

Abstract
Phagocytes such as dendritic cells (DC) and macrophages employ phagocytosis to take up pathogenic bacteria into phagosomes, digest the bacteria and present the bacteria-derived peptide antigens to the adaptive immunity. Hence, efficient antigen presentation depends greatly on a well-regulated phagocytosis process. Lipids, particularly phosphoinositides, are critical components of the phagosomes. Phosphatidylinositol-3,4,5-triphosphate [PI(3,4,5)P3] is formed at the phagocytic cup, and as the phagosome seals off from the plasma membrane, rapid disappearance of PI(3,4,5)P3 is accompanied by high levels of phosphatidylinositol-3-phosphate (PI3P) formation. The sorting nexin (SNX) family consists of a diverse group of Phox-homology (PX) domain-containing cytoplasmic and membrane-associated proteins that are potential effectors of phosphoinositides. We hypothesized that SNX3, a small sorting nexin that contains a single PI3P lipid-binding PX domain as its only protein domain, localizes to phagosomes and regulates phagocytosis in DC. Our results show that SNX3 recruits to nascent phagosomes and silencing of SNX3 enhances phagocytic uptake of bacteria by DC. Furthermore, SNX3 competes with PI3P lipid-binding protein, early endosome antigen-1 (EEA1) recruiting to membranes. Our results indicate that SNX3 negatively regulates phagocytosis in DC possibly by modulating recruitment of essential PI3P lipid-binding proteins of the phagocytic pathways, such as EEA1, to phagosomal membranes.
Keywords: dendritic cells, early endosome antigen-1, phagocytosis, phagosomes, SNX3
Introduction
Phagocytosis of microorganisms by phagocytes is an essential component of the host's defence against infection. Professional antigen-presenting phagocytes such as dendritic cells (DC) and macrophages employ phagocytosis to take up pathogenic bacteria, digest the bacteria and present the bacteria-derived peptide antigens to the adaptive immunity.1 Hence, activation of the immune system depends greatly on the phagocytic process, which is regulated by the membrane sorting and trafficking machinery in the endosomes.2,3
Membrane adaptors, coat proteins, Rab GTPases, soluble N-ethylmaleimide sensitive factor attachment receptors (SNAREs), and phosphoinositides are important players in membrane trafficking.4–6 Notably, many of them have been implicated in the phagocytic pathway. For instance, early stages of phagosome maturation are controlled by Rab5, whereas later fusion events with late endosomes and lysosomes require Rab7.7,8 VAMP-7, an R-SNARE has been shown to be essential for optimal phagocytosis of opsonized particles in macrophages,9 whereas VAMP-8, another R-SNARE, has been shown to negatively regulate phagocytosis of bacteria in DC.10 The enrichment of VAMP-4 on latex-bead-containing phagosomes also suggests a potential role for this R-SNARE in the phagocytic pathway.11 In addition, endoplasmic reticulum-mediated phagocytosis interestingly illustrates the importance of membrane and its components in the phagocytic pathway, albeit controversially.12–14
The sorting nexin (SNX) family consists of a diverse group of Phox-homology (PX) domain-containing cytoplasmic and membrane-associated proteins that are potential effectors of phosphoinositides. The well-characterized PX domain in SNXs has varying affinities to various phosphoinositides and mediates their associations with cellular membranes enriched in phosphoinositides.15,16 SNX1 has been shown to bind to epidermal growth factor receptor and regulates its turnover rate.17 SNX2 and SNX4 have been shown to bind to multiple receptor tyrosine kinases, including receptors for epidermal growth factor, platelet-derived growth factor, insulin, and the long form of the leptin receptor.18 Furthermore, SNX2 and SNX4 together with SNX6 have also been shown to interact with members of the transforming growth factor-β family of receptor serine-threonine kinases.19 These studies have suggested potential roles of SNXs in regulating the endocytosis of receptors. Consistently, the interaction of SNX9 with dynamin and Wiskott–Aldrich syndrome protein has been shown to be important for endocytosis.20,21 Apart from this, SNX3 has been shown to regulate endosomal function,22 and is important for multivesicular body biogenesis as well.23 Furthermore, the elucidation of the retromer complex involving SNX1 and SNX2 has provided greater insight into the importance of SNXs in membrane trafficking.24–27 No work has been carried out to investigate the roles of SNXs in the phagocytic pathway.
We hypothesized that SNX3, a small SNX that contains a single phosphatidylinositol-3-phosphate (PI3P) lipid-binding PX domain as their only protein domain, recruits to phagosomes and regulates phagocytosis in DC.
Materials and methods
Antibodies and cell lines
The antibodies used were purchased commercially from the following manufacturers: rabbit polyclonal anti-EEA1 (where EEA1 is early endosome antigen-1) and anti-Myc, mouse monoclonal anti-EEA1 and rat monoclonal anti-LAMP-1 (Santa Cruz Biotechnology, Dallas, TX), mouse monoclonal anti-c-Myc (Calbiochem, Merck, Boston, MA), rabbit polyclonal anti-Syn16 (SynapticSystem, Goettingen, Germany), and mouse anti-lipopolysaccharide (LPS; AbD Serotec, Oxford, UK). Rabbit polyclonal anti-SNX3 was a generous gift from Wanjin Hong. DyLight™488-, DyLight™649-, FITC- and Cy3-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories. AlexaFluor®647-conjugated anti-rabbit secondary antibody was from Invitrogen. DC2.4 cell line was from Dr Kenneth Rock.28 DC2.4 cells are immortalized mouse bone-marrow-derived DC that display dendritic morphology, express DC-specific markers, MHC molecules, co-stimulatory molecules, and have phagocytic activity as well as antigen-presenting capacity.28 A-431 human skin epithelial carcinoma cells (CRL-1555) were obtained from ATCC (Manassas, VA). Cell lines were cultured at 37° and 5% CO2 in complete Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated fetal bovine serum, 5 mm HEPES solution, 100 μm non-essential amino acids, antibiotic–antimycotic and 2 mm l-glutamine (Gibco, Life Technologies, Grand Island, NY).
Cloning and preparation of GST-proteins
Extraction of plasmid was based on the QIAprep® Spin Miniprep Kit (Qiagen, Valencia, CA). The restriction endonucleases BamHI, EcoRI and XbaI, T4 DNA ligase, Pfu and Taq polymerases were purchased from Promega. The plasmid extracted from Escherichia coli-containing mouse SNX3-ligated pCR4-TOPO vector (MGC: 151262, IMAGE: 40126204, Open Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA), was subjected to PCR using pfu polymerase. Forward primer was designed with an EcoRI restriction sequence and reverse primer with an XbaI sequence. SNX3, forward primer: 5′-GGAATTCCATGGCGGAGACGGTAG-3′ and reverse primer: 5′-GCTCTAGAGCTTAATGGGGATG-3′. The desired DNA band was excised and purified using QIAquick Gel Extraction (Qiagen), and subjected to EcoRI and XbaI digestion. The SNX3 insert was ligated to EcoRI- and XbaI-digested pDMyc plasmid with a double-Myc tag at the 5′-terminal. Transformation was performed and the clones were selected using 200 μg/ml ampicillin. Sequences were confirmed via sequencing.
Cloning of SNX3 and MycEEA1(1277–1411, inclusive of FYVE-domain) into pGEX-KG vector was carried out similarly as described above. EEA1(1277–1411) protein has been shown to bind specifically to PI3P.29 SNX3, forward primer was designed with a BamHI restriction sequence: 5′-CGGGATCCATGGCGGAGACGGTAG-3′ and reverse primer was designed with an EcoRI restriction sequence: 5′-GGAATTCGACTCTAGAGCTCAGGCATG-3′. Myc-EEA1(1277–1411) gene construct (GenBank BC075637.1) was obtained through Gene Synthesis (1st Base). The gene construct was designed with a BamHI restriction sequence at the 5′ end and an EcoRI restriction sequence at the 3′ end. Sequences were confirmed via sequencing. Top10 E. coli containing the respective pGEX-KG-SNX3 or pGEX-KG-Myc-EEA1(1277–1411) vectors were induced for 16 h at room temperature with isopropyl β-d-1-thiogalactopyranoside (Promega, Madison, WI). The GST-proteins were harvested using GSH-Sepharose beads (GE-Health) and columns (Bio-Rad, Hercules, CA), and eluted with 20 mm reduced GSH (Thermo Fisher Scientific, Inc.). GST was cleaved from GST-SNX3 using thrombin (Amersham, GE Healthcare Bio-Sciences Corp., Piscataway, NJ) at 10 μg/ml for 16 h at room temperature. After which, the proteins were dialysed in PBS and free GST was cleared using GSH-Sepharose beads.
Plasmid transfection and selection of stable cells
Transfection was performed with Invitrogen Lipofectamine™ 2000 according to the manufacturer's protocol. At 24–48 h after transfection, Geneticin® (Invitrogen, Life Technologies) was added at 2000 μg/ml to select for cells stably expressing SNX3.
Gene knockdown
Small interfering (si) RNA directed towards mouse SNX3 (catalogue number: LQ-060688-01-0020) and non-targeting siRNA pool (catalogue number: D-001810-10) were purchased from Dharmacon. Transfection was performed using electroporation (BTX ECM830). Briefly, cells were flushed out in PBS and washed once with Opti-MEM (Invitrogen). Cells (5 × 107 cells/ml) were resuspended in 100 μl Opti-MEM, mix with ∼ 760 pmol (∼ 10 μg) of siRNA in a 2-mm gap cuvette (BTX) and electroporated at 300 V for 500 μsecond in a single pulse. After electroporation, cells were transferred to complete DMEM with 10% fetal bovine serum and cultured for at least 36 h before subsequent experiments.
