

Abstract
A balanced immune response requires combating infectious assaults while striving to maintain quiescence towards the self. One of the central players in this process is the pleiotropic cytokine transforming growth factor-β (TGF-β), whose deficiency results in spontaneous systemic autoimmunity in mice. The dominant function of TGF-β is to regulate the peripheral immune homeostasis, particularly in the microbe-rich and antigen-rich environment of the gut. To maintain intestinal integrity, the epithelial cells, myeloid cells and lymphocytes that inhabit the gut secrete TGF-β, which acts in both paracrine and autocrine fashions to activate its signal transducers, the SMAD transcription factors. The SMAD pathway regulates the production of IgA by B cells, maintains the protective mucosal barrier and promotes the balanced differentiation of CD4+ T cells into inflammatory T helper type 17 cells and suppressive FOXP3+ T regulatory cells. While encounters with pathogenic microbes activate SMAD proteins to evoke a protective inflammatory immune response, SMAD activation and synergism with immunoregulatory factors such as the vitamin A metabolite retinoic acid enforce immunosuppression toward commensal microbes and innocuous food antigens. Such complementary context-dependent functions of TGF-β are achieved by the co-operation of SMAD proteins with distinct dominant transcription activators and accessory chromatin modifiers. This review highlights recent advances in unravelling the molecular basis for the multi-faceted functions of TGF-β in the gut that are dictacted by fluid orchestrations of SMADs and their myriad partners.
Keywords: autoimmunity, lymphoid differentiation, transcription factors / gene regulation
Introduction
Animals lacking transforming growth factor-β (TGF-β) are afflicted with a fatal, spontaneous T-cell-mediated multi-organ lymphoproliferative disorder. The disease is similar in severity and scope to that of mice with null mutations in Foxp3, the master transcription factor (TF) specifying CD4+ regulatory T (Treg) cells, and Ctla4, the co-inhibitory receptor on T cells counterbalancing the co-stimulator CD28. TGF-β, FOXP3 and CTLA-4 form the T-cell homeostasis troika and their functional link can be traced to Treg cells, as each is essential for the development or function of the T cells dedicated for immune suppression.1 However, while the function of FOXP3 and CTLA-4 is restricted to T cells, TGF-β is systemically pervasive in maintaining immune quiescence and marshalling appropriate immune responses for optimal pathogen clearance.2 TGF-β functions by activating SMAD transcription factors (TF).3 Cytokines other than TGF-β can also activate SMADs; however, their relevance in the immune system has not been clearly established. This review will focus on recent insights into the TGF-β–SMAD signalling nexus that sets the balance between immune activation to clear pathogens and immune suppression to limit damage to self. We will focus on gut immunity to illustrate the context-dependent orchestration of TGF-β function, because the gut has a central role in shaping the immune network and is arguably the main arena in which TGF-β exerts its overarching influence.
Context-dependent TGF-β–SMAD signal transduction
From the beginning, the Janus-like dual functionality (pro-differentiation, growth-promoting, wound repair contrasted to anti-proliferation and pro-cell death) of TGF-β foreshadowed context and concentration dependency as its main feature, and diverse functions impacting multiple cell types were envisaged.4 However, the emergence of TGF-β as the master regulator of the mammalian immune system was not appreciated until it was observed that mice lacking TGF-β suffer from multi-organ autoimmunity,5,6 primarily as a consequence of deregulated T cells,7,8 While the importance of the TGF family of cytokines (TGF-β, Activin, Nodal and bone morphogenetic proteins in mammals) as tissue morphogens provided the impetus for groundbreaking studies that identified the components of the TGF-β signalling pathway, it was the discovery that Treg cells, and later CD4+ interleukin-17 (IL-17) -producing T (Th17) cells, differentiate under the control of TGF-β,9–11 that spurred intense pursuit of the mechanism by which TGF-β exerts multiple, balanced control over constituents of a cell system to adapt to environmental alterations. Recent advances in understanding SMAD activities in the nucleus are beginning to reveal the molecular basis of the pleiotropic function of TGF-β.
Transforming growth factor-β signals through type I and II transmembrane serine/threonine protein kinase receptors. The binding of the ligand activates the type II receptors, which recruit type I receptors, forming a heterotetrameric complex. The activated type I receptors interact with and phosphorylate SMAD (an acronym from the fusion of Caenorhabditis elegans Sma genes and the Drosophila Mad, Mothers against decapentaplegic) proteins to transduce signals.2 SMADs were first identified as the intracellular signal transducers of TGF-β receptor (TGFβR) in the mid 1990s in studies of inverterbrates,3,12 SMADs are divided into three functional classes: receptor activated (R-SMADs), common mediator (Co-SMADs), and inhibitory (I-SMADs). The R-SMADs are phosphorylated by the activated Type I receptors on their C-terminus. This group includes SMAD1, 2, 3, 5 and 8.13 Phosphorylated R-SMADs form homotrimers and interact with SMAD4, a Co-SMAD that mediates translocation of R-SMADs into the nucleus. R-SMADs and Co-SMAD complexes function as TFs and regulate the expression of genes associated with cell proliferation, survival and differentiation. Their activity is counter-regulated by the I-SMADs, SMAD6/7, which interact with type I receptors and competitively interfere with the activation of R-SMADs.14 SMADs constantly shuttle between the cytoplasm and nucleus but they are retained longer in the nucleus upon activation by TGFβRI. A number of accessory proteins have been identified that mediate the interactions of R-SMADs with their membrane receptors. Examples of such proteins are Disabled-2 (Dab2) and SMAD anchor for receptor activation (SARA, encoded by Zfyve9). Dab2 interacts with SMAD2/3 and promotes their association with TGFβRI and RII.15 In the immune system Dab2 is required for normal Treg cell function, perhaps in settings that require high TGF-β signalling.16 SARA tethers unphosphorylated SMADs to the TGFβRI kinase in the cytoplasm and dissociates from them upon SMAD activation.17 Both Dab2 and SARA are primarily expressed in the myeloid lineage and may not operate in conventional effector lymphocytes (http://www.Immgen.org).
The human genome contains nearly 40 members of the TGF-β family.18 However, the primary experimental focus has been on TGF-β1, one of the three TGF-β proteins that is expressed in most cell types. TGF-β signalling activates SMAD2 and SMAD3. These proteins comprise two N-terminus Mad homology (MH) domains. The MH1 domain is necessary for nuclear import, DNA binding and transcription, while the MH2 domain promotes protein oligomerization, which is required for efficient transcriptional activation. The MH2 domains of SMAD2 and SMAD3 proteins are nearly identical,17 whereas their MH1 domains only share ∼ 66% homology. In the nucleus, the MH1 domains of SMAD3 and SMAD4 recognize the sequence 5′-GTCT-3′ or its reverse complement that form a palindrome; complexes of SMAD3/4 recognize direct or inverted repeats of the SMAD binding element,19,20 In contrast, the elongated MH1 domain of full-length SMAD2 cannot efficiently bind to DNA.21 However, an alternatively spliced form of Smad2 (lacking exon 3) that can bind to DNA exists and this isoform has been shown to functionally mediate most aspects of TGF-β/Nodal signalling.22 The relative proportions of the two forms of SMAD2 have not yet been systematically determined in lymphocytes. Due to their weak binding, SMADs work as oligomeric complexes at the SMAD binding element and require synergistic actions from other TFs. Importantly, unlike some TFs that can bind to relatively unoccupied DNA and directly recruit transcription activation complexes to proximal promoters, SMAD TFs are dependent on chromatin modifiers to assemble the basal transcription machinery.23 Once positioned on the chromatin, SMADs promote further remodelling by recruiting histone-modifying enzymes such as the histone acetylase p300 (which acetylates histone H3), the SWI/SNF component Brg1, and the histone demethylase KDM6B (JMJD3),23,24 or by interacting with TFs that can modulate the activities of chromatin regulators, such as ATF-3,25 HEB/E2A26 and LEF1.27 Furthermore, factors that discriminate histone modification marks can interact with SMAD2/3 and distribute them to discrete chromatin regions. One example of this mode of shuttling involves TRIM33 (Tif1γ), which selectively binds to active H3 modifications,28,29 TRIM33 binds to SMAD2/3 and shuttles them to specific promoters for chromatin remodelling and transcription activation.29,30
The co-operation of chromatin modifiers and TFs in regulating gene transcription underpins the context-dependent function of TGF-β-activated SMADs. System-wide studies to map SMAD docking sites in the genome of diverse cell lineages showed that SMADs (including SMAD3 activation downstream of TGF-β) are co-localized with, and are regulated by, cell-type-specific master TFs,31–33 Hence, a substantial proportion of, if not most, SMAD occupancy and transcriptional modulation in a cell type reflects the global positioning of master TFs and their gene network. The master TFs help in promoting SMAD binding by establishing open chromatin, where SMADs bind to SMAD binding element and form a physical complex with the master TFs. This versatile chromatin docking of SMADs (directed by cell-type-specific master TFs) enables them to mediate expression of a wide variety of unrelated genes in distinct cell types,32,33 The global SMAD genomic occupancy is also likely to evolve with the lymphocyte activational state because the two major activation induced TFs, nuclear factor of activated T cells (NFAT) and activator protein 1 (AP-1), co-operatively control genes that dominantly specify cell fate, such as FOXP3,34,35 and cell survival.36–38
Many TGF-β target genes are redundantly regulated in a given cell type by SMAD2 and SMAD3, but unique targets also exist. For example, SMAD3, but not SMAD2, interacts with the FOXO TF family members to activate transcription of the cell cycle regulator p21 (Cdkn1a).39 Similarly, SMAD3 specifically interacts with vitamin D receptor and its over-expression induces transactivation of the vitamin D receptor-associated target genes.40 Conversely, SMAD2 is specifically necessary for Mmp2 induction in fibroblasts.41 As discussed below, different nuclear SMAD TF complexes mediate lymphoid effector lineage diversification.
