

Abstract
Inflammasomes in innate immune cells mediate the induction of inflammation by sensing microbes and pathogen-associated/damage-associated molecular patterns. Inflammasomes are also known to be involved in the development of some human and animal autoimmune diseases. The Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome is currently the most fully characterized inflammasome, although a limited number of studies have demonstrated its role in demyelinating autoimmune diseases in the central nervous system of humans and animals. Currently, the development of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS), is known to be induced by the NLRP3 inflammasome through enhanced recruitment of inflammatory immune cells in the central nervous system. On the other hand, interferon-β (IFNβ), a first-line drug to treat MS, inhibits NLRP3 inflammasome activation, and ameliorates EAE. The NLRP3 inflammasome is indeed a factor capable of inducing EAE, but it is dispensable when EAE is induced by aggressive disease induction regimens. In such NLRP3 inflammasome-independent EAE, IFN-β treatment is generally not effective. This might therefore be one mechanism that leads to occasional failures of IFN-β treatment in EAE, and possibly, in MS as well. In the current review, we discuss inflammasomes and autoimmunity; in particular, the impact of the NLRP3 inflammasome on MS/EAE, and on IFN-β therapy.
Keywords: cell migration, chemotaxis, experimental autoimmune encephalomyelitis, interferon-β, multiple sclerosis, NLRP3 inflammasome
CNS autoimmune demyelination and inflammasomes
Inflammation induced by innate immune cells plays a critical role in eliciting autoimmunity. Our understanding of the relation between inflammation in the innate immune system and autoimmunity has significantly increased in the past decade as a result of extraordinary progress in analysing pattern-recognition receptors. At the same time, the role of inflammasomes in autoimmunity remains largely unclear; with this being particularly true for autoimmunity in the central nervous system (CNS). Autoimmune responses trigger demyelination in the CNS. Important examples of this phenomenon include multiple sclerosis (MS), neuromyelitis optica (NMO) and acute disseminated encephalomyelitis (ADEM). Although the direct role of inflammasomes in those diseases remains largely unknown, the use of experimental autoimmune encephalomyelitis (EAE), an animal model of MS, has made the impact of inflammasomes on CNS autoimmune demyelinating diseases more apparent.
Inflammasomes process interleukin-1β (IL-1β) and IL-18 maturation in myeloid cells, such as macrophages and dendritic cells (DCs); and, the basic biological function of inflammasomes is shared between humans and mice. Inflammasome is a multi-protein complex. Formation of the complex leads to pro-caspase-1 self-cleavage and generates active caspase-1, which processes pro-IL-1β and pro-IL-18 to mature IL-1β and IL-18, respectively, and induces cell death termed “pyroptosis”. Pyroptosis is distinguished from apoptosis and necrosis by cytoplasmic swelling and activation of caspase-1. Early plasma membrane rupture by pyroptosis1–3 leads to the release of mature IL-1β and IL-18 and other cytoplasmic contents to the extracellular space.4 Inflammasomes are known to sense and are activated by pathogen-associated molecular patterns (PAMPs), as well as damage-associated molecular patterns (DAMPs). The Nod-like receptor (NLR) family pyrin domain containing 3 (NLRP3, also known as NALP3 or CIAS1) inflammasome, is currently the most fully characterized inflammasome. It is known to sense bacteria, fungi, extracellular ATP, amyloid β and uric acid,5–8 as well as various environmental irritants, such as silica, asbestos and alum.7,9–11 In addition to NLRP3, other NLR family members, including NLRP1, NLRC4 (IPAF) and AIM2, are known to have clear physiological functions in vivo upon inflammasome formation;12 however, their involvement in CNS autoimmunity is not clear.
Many excellent reviews are available in the literature that provide information on the detailed functions and structure of inflammasomes. Further discussion on inflammasomes themselves is therefore spared here. Rather, we look to briefly mention several basic features of inflammasomes below to provide a foundation for later discussions in this review, and to highlight selected recent findings considered crucial to the further study of inflammasomes in CNS autoimmune demyelinating diseases.