Phagocytosis assay
Phagocytosis level was measured using flow cytometry, based on the gross mean green fluorescent protein (GFP) -fluorescence detected from cells, after allowing the cells to phagocytose fixed GFP-E. coli in suspension for various time-points. This assay has been validated through its specific detection of the difference in phagocytosis levels between ‘Normal’ and ‘3-MA-treated’ DC (see Supplementary material, Fig. S1B). For the measurement of phagocytosis activity at 30 min and 60 min, cells were harvested from 60-mm dishes by treatment with 0·5 mm EDTA for 10 min, washed three times with DMEM, followed by mixing 2 × 106 cells with 1 × 108 fixed GFP-E. coli in a final volume of 1 ml DMEM, and incubation in a 37° incubator. The cells and bacteria suspension was mixed by gentle tapping every 15 min over the 30-min or 60-min period. After phagocytosis, 1 ml cold PBS was added to the suspension followed by three cycles of spinning down at 400 g for 5 min at 4° and washing with cold PBS. After which, the cell pellet was re-suspended in 2% paraformaldehyde before analysis by flow cytometry. For the detection of extracellular-bound bacteria by LPS staining, cells were re-suspended in 100 μl of staining buffer (5% BSA, 2 mm EDTA, 0·05% NaN3 in 1 × PBS) containing 40 μg/ml mouse monoclonal anti-LPS antibody, and incubated on ice for 60 min. After which, cells were washed and fixed with 2% paraformaldehyde as described above. For the measurement of phagocytosis at 4 min, cells were harvested similarly, but 2 × 106 cells were mixed with 1 × 108 fixed GFP-E. coli in a final volume of 100 μL. The DMEM used was pre-warmed at 37° and tubes containing the cells and bacteria suspension were incubated in a 37° water bath immediately after mixing. The smaller volume increased the chances of cells contacting the bacteria and for phagocytosis to occur within the short period of 4 min. After 4 min, 1 ml cold PBS was added to the suspension, mixed and three replicates of 100 μl each were removed and kept in 2% paraformaldehyde before analysis by flow cytometry. All phagocytosis experiments were carried out with parallel 0-min samples to allow for the subtraction of background signal from the data. ‘Parallel 0-min samples’ refer to DC that were added with the same amount of GFP-E. coli bacteria and transferred to 0° on ice instead of 37°, hence with total inhibition of phagocytosis. ‘Parallel 0 min samples’ served as background readings because of extracellular-bound bacteria and as negative controls for the assay. In addition, since silencing of SNX3 by siRNA transfection did not affect the binding of GFP-E. coli to the cell surface (see Fig. 4b), readings that were possibly contributed by the GFP-E. coli bacteria binding to the surface of cells were not expected to affect the results.
Figure 4.
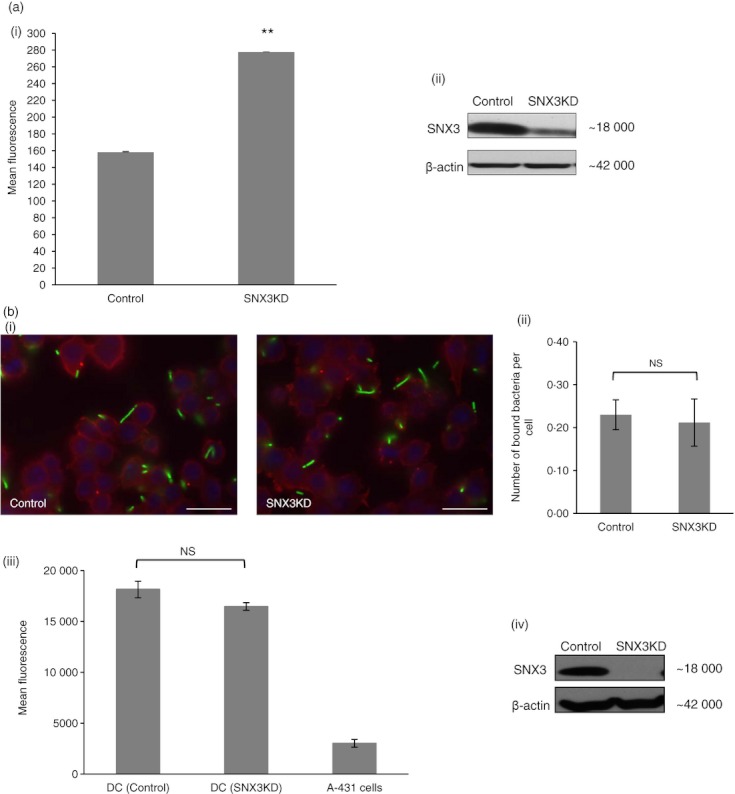
Sorting nexin 3 (SNX3) negatively regulates phagocytosis. DC2.4 cells were transfected by electroporation with either non-targeting small interfering RNA (siRNA) (control) or siRNA directed towards SNX3. Cells were allowed to phagocytose fixed GFP-Esherichia coli for 60 min, before they were fixed and quantified for GFP-fluorescence by flow cytometry (a). Bacteria binding to DC2.4 cells were quantified by manual counting as well as by measurement of fluorescence emitted from bound-GFP-E. coli (b). (a) Knockdown of SNX3 resulted in an increase in phagocytosis at 60 min (panel i). A similar trend was observed in three independent experiments. Immunoblot shows the effect of SNX3KD (panel ii). (b) Binding of GFP-E.coli was not affected in SNX3KD DC. Binding of bacteria is shown in representative pictures (panel i). Both SNX3KD cells and control cells showed similar amounts of GFP-E. coli surface binding (panel ii and iii). A similar trend was observed in two independent experiments for (panel ii) and three independent experiments for (panel iii). Immunoblot showing the effect of SNX3KD for experiment in (panel iii and iv). P-value **< 0·005; NS, not significant.
Phagocytosis kinetics
Cells grown on cover slips were washed twice with cold PBS, and incubated on ice for 1 h in 1·5 ml of complete DMEM containing fixed GFP-E. coli at 5 × 108/ml. After which the cover slips were washed five times with cold PBS before they were transferred to 37° for the respective time periods. The cover slips were washed and fixed on ice at various time-points, and used for immunofluorescence microscopy.
Quantification of bacteria binding
DC2.4 cells, adhered to cover slips, were incubated with 5 × 108/ml GFP-E. coli for 15 min at 37°. Cells were washed thoroughly with cold PBS to remove unbound bacteria, fixed and stained for β-actin. For each cover slip, three counts were conducted. Each count consisted of 10 different random frames, and GFP-E. coli in contact with the cells’ surface actin (bound-bacteria) were counted. At least 300 cells were observed per count. The number of bound-bacteria per-cell was recorded for each count, and an average of this value was obtained from the three counts. Alternatively, the amount of GFP-E. coli binding to cells was measured through the detection of GFP-fluorescence. DC and non-phagocytic A-431 cells were seeded overnight (∼ 16 h) in a 12-well plate, at 1·5 × 106 cells per well. Wells were checked to be 100% confluent with cells before they were washed once with cold PBS and equilibrated to 0° by incubation on ice for 15 min in 500 μL of cold Opti-MEM. After which, wells were tapped dry and 2 × 108 GFP-E. coli in 500 μL of Opti-MEM was added to cells and incubated on ice for 90 min. Wells without the addition of GFP-E. coli served as background controls. After 90 min of bacteria binding, 1 ml cold PBS was added, washed and tapped dry. Cells were then fixed with 500 μl of 4% paraformaldehyde on ice for 20 min. After fixing, 1 ml cold PBS was added, washed and tapped dry, before measurement with an ELISA plate fluorescence reader at Excitation 480 nm/Emission 520 nm (Tecan Infinite™ M200; Tecan US, Inc., Morrisville, NC).
Quantification of EEA1-positive early phagosomes
Early phagosomes were quantified by manual counting under immunofluorescence microscopy. Briefly, cells adhering to glass cover slips were allowed to phagocytose fixed GFP-E. coli for 30 min. After which, the cover slips were washed, fixed and stained for EEA1, an early phagosomal marker.30 For a cover slip of cells, at least three counts were conducted. Each count consisted of 15 different random frames, and phagosomes enriched with EEA1 were counted. At least 150 cells were observed per count. The number of EEA1-positive phagosomes per cell was recorded for each count, and an average of this value was obtained from the three counts. Subsequently, a phagocytosis index was generated by normalizing the value of phagosomes per cell with that of the control.
Lipid-binding assay
The lipid-binding assay had been designed to measure the lipid-binding capacity of GST, GST-MycEEA1 (1277–1411, inclusive of FYVE domain)29, and GST-SNX322. Co-star ELISA plate (96-well) was coated with 0·5 μg of PI3P [Calbiochem and CaymanChem (Ann Arbor, MI)] or phosphatidylinositol-4,5-diphosphate [PI(4,5)P2; Calbiochem] re-suspended in methanol, and air-dried in a 28° incubator. Wells were washed with PBS and blocked with PBS containing 5% BSA for 60 min at room temperature. Subsequent washing was carried out using 0·05% PBS-Tween-20. In an initial lipid-blocking step, 5 μg GST and either GST-SNX3 or GST-MycEEA1 proteins were added, respectively, to the plate and incubated for 60–90 min at room temperature. This step allowed proteins that have affinity to the lipid to bind and block the protein-binding sites on the lipid, hence reducing the binding by another protein to the lipid subsequently. After fixing with 4% paraformaldehyde for 15 min and washing, a second step which involved the addition of either GST-MycEEA1 or SNX-3 (5 μg), was carried out and incubated for 60 min at room temperature. Similar fixation was carried out, followed by the detection of the ‘second step’ lipid-bound proteins using primary (mouse anti-cMyc or rabbit anti-SNX-3) and secondary antibodies (DyLight™488-conjugated anti-mouse antibody or FITC-conjugated anti-rabbit antibody), respectively. The binding capacity of proteins to the lipid was quantified through fluorescence detection using an ELISA plate reader (Tecan Infinite™ M200).