SMADs as molecular relays for sensing commensal microflora
The defining role of the immune system is to respond to pathogens while remaining tolerant to innocuous antigens. This delicate discerning action is especially critical in the gut, which is home to countless commensal microbes, and at the same time, constantly bombarded by food-derived antigens and harmful agents. A cell network in the gut-associated lymphoid tissues (the epithelial border, sub-mucosal lamina propria, isolated lymphoid follicles, Peyer's patches and mesenteric lymph nodes, lymph nodes) consisting of innate and adaptive immune cells and diverse mucosal epithelial cell subsets permits nutrient absorption while remaining poised for inflammatory responses to deal with pathogens. A central intercellular cue directing this network is TGF-β.
The intestinal microflora has been shown to be the primary determinant of gut and immune system homeostasis, central to the overall health of the host.42 Nearly all cells in the gut are modulated by the microbiota. Although no comprehensive studies of tissue distribution of bioactive TGF-β exist, the gut is considered to be rich in TGF-β, with specialized CD103+ dendritic cells as a major source directing T-cell differentiation,43,44 TGF-β-dependent T-cell and B-cell subsets, such as Treg cells generated outside the thymus from naive CD4+ T cells (induced Treg, iTreg45), Th1746 and IgA+ B cells,47 are found predominantly in the gut in pathogen-naive rodents (see below). It has been shown that the components of healthy microbiota stimulate TGF-β production from epithelial cells and phagocytes to drive suppressive iTreg (IL-10+ CTLA− 4hi Helios– FOXP3+ CD4+ T cells, by Clostridium species)48 or Th17 (by segmented filamentous bacteria) cell generation,49,50 likely dependent on the location (colon versus small intestine) and the mode of bacterial–intestinal epithelial cell interactions,48,49 The commensal-dependent TGF-β-induced colonic iTreg cells can arise in the absence of Myd88 (an obligate adaptor for Toll-like receptors) and may be particularly critical during the neonatal period to suppress innate or innate-like T cells such as invariant natural killer T cells51 that can cause diseases similar to human ulcerative colitis. Interleukin-10 has been shown to co-operate with TGF-β to promote iTreg cell generation and the microbiota is also able to modulate IL-10 production from the colonic enterocytes and CD4+ T cells.42 Moreover, given the high turnover of intestinal epithelial cells, the gut is replete with apoptotic cells, and phagocytic ingestion of these dying cells has been shown to be a potent inducer of TGF-β secretion. Concurrent IL-6 production by innate receptor signalling on immune cells52 or high rate of activation-induced T-cell death and phagocytosis,53,54 can promote the generation of Th17 cells that can be further differentiated in the intestine to acquire immune suppressive properties.53 It is important to note that without the microbiota (such as in germ-free mice) functional natural Treg (nTreg) cells are still generated from the thymus,55 while Th17 cells are severely diminished.
Paradoxically, TGF-β expression is increased in most tissues during inflammation. Therefore an obvious question is why this increase does not support immunosuppression and amelioration of disorders. One answer is that inflammatory milieus are intimately linked to disruptions in the normal TGF-β regulatory circuit. Tumour necrosis factor-α and interferon-γ (IFN-γ) made by activated lymphocytes can induce the I-SMAD, SMAD7,56,57 The expression of SMAD7 is increased in patients with Crohn's disease, with a corresponding decrease in the levels of phosphorylated SMAD3.58 Similar situations exist in other autoimmune diseases such as multiple sclerosis.59 CD4+ T cells from patients with Crohn's disease cannot be suppressed by functional Treg cells isolated from healthy patients, but this insensitivity can be reversed by decreasing the amounts of SMAD7 in the pathogenic lymphocytes.60 Mice ectopically expressing Smad7 replicate the desensitized TGF-β signalling in Treg cells observed in human inflammatory disorders,58 although the enhanced Smad7 expression does not lead to a breakdown in global T-cell homeostasis.61 The impaired TGF-β signalling not only disrupts the Treg cell-mediated suppressive loop, but it can lead to pathogenic IFN-γ+ CD4+ T effector cell generation from Th17 cells,62 further amplifying the inflammatory loop. SMAD2 activated by TGF-β also directly influences the homing of T cells to the gut by controlling the expression of integrin αEβ7 (CD103).63 CD103 is expressed on the majority of FOXP3+ nTreg cells and ectopically high expression of SMAD7 decreases CD103 expression on T cells, potentially interfering with the gut tropism of Treg cells.64 Hence, while acute dampening of TGF-β signalling can be beneficial during infections in the gut, chronic inflammation can compromise the TGF-β-mediated suppressive loop, precipitating and/or propagating autoimmunity in the gut.
SMADs mediate IgA production in the gut-associated lymphoid tissues
The importance of TGF-β for gut immunity and homeostais was presaged more than 25 years ago with the finding that TGF-β is critical for the IgA production by LPS-activated B cells,47,65,66 Mucophilic IgA restricts commensal bacterial penetration into the gut mucosa and neutralizes pathogenic bacteria and viruses through the complement cascade.67 Peyer's patches are the sites of germinal centre formation in the small intestine populated mostly with B cells that can produce copious amounts of IgA. SMAD2/3/4 complexes can dock onto the SMAD binding element sequence of the immunoglobulin Cα gene, and in conjunction with multiple co-factors, including RUNX3 and PU.1,68–70 promote germline transcription and switch recombination. Mice deficient in SMAD2 specifically in B cells produce more B cells, but class switching to IgA is reduced.71 Complementary to the Smad2-deficient mice, the loss of SMAD7 in B cells resulted in a decreased cellularity of B cells, but the residual B cells exhibited an increased bias for IgA production, which correlated with the peak SMAD2 phosphorylation after TGF-β stimulation. The absence of SMAD7 enhances the transcription of germline and post-switch Cα transcripts. Although over-expression of SMAD3 and SMAD4 in splenic B cells increased the expression of IgA,72 Smad3-deficient mice were reported to have normal IgA production,73 indicating that in vivo, SMAD2 and SMAD3 perform redundant functions and/or SMAD2 is the dominant activator of IgA class switching.