The multi-protein complex of the NLRP3 inflammasome is comprised of three different proteins; NLRP3, ASC (apoptosis-associated speck like protein containing a caspase recruitment domain), and pro-caspase-1. Other types of inflammasomes have different compositions of proteins, but all have pro-caspase-1; therefore, the release of IL-1β and IL-18 from cells is a major common outcome by all inflammasomes. Pro-caspase-1 must be self-cleaved to become activated caspase-1; it then exerts cytokine maturation and pyroptosis by inflammasomes. (We refer to this stage of inflammasomes as ‘active inflammasomes’ in this review.) In the human NLRP3 inflammasome, a molecule termed CARDINAL (CARD8, TUCAN) is known to be involved.13 However, there is no mouse homologue of human CARDINAL, and CARDINAL is dispensable for IL-1β production in human cells.14 Recent reports showed that there are NLRP proteins that inhibit inflammation. For example, NLRP12 attenuates a non-canonical nuclear factor-κB (NFκB) pathway by interacting with NF-κB-inducing kinase, and the tumour necrosis factor receptor-associated factor (TRAF) 3 in innate immune cells without inflammasome formation.15–17 Importantly, caspase-1 knockout mice, used in early published studies, appear to have been a double-knockout of both caspase-1 and caspase-11 due to the failure to segregate close genetic loci of Casp1 and Casp11 by gene recombination.18 Caspase-1 is still required by ATP-mediated maturation of IL-1β and IL-18 and induction of pyroptosis, but caspase-11 plays a key role when cells are stimulated by cholera toxin B or Escherichia coli, but not ATP stimulation.18
Inflammasomes and autoimmune/autoinflammatory diseases
Before limiting our discussion on inflammasomes to CNS demyelinating diseases, we look to briefly discuss what is generally known about inflammasomes in autoimmune/autoinflammatory diseases. Of the four types of inflammasomes (NLRP1, NLRP3, NLRC4, AIM2), most of the earlier studies were carried out on NLRP3 within the context of autoimmunity. Mutations in the human Nlrp3 locus were found to be associated with rare, inherited cryopyrin-associated periodic syndromes (CAPS); such as Muckle–Wells syndrome (MWS), familial cold-induced autoinflammatory syndrome (FCAS), and chronic infantile neurological cutaneous and articular (CINCA) syndrome.19–22
Involvement of NLRP3 in autoinflammation was demonstrated by using mice expressing the Nlrp3 gene mutation, which corresponds to the MWS-associated Nlrp3 mutation.23 Such mice showed hyperactivation of the NLRP3 inflammasome, as well as increased production of IL-1β and IL-18. Further, they developed skin inflammation characterized by induced IL-17-producing T helper cell (Th17) responses.23 NLRP3 inflammasome also appears to correlate with various human autoimmune diseases. Single nucleotide polymorphisms within the Nlrp3 locus are predisposed to systemic lupus erythematosus (SLE), type 1 diabetes, coeliac disease, Crohn's disease and ulcerative colitis.24–26 In addition, NLRP1 inflammasome is associated with other autoimmune diseases, such as vitiligo, type 1 diabetes and rheumatoid arthritis.25,27,28 On the other hand, involvement of AIM2 and NLRC4 in autoimmune/autoinflammatory diseases remains unclear. Nevertheless, involvement of the AIM2 inflammasome in SLE, for example, may be possible because AIM2 senses DNA, which is a major autoimmune target.29
NLRP3 inflammasome in MS and EAE
A number of reports suggest involvement of the NLRP3 inflammasome in the development of both MS and EAE (Table 1). Increased levels of caspase-1, IL-1β, IL-18 and activators of the NLRP3 inflammasome (ATP, uric acid, cathepsin B) have been reported in MS patients (Table 1). For example, Casp-1 mRNA levels correlate with disease severity in MS patients,30 and caspase-1 protein is highly abundant in MS plaques.31 Further, expression of caspase-1 and IL-18 in peripheral mononuclear cells from MS patients has been found at increased levels compared with those in cells from healthy controls.32 High IL-1β and low IL-1 receptor antagonist (IL-1RA) production has been hypothesized as a predisposition of increased susceptibility and disease progression of MS.33 Patients with MS are also known to express high levels of purine compounds and uric acid in cerebrospinal fluid,34 as well as high serum uric acid levels.35 Increased activity of cathepsin B, which is known to induce NLRP3 inflammasome activation by leaking out from lysozomes,10 was also reported in peripheral blood mononuclear cells, as well as brain cells of MS patients.36,37 The NLRP3 inflammasome is activated by triggering the P2X7 receptor (P2X7R) signalling by extracellular ATP. Sustained activation of P2X7R during EAE appears to cause MS plaque-like lesions, and treatment with P2X7R antagonists ameliorates EAE.38 The same study also suggested that P2X7R signalling is enhanced in normal-appearing axonal tracts of the CNS in MS patients.38 Further, expression of the P2x7r gene is increased in the optic nerve region of MS patients.39 Single nucleotide polymorphisms in the P2x7r locus were found more frequently in MS patients compared with healthy controls.40 Because MS is a multifactorial and heterogeneous disease, the NLRP3 inflammasome may not be involved in the development of all forms of MS. However, these studies strongly suggest the general involvement of the NLRP3 inflammasome in MS progression.
Table 1.
Involvement of Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome in the development of multiple sclerosis and experimental autoimmune encephalomyelitis
| Multiple sclerosis | Experimental autoimmune encephalomyelitis | |||
|---|---|---|---|---|
| Classification | Phenotype | Ref. | Phenotype | Ref. |
| NLRP3 inflammasome component | Increase of Casp1 mRNA in peripheral blood mononuclear cells | 30 | Increase of caspase 1 in peripheral blood mononuclear cells | 45 |
| Increase of caspase 1 in MS plaques and peripheral mononuclear cells | 31,32 | Mild EAE in Casp1−/− mice | 45,46 | |
| Caspase 1 inhibitor attenuates EAE | 45 | |||
| Mild EAE in Nlrp3−/− mice | 41–44 | |||
| Mild EAE in Asc−/− mice | 43 | |||
| NLRP3 inflammasome-mediated cytokine | Increase of IL-1β in CSF | 33,82 | Increase of IL-1β in serum and splenocyte | 41,44 |
| Increase of IL-18 in peripheral mononuclear cells | 32 | IL1R antagonist attenuates EAE | 83 | |
| Increase of IL-18 in serum | 41,44 | |||
| Mild EAE in Il18−/− mice | 41,84 | |||
| NLRP3 inflammasome activator | Increase of P2X7R in optic nerve | 38 | Increase of P2X7R in brain | 38 |
| Increase of P2x7r mRNA in optic nerve | 39 | P2X7R antagonist attenuates EAE | 38 | |
| Relationship of P2x7r gene by SNP analysis | 40 | |||
| Increase of purine compounds in CSF | 34 | |||
| Increase of uric acid in CSF and serum | 34,35 | |||
| Increase of cathepsin B activity in peripheral mononuclear cells and brain | 36,37 | |||
CSF, cerebrospinal fluid; EAE, experimental autoimmune encephalomyelitis IL-1β, interleukin-1β; MS, multiple sclerosis; SNP, single nucleotide polymorphism.
