

Abstract
T helper type 17 (Th17) lymphocytes are found in high frequency in tumour-burdened animals and cancer patients. These lymphocytes, characterized by the production of interleukin-17 and other pro-inflammatory cytokines, have a well-defined role in the development of inflammatory and autoimmune pathologies; however, their function in tumour immunity is less clear. We explored possible opposing anti-tumour and tumour-promoting functions of Th17 cells by evaluating tumour growth and the ability to promote tumour infiltration of myeloid-derived suppressor cells (MDSC), regulatory T cells and CD4+ interferon-γ+ cells in a retinoic acid-like orphan receptor γt (RORγt) -deficient mouse model. A reduced percentage of Th17 cells in the tumour microenvironment in RORγt-deficient mice led to enhanced tumour growth, that could be reverted by adoptive transfer of Th17 cells. Differences in tumour growth were not associated with changes in the accumulation or suppressive function of MDSC and regulatory T cells but were related to a decrease in the proportion of CD4+ T cells in the tumour. Our results suggest that Th17 cells do not affect the recruitment of immunosuppressive populations but favour the recruitment of effector Th1 cells to the tumour, thereby promoting anti-tumour responses.
Keywords: anti-tumour immunity, Rorγt, T helper type 1 cells, T helper type 17 cells
Introduction
Following their characterization as a functionally distinct T helper lineage, T helper type 17 (Th17) cells have become an important focus of cancer immunology in light of several studies that have reported their high frequency in various cancers, including ovarian, breast, colon, prostate and melanoma tumours.1–4 Moreover, it has been demonstrated that tumour-secreted chemokines support the mobilization and expansion of Th17 cells. 5 These findings have sparked tremendous curiosity and efforts to characterize the function of Th17 cells in cancer development. In haematopoietic tumours, the expression of IL-17 was reported to reduce tumour growth rate by promoting the generation of tumour-specific CD8+ cytotoxic T cells.5 In contrast, interleukin-17 (IL-17) over-expression in fibrosarcoma and colon carcinoma promotes tumour growth both in immunocompetent and immunoablated mice, and correlates with increased microvascularity in the tumour, suggesting that IL-17 contributes to angiogenesis.6 By addressing the function of Th17 cells using Il17−/− mice, it has been shown that the absence of this cytokine increases tumour growth and colonization of melanoma and colon carcinoma loci in the lungs and correlates with decreased infiltration of CD4+ and CD8+ T cells.7,8 In contrast, reduced growth of solid melanoma and bladder carcinoma tumours has been observed in Il17−/− mice along with an increased number of interferon-γ+ (IFN-γ+) CD4+ and CD8+ T cells.9 The basis for these contradictory functions associated with IL-17 has not been elucidated. Treatment of mice with tumour-specific Th17 polarized cells has been reported to revert the growth of established solid and lung melanoma, and this is associated with enhanced antigen presentation and activation of tumour-specific CD8+ cytotoxic T cells. However, since this effect depends on IFN-γ, it could be mediated by reprogramming the transferred Th17 cells into Th1 cells.8,10 When Th17 cells are specifically induced at the tumour site with IL-6-transduced pancreatic cancer cells, the enhanced generation of Th17 cells has protective activity against pancreatic tumour growth and improved survival.11 Altogether, the exact functions of Th17 cells and IL-17 in the tumour environment remain to be fully defined.