R-SMADs control adaptive lymphocyte differentiation
CD4+ T-cell subsets play a critical role in the regulation of immune homeostasis. In general, TGF-β is anti-proliferative, and in CD4+ T cells, it suppresses the production of IL-2,74,75 the quintessential autocrine growth factor for T cells made immediately after T-cell activation. SMADs bind to the SMAD binding element in the Il2 promoter and interact with recruited histone methyl transferases, Setb1 and Suv39 h1, to inhibit T-cell receptor (TCR) signal-mediated Il2 transcription.74 In the immune suppressive network, TGF-β is necessary for the generation of nTreg cells, particularly during the neonatal stage76 and iTreg cells that differentiate from naive CD4+ T cells extrathymically. Conversely, TGF-β in association with IL-6 made during inflammation drives the differentiation of naive T cells to non-pathogenic Th17 cells, as opposed to pathogenic Th17 cells that develop with IL-1β and IL-23.77 The TGF-β directly inhibits IFN-γ+ Th1 and IL-4/13+ Th2 cell subset differentiation,10,61 by blocking the expression of TFs Stat4 and Gata3, respectively.78 Further, recently published data show that the induction of HMG box TF SOX4 downstream of the TGF-β–SMAD3 pathway antagonizes the action of GATA3, thereby inhibiting Th2 differentiation.79 TGF-β signalling, in synergy with IL-2 and IL-4, is also critical for the generation of IL-9-producing CD4 T cells (Th9),80,81 IL-9 is a pleiotropic cytokine which can enhance both Treg and Th17 cell differentiation or proliferation.82 Interleukin-9 induction requires co-operation between Notch and TGF-β–SMAD3 signalling pathways.83 Hence, TGF-β is central to assembling an optimal mix of specialized lymphoid effectors for efficient pathogen clearance. The importance of TGF-β–SMAD signalling in promoting balanced immunity in the gut is underscored during infections with the intestinal helminth Heligmosomoides polygyrus.84 The helminth antigens ligate TGF-βR and activate SMAD2/3 to enhance iTreg or Th17 cell generation in the gut. The hijacking of the SMAD pathway by microbes for their survival illustrates the dynamic tuning of immune responses by TGF-β that can dictate life and death for the host and pathogens.
There are several mutually contingent models to account for the context-dependent function of TGF-β in T-cell subset differentiation (Fig. 1). First, different concentrations of bioactive TGF-β may determine the signal outcome. For instance, high concentrations of TGF-β have been associated with iTreg cell differentiation, whereas relatively weak TGF-β signalling favours Th17 cell differentiation.85 Second, T-cell subset lineage master TFs and cytokine-activated TFs can impose a distinct repertoire of nuclear SMAD target gene selectivity such that each T-cell subset contains common as well as unique SMAD-regulated genes,32,33 The Th17 master TF RORγt physically interacts with SMAD2 and SMAD3,86,87 whereas TCR signalling induced NFAT co-operates with SMAD3 to regulate Foxp3 transcription in vitro.34 Moreover, cytokine-activated signal transducers and activators of transcription (STATs) and the negative feedback loop regulators suppressor of cytokine signaling/protein inhibitors of activated STATs (SOCS/PIAS),88–90 have strong modulatory effects on SMAD function. Third, distinct SMAD complexes can have biased activities during T-cell subset diversification, most likely as a result of the integration of the first two processes specifying the context of nuclear SMAD function with unique biophysical features of SMAD complexes.91
Figure 1.
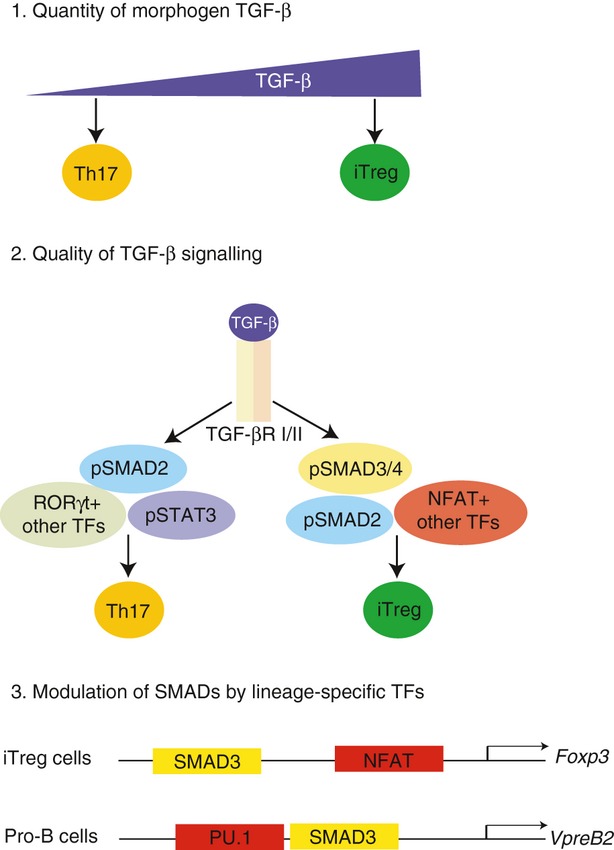
Context dependent mechanisms of action of transfomring growth factor-β (TGF-β) in mediating cell fate specification. 1. Higher concentrations of TGF-β favours inducible regulatory T (iTreg) cell differentiation while at lower concentrations, TGF-β along with interleukin-6 (IL-6) induces T helper type 17 (Th17) cell generation. 2. Differential activation of SMAD2 versus SMAD3 regulates effector T-cell differentiation. Downstream of TGF-βR triggering, R-SMADs and co-SMAD are activated and each may exert unequal control over discrete clusters of TGF-β-modulated genes. For instance, SMAD2/3/4 are all important for iTreg cell differentiation. However, for Th17 cell generation, SMAD2 is necessary while SMAD3/4 are dispensable. 3. Co-operative actions of SMADs at gene loci with context-specific (cell type or state) transcription factors (TFs) can account for variable sets of genes controlled by TGF-β. For example, during iTreg cell differentiation, SMAD3 binds to SMAD binding elements in the Foxp3 gene enhancer in concert with the T-cell activation induced TF NFAT, which binds at the promoter region of Foxp3, to induce the co-regulated target gene. In pro-B cells, SMAD3 co-occupies the genome with PU.1 to regulate the transcription of B-cell-specific genes such as Vpreb2.
T-cell defects in mice lacking SMADs
To dissect how TGF-β controls the activation, differentiation and homeostasis of T cells, SMAD-deficient mice have been investigated. Mice lacking SMAD2/3 (double knockout, DKO) in αβ T cells (CD4 promoter-regulated Cre transgene crossed to Smad2 fl/fl Smad3−/− mice, Cd4Cre:DKO, the deletion starting in CD4+ CD8+ double-positive thymocytes) developed a fatal lymphoproliferative disorder similar to the disease in Tgfb−/− mice,92,93 confirming that the TGF-β-mediated T-cell homeostasis is dependent on the conventional TGF-β signalling, and the alternative pathways involving Tripartite motif-containing 33 (TRIM33),30 TNF receptor associated factor 6 (TRAF6)-TGF-beta activated kinase 1 (TAK1)94 or mitogen-activated protein kinase13 cannot compensate effectively. However, the deregulated T cells in DKO mice arise as a result of impairments in conventional T cells, not nTreg cells that are normal in number and function. Rather, nTreg cell production is regulated by TAK1 in the presence95 or absence96 of SMAD2/3. Interestingly, when SMAD2/3 were rendered non-functional starting in T-cell precursors (using Lck-Cre), rather than in double-positive thymocytes, nTreg cells were severely impacted.97 This result raises a provocative possibility that nTreg cell differentiation is initiated or programmed in thymic T precursor cells by SMAD2/3 activating factors, most likely Activin or TGF-β.