The critical role of the NLRP3 inflammasome in EAE has recently become clear.41–44 NLRP3 inflammasome induces demyelination as indicated using the chemically induced demyelinating disease and EAE models.42 A subsequent study showed that the NLRP3 inflammasome induces EAE progression by enhancing chemotactic migration of T helper cells, and antigen-presenting cells (APCs) into the CNS.43 Nlrp3−/− mice were characterized by being resistant to EAE and to reduction in both Th1 and Th17 cells in the peripheral lymphoid tissues, as well as in the spinal cord.41,43 It appears that the NLRP3 inflammasome has the most critical impact on EAE among all inflammasomes, because of a similar phenotype between Nlrp3−/− and Asc−/− mice in their resistance to EAE.43 Caspase-1-deficient mice (which may have also been lacking caspase-1118) are resistant to EAE, supporting the involvement of inflammasomes (most probably, NLRP3 inflammasome) in EAE pathogenicity.45,46
Correlation between the NLRP3 inflammasome and EAE development was also suggested by a number of other studies demonstrating enhanced levels of caspase-1, IL-1β, IL-18 and ATP during EAE development (Table 1). Other than NLRP3, NLRP1 is the only inflammasome NLR protein reported in the context of EAE for its intra-axonal accumulation,47 but involvement of the NLRP1 inflammasome in EAE is not yet known.
Mechanism of NLRP3 inflammasome-mediated EAE progression
A major function of the NLRP3 inflammasome is the maturation and secretion of IL-1β and IL-18. It is known that IL-1β plays a role in demyelination,48 breakdown of blood–brain barrier (BBB),48,49 microglia activation49 and promotion of IL-17 expression both by CD4+ T and γδT cells.50,51 The outcome from these responses is the enhancement of EAE progression. Interleukin-18 is also known to promote IL-17 production by CD4 T+ cells, as well as γδT cells,52 and exacerbates demyelination.42 Attenuated Th17 (and Th1) responses were originally considered to be a major underlying mechanism for the resistance of NLRP3 inflammasome-deficient mice against EAE.41,52 However, it now appears that the lack of the NLRP3 inflammasome (in APCs) disables T helper cells and APCs in migrating to the CNS. This inability to migrate cells to the CNS is a major cause of resistance against EAE in Asc−/− and Nlrp3−/− mice.43 Interestingly, T cells primed by NLRP3 inflammasome-deficient APCs do not migrate into the CNS, but are encephalitogenic, only lacking chemotactic ability.43 Therefore, when directly transferred into the CNS, transfer of T cells primed by NLRP3 inflammasome-deficient APCs is able to induce EAE.43 This result strongly suggests that cell migration is one of the most critical factors for the NLRP3 inflammasome in exerting an effect on EAE progression.
The cell migration mechanism was explained with IL-1β and IL-18, which are processed by the NLRP3 inflammasome and up-regulate expression of chemokines and their receptors both in T helper cells and APCs. Total T helper cells (as well as Th17 and Th1 cells) from immunized Asc−/− and Nlrp3−/− mice express low levels of CCR2, CXCR6 and osteopontin, which are critical to MS and EAE progression.53–62 Without the NLRP3 inflammasome, APCs also reduce expression of chemokines and their receptors, such as CCL7/MCP3 (CCR2 ligand), CCL8/MCP2 (CCR2 ligand), CXCL16 (CXCR6 ligand) and α4β1 integrin (osteopontin receptor).43 The NLRP3 inflammasome induces expression of molecules that enhance cell migration by providing IL-1β and IL-18. Intriguingly, those molecules are matching pairs of chemokines and their receptors between T cells and APCs (Fig. 1).
Figure 1.
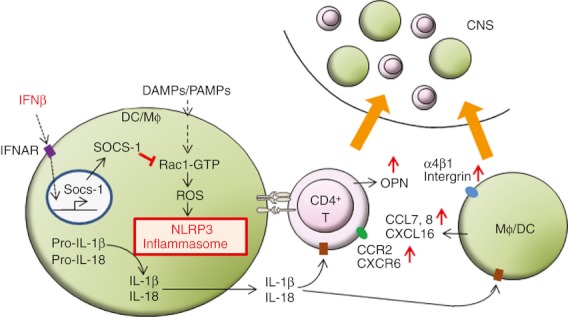
Schematic diagram: Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome induces experimental autoimmune encephalomyelitis (EAE) development by cell recruitment to the central nervous system (CNS), but interferon-β (IFN-β) inhibits NLRP3 inflammasome activity. Active NLRP3 inflammasome in antigen-presenting cells [APCs; macrophages (Mϕ) and dendritic cells (DC)] processes maturation of IL-1β and IL-18, which are detected by CD4+ and APCs, in paracrine and autocrine fashions, respectively. The cytokines induce expression of genes encoding migration-related proteins in T cells and APCs in the peripheral lymphoid organs, resulting in enhancement of cell migration into the CNS; and this causes development of EAE. On the other hand, IFN-β inhibits activation of the NLRP3 inflammasome in APCs through IFN-I receptor (IFNAR) signalling induction of Suppressor Of Cytokine Signalling-1 (SOCS1). SOCS1 down-regulates Rac1-GTP (active Rac1) and reactive oxygen species (ROS) generation, resulting in inhibition of NLRP3 inflammasome activation. DAMP, damage-associated molecular patterns; OPN, osteopontin; PAMP, pathogen-associated molecular patterns.