Inflammatory responses play an important role in different stages of cancer development. Cross-talk between the tumour stroma and immune cells can subvert the protective anti-tumour response into signals that enable tumour growth, angiogenesis and metastasis.12 Myeloid-derived suppressor cells (MDSC) are a heterogeneous population composed of myeloid progenitor cells and immature myeloid cells that strongly suppress T-cell proliferation and are greatly expanded in human and mouse tumours.13 Recent studies have suggested that chronic inflammation promotes tumour progression by down-regulating anti-tumour responses through the induction of MDSC by pro-inflammatory cytokines IL-6 and IL-1β present in the tumour environment14 as well as other inflammatory mediators induced in various cell types by IL-17.15–20 These MDSC can in turn promote the development of regulatory T (Treg) cells in vivo in tumour-bearing mice.21 Moreover, two studies reported an effect of IL-17 in promoting the recruitment of immunosuppressive populations to the tumour microenvironment. The first study showed that the development of tumours was inhibited in IL-17R-deficient mice or after IL-17 neutralization using antibodies, whereas systemic administration of IL-17 promoted tumour growth. This study also demonstrated that IL-17 promotes tumour progression by recruiting MDSC and by reducing CD8+ T-cell infiltration in the tumour microenvironment.22 A second study showed that IL-17 mediates Treg cell infiltration by up-regulating CCL17 and CCL22 chemokines and Treg cell activity by up-regulating CD39 and CD73 expression on these cells.23
In the current study, we examined the effect of endogenous Th17 cells in the development of intradermal B16.F10 melanoma using the Rorγtgfp/gfp mouse model, which has impaired Th17 differentiation. We confirmed that Rorγtgfp/gfp mice have a reduced frequency of Th17 cells in the tumour as well as undetectable levels of IL-17. Importantly, Rorγtgfp/gfp mice show enhanced tumour growth compared with wild-type (WT) mice. Further comparison of different populations in the tumour environment revealed that the enhanced tumour growth did not correlate with a higher frequency of immunosuppressive MDSC and Treg cells; however, retinoic acid-like orphan receptor γt (RORγt) -deficient mice show a significant decrease in the percentage of total CD4+ T cells, and an increase in the percentage of CD8+ T cells.
Our results support the idea that Th17 cells may provide protection against tumour growth by promoting the recruitment of Th1 cells into the tumour.
Materials and methods
Mice
C57BL/6 and Rorγtgfp/gfp mice were purchased from Jackson Laboratory (Bar Harbor, ME) and kept in an animal facility under standard housing guidelines. The animal work was carried out under institutional regulations of Fundacion Ciencia y Vida and the Facultad de Ciencias, Universidad de Chile, and was locally approved by an ethics review committee. Before conducting experiments, the genotype of 21-day-old Rorγtgfp/gfp and Rorγt+/gfp pups was tested with DNA isolated from the tail through conventional PCR, using Platinum PCR Supermix (Invitrogen, Carlsbad, CA) and the following primers (Invitrogen): forward 5′-CCC CCT GCC CAG AAA CAC T-3′; reverse 5′-GGA TGC CCC CAT TCA CTT ACT TCT-3′; mutant reverse 5′-CGG ACA CGC TGA ACT TGT GG-3′. PCR products were 174 bp in WT mice, 241 bp in Rorγtgfp/gfp homozygous mice, and 174 and 241 bp in Rorγtgfp/+ heterozygous mice.
Tumour cells
B16.F10 murine melanoma cells were obtained from the American Type Culture Collection (Manassas, VA). B16.OVA murine melanoma cells were kindly provided by Dr Randolph Noelle (King's College London, UK). Hepa 1-6 cells and MB49 cells were kindly provided by Dr Luis Burzio (Fundacion Ciencia & Vida) and Dr Maria Ines Becker (Biosonda, Santiago, Chile), respectively. A total of 0·24 × 106 cells (B16.F10 and B16.OVA), 0·3 × 106 cells (MB49) and 2 × 106 cells (Hepa 1-6) (> 95% viability assessed by Trypan blue exclusion) were injected in 50 μl PBS in the intradermal layer of the right flank of mice that were previously shaved and anaesthetized with sevofluorane (Abbott Laboratories, Buenos Aires, Argentina). Tumours became visible at day 10 and the size was measured every 2–3 days. Two perpendicular measurements were made with a caliper and the tumour area was estimated as the product of both measurements.