To define unique functions of SMAD2 and SMAD3, mice lacking individual SMADs were used. The first line of Smad3 mutant mice was generated by the disruption of exon 2. These mice were viable but developed metastatic colorectal cancer that spread to the lymph nodes.98 The role of SMAD3 in the immune response was examined in mice in which exon 8 of the Smad3 gene was excised. The deletion of this exon, which encodes for the C-terminus of the SMAD3 protein required for the interaction with TGF-β receptors, resulted in enlarged peripheral lymph nodes because of hyperactive T cells that were unresponsive to TGF-β.73 Symptomatic Smad3 mutant mice die between 1 and 3 months of age as the result of a wasting syndrome that is associated with the formation of pyogenic abscesses within the walls of the stomach and intestine. However, in another line generated by removing the exon 1, immune homeostasis was maintained,86,99 The exact source of the discrepancy in phenotypes has not been pinpointed. Similar to T cells from DKO mice, Smad3-deficient T cells are defective in iTreg cell differentiation with altered induction of iTreg cell-specific genes.86 The iTreg cells are required particularly for the maintenance of mucosal tolerance in the gastrointestinal tract and lungs.100 Other cytokines and morphogens can enhance or inhibit the differentiation of iTreg cells by modulating the activity of SMAD3. For instance, the vitamin A metabolite retinoic acid and Notch signalling synergize with TGF-β to induce Foxp3 while other cytokines inhibit iTreg cell differentiation.88,101,102 Retinoic acid is prevalent in the gut, it drives iTreg cell differention,101,103 by increasing the phosphorylation of SMAD3,104 as well as facilitating its docking at the CNS1 region of the Foxp3 enhancer.88 Similarly, it was shown that pSMAD3 can interact with the intracellular domain of Notch in T cells and this interaction facilitates nuclear translocation of pSMAD3,83,105 Conversely, cytokines such as IL-27 induce activated STAT1 and STAT3 to inhibit the binding of pSMAD3 at the Foxp3 enhancer.88,106
In contrast to defective iTreg cell induction, SMAD3-deficient T cells have an enhanced propensity to develop into Th17 cells in the presence of TGF-β and IL-6.86 SMAD3 can physically interact with RORγt and potentially inhibit RORγt-dependent transcriptional activation of the Il17 gene.86 One of the functions of IL-6 in Th17 cultures is to inhibit the phosphorylation of SMAD3,107 consistent with a negative role of SMAD3 in Th17 cell differentiation, possibly as a consequence of its ability to promote Foxp3 transcription during iTreg cell differentiation. To complete this regulatory loop, FOXP3 in turn has also been shown to be a negative regulator of RORγt, by direct protein interactions.85,108
These results suggested a biased function of SMAD3 in iTreg cell generation and raised the possibility that the alternate R-Smad, SMAD2, would counterbalance SMAD3 and promote TGF-β-regulated Th17 cell generation. In support of this, Smad2-deficient T cells exhibit significant defects in their differentiation to Th17 lineage cells in vitro and in vivo.87,92,109 Infection of Smad2-deficient mice with Citrobacter rodentium showed a diminished ability of T cells to produce IL-17.109 Similarly, Smad2-deficient mice exhibited delayed onset of experimental autoimmune encephalitis.87 SMAD2 regulates Il6ra expression and STAT3 phosphorylation in T cells. It has also been shown that SMAD2 interacts with RORγt, but unlike SMAD3, this complex enhances IL-17 production in T cells.87 Similar to SMAD3, SMAD2 is also required for the development of FOXP3+ iTreg cells.86,87,109 While SMAD3 has been shown to interact with NFAT at the Foxp3 gene enhancer in vitro, recruitment of SMAD2 to the Foxp3 locus was not observed.34 Recent data indicated that in vivo SMAD3 binding to the Foxp3 gene enhancer was necessary for iTreg cell generation, but not for nTreg cell differentiation.110 Therefore, how and to what extent SMAD2 and SMAD3 mediate induction of Foxp3 in vivo will require additional studies.
Although, Smad2 conditional KO (CKO) mice do not suffer from spontaneous autoimmunity, administration of the colitogenic compound dextran sodium sulphate induces more extensive intestinal damage in Smad2-deficient mice relative to wild-type mice.92 Also, in the bone marrow chimeric system, transfer of Smad2-deficient bone marrow cells into irradiated Rag1−/− recipients, with or without passenger wild-type cells, induced massive T-cell-mediated systemic autoimmunity (unpublished results,109). The latter finding reinforces the importance of TGF-β in regulating self-reactivity of peripheral T cells under lymphopenic conditions111 and demonstrates that SMAD2 may be functionally relevant in this process.
Selective function of SMAD4 in TGF-β signalling in the immune system
Since SMAD2 and SMAD3 use SMAD4 for translocation to the nucleus, it was expected that the absence of SMAD4 would phenocopy mice lacking SMAD2/3. However, aggregate results from several independently generated lines of Smad4 mutants suggest a more nuanced function of SMAD4 in controlling immune homeostasis. T-cell development and peripheral homeostasis are relatively normal in mice with Smad4 deficiency in T cells, but iTreg cell generation is inefficient.108,112 The deletion of Smad4 using Lck-Cre or GranzymeB-Cre (which are initiated in double-negative thymic precursors or activated T cells, respectively) renders mice susceptible to gastroduodenal lesions with age, characterized by an increased production of pro-inflammatory cytokines in the intestinal lamina propria, including IL-1β, IL-6 and IL-17.112 This inflammatory phenotype was not noted in Cd4Cre:Smad4 fl/fl mice.108 In BALB/c strains, the absence of SMAD4 in T cells results in gastrointestinal cancer, attributed to a deviated differentiation of CD4 T cells that secrete Th2-related cytokines IL-5, IL-6 and IL-13, which are postulated to interfere with tumour surveillance.113 Currently it is difficult to integrate these observations because the mouse models have different genetic backgrounds and the analyses were performed using mice of substantially different ages. Minimally, the results suggest that SMAD4 is non-redundant for only select paths of the TGF-β-regulated gene circuit, including FOXP3 induction in naive CD4+ T cells, suppression of Th2 differentation and control of cell proliferation (particularly the intestinal epithelial cells).13,114 This selectivity of SMAD4 is a recurring theme in organogenesis in various species115 and is not restricted to the immune system. It is currently unknown whether a functional homologue of SMAD4 exists that can shuttle SMAD2/3 into the nucleus. However, it has been shown that in the nucleus, SMAD2 and SMAD3 can function independent of SMAD4. For instance, in erythroid cells TRIM33 (TIF1γ) has been shown to compete with SMAD4 for interacting with SMAD2/3.30 TRIM33-SMAD2/3 complexes are also observed in T cells, although SMAD2/3 are not obligatory for TIF1γ function downstream of TGF-β stimulation.116
SMADs modulate the function of gut-associated lymphoid tissue innate lymphocytes
Intestinal intraepithelial lymphocytes (i-IELs) are instrumental for maintaining the integrity of the mucosal barrier,117,118 modulating inflammatory responses by secreting TGF-β,119,120 and inducing production/release of microbial peptides by epithelial cells.118,121 Intestinal IELs are distributed across the epithelial barrier in a sentry formation, one IEL for every five to ten epithelial cells, and are enriched for cells expressing CD8αα homodimers (distinct from CD8αβ heterodimers expressed by circulating T cells), with a marginal bias for TCR-γδ+ cells for most laboratory mouse strains.
