phosphoinositol activation on native heteromeric TRPC1/C5 channels in vascular smooth muscle: comparative study of wild-type and TRPC1–/– mice. FASEB J. 2012;26:409–419. doi: 10.1096/fj.11-185611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles JR, Bartol TM. Computational Neuroscience: Realistic Modeling for Experimentalists. Boca Raton: CRC Press; 2001. Monte Carlo methods for simulating realistic synaptic microphysiology using MCell; pp. 87–127. [Google Scholar]
- Stiles JR, Van Helden D, Bartol TM, Jr, Salpeter EE, Salpeter MM. Miniature endplate current rise times less than 100 microseconds from improved dual recordings can be modeled with passive acetylcholine diffusion from a synaptic vesicle. Proc Natl Acad Sci U S A. 1996;93:5747–5752. doi: 10.1073/pnas.93.12.5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swärd K, Dreja K, Lindqvist A, Persson E, Hellstrand P. Influence of mitochondrial inhibition on global and local [Ca2+]i in rat tail artery. Circ Res. 2002;90:792–799. doi: 10.1161/01.res.0000015214.40360.84. [DOI] [PubMed] [Google Scholar]
- Szado T, Kuo K-H, Bernard-Helary K, Poburko D, Lee CH, Seow C, Ruegg UT, van Breemen C. Agonist-induced mitochondrial Ca2+ transients in smooth muscle. FASEB J. 2003;17:28–37. doi: 10.1096/fj.02-0334com. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Murakami M, Watanabe H, Hasegawa H, Ohba T, Munehisa Y, Nobori K, Ono K, Iijima T, Ito H. Essential role of the N-terminus of murine Orai1 in store-operated Ca2+ entry. Biochem Biophys Res Commun. 2007a;356:45–52. doi: 10.1016/j.bbrc.2007.02.107. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Watanabe H, Murakami M, Ono K, Munehisa Y, Koyama T, Nobori K, Iijima T, Ito H. Functional role of stromal interaction molecule 1 (STIM1) in vascular smooth muscle cells. Biochem Biophys Res Commun. 2007b;361:934–940. doi: 10.1016/j.bbrc.2007.07.096. [DOI] [PubMed] [Google Scholar]
- Takeda Y, Nystoriak MA, Nieves-Cintrón M, Santana LF, Navedo MF. Relationship between Ca2+ sparklets and sarcoplasmic reticulum Ca2+ load and release in rat cerebral arterial smooth muscle. Am J Physiol Heart Circ Physiol. 2011;301:H2285–H2294. doi: 10.1152/ajpheart.00488.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima H, Yamazawa T, Ikemoto T, Takekura H, Nishi M, Noda T, Iino M. Ca2+-induced Ca2+ release in myocytes from dyspedic mice lacking the type-1 ryanodine receptor. EMBO J. 1995;14:2999–3006. doi: 10.1002/j.1460-2075.1995.tb07302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Breemen C. Calcium requirement for activation of intact aortic smooth muscle. J Physiol. 1977;272:317–329. doi: 10.1113/jphysiol.1977.sp012046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Breemen C, Chen Q, Laher I. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. Trends Pharmacol Sci. 1995;16:98–105. doi: 10.1016/s0165-6147(00)88990-7. [DOI] [PubMed] [Google Scholar]
- Verboomen H, Wuytack F, De Smedt H, Himpens B, Casteels R. Functional difference between SERCA2a and SERCA2b Ca2+ pumps and their modulation by phospholamban. Biochem J. 1992;286:591–595. doi: 10.1042/bj2860591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamhoff BR, Bowles DK, Dietz NJ, Hu Q, Sturek M. Exercise training attenuates coronary smooth muscle phenotypic modulation and nuclear Ca2+ signaling. Am J Physiol Heart Circ Physiol. 2002;283:H2397–H2410. doi: 10.1152/ajpheart.00371.2001. [DOI] [PubMed] [Google Scholar]
- Wilson DP, Sutherland C, Walsh MP. Ca2+ activation of smooth muscle contraction: evidence for the involvement of calmodulin that is bound to the triton insoluble fraction even in the absence of Ca2+ J Biol Chem. 2002;277:2186–2192. doi: 10.1074/jbc.M110056200. [DOI] [PubMed] [Google Scholar]
- Wray S, Burdyga T. Sarcoplasmic reticulum function in smooth muscle. Physiol Rev. 2010;90:113–178. doi: 10.1152/physrev.00018.2008. [DOI] [PubMed] [Google Scholar]
- Zahler R, Zhang ZT, Manor M, Boron WF. Sodium kinetics of Na,K-ATPase α isoforms in intact transfected HeLa cells. J Gen Physiol. 1997;110:201–213. doi: 10.1085/jgp.110.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]