Type 1 interferons and the NLRP3 inflammasome
Type 1 interferons (IFN-I), such as IFN-α and IFN-β, are involved in various aspects of immune responses. IFN-β has been used for more than 15 years as a first-line treatment for MS, and also markedly attenuates EAE development. Previous studies have shown that IFN-β suppresses the production of IL-1β through reduction of pro-IL-1β via the autocrine effect of IL-10.63 More recent reports showed involvement of nitric oxide and the degradation of active Rac1 in IFN-I-mediated suppression of NLRP3 inflammasome activity.44,64 In the latter mechanism, ligation of the IFN-I receptor (IFNAR) by IFN-I induces association of Suppressor Of Cytokine Signalling-1 (SOCS1) with active Rac1, leading to ubiquitination and degradation of active Rac1.44 Consequently, the reduction of active Rac1 decreases generation of reactive oxygen species (ROS) by mitochondria, and NLRP3 inflammasome activity is down-regulated accordingly (Fig. 1).44 The NLRP3 inflammasome itself does not exert a feedback effect on upstream effector molecules in the IFNAR–NLRP3 axis, such as SOCS1, Vav1, activated Rac1 and ROS.44 Signalling by IFNAR also does not affect expression of Nlrp3, Asc, Casp-1, Txnip, or the abundance of P2X7R. Hence, IFNAR signalling appears to have a direct impact on suppression of the NLRP3 inflammasome through SOCS1, Rac1 and ROS.44 The mechanism by which IFNAR signalling suppresses NLRP3 inflammasome is connected to reduced expression of cellular chemotaxis, which was described in the previous section, eventually to ameliorate EAE (Fig. 1).
Mechanisms that ameliorate MS and EAE by IFN-β
In addition to targeting the NLRP3 inflammasome, IFN-β has multiple functions to ameliorate MS and EAE. For example, IFN-β suppresses the Th17 cell response in both MS and EAE by regulating the expression of cytokines, such as IL-4, IL-10 and IL-27.62,65–69 In particular, expression of IL-27, which negatively regulates Th17 responses, is induced by IFNAR signalling.62,65,70 How IL-27 expression is induced upon IFNAR stimulation is not entirely clear, but intracellular osteopontin (iOPN) appears to mediate IL-27 induction upon IFNAR stimulation.62 Interferon-β is also known to inhibit T-cell activation via down-regulation of the MHC II co-stimulatory molecules as well as cell adhesion molecules in APCs.66,71 At the same time, IFN-β induces T cell death by down-regulating the anti-apoptosis protein FLIP (FLICE-inhibitory protein),72 and by up-regulating TRAIL (tumour necrosis factor-related apoptosis inducing ligand) in MS.73 Interferon-β treatment expands regulatory T cells by induction of glucocorticoid-induced tumour necrosis factor receptor ligand (GITRL) expression in MS patients,74 in addition to down-regulating very late antigen-4 (VLA4) expression on effector T cells so as to limit T cell trafficking to the CNS.75 Other studies showed that IFN-β treatment decreases expression of matrix metalloprotease-9 (MMP-9), which plays a key role in the disruption of BBB by destabilizing tight junctions and increases expression of MMP-9 inhibitor, tissue inhibitor of matrix metalloproteinase-1 (TIMP-1), in MS patients.76,77 In summary, IFNAR signalling has impacts on various biological responses to ameliorate both EAE and MS. Importantly, however, a cell-specific IFNAR deletion model using the Cre-lox system showed that IFNAR on myeloid cells, and not on CD4+ T cells, exerts the functional outcomes of EAE amelioration.66
A subtype of EAE that develops without the NLRP3 inflammasome
EAE can be induced both actively and passively. Active EAE is induced by autoantigen immunization, whereas passive EAE is induced by the adoptive transfer of encephalitogenic T cells. Although the NLRP3 inflammasome is activated in both active and passive EAE,44 Asc−/− and Nlrp3−/− mice can develop severe EAE if the active EAE induction regimen is aggressive.44 In active EAE induction, autoantigen emulsified in complete Freund's adjuvant (CFA) plus injections of pertussis toxin is used. To induce EAE in Asc−/− and Nlrp3−/− mice, increased dosages of heat-killed Mycobacterium tuberculosis (Mtb) in CFA alone are sufficient.44 A similar observation was reported in a study using Casp1−/− mice, in which disease susceptibility is associated with repeated immunization, and high dosages or high MHC-binding affinity of antigen peptides.45 These studies suggest the presence of an NLRP3 inflammasome-independent pathway in progression of EAE. In addition, the studies cited herein suggest that dosages of adjuvant and/or the abundance of high-affinity antigen shift EAE to an NLRP3 inflammasome-independent disease.
Two earlier reports on NLRP3 inflammasome in EAE showed important but contrasting results. One showed susceptibility of Nlrp3−/− mice to EAE,78 while the other showed resistance of Nlrp3−/− mice.41 As a result, the requirement of NLRP3 inflammasome in EAE was considered to be controversial, wherein the “basis for these conflicting data” was said to be unknown.79 Here, we assume that the two distinct results reflect two different subtypes of EAE: NLRP3 inflammasome-dependent and -independent. The EAE induced in Asc−/− and Nlrp3−/− mice are clearly NLRP3 inflammasome-independent. However, in wild-type mice, two subtypes of EAE, NLRP3 inflammasome-dependent and -independent, may be occasionally occurring at the same time, particularly when disease induction is not aggressive enough. In other words, the two subtypes are not mutually exclusive during EAE development. Depending on the triggers of the disease, and the genetic environment at hand, it is possible that the balance between the two subtypes may be altered. We have therefore shown that aggressive immunization induces NLRP3 inflammasome-independent EAE.44 We must then ask: What is the equivalent to such NLRP3 inflammasome-independent EAE in human disease? If there is NLRP3 inflammasome-independent MS, it might be caused by intensive stimulation on innate immune cells, or by other factors that provide strong autoantigen affinity to T cells. This, we believe, is an important and intriguing possibility.