The TGF-β signalling is not necessary for the development of γδ IELs, and these rely instead on TCF1, the nuclear effector of canonical WNT signalling.122 However, γδ IELs are a potent and unique source of TGFβ-3 (www.Immgen.org). Given their unique developmental requirements,123,124 their dependence on Aryl hydrocarbon receptor signalling for proper maintenance and function,121 and the known function of TGF-β3 in wound healing and epithelial cell turnover,125 it is likely that γδ IELs elaborate unique aspects of TGF-β function in the gut. In contrast to γδ IELs, the development of TCR-αβ+ CD8αα+ IELs is dependent on TGF-β–SMAD signalling.126 Mice deficient in TGF-β signalling or Smad3 are depleted of αβ IELs in the intestine and their precursors in the thymus, in part through enhanced apoptosis, although impaired trafficking of IELs may also contribute to the loss, as TGF-β signalling controls gut homing CD103 integrin expression.64 It has been shown that TGF-β signalling is an arbiter of cell survival in the immune system by setting the balance of the apoptosis-regulating proteins Bcl-xl and Bim,127,128 For instance, TGF-β signalling protects TCR-αβ+ i-IEL precursors and nTreg cells from apoptosis in the thymus.129 However, in other cells, TGF-β signalling can promote apoptosis. In persistent lymphocytic choriomeningitis virus infections, TGF-β–SMAD2 signal transduction is enhanced in virus-specific CD8 T cells, which leads to the increased expression of Bim and subsequent cell death.130 The cause of these opposing outcomes of TGF-β signalling is undetermined, but is likely to reflect distinct chromatin states and co-factor availabilities in cells of different maturity, in distinct tissues and at different phases of activation.
SMADs permit controlled activation of intestinal epithelial cells
One function of IL-17 is to elicit epithelial cell secretion of inflammatory chemokines, including CXCL16 that has been implicated as a permissive and necessary factor for colitis induction.51 Activation of the single layer of epithelial cells of the intestinal barrier by pathogens and cytokines is critical for host defence and regulated by TGF-β, which maintains a basal suppressed state and inhibits proliferation and transformation.131 Upon damage or breach to the barrier, epithelial cells induce TGF-β and epidermal growth factor for wound repair,132,133 Epithelial cells impaired in TGF-βR signalling are hypersensitive to chemically induced damage and are impaired in re-epithelialization of the barrier post-injury, leading to more pronounced weight loss and fatality.133
Although TGF-β is the central conductor of gut homeostasis, its function relies to a significant extent on co-operation with other immunomodulatory factors, of which IL-10 stands out. Il10−/− mice develop spontaneous rectal prolapses, dependent on the gut flora. Upon mono-colonization with the Gram-positive gut commensal Enterococcus faecalis, Il10−/− mice show heightened inflammatory responses characterized by increased Toll-like receptor 2 signalling in the intestinal epithelial cells,134,135 Normally, this innate signalling is counter-balanced by TGF-β–SMAD2 activation,134,136 but in the absence of IL-10, SMAD2 activation was not observed. It is unclear how IL-10 potentiates TGF-β signalling at the molecular level, but in vitro studies have shown that IL-10 can induce TGF-β expression in T cells.137 Reciprocally, it has been shown that TGF-β–SMAD4 signalling increases the IL-10 secretion necessary for preventing bleomycin-induced fibrosis in mice.138 Hence, synergistic actions of autocrine and paracrine TGF-β and IL-10, as well as a multitude of other suppressive factors139 on epithelial cells dampen the inflammatory responses of intestinal epithelial cells.
Future perspectives
The molecular basis for context-dependent activity of the TGF-β–SMAD regulatory network is coming into a sharper focus, but much still remains blurred. Data so far indicate that while SMAD2 and SMAD3 are redundantly required downstream of TGF-β for systemic homeostasis, each has unique function in controlling regulatory and effector lymphocytes. In assessing the relative contribution of SMAD2 versus SMAD3 in maintaining T-cell homeostasis it is instructive to note that while the Smad2/3 DKO phenocopies Tgfb−/− mice, Smad3−/− mice are associated with more localized inflammatory disorder, indicating robust functionality of the SMAD2/4 complex downstream of TGF-β. Identifying the context (concentration and source of TGF-β, the state of responding cells, impact of other cytokines and tissue niches) in which distinct SMAD complexes are functionally dominant and how they affect target gene landscape will require further studies. This has clinical importance given that most human intestinal tumours are associated with mutations in Smad2/4 genes, but Smad3 alterations are rare.140 Studies to date have focused on SMAD function in the adaptive arm of the immune system, but more effort needs to be applied to map the SMAD-regulated gene networks in innate and innate-like lymphoid effector cells, which are prevalent in mucosal tissues141 as well as in the epithelial cell barrier that forms the first line of defence against pathogens. As the quintessential tonic suppressor of resting haematopoietic cells, SMAD complexes are likely to be ubiquitous at basal states and must be redistributed genomically to permit transitions to several possible active states. Deciphering the detailed dynamics involved in this process, particularly the role of myriad activation induced co-factors downstream of the sensors of altered extracellular milieu (such as TCR, cytokine receptors and Toll-like receptors) in modifying SMAD activities, will be a major challenge for the field.
Acknowledgments
We thank M. Bix, K. Narayan and N. Jain for comments on the manuscript. The work was supported by National Institutes of Health grants AI083505 and CA100382 to J.K.
Disclosures
The authors have no financial disclosures or competing interests.
References
- 1.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Li MO, Flavell RA. TGF-β: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 4.Wahl SM. Transforming growth factor β: the good, the bad, and the ugly. J Exp Med. 1994;180:1587–90. doi: 10.1084/jem.180.5.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–9. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kulkarni AB, Huh CG, Becker D, et al. Transforming growth factor β1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770–4. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-β1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26:579–91. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 8.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-β receptor. Immunity. 2006;25:441–54. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 9.Fantini MC, Becker C, Tubbe I, Nikolaev A, Lehr HA, Galle P, Neurath MF. Transforming growth factor β induced FoxP3+ regulatory T cells suppress Th1 mediated experimental colitis. Gut. 2006;55:671–80. doi: 10.1136/gut.2005.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-β induces development of the TH17 lineage. Nature. 2006;441:231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 11.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 12.Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-β responses. Cell. 1998;95:737–40. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- 13.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 14.Schmierer B, Hill CS. TGFβ-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–82. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 15.Hocevar BA, Smine A, Xu XX, Howe PH. The adaptor molecule Disabled-2 links the transforming growth factor β receptors to the Smad pathway. EMBO J. 2001;20:2789–801. doi: 10.1093/emboj/20.11.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain N, Nguyen H, Friedline RH, et al. Cutting edge: Dab2 is a FOXP3 target gene required for regulatory T cell function. J Immunol. 2009;183:4192–6. doi: 10.4049/jimmunol.0902041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown KA, Pietenpol JA, Moses HL. A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF-β signaling. J Cell Biochem. 2007;101:9–33. doi: 10.1002/jcb.21255. [DOI] [PubMed] [Google Scholar]
- 18.Feng XH, Derynck R. Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–93. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 19.Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-β signaling. Cell. 1998;94:585–94. doi: 10.1016/s0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]