Bone marrow (BM) chimeras
C57BL/6 mice were lethally irradiated (900 cGy) and reconstituted with 20 × 106 total BM cells through tail vein injection. Donor bone marrow cells were obtained from the femur and tibia of C57BL/6 and Rorγtgfp/gfp mice. Irradiated mice were allowed to recover for 5 weeks before inoculation with B16.F10 cells.
Isolation of mononuclear cells from solid tumours
To examine tumour-infiltrating cells, whole tumours were dissected and disaggregated mechanically. Minced tissues were resuspended in 5 ml Hanks' balanced salt solution + 5% fetal calf serum (FCS) and digested in the presence of 1 mg/ml Collagenase D (Roche, Mannheim, Germany) and 25 μg/ml DNase I (Roche) for 30 min at 37° with constant agitation. The cell suspension was filtered with a 70-μm cell strainer (BD Falcon, Franklin Lakes, NJ). Red blood cells were lysed in a hypotonic ammonium chloride solution. Leucocytes were resuspended in 40% Percoll (GE Healthcare, Uppsala, Sweden) and gently layered over 70% Percoll. The gradient was centrifuged at 750 g for 20 min at room temperature. Mononuclear cells were collected from the interphase and were washed and resuspended in RPMI + 10% FCS.
Flow cytometry and antibodies
The following antibodies were purchased from BD Pharmingen (Franklin Lakes, NJ): CD4 (clone L3T4), CD8 (clone 53-6.7), H2-Kb (clone AF6-88.5), H2-Kd (clone SF1-1.1), CD11c, IAb (clone 25-9-17), CD19 (clone 1D3), CTLA-4 (clone UC10-4F10-11), PD-L1 (MIH5) and CD49b (clone DX5). The following antibodies were purchased from eBioscience (San Diego, CA): CD16/32 (clone 93), CD4 (clone GK1.5), CD25 (clone PC61.5), CD11c (clone N418), CD39 (clone 24DMS1), CD45.2 (clone 104), CD62L (clone MEL-14), GARP (clone YGIC86), Gr-1 (clone RB6-8C5), Foxp3 (clone FJK-16s), IFN-γ (XMG1.2) and IL-17 (clone eBio17B7). The following antibodies were purchased from Biolegend (San Diego, CA): CD3 (clone 17A2), NK1.1 (clone PK136), CD49d (clone 9C10), CD11b (clone M1/70), CD45.1 (clone A20) and B220 (clone RA3-6B2).
For intracellular detection of cytokines, cells were stimulated for 4 hr at 37° and 5% CO2 with 0·25 µm PMA (Sigma-Aldrich, St Louis, MO) and 1 µg/ml Ionomycin (Sigma-Aldrich) in the presence of Golgi Plug (BD Biosciences). Cells were surface-stained with anti-CD3 and anti-CD4 antibodies in PBS + 2% FCS for 20 min at 4°. Cells were resuspended in a fixation/permeabilization solution (Cytofix/Cytoperm; BD Pharmingen) and incubated with anti-IFN-γ and anti-IL-17 antibodies for 30 min at 4°. For Foxp3 detection, unstimulated cells were surface stained with antibodies against CD4 and CD25 and permeabilized with Foxp3 Staining Buffer (eBioscience) and incubated with anti-Foxp3 antibody or isotype control for 30 min at 4°. Cells were then washed with permeabilization buffer and resuspended in PBS + 2% FCS for FACS analysis (FACSCanto II; BD Bioscience). In some cases, propidium iodide (Calbiochem, La Jolla, CA) was used to discard dead cells from the analysis. Analysis of FACS data was performed using the flowjo software (Tree Star Inc., Ashland, OR).