NLRP3 inflammasome and efficacy of IFN-β
Although IFN-β is a first-line drug to treat MS, it has been found that one-third of patients do not respond to IFN-β treatment.80 Is IFN-β still effective without activated NLRP3 inflammasome, which is a target of IFN-β? This question was addressed in NLRP3 inflammasome-independent EAE.44 Results suggest that IFN-β was not effective in treating EAE in Asc−/− and Nlrp3−/− mice.44 When EAE is induced in wild-type mice by a moderately aggressive regimen, the efficacy of IFN-β is partial.44 On the other hand, very aggressive EAE induction (for example, repeated immunization with high dosages of heat-killed Mtb) completely abrogates IFN-β efficacy in wild-type mice (Inoue et al., unpublished data). Hence, EAE induced by moderately aggressive immunization may develop as a mixture of two EAE subtypes; NLRP3 inflammasome-dependent and -independent. When two subtypes of EAE are ongoing, it may be possible that IFN-β efficacy correlates with levels of NLRP3 inflammasome dependency in EAE development. Although two subtypes of EAE may be occurring simultaneously within some of the disease in WT mice, the findings are summarized as follows: NLRP3 inflammasome-dependent EAE is a disease that responds to IFN-β treatment, whereas NLRP3 inflammasome-independent EAE is a disease that is resistant to IFN-β (Fig. 2).
Figure 2.
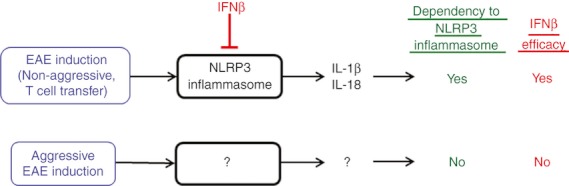
Two subtypes of experimental autoimmune encephalomyelitis (EAE). EAE induced by conventional disease induction regimens or adoptive transfer of activated T cells is accompanied with active Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, which is a target of interferon-β (IFN-β), and expression of interleukin-1β (IL-1β) and IL-18. IFN-β is effective in such EAE. In contrast, aggressive disease induction regimens can trigger NLRP3 inflammasome-independent EAE, which is not ameliorated by IFN-β treatment. It is not clear what mediates EAE progression, instead of the NLRP3 inflammasome. The two subtypes are not mutually exclusive.
Previous studies have shown that passive EAE induced by Th17 cell transfer is resistant to IFN-β treatment, whereas the disease induced by Th1 cells responds to IFN-β treatment.81 The result is counterintuitive because IFN-β inhibits Th17 responses;62,65 and it will be of great interest to understand why Th17-mediated EAE cannot be treated by IFN-β. Activation status of the NLRP3 inflammasome is not known in the Th17-mediated EAE model, but the result (resistance of Th17-mediated passive EAE to IFN-β) does not conflict with IFN-β resistance in NLRP3 inflammasome-independent EAE. This is because the Th17 response itself is not the reason for NLRP3 inflammasome-dependent EAE progression.44 Further studies will be necessary to determine whether or not these two types of IFN-β-resistant EAE (Th17-type EAE and NLRP3 inflammasome-independent EAE) share the same mechanism.
Clinical implication of the NLRP3 inflammasome and its inhibition by IFN-β
It is currently unknown whether NLRP3 inflammasome-independent MS exists. It is also not known what type of event is an equivalent of ‘aggressive immunization’ in MS. However, if the current findings on the correlation between NLRP3 inflammasome activation and response to IFN-β in EAE can be applied to MS, it might be possible to predict MS patients who do not respond well to IFN-β therapy. For example, the activation status of the NLRP3 inflammasome might be a prediction marker. Or, it might be possible to identify prediction markers by screening molecules that show altered expression in NLRP3 inflammasome-independent EAE. It is also possible to test such molecules for prognosis markers, or even as molecular targets of selected treatment(s).