- 20.Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–7. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- 21.Dennler S, Huet S, Gauthier JM. A short amino-acid sequence in MH1 domain is responsible for functional differences between Smad2 and Smad3. Oncogene. 1999;18:1643–8. doi: 10.1038/sj.onc.1202729. [DOI] [PubMed] [Google Scholar]
- 22.Dunn NR, Koonce CH, Anderson DC, Islam A, Bikoff EK, Robertson EJ. Mice exclusively expressing the short isoform of Smad2 develop normally and are viable and fertile. Genes Dev. 2005;19:152–63. doi: 10.1101/gad.1243205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ross S, Cheung E, Petrakis TG, Howell M, Kraus WL, Hill CS. Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J. 2006;25:4490–502. doi: 10.1038/sj.emboj.7601332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim SW, Yoon SJ, Chuong E, Oyolu C, Wills AE, Gupta R, Baker J. Chromatin and transcriptional signatures for Nodal signaling during endoderm formation in hESCs. Dev Biol. 2011;357:492–504. doi: 10.1016/j.ydbio.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 25.Kang Y, Chen CR, Massague J. A self-enabling TGFβ response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell. 2003;11:915–26. doi: 10.1016/s1097-2765(03)00109-6. [DOI] [PubMed] [Google Scholar]
- 26.Yoon SJ, Wills AE, Chuong E, Gupta R, Baker JC. HEB and E2A function as SMAD/FOXH1 cofactors. Genes Dev. 2011;25:1654–61. doi: 10.1101/gad.16800511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Labbe E, Letamendia A, Attisano L. Association of Smads with lymphoid enhancer binding factor 1/T cell-specific factor mediates cooperative signaling by the transforming growth factor-β and wnt pathways. Proc Natl Acad Sci USA. 2000;97:8358–63. doi: 10.1073/pnas.150152697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agricola E, Randall RA, Gaarenstroom T, Dupont S, Hill CS. Recruitment of TIF1γ to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol Cell. 2011;43:85–96. doi: 10.1016/j.molcel.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 29.Xi Q, Wang Z, Zaromytidou AI, et al. A poised chromatin platform for TGF-β access to master regulators. Cell. 2011;147:1511–24. doi: 10.1016/j.cell.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He W, Dorn DC, Erdjument-Bromage H, Tempst P, Moore MA, Massague J. Hematopoiesis controlled by distinct TIF1γ and Smad4 branches of the TGFβ pathway. Cell. 2006;125:929–41. doi: 10.1016/j.cell.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 31.Mizutani A, Koinuma D, Tsutsumi S, et al. Cell type-specific target selection by combinatorial binding of Smad2/3 proteins and hepatocyte nuclear factor 4α in HepG2 cells. J Biol Chem. 2011;286:29848–60. doi: 10.1074/jbc.M110.217745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mullen AC, Orlando DA, Newman JJ, et al. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell. 2011;147:565–76. doi: 10.1016/j.cell.2011.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trompouki E, Bowman TV, Lawton LN, et al. Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell. 2011;147:577–89. doi: 10.1016/j.cell.2011.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008;9:194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- 35.Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, Tone M, Chen YH. Development of Foxp3+ regulatory T cells is driven by the c-Rel enhanceosome. Immunity. 2009;31:932–40. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Feng XH, Derynck R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-β-induced transcription. Nature. 1998;394:909–13. doi: 10.1038/29814. [DOI] [PubMed] [Google Scholar]
- 37.Verrecchia F, Tacheau C, Schorpp-Kistner M, Angel P, Mauviel A. Induction of the AP-1 members c-Jun and JunB by TGF-β/Smad suppresses early Smad-driven gene activation. Oncogene. 2001;20:2205–11. doi: 10.1038/sj.onc.1204347. [DOI] [PubMed] [Google Scholar]
- 38.Yamamura Y, Hua X, Bergelson S, Lodish HF. Critical role of Smads and AP-1 complex in transforming growth factor-β-dependent apoptosis. J Biol Chem. 2000;275:36295–302. doi: 10.1074/jbc.M006023200. [DOI] [PubMed] [Google Scholar]
- 39.Seoane J, Le HV, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–23. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 40.Yanagisawa J, Yanagi Y, Masuhiro Y, et al. Convergence of transforming growth factor-β and vitamin D signaling pathways on SMAD transcriptional coactivators. Science. 1999;283:1317–21. doi: 10.1126/science.283.5406.1317. [DOI] [PubMed] [Google Scholar]
- 41.Piek E, Ju WJ, Heyer J, et al. Functional characterization of transforming growth factor β signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem. 2001;276:19945–53. doi: 10.1074/jbc.M102382200. [DOI] [PubMed] [Google Scholar]
- 42.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–18. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 43.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–64. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamazaki S, Bonito AJ, Spisek R, Dhodapkar M, Inaba K, Steinman RM. Dendritic cells are specialized accessory cells along with TGF-β for the differentiation of Foxp3+ CD4+ regulatory T cells from peripheral Foxp3 precursors. Blood. 2007;110:4293–302. doi: 10.1182/blood-2007-05-088831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25– naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor-β1 promotes in vivo Th17 cell differentiation. Immunity. 2011;34:396–408. doi: 10.1016/j.immuni.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stavnezer J, Kang J. The surprising discovery that TGF β specifically induces the IgA class switch. J Immunol. 2009;182:5–7. doi: 10.4049/jimmunol.182.1.5. [DOI] [PubMed] [Google Scholar]
- 48.Atarashi K, Tanoue T, Shima T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–41. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–98. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31:677–89. doi: 10.1016/j.immuni.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 51.Olszak T, An D, Zeissig S, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–93. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Torchinsky MB, Garaude J, Martin AP, Blander JM. Innate immune recognition of infected apoptotic cells directs TH17 cell differentiation. Nature. 2009;458:78–82. doi: 10.1038/nature07781. [DOI] [PubMed] [Google Scholar]
- 53.Esplugues E, Huber S, Gagliani N, et al. Control of TH17 cells occurs in the small intestine. Nature. 2011;475:514–8. doi: 10.1038/nature10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perruche S, Zhang P, Liu Y, Saas P, Bluestone JA, Chen W. CD3-specific antibody-induced immune tolerance involves transforming growth factor-β from phagocytes digesting apoptotic T cells. Nat Med. 2008;14:528–35. doi: 10.1038/nm1749. [DOI] [PubMed] [Google Scholar]
- 55.Chinen T, Volchkov PY, Chervonsky AV, Rudensky AY. A critical role for regulatory T cell-mediated control of inflammation in the absence of commensal microbiota. J Exp Med. 2010;207:2323–30. doi: 10.1084/jem.20101235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ulloa L, Doody J, Massague J. Inhibition of transforming growth factor-β/SMAD signalling by the interferon-γ/STAT pathway. Nature. 1999;397:710–3. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- 57.Bitzer M, von Gersdorff G, Liang D, Dominguez-Rosales A, Beg AA, Rojkind M, Bottinger EP. A mechanism of suppression of TGF-β/SMAD signaling by NF-kappa B/RelA. Genes Dev. 2000;14:187–97. [PMC free article] [PubMed] [Google Scholar]
- 58.Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J Clin Investig. 2001;108:601–9. doi: 10.1172/JCI12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kleiter I, Song J, Lukas D, et al. Smad7 in T cells drives T helper 1 responses in multiple sclerosis and experimental autoimmune encephalomyelitis. Brain. 2010;133(Pt 4):1067–81. doi: 10.1093/brain/awq039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fantini MC, Rizzo A, Fina D, et al. Smad7 controls resistance of colitogenic T cells to regulatory T cell-mediated suppression. Gastroenterology. 2009;136:1308–16. doi: 10.1053/j.gastro.2008.12.053. e1–3. [DOI] [PubMed] [Google Scholar]
- 61.Nakao A, Miike S, Hatano M, Okumura K, Tokuhisa T, Ra C, Iwamoto I. Blockade of transforming growth factor β/Smad signaling in T cells by overexpression of Smad7 enhances antigen-induced airway inflammation and airway reactivity. J Exp Med. 2000;192:151–8. doi: 10.1084/jem.192.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, Strober W. TGF-β1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol. 2004;172:834–42. doi: 10.4049/jimmunol.172.2.834. [DOI] [PubMed] [Google Scholar]