Generation of Th17 and Th1-polarized cells
Antigen presenting cells (APC) were purified from spleens of WT and Rorγtgfp/gfp mice. The spleen was mechanically disaggregated and resuspended in 5 ml RPMI + 10% FCS. Tissues were digested in the presence of Collagenase D (1 mg/ml) and DNase I (25 μg/ml) for 45 min at 37° with constant agitation. Cell suspensions were filtered with a 70-μm cell strainer (BD Falcon). Red blood cells were lysed in a hypotonic ammonium chloride solution. The APC were positively selected using anti-CD11c MACS (clone n418; Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer's instructions. The purity of APC in the positive fraction was confirmed by FACS analysis, which contained CD11c+ dendritic cells (70%) and CD19+ B220+ B cells (30%).
CD4+ T cells were purified from spleens of WT, Rorγtgfp/gfp or OT-II mice. The spleen was perfused with RPMI + 10% FCS. Red blood cells were lysed in a hypotonic ammonium chloride solution. CD4+ T cells were positively selected using anti-CD4 MACS (clone L3T4; Miltenyi Biotec) following the manufacturer's instructions. The purity of CD4+ T cells contained in the positive fraction was confirmed by FACS analysis (> 80%). CD4+ T cells and APC were co-cultured in a 5 : 1 ratio (0·12 × 106 CD4+ T cells/well) in a 96-well round bottom microplate (Orange Scientific, Braine-l'Alleud, Belgium). T cells were stimulated with 1 μg/ml α-CD3 antibody (eBioscience) in the presence or absence of Th17 polarizing conditions (Th17 PC): 20 ng/ml IL-6 (R&D Systems, Minneapolis, MN), 5 ng/ml transforming growth factor-β, 10 ng/ml IL-1β (eBioscience) and 5 μg/ml α-IFN-γ (Biolegend); or Th1 polarizing conditions (Th1 PC): 10 ng/ml IL-2 and 10 ng/ml IL-12 (R&D Systems). After 7 days of culture, intracellular cytokines were analysed as described in the Flow Cytometry section.
Cytokine secretion measurement
To determine cytokine secretion by splenocytes and tumour-infiltrating lymphocytes, cells were activated for 4 hr with 0·25 µm PMA (Sigma Aldrich) and 1 µg/ml ionomycin (Sigma Aldrich). Following activation the supernatants were harvested and analysed using the mouse Th1/Th2/Th17 CBA Kit (BD Biosciences), following the manufacturer's instructions.
Statistical analysis
Data are presented as mean ± SEM. Differences between WT and Rorγtgfp/gfp mice were determined with a Student's t-test. Where indicated, differences were analysed using one-way analysis of variance paired with Bonferroni post-tests. Statistical analysis and graphs were obtained with graphpad prism (GraphPad Software Inc., La Jolla, CA).
Results
Rorγtgfp/gfp mice present enhanced tumour growth and decreased percentage of tumour-infiltrating Th17 cells
To evaluate the effect of Th17 cells in tumour progression, we used a mouse model with impaired Th17 differentiation. Homozygous Rorγtgfp/gfp mice do not express the RORγt transcription factor required for the polarization of naive CD4+ T cells into IL-17-producing CD4+ T cells.24 To address the effect of Th17 cells in tumour development, we challenged WT and Rorγtgfp/gfp with a solid melanoma tumour by an intradermal injection of B16.F10 cells. After 3 weeks of monitoring tumour growth, results showed that tumour growth was significantly enhanced in Th17-deficient Rorγtgfp/gfp mice (Fig. 1a). Similar results were obtained when we used MB-49 and Hepa 1-6 cells as other models of solid tumours (see Supplementary material, Fig. S1). To confirm that the reduction in the percentage of Th17 cells was the cause of the enhanced tumour growth, we compared the rate of tumour growth in Rorγtgfp/gfp mice that were adoptively transferred with Th17 cells generated in vitro versus untreated Rorγtgfp/gfp mice and WT mice. For this experiment, mice were challenged with ovalbumin (OVA) -expressing B16.F10 cells and then injected with 1 × 106 OVA-specific Th17 or Th1 cells generated from OT-II CD4+ T cells. Our results show that tumour growth was significantly reduced in Rorγtgfp/gfp mice that received Th17 cells or Th1 cells compared with control Rorγtgfp/gfp mice; however, the transfer of Th17 cells had a more dramatic effect on tumour growth than the transfer of Th1 cells (Fig. 1b). Moreover, we observed that upon transfer to Rorγtgfp/gfp or C57BL/6 mice, almost 50% of in vitro-generated Th17 cells produce IFN-γ within the tumour (see Supplementary material, Fig. S2), a fact that suggests that the effect of transferred Th17 cells on tumour growth may be at least partially mediated by IFN-γ production. This result confirms that enhanced tumour growth observed in Rorγtgfp/gfp mice is the result of a reduced frequency of Th17 cells, suggesting that these cells may have important anti-tumour functions.