References
- 1.Kepp O, Galluzzi L, Zitvogel L, Kroemer G. Pyroptosis – a cell death modality of its kind? Eur J Immunol. 2010;40:627–30. doi: 10.1002/eji.200940160. [DOI] [PubMed] [Google Scholar]
- 2.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–16. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamkanfi, M Emerging inflammasome effector mechanisms. Nat Rev Immunol. 2011;11:213–20. doi: 10.1038/nri2936. [DOI] [PubMed] [Google Scholar]
- 5.Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 6.Gross O, Poeck H, Bscheider M, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459:433–6. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- 7.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 8.Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol. 2008;9:857–65. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–56. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–5. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 12.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 13.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity. 2004;20:319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 14.Allen IC, Scull MA, Moore CB, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–65. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiorentino L, Stehlik C, Oliveira V, Ariza ME, Godzik A, Reed JC. A novel PAAD-containing protein that modulates NF-κB induction by cytokines tumor necrosis factor-α and interleukin-1β. J Biol Chem. 2002;277:35333–40. doi: 10.1074/jbc.M200446200. [DOI] [PubMed] [Google Scholar]
- 16.Ye Z, Lich JD, Moore CB, Duncan JA, Williams KL, Ting JP. ATP binding by monarch-1/NLRP12 is critical for its inhibitory function. Mol Cell Biol. 2008;28:1841–50. doi: 10.1128/MCB.01468-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allen IC, Wilson JE, Schneider M, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity. 2012;36:742–54. doi: 10.1016/j.immuni.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 19.Arostegui JI, Aldea A, Modesto C, et al. Clinical and genetic heterogeneity among Spanish patients with recurrent autoinflammatory syndromes associated with the CIAS1/PYPAF1/NALP3 gene. Arthritis Rheum. 2004;50:4045–50. doi: 10.1002/art.20633. [DOI] [PubMed] [Google Scholar]
- 20.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nat Genet. 2001;29:301–5. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dode C, Le Du N, Cuisset L, et al. New mutations of CIAS1 that are responsible for Muckle–Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet. 2002;70:1498–506. doi: 10.1086/340786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feldmann J, Prieur AM, Quartier P, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71:198–203. doi: 10.1086/341357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. 2009;30:860–74. doi: 10.1016/j.immuni.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pontillo A, Girardelli M, Kamada AJ, Pancotto JA, Donadi EA, Crovella S, Sandrin-Garcia P. Polymorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity. 2012;45:271–8. doi: 10.3109/08916934.2011.637532. [DOI] [PubMed] [Google Scholar]
- 25.Pontillo A, Brandao L, Guimaraes R, Segat L, Araujo J, Crovella S. Two SNPs in NLRP3 gene are involved in the predisposition to type-1 diabetes and celiac disease in a pediatric population from northeast Brazil. Autoimmunity. 2010;43:583–9. doi: 10.3109/08916930903540432. [DOI] [PubMed] [Google Scholar]
- 26.Villani AC, Lemire M, Louis E, et al. Genetic variation in the familial Mediterranean fever gene (MEFV) and risk for Crohn's disease and ulcerative colitis. PLoS ONE. 2009;4:e7154. doi: 10.1371/journal.pone.0007154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glinskii AB, Ma J, Ma S, Grant D, Lim CU, Sell S, Glinsky GV. Identification of intergenic trans-regulatory RNAs containing a disease-linked SNP sequence and targeting cell cycle progression/differentiation pathways in multiple common human disorders. Cell Cycle. 2009;8:3925–42. doi: 10.4161/cc.8.23.10113. [DOI] [PubMed] [Google Scholar]
- 28.Sui J, Li H, Fang Y, et al. NLRP1 gene polymorphism influences gene transcription and is a risk factor for rheumatoid arthritis in Han Chinese. Arthritis Rheum. 2012;64:647–54. doi: 10.1002/art.33370. [DOI] [PubMed] [Google Scholar]
- 29.Veeranki S, Choubey D. Systemic lupus erythematosus and increased risk to develop B cell malignancies: role of the p200-family proteins. Immunol Lett. 2010;133:1–5. doi: 10.1016/j.imlet.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furlan R, Filippi M, Bergami A, et al. Peripheral levels of caspase-1 mRNA correlate with disease activity in patients with multiple sclerosis; a preliminary study. J Neurol Neurosurg Psychiatry. 1999;67:785–8. doi: 10.1136/jnnp.67.6.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ming X, Li W, Maeda Y, Blumberg B, Raval S, Cook SD, Dowling PC. Caspase-1 expression in multiple sclerosis plaques and cultured glial cells. J Neurol Sci. 2002;197:9–18. doi: 10.1016/s0022-510x(02)00030-8. [DOI] [PubMed] [Google Scholar]
- 32.Huang WX, Huang P, Hillert J. Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult Scler. 2004;10:482–7. doi: 10.1191/1352458504ms1071oa. [DOI] [PubMed] [Google Scholar]
- 33.de Jong BA, Huizinga TW, Bollen EL, et al. Production of IL-1β and IL-1Ra as risk factors for susceptibility and progression of relapse-onset multiple sclerosis. J Neuroimmunol. 2002;126:172–9. doi: 10.1016/s0165-5728(02)00056-5. [DOI] [PubMed] [Google Scholar]