Cell fractionation
Cell fractionation was carried out similarly to previous descriptions.31 Cells were homogenized and fractionated by ultracentrifugation to separate the membrane from the cytosol. For different samples such as SNX3KD cells and control cells, equal numbers of cells were used for the homogenization. Cells were suspended in 500 μl homogenizing buffer (8·6% sucrose, 25 mm HEPES, 5 mm MgCl, 1 mm PMSF) and incubated on ice for 40 min before being subjected to an upward–downward mechanical shearing in a manual homogenizer (Bellco, Vineland, NJ) for 20 min. The homogenate was observed under a light microscope to contain at least 95% broken cells and released nuclei, before they were subjected to two spun down at 100 gfor 5 min at 4° and 400 g for 5 min at 4° to clear off nuclei and intact cells. The cleared homogenate (400 μl) was then centrifuged for 90 min at 150 000 g using the Optima™ Max-XP (Beckman Coulter, Brea, CA) ultracentrifuge and MLA-130 rotor. After ultracentrifugation, the cytosol fraction was separated from the membrane pellet by careful aspiration. Both the cytosol and membrane fractions were then added with 20% CHAPS, protease inhibitors, PMSF and PBS, to a final volume of 500 μl with a final concentration of 1% CHAPS. In addition, the membrane fraction was subjected to light sonication of three cycles of a 5-second pulse at 10 Amp/20 seconds rest on ice. Both membrane and cytosol fractions were incubated on ice in lysis buffer for at least 2 h. Both fractions were then spun at 16 000 g for 15 min at 4° using a table-top centrifuge to clear off insoluble materials. The supernatant were aspirated and protein quantification was carried out before Western blot analysis.
Western blot analysis
Proteins resolved by SDS–PAGE were electroblotted to a nitrocellulose membrane (Bio-Rad) and incubated overnight at 4° with blocking buffer (PBS containing 5% skim milk and 0·05% Tween-20). Nitrocellulose membranes were then incubated with primary/secondary antibodies in blocking buffer, washed with PBS containing 0·05% Tween-20 and processed using the Supersignal Chemiluminescent Kit (Pierce; Thermo Fisher Scientific, Inc.) according to the manufacturer's recommendation. Quantification of band intensities in immunoblots was carried out using the ImageJ software.
Immunofluorescence microscopy
Cells grown on cover slips were washed once with PBS, and then fixed with 4% paraformaldehyde for 30 min on ice or for 20 min at room temperature, after which they were washed thrice with PBS, followed by twice with 0·1% saponin in PBS. The cells were permeabilized in 0·1% saponin at room temperature and incubated with primary antibody for 1 h. After washing with 0·1% saponin in PBS, secondary antibody was then added and the cover slips were incubated in the dark for 1 h at room temperature. Cells on cover slips were washed and mounted on glass slides using Vectashield® mounting medium with DAPI. Alternatively, cells were fixed with cold methanol at − 20° for 4 min and washed extensively with PBS before labelling them with primary and secondary antibodies. Confocal images were taken using Leica TCS SP5/Leica Application Suite Advanced Fluorescence version 2.1 software. The conventional fluorescent images were taken using Olympus BX-60 digital microscope/ImagePro Plus software.
Phagosome enrichment
Enrichment of phagosomes was carried out as previously described,10 with some minor changes. DC2.4 cells were allowed to phagocytose GFP-E. coli in suspension at 5 × 108 bacteria per 5 × 106 cells for 30 min at 37°. A mock sample (all conditions remained identical, except without the addition of bacteria, i.e. no phagocytosis) was run in parallel. To remove extracellular bacteria after phagocytosis, cells were thoroughly washed with 3 ×10 ml cold PBS in 15-ml centrifuge tubes and 3 × 1 ml cold PBS in 1·5-ml centrifuge tubes. Cells were spun down at 100 g for 5 min at 4° during all washes. The cells were then re-suspended in 500 μl of homogenizing buffer (8·6% sucrose, 25 mm HEPES, 5 mm MgCl2, 1 mm PMSF) and subjected to an upward–downward mechanical shearing in a manual homogenizer (Bellco) for 20 min. Homogenates were checked under a light microscope to contain at least 95% broken cells. The homogenate (∼ 300 μl) from each sample was removed and topped up to 1 ml with homogenizing buffer. After which, the homogenates were spun down at 100 g for 5 min at 4°. Supernatant containing the less dense phagosomes and membrane debris were carefully aspirated, leaving behind larger membrane debris and nuclei in the pellet. This was repeated at least twice until the supernatant was ∼ 100% cleared from nuclei as observed under microscopic examination. Subsequently, the supernatant (∼ 800 μl) was carefully layered on 62% sucrose (3 ml), and centrifuged at 18 000 g for 45 min at 4° using Beckman Optima™ Max-XP (MLS-50 rotor). The lighter membranes remained on top of the 62% sucrose, while the membrane fraction at the bottom was enriched with heavier phagosomes. The lighter membranes and sucrose were carefully removed, and lysis buffer (containing 1% CHAPS) was added to the membrane fraction and incubated on ice for 60 min. Subsequently, the lysate were treated in a similar way to that in Western blot analysis.
Statistical analysis
The statistical significance of the data were tested with an unpaired Student's t-test, and calculated goodness-of-fit value (P-value) is indicated in the figures. Differences were considered significant if P < 0·05 (*) and P < 0·005 (**).
Results
SNX3 is localized to the endosomes and phagosomes in DC
SNX3 was previously shown to associate with the early endosomes and intermediate structures in A-431 cells.22 To determine the intracellular localization of SNX3 in DC, DC2.4 cells were fixed, permeabilized and double labelled with antibodies specific for EEA1 and SNX3 before analysis by indirect immunofluorescence microscopy. Rab5 effector molecule EEA1 is a tethering molecule known to localize to the early endosomes.32 As shown in Fig. 1(a, panel i–iii), SNX3 and EEA1 were dispersed throughout the cell in punctate membrane structures depicting the endosomes. SNX3 and EEA1 co-localize partially on endosomes. SNX3's localization to the endosomes suggests that it may potentially play essential roles in regulating important functions such as phagocytosis and antigen presentation in DC.
Figure 1.
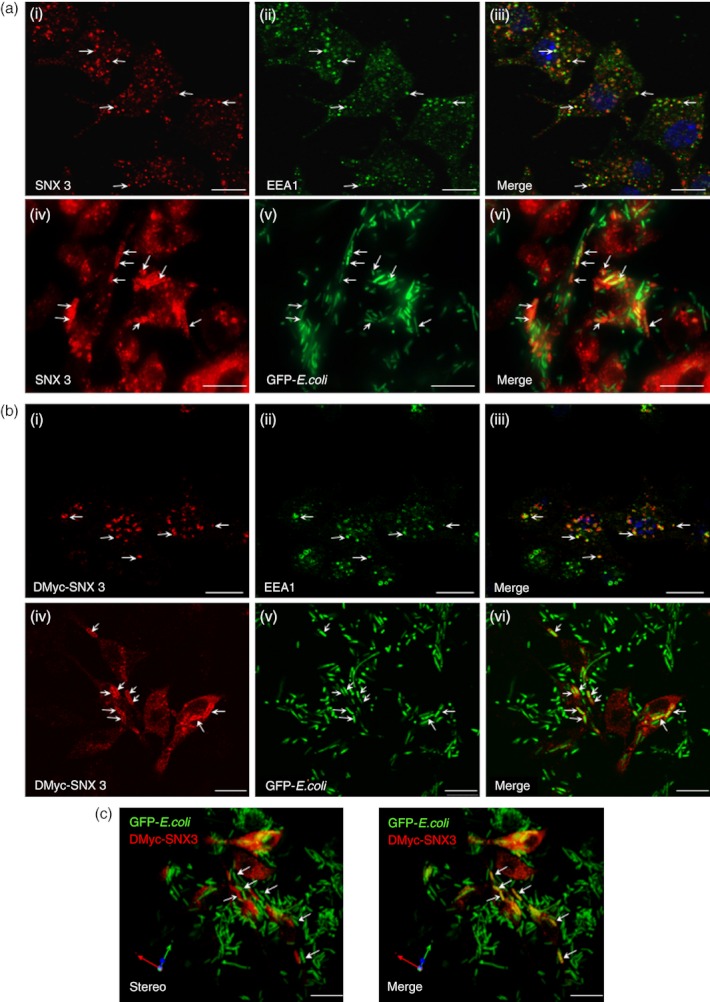
Sorting nexin 3 (SNX3) is localized to the endosomes and phagosomes in dendritic cells (DC). DC2.4 cells (a) or DC2.4 cells over-expressing DMyc-SNX3 (b and c), were fixed and labelled with anti-EEA1 primary antibody (Green), and co-stained with either anti-SNX3 (a, panel i–iii, red) or anti-Myc (b, panel i–iii, red) antibodies. After phagocytosis, cells were washed, fixed and singly stained with antibody against SNX3 (a, panel iv–vi) or Myc (b, panel vi–vi and c), followed by Cy3-conjugated anti-rabbit IgG. (a, panel i–iii) Endogenous SNX3 partially co-localizes with early endosome antigen-1 (EEA1) at endosomes. (a, panel iv–vi) Endogenous SNX3 is recruited to phagosomes containing GFP-Escherichia coli. (b, panel i–iii) DMyc-SNX3 co-localizes with EEA1 at endosomes, showing similar localization to endogenous SNX3. (b, panel iv–vi) DMyc-SNX3 is recruited to phagosomes containing GFP-E. coli. (c) Stereo three-dimensional rendering of collected confocal images showing the recruitment of DMyc-SNX3 (red) to the GFP-E. coli containing phagosomes (green/yellow). Arrows indicate SNX3- and EEA1-positive endosomes, and SNX3-positive phagosomes, respectively. Scale bars: 10 μm.