Figure 1.

Reduced frequency of endogenous T helper type 17 (Th17) cells and enhanced tumour growth in Rorγtgfp/gfp mice. Tumour growth curves of wild-type (WT) and Rorγtgfp/gfp mice injected with B16.F10 cells (n = 5) (a). Tumour growth curves of wild-type (WT) and Rorγtgfp/gfp mice injected with B16.F10 cells that received PBS, 1 × 106 in vitro-generated Th17 or Th1 cells (n = 4) (b). Frequency of Th17 cells was monitored in tumour-free and tumour-bearing heterozygous Rorγtgfp/+ mice by analysing Rorγt-GFP+ cells in spleen, peripheral lymph node (PLN) or tumour-draining lymph nodes (TdLN) by FACS (c). Cells from tumour and lamina propria were isolated from tumour-bearing WT and Rorγtgfp/gfp mice, activated for 4 hr with PMA plus ionomycin in the presence of Brefeldin A and then stained to detect intracellular cytokines as described in the Materials and methods section. Dot plots show interleukin-17+ (IL-17+) cells in a CD3+ CD4+ gate (d). Percentage of tumour-infiltrating IL-17+ cells in a CD3+ CD4+ gate from WT and Rorγtgfp/gfp mice (e). Splenocytes and tumour-infiltrating lymphocytes from WT and Rorγtgfp/gfp mice were stimulated for 4 hr with PMA plus ionomycin and cytokine concentrations in the supernatant were determined using the Th1/Th2/Th17 CBA kit. (f). Values expressed represent the mean ± SEM of at least three independent experiments *P < 0·05, **P < 0·01, ***P < 0·0001.
Since it has been described that Th17 cells are expanded in tumour-bearing mice and humans and are enriched in tumour tissues compared with blood and lymphoid organs,1–4 we confirmed the expansion of Th17 cells upon a challenge with B16.F10 melanoma cells in Rorγtgfp/+ heterozygous mice. Indeed, we found an increase in the CD4+ Rorγt-GFP+ population in tumour-bearing mice compared with mice without a tumour (Fig. 1c). Next, we compared the percentage of Th17 cells found in the spleen, tumour and lamina propria of Rorγtgfp/gfp mice and WT mice. Intracellular staining of IL-17 in splenocytes from WT and Rorγtgfp/gfp mice was not significantly detected as compared with isotype control staining (data not shown). Notably, a significant decrease in the percentage of tumour-infiltrating Th17 cells was found in Rorγtgfp/gfp mice compared with WT mice (Fig. 1d–e). The percentage of Th17 cells in the lamina propria, a site where these cells are particularly enriched in WT mice, was dramatically diminished in Rorγtgfp/gfp mice (Fig. 1d). Accordingly, upon stimulation with PMA plus ionomycin, the levels of IL-17 produced by tumour-infiltrating lymphocytes in Rorγtgfp/gfp mice were consistently below detection levels (Fig. 1f), indicating that both tumour-infiltrating Th17 cells as well as IL-17 production are compromised in Rorγtgfp/gfp mice. With the exception of an increase in the level of IL-6 in Rorγtgfp/gfp tumour tissue, no other changes in cytokine profile were observed between WT and Rorγtgfp/gfp mice (Fig. 1f). To confirm a defect in the differentiation of Th17 cells in Rorγtgfp/gfp mice we also tested whether we could induce the in vitro Th17 differentiation of CD4+ T cells isolated from Rorγtgfp/gfp mice. Our results indicate that the percentage of naive CD4+ T cells that differentiate into IL-17-producing cells in Rorγtgfp/gfp mice was dramatically reduced compared with those from WT mice whereas Th1 differentiation was not affected (See Supplementary material, Fig. S3), confirming that CD4+ T cells from Rorγtgfp/gfp mice cannot differentiate to Th17 cells but can differentiate into other Th cell subsets.