- 34.Amorini AM, Petzold A, Tavazzi B, et al. Increase of uric acid and purine compounds in biological fluids of multiple sclerosis patients. Clin Biochem. 2009;42:1001–6. doi: 10.1016/j.clinbiochem.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 35.Liu B, Shen Y, Xiao K, Tang Y, Cen L, Wei J. Serum uric acid levels in patients with multiple sclerosis: a meta-analysis. Neurol Res. 2012;34:163–71. doi: 10.1179/1743132811Y.0000000074. [DOI] [PubMed] [Google Scholar]
- 36.Bever CT, Jr, Panitch HS, Johnson KP. Increased cathepsin B activity in peripheral blood mononuclear cells of multiple sclerosis patients. Neurology. 1994;44:745–8. doi: 10.1212/wnl.44.4.745. [DOI] [PubMed] [Google Scholar]
- 37.Bever CT, Jr, Garver DW. Increased cathepsin B activity in multiple sclerosis brain. J Neurol Sci. 1995;131:71–3. doi: 10.1016/0022-510x(95)00039-5. [DOI] [PubMed] [Google Scholar]
- 38.Matute C, Torre I, Perez-Cerda F, et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci. 2007;27:9525–33. doi: 10.1523/JNEUROSCI.0579-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vallejo-Illarramendi A, Domercq M, Perez-Cerda F, Ravid R, Matute C. Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol Dis. 2006;21:154–64. doi: 10.1016/j.nbd.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 40.Oyanguren-Desez O, Rodriguez-Antiguedad A, Villoslada P, Domercq M, Alberdi E, Matute C. Gain-of-function of P2X7 receptor gene variants in multiple sclerosis. Cell Calcium. 2011;50:468–72. doi: 10.1016/j.ceca.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Gris D, Ye Z, Iocca HA, et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol. 2010;185:974–81. doi: 10.4049/jimmunol.0904145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jha S, Srivastava SY, Brickey WJ, et al. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci. 2010;30:15811–20. doi: 10.1523/JNEUROSCI.4088-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2012;109:10480–5. doi: 10.1073/pnas.1201836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inoue M, Williams KL, Oliver T, Vandenabeele P, Rajan JV, Miao EA, Shinohara ML. Interferon-β therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal. 2012;5:ra38. doi: 10.1126/scisignal.2002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Furlan R, Martino G, Galbiati F, et al. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol. 1999;163:2403–9. [PubMed] [Google Scholar]
- 46.Ahmed Z, Doward AI, Pryce G, et al. A role for caspase-1 and -3 in the pathology of experimental allergic encephalomyelitis: inflammation versus degeneration. Am J Pathol. 2002;161:1577–86. doi: 10.1016/S0002-9440(10)64436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soulika AM, Lee E, McCauley E, Miers L, Bannerman P, Pleasure D. Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis. J Neurosci. 2009;29:14965–79. doi: 10.1523/JNEUROSCI.3794-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alvarez JI, Dodelet-Devillers A, Kebir H, et al. The Hedgehog pathway promotes blood–brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727–31. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- 49.Ferrari CC, Depino AM, Prada F, et al. Reversible demyelination, blood–brain barrier breakdown, and pronounced neutrophil recruitment induced by chronic IL-1 expression in the brain. Am J Pathol. 2004;165:1827–37. doi: 10.1016/S0002-9440(10)63438-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–91. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–41. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 52.Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KH. Caspase-1-processed cytokines IL-1β and IL-18 promote IL-17 production by γδ and CD4 T cells that mediate autoimmunity. J Immunol. 2011;186:5738–48. doi: 10.4049/jimmunol.1003597. [DOI] [PubMed] [Google Scholar]
- 53.Matejuk A, Vandenbark AA, Burrows GG, Bebo BF, Jr, Offner H. Reduced chemokine and chemokine receptor expression in spinal cords of TCR BV8S2 transgenic mice protected against experimental autoimmune encephalomyelitis with BV8S2 protein. J Immunol. 2000;164:3924–31. doi: 10.4049/jimmunol.164.7.3924. [DOI] [PubMed] [Google Scholar]
- 54.Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–80. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim JV, Jiang N, Tadokoro CE, Liu L, Ransohoff RM, Lafaille JJ, Dustin ML. Two-photon laser scanning microscopy imaging of intact spinal cord and cerebral cortex reveals requirement for CXCR6 and neuroinflammation in immune cell infiltration of cortical injury sites. J Immunol Methods. 2010;352:89–100. doi: 10.1016/j.jim.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chabas D, Baranzini SE, Mitchell D, et al. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science. 2001;294:1731–5. doi: 10.1126/science.1062960. [DOI] [PubMed] [Google Scholar]
- 57.Sorensen TL, Sellebjerg F. Distinct chemokine receptor and cytokine expression profile in secondary progressive MS. Neurology. 2001;57:1371–6. doi: 10.1212/wnl.57.8.1371. [DOI] [PubMed] [Google Scholar]
- 58.Misu T, Onodera H, Fujihara K, et al. Chemokine receptor expression on T cells in blood and cerebrospinal fluid at relapse and remission of multiple sclerosis: imbalance of Th1/Th2-associated chemokine signaling. J Neuroimmunol. 2001;114:207–12. doi: 10.1016/s0165-5728(00)00456-2. [DOI] [PubMed] [Google Scholar]
- 59.Shinohara ML, Jansson M, Hwang ES, Werneck MB, Glimcher LH, Cantor H. T-bet-dependent expression of osteopontin contributes to T cell polarization. Proc Natl Acad Sci USA. 2005;102:17101–6. doi: 10.1073/pnas.0508666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jansson M, Panoutsakopoulou V, Baker J, Klein L, Cantor H. Cutting edge: attenuated experimental autoimmune encephalomyelitis in eta-1/osteopontin-deficient mice. J Immunol. 2002;168:2096–9. doi: 10.4049/jimmunol.168.5.2096. [DOI] [PubMed] [Google Scholar]