Phagocytosis is one of the important processes of antigen uptake in the antigen presentation pathway, and is closely associated with the endosomal membrane trafficking pathway.33 To examine if SNX3 recruits to phagosomes in DC, DC2.4 cells were allowed to phagocytose chemically fixed GFP-E. coli, fixed, stained with antibody specific for endogenous SNX3 and analysed by immunofluorescence microscopy. GFP-E. coli bacteria (instead of latex beads) were used in this experiment to maintain the microbiological relevance of this study. To prevent unwanted bacterial cell division during incubation with DC, GFP-E. coli were chemically fixed with 4% paraformaldehyde. All antibodies used were ‘pre-cleaned’ by incubating with high concentrations of the fixed GFP-E. coli to prevent the antibodies from non-specifically binding to bacteria during immunofluorescence staining. As shown in Fig. 1(a, panel iv–vi), endogenous SNX3 (red) co-localizes with the green GFP-E. coli (yellow), suggesting that SNX3 recruits to GFP-E. coli-positive membrane compartments depicting the phagosomes. Consistently, double c-Myc-tagged SNX3 (DMyc-SNX3) proteins, like endogenous SNX3, were also localized to the endosomes (Fig. 1b, panel i–iii) and recruited to the GFP-E. coli-positive phagosomes (Fig. 1b, panel iv–vi). Stereo three-dimensional rendering images in Fig. 1(c) further confirmed the recruitment of SNX3 to phagosomal membranes in DC. It is worth noting that not all the phagosomes were SNX3-positive, inferring that the recruitment of SNX3 to phagosomes was dependent on the maturity of the phagosomes.
SNX3 preferentially recruits to early phagosomes
Phagosomes acquire new components through sequential fusions with endosomes, and ultimately form the phagolysosomes when they undergo fusion with lysosomes.33 Hence, it will be interesting to determine the phagosomal maturation stage at which SNX3 recruits to phagosomes, and thereby draw insight into the role which SNX3 plays during phagocytic uptake of bacteria in DC. The DC2.4 cells were pre-incubated with GFP-E. coli for 1 h on ice (0°), washed and allowed to internalize the GFP-E. coli at 37° for various times. As shown in Fig. 2(a, panel v–viii), SNX3 was detected on phagosomes 8 min after phagocytosis. SNX3 remained associated with phagosomes until 30 min, albeit at a diminished level (Fig. 2a, panel ix–xvi). SNX3-positive phagosomes were not observed beyond the 30-min time point (Fig. 2a, panel xvii–xx). A corresponding staining with antibody against LAMP-1, a marker of late phagosome/phagolysosome,34 at 30 min and 60 min, showed that the SNX3-negative phagosomes (GFP-E. coli-positive) were enriched with LAMP-1 (Fig. 2b). This confirmed that SNX3 does not associate with late phagosomes/phagolysosomes, but preferentially associates with the early phagosomes, similar to VAMP-3 as previously reported.10
Figure 2.
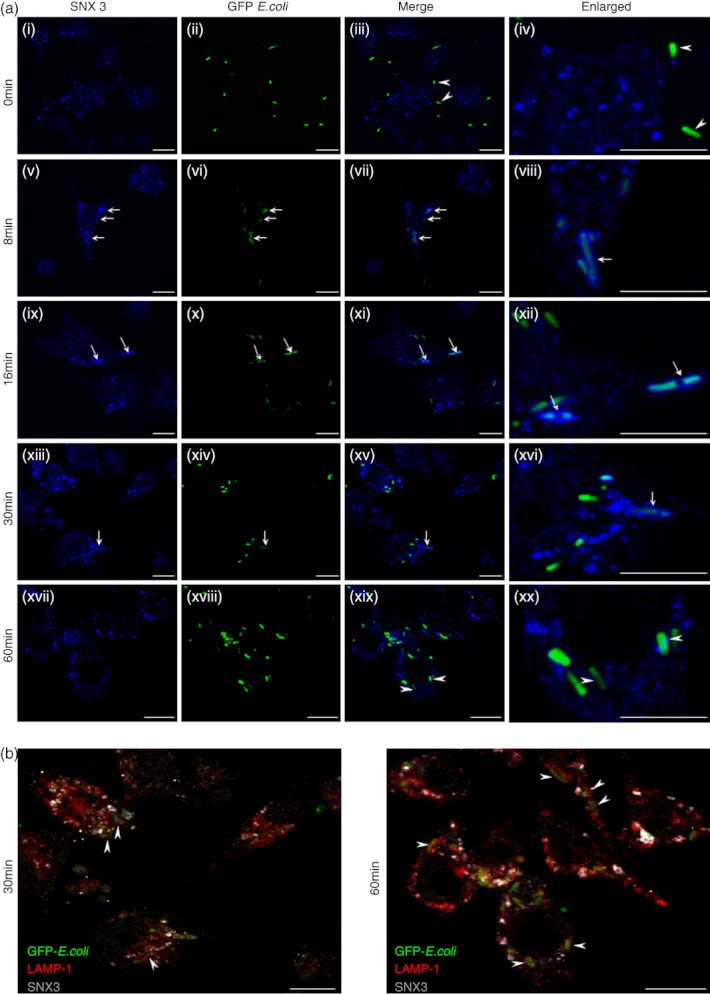
Sorting nexin 3 (SNX3) preferentially recruits to the early phagosomes. Cells were incubated with GFP-Escherichia coli on ice for 1 hour before they were washed and transferred to 37°. After which, cells were washed and fixed at various time-points, followed by staining with respective antibodies. Cells were stained with antibody against SNX3 (polyclonal, a, panel i–xx), followed by Alexa Fluor®647-conjugated anti-rabbit IgG. In addition to antibody against SNX3, cells were correspondingly stained with antibody against LAMP-1 (monoclonal), followed by Cy3-conjugated anti-rat IgG (b, a, panel i–xx). (a) The kinetic assay showing the recruitment of SNX3 to the early phagosomes at 8-, 16- and 30-min (panel v–xvi), but not the late phagosomes at 60-min (panel xvii–xx). Enlarged images of selected regions were shown for 0 min with arrowheads pointing to bacteria outside of cell (panel iv), for 8 to 30 min with arrows pointing to SNX3 enriched phagosomes (panel viii, xii and xvi), and for 60 min with arrowheads pointing to phagosomes devoid of SNX3 (panel xx). (b) Merged images from 30-min and 60-min kinetics, showing LAMP-1-positive late phagosomes/phagolysosomes were not enriched with SNX3. Arrow heads point to LAMP-1 and GFP-E.coli-positive late phagosomes/phagolysosomes which are devoid of SNX3. Scale bars: 10 μm.
SNX3 recruits to β-actin-positive nascent phagosomes
To further characterize the recruitment of SNX3 to early phagosome, DC2.4 cells, adhered to cover slip, were incubated on ice with fixed GFP-E. coli for 1 h. After washing away unbound bacteria, the cover slips were transferred to pre-warmed (37°) DMEM for 4 min and 8 min, before they were washed, fixed and stained for the respective proteins of interest. Stereo three-dimensional rendering images in Fig. 3(a) show the enrichment of β-actin (red) on phagocytic cups (white and red arrows). Figure 3(a, panel iii), clearly shows the exposed part of the GFP-E. coli bacteria (green arrow) on the phagocytic cup. Interestingly, we consistently observed recruitment of SNX3 (white arrowhead) to β-actin-positive (white arrow) complete phagosomes at 4 min of phagocytosis (Fig. 3b, panel i–iii). Figure 3(b, panel iv) shows the enlarged XZ projection image of the SNX3/β-actin-positive complete phagosome. In this study, with SNX3 as an additional phagosomal marker, it is possible to distinguish between the phagocytic cups, the nascent phagosomes and the early phagosomes stage. Figure 3(c) highlights the different stages of phagocytosis that have been observed in the phagocytosis kinetics study at 4 and 8 min. Arrows (ii) and (iii) point to phagocytic cups during phagocytic cup extension at 4 min [see schematic, Fig. 3d, (ii) and (iii)]. Arrow (iv) points to a nascent phagosome that was completely enclosed by β-actin-positive and SNX3-positive membrane at 4 min [see schematic, Fig. 3d(iv)]. Arrow (v) indicates a nascent phagosome with full SNX3 enrichment and diminishing β-actin recruitment, representing a stage where the nascent phagosome is separating from the plasma membrane at 4 min [see schematic, Fig. 3d(v)]. Arrows (vi) point to early phagosomes that were devoid of β-actin, but fully enriched with SNX3, maturing and moving away from the cell periphery at 8 min [see schematic, Fig. 3d(vi)]. Taking together, based on the labelling of SNX3 and β-actin, it is possible to classify β-actin+ SNX3− GFP-E.coli+ structures as phagocytic cups, β-actin+ SNX3+ GFP-E. coli+ structures as nascent phagosomes, and β-actin− SNX3+ GFP-E.coli+ as early phagosomes. According to Tsuboi and Meerloo, β-actin is absent from complete phagosomes.35 It is possible that the authors were referring to the β-actin-negative early phagosomes.
Figure 3.

Sorting nexin 3 (SNX3) recruits to β-actin-positive nascent phagosomes. (a) β-Actin is a marker for phagocytic cup. DC2.4 cells were pre-incubated with fixed GFP-Escherichia coli on ice for 1 h, after which they were washed to remove extracellular unbound GFP-E. coli. Cells were allowed to phagocytose the GFP-E. coli for 4 min at 37° before they were washed and fixed on ice. Cells were stained with antibody against β-actin (monoclonal), followed by Cy3-conjugated anti-mouse IgG. Stereo three-dimensional rendering of collected confocal images showing the recruitment of β-actin (red) to the GFP-E. coli-containing phagocytic cups (white and red arrows). (b) β-Actin and SNX3 co-localize at the nascent phagosomes. Following the same procedure for GFP-E. coli uptake, cells were co-stained with antibody against β-actin (monoclonal) and SNX3 (polyclonal), followed by Cy3-conjugated anti-mouse IgG and AlexaFluor®647-conjugated anti-rabbit IgG. (Panel i–iii) Confocal images of the co-localization of β-actin (red) and SNX3 (blue) at the nascent phagosomes. Arrowhead points to the complete SNX3 enrichment and arrow points to the partial β-actin enrichment at the nascent phagosome. (Panel iv) a stereo three-dimensional rendering and X–Z projection of the confocal images showing SNX3 localized to the whole length of the nascent phagosome (blue arrows) and β-actin localized to part of the nascent phagosome (red arrow). (c) Confocal images showing the complete enrichment of SNX3 to nascent phagosomes/early phagosomes, as well as the differential enrichment of β-actin to phagocytic cups/nascent phagosomes at different stages of phagocytosis. Arrowheads point to SNX3-positive nascent phagosomes/early phagosomes and arrows indicate β-actin-positive phagocytic cups/nascent phagosomes. Roman numerals (ii) – (vi) indicate the different stages of phagocytosis represented, corresponding to Fig. 3(d). Enlarged images are shown in corresponding panels (ii) – (vi). (d) A schematic showing the different stages during phagosome synthesis, namely phagocytic cup initiation (i), phagocytic cup extension [(ii) and (iii)], transitional closure (iv) and nascent phagosome separation from the plasma membrane [(v) and (vi)]. Scale bars: 10 μm.