- 64.Suzuki R, Nakao A, Kanamaru Y, Okumura K, Ogawa H, Ra C. Localization of intestinal intraepithelial T lymphocytes involves regulation of alphaEβ7 expression by transforming growth factor-β. Int Immunol. 2002;14:339–45. doi: 10.1093/intimm/14.4.339. [DOI] [PubMed] [Google Scholar]
- 65.Coffman RL, Lebman DA, Shrader B. Transforming growth factor β specifically enhances IgA production by lipopolysaccharide-stimulated murine B lymphocytes. J Exp Med. 1989;170:1039–44. doi: 10.1084/jem.170.3.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sonoda E, Matsumoto R, Hitoshi Y, et al. Transforming growth factor β induces IgA production and acts additively with interleukin 5 for IgA production. J Exp Med. 1989;170:1415–20. doi: 10.1084/jem.170.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Macpherson AJ, McCoy KD, Johansen FE, Brandtzaeg P. The immune geography of IgA induction and function. Mucosal Immunol. 2008;1:11–22. doi: 10.1038/mi.2007.6. [DOI] [PubMed] [Google Scholar]
- 68.Shi MJ, Park SR, Kim PH, Stavnezer J. Roles of Ets proteins, NF-κB and nocodazole in regulating induction of transcription of mouse germline Ig α RNA by transforming growth factor-β 1. Int Immunol. 2001;13:733–46. doi: 10.1093/intimm/13.6.733. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Y, Derynck R. Transcriptional regulation of the transforming growth factor-β -inducible mouse germ line Ig α constant region gene by functional cooperation of Smad, CREB, and AML family members. J Biol Chem. 2000;275:16979–85. doi: 10.1074/jbc.M001526200. [DOI] [PubMed] [Google Scholar]
- 70.Lin YC, Stavnezer J. Regulation of transcription of the germ-line Ig α constant region gene by an ATF element and by novel transforming growth factor-β 1-responsive elements. J Immunol. 1992;149:2914–25. [PubMed] [Google Scholar]
- 71.Klein J, Ju W, Heyer J, et al. B cell-specific deficiency for Smad2 in vivo leads to defects in TGF-β-directed IgA switching and changes in B cell fate. J Immunol. 2006;176:2389–96. doi: 10.4049/jimmunol.176.4.2389. [DOI] [PubMed] [Google Scholar]
- 72.Park SR, Lee JH, Kim PH. Smad3 and Smad4 mediate transforming growth factor-β1-induced IgA expression in murine B lymphocytes. Eur J Immunol. 2001;31:1706–15. doi: 10.1002/1521-4141(200106)31:6<1706::aid-immu1706>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 73.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 1999;18:1280–91. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wakabayashi Y, Tamiya T, Takada I, et al. Histone 3 lysine 9 (H3K9) methyltransferase recruitment to the interleukin-2 (IL-2) promoter is a mechanism of suppression of IL-2 transcription by the transforming growth factor-β-Smad pathway. J Biol Chem. 2011;286:35456–65. doi: 10.1074/jbc.M111.236794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tzachanis D, Freeman GJ, Hirano N, van Puijenbroek AA, Delfs MW, Berezovskaya A, Nadler LM, Boussiotis VA. Tob is a negative regulator of activation that is expressed in anergic and quiescent T cells. Nat Immunol. 2001;2:1174–82. doi: 10.1038/ni730. [DOI] [PubMed] [Google Scholar]
- 76.Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9:632–40. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 77.Ghoreschi K, Laurence A, Yang XP, et al. Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature. 2010;467:967–71. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Das J, Ren G, Zhang L, et al. Transforming growth factor β is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med. 2009;206:2407–16. doi: 10.1084/jem.20082286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kuwahara M, Yamashita M, Shinoda K, et al. The transcription factor Sox4 is a downstream target of signaling by the cytokine TGF-β and suppresses TH2 differentiation. Nat Immunol. 2012;13:778–86. doi: 10.1038/ni.2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schmitt E, Germann T, Goedert S, et al. IL-9 production of naive CD4+ T cells depends on IL-2, is synergistically enhanced by a combination of TGF-β and IL-4, and is inhibited by IFN-γ. J Immunol. 1994;153:3989–96. [PubMed] [Google Scholar]
- 81.Dardalhon V, Awasthi A, Kwon H, et al. IL-4 inhibits TGF-β-induced Foxp3+ T cells and, together with TGF-β, generates IL-9+ IL-10+ Foxp3− effector T cells. Nat Immunol. 2008;9:1347–55. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Elyaman W, Bradshaw EM, Uyttenhove C, et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc Natl Acad Sci USA. 2009;106:12885–90. doi: 10.1073/pnas.0812530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Elyaman W, Bassil R, Bradshaw EM, et al. Notch receptors and Smad3 signaling cooperate in the induction of interleukin-9-producing T cells. Immunity. 2012;36:623–34. doi: 10.1016/j.immuni.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grainger JR, Smith KA, Hewitson JP, et al. Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-β pathway. J Exp Med. 2010;207:2331–41. doi: 10.1084/jem.20101074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou L, Lopes JE, Chong MM, et al. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature. 2008;453:236–40. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martinez GJ, Zhang Z, Chung Y, Reynolds JM, Lin X, Jetten AM, Feng XH, Dong C. Smad3 differentially regulates the induction of regulatory and inflammatory T cell differentiation. J Biol Chem. 2009;284:35283–6. doi: 10.1074/jbc.C109.078238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martinez GJ, Zhang Z, Reynolds JM, et al. Smad2 positively regulates the generation of Th17 cells. J Biol Chem. 2010;285:29039–43. doi: 10.1074/jbc.C110.155820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xu L, Kitani A, Stuelten C, McGrady G, Fuss I, Strober W. Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity. 2010;33:313–25. doi: 10.1016/j.immuni.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Long J, Matsuura I, He D, Wang G, Shuai K, Liu F. Repression of Smad transcriptional activity by PIASy, an inhibitor of activated STAT. Proc Natl Acad Sci USA. 2003;100:9791–6. doi: 10.1073/pnas.1733973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qin H, Wang L, Feng T, et al. TGF-β promotes Th17 cell development through inhibition of SOCS3. J Immunol. 2009;183:97–105. doi: 10.4049/jimmunol.0801986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gronroos E, Kingston IJ, Ramachandran A, Randall RA, Vizan P, Hill CS. Transforming growth factor β inhibits bone morphogenetic protein-induced transcription through novel phosphorylated Smad1/5-Smad3 complexes. Mol Cell Biol. 2012;32:2904–16. doi: 10.1128/MCB.00231-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Takimoto T, Wakabayashi Y, Sekiya T, et al. Smad2 and Smad3 are redundantly essential for the TGF-β-mediated regulation of regulatory T plasticity and Th1 development. J Immunol. 2010;185:842–55. doi: 10.4049/jimmunol.0904100. [DOI] [PubMed] [Google Scholar]
- 93.Gorelik L, Flavell RA. Abrogation of TGFβ signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–81. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 94.Sorrentino A, Thakur N, Grimsby S, et al. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10:1199–207. doi: 10.1038/ncb1780. [DOI] [PubMed] [Google Scholar]
- 95.Wan YY, Chi H, Xie M, Schneider MD, Flavell RA. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat Immunol. 2006;7:851–8. doi: 10.1038/ni1355. [DOI] [PubMed] [Google Scholar]
- 96.Gu AD, Wang Y, Lin L, Zhang SS, Wan YY. Requirements of transcription factor Smad-dependent and -independent TGF-β signaling to control discrete T-cell functions. Proc Natl Acad Sci USA. 2012;109:905–10. doi: 10.1073/pnas.1108352109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lu L, Wang J, Zhang F, et al. Role of SMAD and non-SMAD signals in the development of Th17 and regulatory T cells. J Immunol. 2010;184:4295–306. doi: 10.4049/jimmunol.0903418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703–14. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]
- 99.Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. Targeted disruption of Smad3 reveals an essential role in transforming growth factor β-mediated signal transduction. Mol Cell Biol. 1999;19:2495–504. doi: 10.1128/mcb.19.4.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, Umetsu DT, Rudensky AY. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature. 2012;482:395–9. doi: 10.1038/nature10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–60. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 102.Samon JB, Champhekar A, Minter LM, et al. Notch1 and TGFβ1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood. 2008;112:1813–21. doi: 10.1182/blood-2008-03-144980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–85. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, Kuchroo VK. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-β-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol. 2008;181:2277–84. doi: 10.4049/jimmunol.181.4.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Asano N, Watanabe T, Kitani A, Fuss IJ, Strober W. Notch1 signaling and regulatory T cell function. J Immunol. 2008;180:2796–804. doi: 10.4049/jimmunol.180.5.2796. [DOI] [PubMed] [Google Scholar]