- 61.Hur EM, Youssef S, Haws ME, Zhang SY, Sobel RA, Steinman L. Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat Immunol. 2007;8:74–83. doi: 10.1038/ni1415. [DOI] [PubMed] [Google Scholar]
- 62.Shinohara ML, Kim JH, Garcia VA, Cantor H. Engagement of the type I interferon receptor on dendritic cells inhibits T helper 17 cell development: role of intracellular osteopontin. Immunity. 2008;29:68–78. doi: 10.1016/j.immuni.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guarda G, Braun M, Staehli F, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–23. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 64.Hernandez-Cuellar E, Tsuchiya K, Hara H, et al. Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J Immunol. 2012;189:5113–7. doi: 10.4049/jimmunol.1202479. [DOI] [PubMed] [Google Scholar]
- 65.Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118:1680–90. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prinz M, Schmidt H, Mildner A, et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity. 2008;28:675–86. doi: 10.1016/j.immuni.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 67.Ramgolam VS, Markovic-Plese S. Interferon-β inhibits Th17 cell differentiation in patients with multiple sclerosis. Endocr Metab Immune Disord Drug Targets. 2010;10:161–7. doi: 10.2174/187153010791213029. [DOI] [PubMed] [Google Scholar]
- 68.Ramgolam VS, Sha Y, Jin J, Zhang X, Markovic-Plese S. IFN-β inhibits human Th17 cell differentiation. J Immunol. 2009;183:5418–27. doi: 10.4049/jimmunol.0803227. [DOI] [PubMed] [Google Scholar]
- 69.Martin-Saavedra FM, Gonzalez-Garcia C, Bravo B, Ballester S. Beta interferon restricts the inflammatory potential of CD4+ cells through the boost of the Th2 phenotype, the inhibition of Th17 response and the prevalence of naturally occurring T regulatory cells. Mol Immunol. 2008;45:4008–19. doi: 10.1016/j.molimm.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 70.Pirhonen J, Siren J, Julkunen I, Matikainen S. IFN-α regulates Toll-like receptor-mediated IL-27 gene expression in human macrophages. J Leukoc Biol. 2007;82:1185–92. doi: 10.1189/jlb.0307157. [DOI] [PubMed] [Google Scholar]
- 71.Yong VW, Chabot S, Stuve O, Williams G. Interferon β in the treatment of multiple sclerosis: mechanisms of action. Neurology. 1998;51:682–9. doi: 10.1212/wnl.51.3.682. [DOI] [PubMed] [Google Scholar]
- 72.Sharief MK, Semra YK, Seidi OA, Zoukos Y. Interferon-β therapy downregulates the anti-apoptosis protein FLIP in T cells from patients with multiple sclerosis. J Neuroimmunol. 2001;120:199–207. doi: 10.1016/s0165-5728(01)00422-2. [DOI] [PubMed] [Google Scholar]
- 73.Arbour N, Rastikerdar E, McCrea E, Lapierre Y, Dorr J, Bar-Or A, Antel JP. Upregulation of TRAIL expression on human T lymphocytes by interferon β and glatiramer acetate. Mult Scler. 2005;11:652–7. doi: 10.1191/1352458505ms1222oa. [DOI] [PubMed] [Google Scholar]
- 74.Chen M, Chen G, Deng S, Liu X, Hutton GJ, Hong J. IFN-β induces the proliferation of CD4+ CD25+ Foxp3+ regulatory T cells through upregulation of GITRL on dendritic cells in the treatment of multiple sclerosis. J Neuroimmunol. 2012;242:39–46. doi: 10.1016/j.jneuroim.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 75.Muraro PA, Leist T, Bielekova B, McFarland HF. VLA-4/CD49d downregulated on primed T lymphocytes during interferon-β therapy in multiple sclerosis. J Neuroimmunol. 2000;111:186–94. doi: 10.1016/s0165-5728(00)00362-3. [DOI] [PubMed] [Google Scholar]
- 76.Boz C, Ozmenoglu M, Velioglu S, Kilinc K, Orem A, Alioglu Z, Altunayoglu V. Matrix metalloproteinase-9 (MMP-9) and tissue inhibitor of matrix metalloproteinase (TIMP-1) in patients with relapsing-remitting multiple sclerosis treated with interferon β. Clin Neurol Neurosurg. 2006;108:124–8. doi: 10.1016/j.clineuro.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 77.Comabella M, Rio J, Espejo C, et al. Changes in matrix metalloproteinases and their inhibitors during interferon-β treatment in multiple sclerosis. Clin Immunol. 2009;130:145–50. doi: 10.1016/j.clim.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 78.Shaw PJ, Lukens JR, Burns S, Chi H, McGargill MA, Kanneganti TD. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:4610–4. doi: 10.4049/jimmunol.1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hanamsagar R, Hanke ML, Kielian T. Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol. 2012;33:333–42. doi: 10.1016/j.it.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arnason BG. Immunologic therapy of multiple sclerosis. Annu Rev Med. 1999;50:291–302. doi: 10.1146/annurev.med.50.1.291. [DOI] [PubMed] [Google Scholar]
- 81.Axtell RC, de JongBA, Boniface K, et al. T helper type 1 and 17 cells determine efficacy of interferon-β in multiple sclerosis and experimental encephalomyelitis. Nat Med. 2010;16:406–12. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kantarci OH, Atkinson EJ, Hebrink DD, McMurray CT, Weinshenker BG. Association of two variants in IL-1beta and IL-1 receptor antagonist genes with multiple sclerosis. J Neuroimmunol. 2000;106:220–7. doi: 10.1016/s0165-5728(00)00202-2. [DOI] [PubMed] [Google Scholar]
- 83.Badovinac V, Mostarica-Stojkovic M, Dinarello CA, Stosic-Grujicic S, Interleukin-1 receptor antagonist suppresses experimental autoimmune encephalomyelitis (EAE) in rats by influencing the activation and proliferation of encephalitogenic cells. J Neuroimmunol. 1998;85:87–95. doi: 10.1016/s0165-5728(98)00020-4. [DOI] [PubMed] [Google Scholar]
- 84.Shi FD, Takeda K, Akira S, Sarvetnick N, Ljunggren HG. IL-18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN-gamma by NK cells. J Immunol. 2000;165:3099–3104. doi: 10.4049/jimmunol.165.6.3099. [DOI] [PubMed] [Google Scholar]