Depletion of SNX3 enhances phagocytosis at the early stage
Specific recruitment to the nascent/early phagosomes suggests that SNX3 could potentially have a role in regulating the phagocytic pathway. To determine the role of SNX3 in phagocytosis, DC2.4 cells were transfected with non-targeting siRNA (control) or siRNA directed towards SNX3 (SNX3KD), and allowed to phagocytose GFP-E. coli for 60 min at 37°. The level of phagocytosis was measured by quantifying the GFP-fluorescence from the ingested E. coli using flow cytometry. As shown in Fig. 4(a), silencing of SNX3 enhances phagocytosis. Immunoblot in Fig. 4(a, panel ii) shows the silencing of SNX3 expression by SNX3-specific siRNA.
SNX3 has been implicated in the modulation of surface receptors such as transferrin receptors in A431 cells.22 It is possible that the increase in phagocytosis activity observed in SNX3KD DC could be the result of an increase in binding of bacteria through an increase in surface expression of phagocytic receptors. However, using immunofluorescence and manual counting of surface-bound GFP-E. coli, we did not observe significant variation in the number of bacteria binding to the surface of control and SNX3KD DC (Fig. 4b, panel i and ii). To further confirm our observation, surface bound GFP-E. coli bacteria were quantified using a fluorescence reader. As shown in Fig. 4(b, panel iii and iv), non-phagocytic A-431 cells showed very low binding of GFP-E. coli as compared with DC, whereas SNX3KD DC and control DC showed no significant difference in the binding of bacteria. Hence, taken together, our results in Fig. 4(b) suggested that the up-regulation of phagocytosis in SNX3KD DC was unlikely to be the result of an increase in bacteria binding to the cell surface. Indeed, when the GFP signals were normalized against the extracellular-bound bacteria/LPS signals, after 60 min of phagocytosis, we consistently detected a higher level of phagocytosis in SNX3KD DC (see Supplementary material, Fig. S1A).
We next determined the possibility that SNX3 could regulate phagocytosis at the early stage of the phagocytic pathway. Indeed, when cells were allowed to phagocytose fixed GFP-E. coli for a shorter time of 30 min, SNX3KD DC consistently showed a greater phagocytic uptake of fixed GFP-E. coli (Fig. 5a). As expected, quantification of EEA1-positive early phagosomes after 30 min of phagocytosis in DC2.4 cells transfected with either non-targeting siRNA (control) or SNX3-specific siRNA (SNX3KD) showed that silencing of SNX3 elevates EEA1-positive early phagosome formation by approximately 1·6–fold (Fig. 5b). Hence, for every 10 phagosomes produced in a control DC, an SNX3KD DC would produce about 16 phagosomes. These data suggest that SNX3 has a role in negatively regulating the formation of phagosomes, which is likely to occur before 4 min of phagocytic uptake as we have observed the recruitment of SNX3 to phagosomes as early as 4 min (Fig. 3). Indeed, as shown in Fig. 5(c), phagocytic uptake was increased at as early as 4 min in SNX3KD DC.
Figure 5.
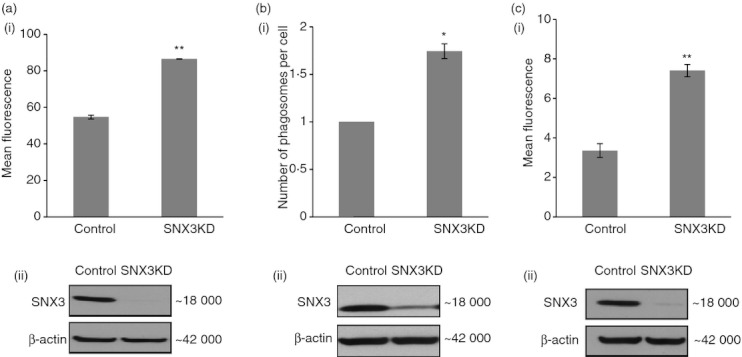
Sorting nexin 3 (SNX3) regulates phagocytosis at the early stage of phagocytic uptake. (a) Increased phagocytosis occurred within 30 min in SNX3KD dendritic cell (DC). A similar trend was observed in three independent experiments. (b) An increased number of early endosome antigen-1 (EEA1) -positive early phagosomes was detected in SNX3KD DC within 30 min of phagocytosis. The graph shows that SNX3KD DC had a higher number of phagosomes per cell, after normalizing with respect to control cells (see Materials ans methods). a total of 794 cells and 583 EEA1-positive phagosomes from control sample and 572 cells and 708 EEA1-positive phagosomes from SNX3KD sample were observed. A similar trend was observed in two independent experiments. (c) Increase in phagocytosis was detected as early as 4 min in SNX3KD DC. A similar trend was observed in three independent experiments. Immunoblots show the reduced protein expression of SNX3 in SNX3KD cells for each experiment. P-value **< 0·005, *< 0·05.
SNX3 competes with EEA1 for binding to phagosomal membrane
Besides a PX domain, SNX3 is not known to have other regulatory domains. This led us to postulate that SNX3 could function as small regulatory molecules, which can compete with another essential lipid-binding molecule in binding to PI3P lipids on phagosomes. SNX3 could potentially block larger PI3P lipid-binding proteins with multiple regulatory domains from recruiting to membranes. One of the potential large PI3P lipid-binding proteins is EEA1. EEA1 has a FYVE domain that allows it to bind to PI3P-rich phagosomes.36 It is an important tethering molecule that can be potentially important for membrane dynamic fusions and remodelling.37 Furthermore, EEA1 has been implicated in modulating phagosomal biogenesis and maturation.30 First, we proceeded to determine the presence of EEA1 at the nascent phagosomes. As shown in Fig. 6(a, panel i–iii), EEA1 was partially recruited to a nascent phagosome (white arrowhead) which had a partial β-actin enrichment (white arrow). To determine the recruitment of both SNX3 and EEA1 to the same nascent phagosome, DC2.4 cells and DC2.4 cells stably expressing DMyc-SNX3, were allowed to phagocytose fixed GFP-E. coli for 15 min, washed, fixed and co-stained with primary antibodies against EEA1 and SNX3 or c-Myc. Both EEA1 (red) and SNX3 (blue) were observed to co-localize on phagosomes (Fig. 6b, panel i–iv, arrow head and arrow). Similar observation that EEA1 also co-localized with DMyc-SNX3 on phagosomes (Fig. 6b, panel v–viii, arrow heads and arrows) confirmed that both EEA1 and SNX3 are enriched at nascent phagosomes. To further confirm the recruitment of SNX3 and EEA1 to the phagosomes, DC2.4 cells were incubated at 37° for 30 min with GFP-E. coli (30-min phagocytosis) or without GFP-E. coli (Mock). Cells were fractionated, phagosome-enriched fractions were extracted with lysis buffer, and equal amounts of proteins were analysed by Western blotting. As shown in Fig. 6(c), higher amount of SNX3 and EEA1 were detected in the phagosome-enriched fractions derived from DC2.4 cells incubated with GFP-E. coli (30-min phagocytosis) as compared with control DC2.4 cells (Mock, without incubation with GFP-E. coli). The presence of EEA1 and SNX3 in the phagosome-enriched fractions derived from control DC2.4 cells (Mock) could be contributed by the presence of ‘E. coli-free phagosomes’ which contain other large particulates possibly derived from cell debris.
Figure 6.