- 106.Huber M, Steinwald V, Guralnik A, Brustle A, Kleemann P, Rosenplanter C, Decker T, Lohoff M. IL-27 inhibits the development of regulatory T cells via STAT3. Int Immunol. 2008;20:223–34. doi: 10.1093/intimm/dxm139. [DOI] [PubMed] [Google Scholar]
- 107.Hu W, Troutman TD, Edukulla R, Pasare C. Priming microenvironments dictate cytokine requirements for T helper 17 cell lineage commitment. Immunity. 2011;35:1010–22. doi: 10.1016/j.immuni.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang XO, Nurieva R, Martinez GJ, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Malhotra N, Robertson E, Kang J. SMAD2 is essential for TGF β-mediated Th17 cell generation. J Biol Chem. 2010;285:29044–8. doi: 10.1074/jbc.C110.156745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–12. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang N, Bevan MJ. TGF-β signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat Immunol. 2012;13:667–73. doi: 10.1038/ni.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hahn JN, Falck VG, Jirik FR. Smad4 deficiency in T cells leads to the Th17-associated development of premalignant gastroduodenal lesions in mice. J Clin Investig. 2011;121:4030–42. doi: 10.1172/JCI45114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim BG, Li C, Qiao W, et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature. 2006;441:1015–9. doi: 10.1038/nature04846. [DOI] [PubMed] [Google Scholar]
- 114.Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor β (TGF-β) target genes and distinguishes TGF-β-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol. 2005;25:8108–25. doi: 10.1128/MCB.25.18.8108-8125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chu GC, Dunn NR, Anderson DC, Oxburgh L, Robertson EJ. Differential requirements for Smad4 in TGFβ-dependent patterning of the early mouse embryo. Development. 2004;131:3501–12. doi: 10.1242/dev.01248. [DOI] [PubMed] [Google Scholar]
- 116.Doisne JM, Bartholin L, Yan KP, et al. iNKT cell development is orchestrated by different branches of TGF-β signaling. J Exp Med. 2009;206:1365–78. doi: 10.1084/jem.20090127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–73. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shui JW, Larange A, Kim G, Vela JL, Zahner S, Cheroutre H, Kronenberg M. HVEM signalling at mucosal barriers provides host defence against pathogenic bacteria. Nature. 2012;488:222–5. doi: 10.1038/nature11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mennechet FJ, Kasper LH, Rachinel N, Minns LA, Luangsay S, Vandewalle A, Buzoni-Gatel D. Intestinal intraepithelial lymphocytes prevent pathogen-driven inflammation and regulate the Smad/T-bet pathway of lamina propria CD4+ T cells. Eur J Immunol. 2004;34:1059–67. doi: 10.1002/eji.200324416. [DOI] [PubMed] [Google Scholar]
- 120.Bhagat G, Naiyer AJ, Shah JG, Harper J, Jabri B, Wang TC, Green PH, Manavalan JS. Small intestinal CD8+TCRγδ+NKG2A+ intraepithelial lymphocytes have attributes of regulatory cells in patients with celiac disease. J Clin Investig. 2008;118:281–93. doi: 10.1172/JCI30989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, Wilhelm C, Veldhoen M. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147:629–40. doi: 10.1016/j.cell.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 122.Ohteki T, Wilson A, Verbeek S, MacDonald HR, Clevers H. Selectively impaired development of intestinal T cell receptor γδ+ cells and liver CD4+ NK1+ T cell receptor αβ+ cells in T cell factor-1-deficient mice. Eur J Immunol. 1996;26:351–5. doi: 10.1002/eji.1830260213. [DOI] [PubMed] [Google Scholar]
- 123.Narayan K, Sylvia KE, Malhotra N, et al. Intrathymic programming of effector fates in three molecularly distinct γδ T cell subtypes. Nat Immunol. 2012;13:511–8. doi: 10.1038/ni.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lambolez F, Arcangeli ML, Joret AM, Pasqualetto V, Cordier C, Di Santo JP, Rocha B, Ezine S. The thymus exports long-lived fully committed T cell precursors that can colonize primary lymphoid organs. Nat Immunol. 2006;7:76–82. doi: 10.1038/ni1293. [DOI] [PubMed] [Google Scholar]
- 125.Mysorekar IU, Lorenz RG, Gordon JI. A gnotobiotic transgenic mouse model for studying interactions between small intestinal enterocytes and intraepithelial lymphocytes. J Biol Chem. 2002;277:37811–9. doi: 10.1074/jbc.M205300200. [DOI] [PubMed] [Google Scholar]
- 126.Konkel JE, Maruyama T, Carpenter AC, et al. Control of the development of CD8αα+ intestinal intraepithelial lymphocytes by TGF-β. Nat Immunol. 2011;12:312–9. doi: 10.1038/ni.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chipuk JE, Bhat M, Hsing AY, Ma J, Danielpour D. Bcl-xL blocks transforming growth factor-β1-induced apoptosis by inhibiting cytochrome c release and not by directly antagonizing Apaf-1-dependent caspase activation in prostate epithelial cells. J Biol Chem. 2001;276:26614–21. doi: 10.1074/jbc.M100913200. [DOI] [PubMed] [Google Scholar]
- 128.Wildey GM, Howe PH. Runx1 is a co-activator with FOXO3 to mediate transforming growth factor β (TGFβ)-induced Bim transcription in hepatic cells. J Biol Chem. 2009;284:20227–39. doi: 10.1074/jbc.M109.027201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ouyang W, Beckett O, Ma Q, Li MO. Transforming growth factor-β signaling curbs thymic negative selection promoting regulatory T cell development. Immunity. 2010;32:642–53. doi: 10.1016/j.immuni.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-β signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity. 2009;31:145–57. doi: 10.1016/j.immuni.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Massague J, Blain SW, Lo RS. TGFβ signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 132.Dignass AU, Stow JL, Babyatsky MW. Acute epithelial injury in the rat small intestine in vivo is associated with expanded expression of transforming growth factor α and β. Gut. 1996;38:687–93. doi: 10.1136/gut.38.5.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Beck PL, Rosenberg IM, Xavier RJ, Koh T, Wong JF, Podolsky DK. Transforming growth factor-β mediates intestinal healing and susceptibility to injury in vitro and in vivo through epithelial cells. Am J Pathol. 2003;162:597–608. doi: 10.1016/s0002-9440(10)63853-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ruiz PA, Shkoda A, Kim SC, Sartor RB, Haller D. IL-10 gene-deficient mice lack TGF-β/Smad signaling and fail to inhibit proinflammatory gene expression in intestinal epithelial cells after the colonization with colitogenic Enterococcus faecalis. J Immunol. 2005;174:2990–9. doi: 10.4049/jimmunol.174.5.2990. [DOI] [PubMed] [Google Scholar]
- 135.Shkoda A, Ruiz PA, Daniel H, Kim SC, Rogler G, Sartor RB, Haller D. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology. 2007;132:190–207. doi: 10.1053/j.gastro.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 136.Naiki Y, Michelsen KS, Zhang W, Chen S, Doherty TM, Arditi M. Transforming growth factor-β differentially inhibits MyD88-dependent, but not TRAM- and TRIF-dependent, lipopolysaccharide-induced TLR4 signaling. J Biol Chem. 2005;280:5491–5. doi: 10.1074/jbc.C400503200. [DOI] [PubMed] [Google Scholar]
- 137.Zhou P, Miller G, Seder RA. Factors involved in regulating primary and secondary immunity to infection with Histoplasma capsulatum: TNF-α plays a critical role in maintaining secondary immunity in the absence of IFN-γ. J Immunol. 1998;160:1359–68. [PubMed] [Google Scholar]
- 138.Kitani A, Fuss I, Nakamura K, Kumaki F, Usui T, Strober W. Transforming growth factor (TGF)- β1-producing regulatory T cells induce Smad-mediated interleukin 10 secretion that facilitates coordinated immunoregulatory activity and amelioration of TGF-β1-mediated fibrosis. J Exp Med. 2003;198:1179–88. doi: 10.1084/jem.20030917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fava F, Danese S. Intestinal microbiota in inflammatory bowel disease: friend or foe? World J Gastroenterol. 2011;17:557–66. doi: 10.3748/wjg.v17.i5.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xu Y, Pasche B. TGF-β signaling alterations and susceptibility to colorectal cancer. Hum Mol Genet. 2007;16(Spec No 1):R14–20. doi: 10.1093/hmg/ddl486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. 2011;12:21–7. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]