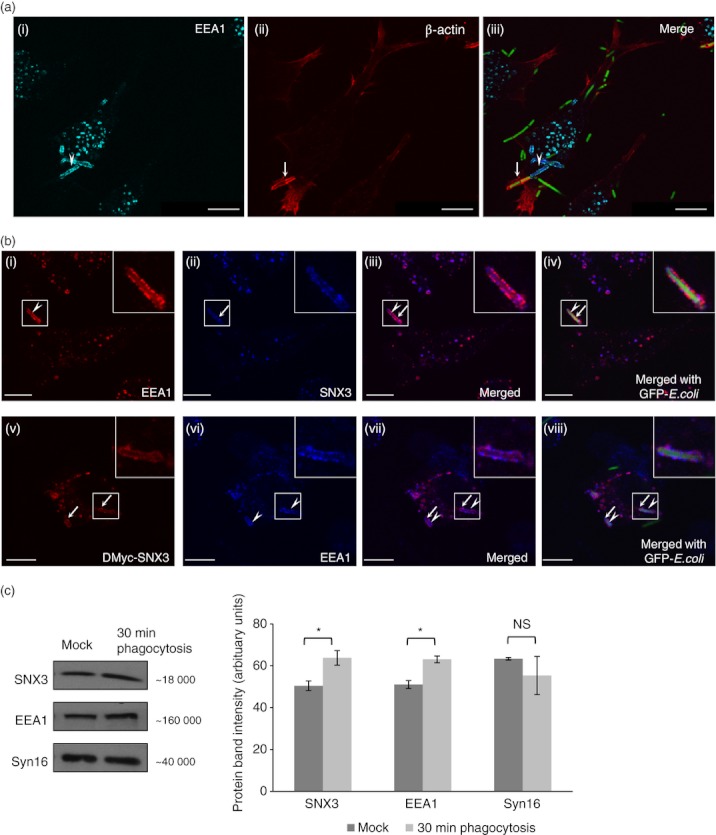
Sorting nexin 3 (SNX3) co-localizes with early endosome antigen-1 (EEA1) on nascent phagosomes. DC2.4 cells were allowed to phagocytose fixed GFP-Escherichia coli at 37° before staining with antibodies against β-actin (monoclonal) and EEA1 (polyclonal, a), or with antibodies against Myc (monoclonal) or SNX3 (polyclonal) and EEA1 (monoclonal or polyclonal, b), followed by respective Cy3-conjugated anti-mouse IgG and AlexaFluor®647-conjugated anti-rabbit IgG. (a) Confocal images showing the co-localization of β-actin (red) and EEA1 (cyan) at the nascent phagosomes. Arrowhead points to the partial enrichment of EEA1 and arrow points to the partial enrichment of β-actin at the nascent phagosome. (b, panel i–iv) Confocal images showing the enrichment of EEA1 (red, arrow head) and SNX3 (blue, arrow) to phagosomes. (b, panel v–viii) Confocal images showing the enrichment of EEA1 (blue, arrow heads) and DMyc-SNX3 (red, arrows) to phagosomes. Inserts are 2·5 times enlargement of the boxed regions. Scale bars: 10 μm. (c) DC2.4 cells were incubated at 37° for 30 min with GFP-E. coli (30-min phagocytosis) or without GFP-E. coli (Mock). After which, phagosome-enriched fractions for both samples were obtained, treated with lysis buffer, and equal amount of proteins were loaded for Western blotting. Comparing ‘30-min phagocytosis’ with ‘Mock’, a significantly greater amount of SNX3 and EEA1 was observed for ‘30-min phagocytosis’, indicating their recruitment to phagosomes. This was not observed in Syn16, which is not known to enrich in phagosomes. A similar trend was observed in two independent experiments. P-value *< 0·05.
To determine whether SNX3 competes with EEA1 binding to PI3P lipids, we carried out in vitro lipid-binding assays to measure the lipid-binding capacities of GST-MycEEA1 (with the FYVE-domain) and SNX3 in the presence or absence of blocking by their counterparts, GST-SNX3 and GST-MycEEA1, respectively. As shown in Fig. 7(a, panel i), when GST-SNX3 was allowed to bind PI3P before the introduction of GST-MycEEA1, it resulted in an approximately 40% drop in the binding of GST-MycEEA1 to PI3P compared with the ‘without GST-SNX3 blocking’. No significant changes in PI3P binding of GST-MycEEA1 was observed when blocking was done using GST. A reciprocal effect (an approximately 30% drop in the binding of SNX3 to PI3P) was observed when GST-MycEEA1 was allowed to bind PI3P before SNX3 (Fig. 7a, panel ii). The binding of GST-MycEEA1 and SNX3 was specific to PI3P (Fig. 7a, panel iii) and the identities of the various proteins used were confirmed by Western blotting (Fig. 7a, panel iv). Our results show that SNX3 and EEA1 compete with each other for binding to PI3P lipids.
Figure 7.

Sorting nexin 3 (SNX3) competes with early endosome antigen-1 (EEA1) for binding to membrane. (a) SNX3 and GST-MycEEA1(1277-1411) mitigate each other's binding capacity to phosphatidylinositol-3-phosphate (PI3P). In (panel i), PI3P lipid was blocked with GST-SNX3 or GST before the introduction of GST-MycEEA1(1277-1411). Binding of GST-MycEEA1(1277-1411) to lipid was detected by anti-cMyc primary antibody followed by DyLight™488-conjugated secondary antibody. In (panel ii), a reciprocal experiment involving the blocking with GST-MycEEA1(1277-1411) or GST before the introduction of SNX3. Binding of SNX3 to lipid was detected by anti-SNX3 primary antibody followed by FITC-conjugated secondary antibody. Lipid-binding capacities were expressed as a percentage to normal lipid binding, without blocking. Error bars represent SEM from three separate experiments. (Panel iii) shows that GST-MycEEA1(1277-1411) and SNX3 bind specifically to PI3P and not other lipid such as PI(4,5)P2. (Panel iv) shows the immunoblots that confirm the identities of the various proteins used in the lipid-binding assays. (b) Over-expression of SNX3 resulted in diminished EEA1 enrichment to membrane. DC2.4 cells stably expressing DMyc-SNX3 and control cells were homogenized, and membrane and cytosolic fractions were obtained by ultracentrifugation. Total protein was quantified for the membrane and cytosolic fractions and 10 μg of protein was loaded for SDS–PAGE. A representative immunoblot of EEA1 was shown (panel i). Dendritic cells expressing DMyc-SNX3 show a lower EEA1 membrane enrichment index (panel ii). A similar trend was observed in three independent experiments. (c) Depletion of SNX3 by small-interfering RNA transfection resulted in an increased EEA1 enrichment to membrane. SNX3KD DC2.4 cells and corresponding control cells were homogenized and the membrane fraction was obtained by ultracentrifugation. Membrane proteins (30 μg) from the membrane fraction were loaded for SDS–PAGE, and the representative immunoblot of EEA1 indicated a greater amount of EEA1 in the membrane fraction from SNX3KD cells (panels i and ii). A similar trend was observed in three independent experiments. The corresponding immunoblot of SNX3 showed the depletion of SNX3 in SNX3KD cells (panel iii). P-value** < 0·005, P-value *< 0·05; NS, not significant.
To determine whether SNX3 competes with EEA1 binding to membranes, membrane and cytosol fractions were isolated from stable DC2.4 clones, control DC (pDMyc vector-transfected) and DMyc-SNX3 expressing DC (pDMyc-SNX3-transfected). After extraction in 1% CHAPS, soluble proteins were quantified and equal amount of proteins from both fractions (membrane and cytosol) were separated by SDS–PAGE and analysed by Western blotting. In Fig. 7(b, panel i), a representative Western blot showed lower amount of EEA1 in the membrane fraction derived from DMyc-SNX3-expressing DC2.4 compared with control. Consistently, the cytosol fraction derived from DMyc-SNX3-expressing DC showed a higher amount of EEA1. As such, when the membrane enrichment indices were analysed by dividing the band intensities of the membrane fraction with that of the cytosol fraction, DMyc-SNX3 expressing DC showed significantly lower membrane enrichment of EEA1 (Fig. 7b, panel ii). Hence, our results suggest that DMyc-SNX3 competes with EEA1 in recruiting to membranes. Corresponding immunoblots show the level of Syn16 (an integral membrane protein), DMyc-SNX3, and endogenous SNX3 proteins in the membrane and cytosol fractions derived from control and DMyc-SNX3 expressing DC2.4 cells. Immunoblot for Syn16 indicated minimal membrane contamination of the cytosolic fraction by membranes, and the immunoblot for SNX3 showed the presence of DMyc-SNX3 protein (∼ 22 000 molecular weight) in addition to the endogenous protein (∼ 18 000 molecular weight) for DC2.4 cells stably expressing DMyc-SNX3.
To further confirm our observation, membrane and cytosol fractions were isolated from either control (non-targeting siRNA transfected) or SNX3KD (SNX3-specific siRNA transfected) DC2.4 cells, separated on SDS–PAGE and analysed by Western blotting to examine the recruitment of EEA1 to membrane. As shown in Fig. 7(c, panel i), depletion of SNX3 by siRNA knockdown enhanced recruitment of EEA1 to the membrane. Graphical representation of the band intensities shows that the increased in enrichment of EEA1 to membrane in SNX3KD cells was significant (Fig. 7c, panel ii). Western blot in Fig. 7(c, panel iii) shows the silencing of SNX3 expression by SNX3-specific siRNA. It is conceivable that silencing of SNX3 increases EEA1 enrichment on membrane. It is possible that in the absence of SNX3, more free membrane PI3P are available for binding to EEA1, assuming that the amount of Rab5/EEA1 in the control and SNX3KD cells remains the same. However, we did not determine whether silencing of SNX3 changes the composition of lipids on membrane.
Discussion
The site of nascent phagosome synthesis and phagocytosis is characterized by a high level of membrane remodelling and dynamics.38 This study, for the first time, has established a role for SNX3 in the regulation of phagocytosis in DC (Fig. 8). Our results suggest that depletion of SNX3 from the phagosomes frees up more PI3P lipid-binding sites for the recruitment of other PI3P lipid-binding proteins such as EEA1 (in this study), which potentially enhances phagosome biogenesis and phagocytosis.
Figure 8.
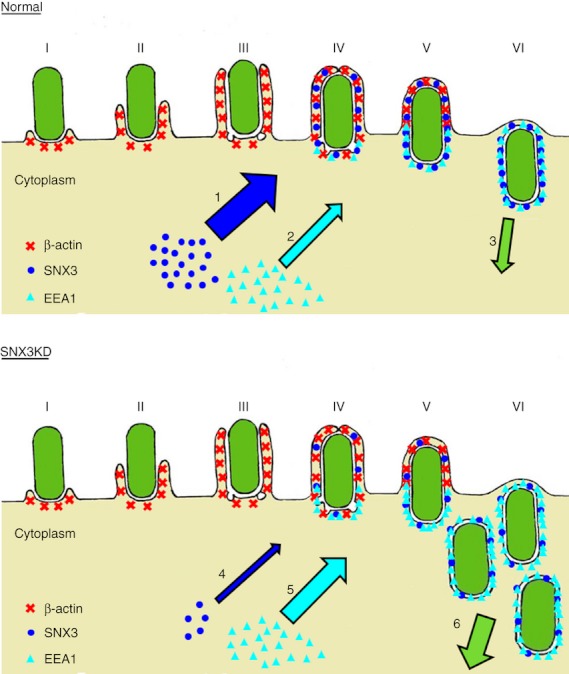
Stages in phagosome formation and a model for the regulation of phagocytosis by sorting nexin 3 (SNX3). Phagocytic cup initiation (I), phagocytic cup extension [(II) and (III)], transitional closure (IV) and nascent phagosome separation from the plasma membrane [(V) and (VI)] are illustrated with emphasis on the recruitment of β-actin (red), SNX3 (blue) and early endosome antigen-1 (EEA1) (cyan). Under normal condition, normal level of SNX3 recruitment to nascent phagosomes (1) keeps the level of EEA1 recruitment to nascent phagosomes in control (2), resulting in a well-regulated input of membrane materials and phagosome formation (3). With the depletion of SNX3 through small-interfering RNA knockdown, a drastic decrease in SNX3 recruitment to nascent phagosomes (4) reduces the competition with EEA1 for binding to the phagosomal membrane, hence an increase in EEA1 recruitment to nascent phagosomes (5). This in turn results in an increase or dysregulated input of membrane materials to the nascent phagosomes, which accelerates the synthesis of phagosomes (6). Thickness of the arrows in the illustration is proportional to the magnitude of the various events described.
Our results show that SNX3 recruits not to the late phagosomes but to the nascent phagosomes. SNX3 was previously shown to recruit to the salmonella-containing vesicles (SCV) in Salmonella enterica-infected non-phagocytic HeLa cells.39 In Braun's study,39 SNX3 was reported to localize to salmonella-induced tubules and had a role in regulating the maturation of SCVs. Consistently, SNX3 has been shown to associate with tubular–vesicular structures and tubular extensions of the early endosomes in A-431 cells.22 It is worth noting that the SCVs are not products of the phagocytosis process because HeLa cells (epithelial origins) have not been reported previously to have the ability to phagocytose bacteria. Hence, before this study, the role of SNX3 along the phagocytic pathway in phagocytes such as DC and macrophages was unclear.
It is noteworthy that our understanding of the progression of phagocytic uptake, particularly at the early stage of phagocytosis, has been improved through the phagocytosis kinetics and immunofluorescent staining of β-actin and SNX3 undertaken in this study. Phagocytic cups, nascent phagosomes and early phagosomes have been distinguished by the differential enrichment of β-actin and SNX3 as observed through immunofluorescence. Interestingly, during the course of preparing this manuscript, Michal Bohdanowicz and his co-workers reported a phase, after complete phagosome formation, in which actin transiently surrounded the entire phagosome and asymmetrically dissociated to form an actin ‘comet tail’, likely to drive phagosome displacement.40 Contrary to this study, we did not observe phenomena that resemble the actin ‘comet tail’ on complete phagosomes. Our observations when put together, suggest a smooth transition from phagocytic cup initiation to complete nascent phagosome synthesis (Fig. 3). The different modes of phagocytic uptake could be a main reason for this discrepancy, as it was mentioned that the actin ‘comet tail’ events were exclusive to CR3-mediated uptake. Following the immunofluorescence study at the early stage of phagocytosis was the discovery that silencing of SNX3 enhanced phagocytosis as early as 4 min post-exposure to bacteria.
At the early stage of phagocytosis (as early as 4 min), the synthesis of phagosomes is likely to be regulated at two phases. First, the phagocytic cup formation phase, where actin filaments concentrate at the site of phagocytosis to push the plasma membrane into a cup-shaped extension, and the second nascent phagosome phase, where the phagosome is sealing off from the plasma membrane. Tsuboi and Meerloo reported that Wiskott–Aldrich syndrome protein was a key regulator of the phagocytic cup formation.35 Interestingly, these authors reported the use of 3-methyladenine (3-MA) to inhibit phagosome formation without affecting the formation of phagocytic cups. The 3-MA is a PtdIns3 kinase inhibitor that can inhibit the production of PI3P. SNX3 is an established effector for PI3P lipids. As 3-MA treatment did not affect phagocytic cup formation, it was not likely for SNX3 to regulate phagocytic cup synthesis during phagocytosis. Absence of SNX3 on phagocytic cups and preferential recruitment of SNX3 to nascent and early phagosomes are consistent with the 3-MA study.
How does SNX3 regulate phagosome synthesis at the nascent phagosomes in DC? Besides the PX domain, SNX3 has no other prominent functional domains, suggesting that SNX3's regulatory role might be linked to the function(s) of another protein on the nascent phagosome. Interestingly, we have observed the recruitment of Rab5 effector EEA1 to the nascent phagosome. EEA1 is an important tethering protein that has been shown in a previous study to be essential for the maturation of early phagosomes through the acquisition of lysobisphophatidic acid.30 In the study, microinjection of EEA1-specific and PI-3K hVPS34-specific antibodies into macrophages reduced the acquisition of late endocytic markers by latex bead phagosomes, demonstrating an essential role of Rab5 effectors in phagosomal biogenesis.30 SNX3 and EEA1 share a common target, PI3P, for binding to membrane. Consistently, we have confirmed that SNX3 and EEA1 could mitigate each other's PI3P binding capacity in an in vitro lipid binding assay. As such, when it was observed that SNX3 and EEA1 were enriched on the same phagosomal membrane, and ectopic expression of SNX3 resulted in lower EEA1 enrichment to membrane while depletion of SNX3 caused an increase in EEA1 enrichment to membrane, it became conceivable that SNX3 competes with EEA1 for binding to the phagosomal membrane. As EEA1 also localizes to the nascent phagosomes, it is possible that other than functioning at the early to late phagosomal maturation stage during phagocytosis, EEA1 also plays a role in nascent phagosome biogenesis during phagocytosis in DC. In this context, it is possible that SNX3 regulates phagocytosis by regulating EEA1 recruitment to the nascent phagosomes. Silencing of SNX-3 possibly frees up more PI3P lipid-binding sites on nascent phagosomes for the recruitment of EEA1, which potentially enhances nascent phagosome biogenesis. Furthermore, knocking-down SNX-3 may also expose more PI3P lipid-binding sites on the early phagosomes for the recruitment of EEA-1 and promote maturation of early phagosomes to the late phagosomes/phagolysosomes. As EEA1 is not detected in the late phagosomes/phagolysosomes, it is possible that EEA1 is released from the early phagosomes into the cytoplasm during maturation to the late phagosomes/phagolysosomes. Hence, depletion of SNX3 may possibly promote the turnover of EEA1 molecules from early phagosomes, releasing them for binding to more freed up PI3P lipid-binding sites on the nascent phagosomes, and may stimulate their biogenesis. Further study along this line may shed more light on this interesting model.
Our results show that silencing of SNX3 in DC enhances phagocytosis, highlighting the role of SNX3 in controlling phagocytosis. A well-regulated phagocytic pathway is essential to DC. A dysregulated and excessive phagocytosis may exhaust cellular membrane materials and compromise other cellular functions such as secretion of cytokines. Particularly in the context of antigen presentation, a well-regulated phagocytic pathway is linked to efficient antigen processing and loading into MHC molecules.2 The phosphatidylinositol-3,4,5-triphosphate [PI(3,4,5)P3] and PI3P phosphoinositides and their effectors have been widely discussed in the area of phagocytosis regulation. Other than SNX3 and EEA1, many potential regulators of phagocytosis may well exist among the many PI3P-binding proteins.37 One example is another small SNX, SNX12, which is four amino acids longer than SNX3 and is about 77% identical to SNX3. It is possible that SNX12 may share a similar mechanistic relationship to that observed between SNX3 and EEA1 to regulate the phagocytic pathway in DC. Interestingly, during the preparation of this manuscript, Martin Harterink's group showed that SNX3 forms a complex with VPS26, VPS29 and VPS35 that is essential for the trafficking of the Wnt sorting receptor, Wntless.41 The group suggested a role for SNX3 in modulating the formation of retromer complexes through its competition with SNX1/SNX2 and SNX5/SNX6 for binding with the VPS26–VPS29–VPS35 trimeric complex. This is in line with the outcome of our present study where small sorting nexins may play important roles in regulating membrane trafficking in the endocytic/phagocytic pathways by competing with PI3P lipid-binding proteins to membrane.
Collectively, our results show for the first time that small SNXs (SNX3 in this study) which contain a single PX domain as their only protein domain can function as effective regulators of phagocytosis in phagocytes. Hence, phagocytes could potentially modulate their phagocytosis activity in response to pathogenic bacterial infection by regulating protein expression of SNX3, SNX12, and possibly other SNXs. Consistently, it was reported recently that expression and functions of SNXs in cells were modulated in response to infection by S. enterica.39,42,43 Hence, regulation of SNX expression and functions could be an effective mechanism used by activated phagocytes to keep pathogenic bacterial infections under control by enhancing the rate of phagocytic bacterial ingestion. Apart from understanding the regulation of phagocytosis, the potential mechanistic relationship between SNX3 and EEA1 observed in this study may pave the way for understanding of the roles of small SNXs in controlling the recruitment of PI3P effectors that govern the endosomal membrane/protein trafficking pathways in mammalian cells.
Acknowledgments
We thank Dr Kevin Tan Shyong-wei (Department of Microbiology, NUS, Singapore) for the generous gift of GFP-E. coli, Dr Kenneth Rock for the DC2.4 cell line, Dr Wanjin Hong (IMCB, Singapore) for the SNX3 antibody, and members of the Membrane Trafficking and Immunoregulation Research Laboratory for critical reading of the manuscript. This work was supported by research grants from the Academic Research Council-Ministry of Education, Singapore (R182-000-099-112 to S.H.W) and the Singapore National Medical Research Council (R182-000-137-213 to S.H.W).
Glossary
- DC
dendritic cells
- DMEM
Dulbecco's modified Eagle's medium
- GFP
green fluorescence protein
- LPS
lipopolysaccharide
- PI3P
Phosphatidylinositol-3-phosphate
- siRNA
small interfering RNA
- SNARE
soluble N-ethylmaleimide sensitive factor attachment receptors
- SNX3
Sorting nexin 3
- SNX3KD
Sorting nexin 3 knockdown
Disclosure
The authors report that they have no interests to disclose.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Validation of the phagocytosis assay.
References
- 1.Burgdorf S, Kurts C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol. 2008;20:89–95. doi: 10.1016/j.coi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Savina A, Amigorena S. Phagocytosis and antigen presentation in dendritic cells. Immunol Rev. 2007;219:143–56. doi: 10.1111/j.1600-065X.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- 3.Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9:781–95. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin TF. Phosphoinositide lipids as signaling molecules: common themes for signal transduction, cytoskeletal regulation, and membrane trafficking. Annu Rev Cell Dev Biol. 1998;14:231–64. doi: 10.1146/annurev.cellbio.14.1.231. [DOI] [PubMed] [Google Scholar]
- 5.Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–66. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- 6.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–25. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 7.Vieira OV, Bucci C, Harrison RE, et al. Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol Cell Biol. 2003;23:2501–14. doi: 10.1128/MCB.23.7.2501-2514.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts EA, Chua J, Kyei GB, Deretic V. Higher order Rab programming in phagolysosome biogenesis. J Cell Biol. 2006;174:923–9. doi: 10.1083/jcb.200603026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braun V, Fraisier V, Raposo G, Hurbain I, Sibarita JB, Chavrier P, Galli T, Niedergang F. TI-VAMP/VAMP7 is required for optimal phagocytosis of opsonised particles in macrophages. EMBO J. 2004;23:4166–76. doi: 10.1038/sj.emboj.7600427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho YH, Cai DT, Wang CC, Huang D, Wong SH. Vesicle-associated membrane protein-8/endobrevin negatively regulates phagocytosis of bacteria in dendritic cells. J Immunol. 2008;180:3148–57. doi: 10.4049/jimmunol.180.5.3148. [DOI] [PubMed] [Google Scholar]
- 11.Shui W, Sheu L, Liu J, et al. Membrane proteomics of phagosomes suggests a connection to autophagy. Proc Natl Acad Sci U S A. 2008;105:16952–7. doi: 10.1073/pnas.0809218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gagnon E, Duclos S, Rondeau C, et al. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. 2002;110:119–31. doi: 10.1016/s0092-8674(02)00797-3. [DOI] [PubMed] [Google Scholar]
- 13.Becker T, Volchuk A, Rothman JE. Differential use of endoplasmic reticulum membrane for phagocytosis in J774 macrophages. Proc Natl Acad Sci U S A. 2005;102:4022–6. doi: 10.1073/pnas.0409219102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Touret N, Paroutis P, Terebiznik M, et al. Quantitative and dynamic assessment of the contribution of the ER to phagosome formation. Cell. 2005;123:157–70. doi: 10.1016/j.cell.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 15.Worby CA, Dixon JE. Sorting out the cellular functions of sorting nexins. Nat Rev Mol Cell Biol. 2002;3:919–31. doi: 10.1038/nrm974. [DOI] [PubMed] [Google Scholar]
- 16.Carlton J, Bujny M, Rutherford A, Cullen P. Sorting nexins–unifying trends and new perspectives. Traffic. 2005;6:75–82. doi: 10.1111/j.1600-0854.2005.00260.x. [DOI] [PubMed] [Google Scholar]
- 17.Kurten RC, Cadena DL, Gill GN. Enhanced degradation of EGF receptors by a sorting nexin, SNX1. Science. 1996;272:1008–10. doi: 10.1126/science.272.5264.1008. [DOI] [PubMed] [Google Scholar]
- 18.Haft CR, de la Luz Sierra M, Barr VA, Haft DH, Taylor SI. Identification of a family of sorting nexin molecules and characterization of their association with receptors. Mol Cell Biol. 1998;18:7278–87. doi: 10.1128/mcb.18.12.7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parks WT, Frank DB, Huff C, et al. Sorting nexin 6, a novel SNX, interacts with the transforming growth factor-β family of receptor serine-threonine kinases. J Biol Chem. 2001;276:19332–9. doi: 10.1074/jbc.M100606200. [DOI] [PubMed] [Google Scholar]
- 20.Soulet F, Yarar D, Leonard M, Schmid SL. SNX9 regulates dynamin assembly and is required for efficient clathrin-mediated endocytosis. Mol Biol Cell. 2005;16:2058–67. doi: 10.1091/mbc.E04-11-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Badour K, McGavin MK, Zhang J, et al. Interaction of the Wiskott–Aldrich syndrome protein with sorting nexin 9 is required for CD28 endocytosis and cosignaling in T cells. Proc Natl Acad Sci U S A. 2007;104:1593–8. doi: 10.1073/pnas.0610543104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y, Hortsman H, Seet L, Wong SH, Hong W. SNX3 regulates endosomal function through its PX-domain-mediated interaction with PtdIns(3)P. Nat Cell Biol. 2001;3:658–66. doi: 10.1038/35083051. [DOI] [PubMed] [Google Scholar]
- 23.Pons V, Luyet PP, Morel E, Abrami L, van der Goot FG, Parton RG, Gruenberg J. Hrs and SNX3 functions in sorting and membrane invagination within multivesicular bodies. PLoS Biol. 2008;6:e214. doi: 10.1371/journal.pbio.0060214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seaman MN. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol. 2004;165:111–22. doi: 10.1083/jcb.200312034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffin CT, Trejo J, Magnuson T. Genetic evidence for a mammalian retromer complex containing sorting nexins 1 and 2. Proc Natl Acad Sci U S A. 2005;102:15173–7. doi: 10.1073/pnas.0409558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bujny MV, Popoff V, Johannes L, Cullen PJ. The retromer component sorting nexin-1 is required for efficient retrograde transport of Shiga toxin from early endosome to the trans Golgi network. J Cell Sci. 2007;12:2010–21. doi: 10.1242/jcs.003111. [DOI] [PubMed] [Google Scholar]
- 27.Rojas R, Kametaka S, Haft CR, Bonifacino JS. Interchangeable but essential functions of SNX1 and SNX2 in the association of retromer with endosomes and the trafficking of mannose 6-phosphate receptors. Mol Cell Biol. 2007;27:1112–24. doi: 10.1128/MCB.00156-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158:2723–30. [PubMed] [Google Scholar]
- 29.Patki V, Lawe DC, Corvera S, Virbasius JV, Chawla A. A functional PtdIns(3)P-binding motif. Nature. 1998;394:433–4. doi: 10.1038/28771. [DOI] [PubMed] [Google Scholar]
- 30.Fratti RA, Backer JM, Gruenberg J, Corvera S, Deretic V. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J Cell Biol. 2001;154:631–44. doi: 10.1083/jcb.200106049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiow KH, Tan Y, Chua RY, Huang D, Ng ML, Torta F, Wenk MR, Wong SH. SNX3-dependent regulation of epidermal growth factor receptor (EGFR) trafficking and degradation by aspirin in epidermoid carcinoma (A-431) cells. Cell Mol Life Sci. 2012;69:1505–21. doi: 10.1007/s00018-011-0887-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson JM, de Hoop M, Zorzi N, Toh BH, Dotti CG, Parton RG. EEA1, a tethering protein of the early sorting endosome, shows a polarized distribution in hippocampal neurons, epithelial cells, and fibroblasts. Mol Biol Cell. 2000;11:2657–71. doi: 10.1091/mbc.11.8.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol. 2006;7:1029–35. doi: 10.1038/ni1006-1029. [DOI] [PubMed] [Google Scholar]
- 34.Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007;26:313–24. doi: 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsuboi S, Meerloo J. Wiskott–Aldrich syndrome protein is a key regulator of the phagocytic cup formation in macrophages. J Biol Chem. 2007;282:34194–203. doi: 10.1074/jbc.M705999200. [DOI] [PubMed] [Google Scholar]
- 36.Kutateladze TG. Mechanistic similarities in docking of the FYVE and PX domains to phosphatidylinositol 3-phosphate containing membranes. Prog Lipid Res. 2007;46:315–27. doi: 10.1016/j.plipres.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gillooly DJ, Simonsen A, Stenmark H. Phosphoinositides and phagocytosis. J Cell Biol. 2001;155:15–7. doi: 10.1083/jcb.200109001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee WL, Mason D, Schreiber AD, Grinstein S. Quantitative analysis of membrane remodeling at the phagocytic cup. Mol Biol Cell. 2007;18:2883–92. doi: 10.1091/mbc.E06-05-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braun V, Wong A, Landekic M, Hong WJ, Grinstein S, Brumell JH. Sorting nexin 3 (SNX3) is a component of a tubular endosomal network induced by Salmonella and involved in maturation of the Salmonella-containing vacuole. Cell Microbiol. 2010;12:1352–67. doi: 10.1111/j.1462-5822.2010.01476.x. [DOI] [PubMed] [Google Scholar]
- 40.Bohdanowicz M, Cosio G, Backer JM, Grinstein S. Class I and class III phosphoinositide 3-kinases are required for actin polymerization that propels phagosomes. J Cell Biol. 2010;191:999–1012. doi: 10.1083/jcb.201004005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harterink M, Port F, Lorenowicz MJ, et al. A SNX3-dependent retromer pathway mediates retrograde transport of the Wnt sorting receptor Wntless and is required for Wnt secretion. Nat Cell Biol. 2011;13:914–23. doi: 10.1038/ncb2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bujny MV, Ewels PA, Humphrey S, Attar N, Jepson MA, Cullen PJ. Sorting nexin-1 defines an early phase of Salmonella-containing vacuole-remodeling during Salmonella infection. J Cell Sci. 2008;12:2027–36. doi: 10.1242/jcs.018432. [DOI] [PubMed] [Google Scholar]
- 43.Shi L, Chowdhury SM, Smallwood HS, et al. Proteomic investigation of the time course responses of RAW 264.7 macrophages to infection with Salmonella enterica. Infect Immun. 2009;77:3227–33. doi: 10.1128/IAI.00063-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.