In addition to its expression in Th17 cells, the RORγt transcription factor is expressed during fetal life and is required for the generation of lymphoid tissue inducer cells. Therefore, lymphoid tissue inducer cells as well as lymph nodes and Peyer's patches are absent in Rorγtgfp/gfp mice.25 To demonstrate that the enhanced tumour growth observed in Rorγtgfp/gfp mice is a result of the lack of Th17 cells and not of the lack of secondary lymphoid organs, we performed bone marrow transplants from WT or Rorγtgfp/gfp donors to WT mice and tumour progression was evaluated after B16.F10 injection. In agreement with our previous findings that suggest that Th17 cells are essential in mediating anti-tumour responses, mice that received bone marrow from Rorγtgfp/gfp donors showed enhanced tumour growth compared with the group that received bone marrow from WT donors (Fig. 2a). Maximum tumour growth 3 weeks after injection observed in mice transplanted with Rorγtgfp/gfp bone marrow (82·2 ± 9·9 mm2) was less severe than in Rorγtgfp/gfp mice (162·3 ± 30·7 mm2).
Figure 2.
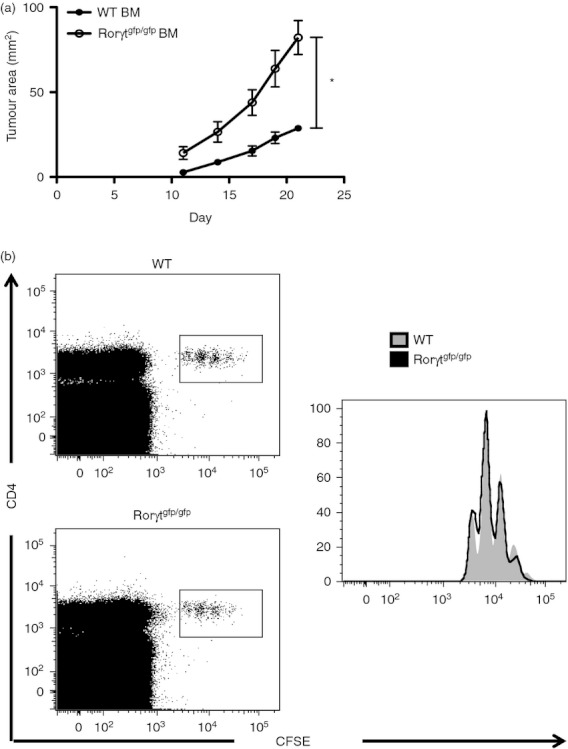
T helper type 17 (Th17) cell deficiency contributes to the enhanced tumour growth in Rorγtgfp/gfp mice. Tumour growth curves of transplanted wild-type (WT) mice that received bone marrow from WT or Rorγtgfp/gfp mice that were injected with B16.F10 cells (n = 5) (a). Wild-type mice were transferred with CFSE-stained OT-II CD4+ T cells and 24 hr later, were challenged intraperitoneally with ovalbumin (OVA; 1 mg) + LPS (50 μg). Four days later, cell proliferation was analysed in splenocytes as CFSE dilution (b). (*P < 0·05)
Next, we investigated the immunocompetence of Rorγtgfp/gfp mice by measuring the ability to mount a response against a foreign antigen. For this, WT and Rorγtgfp/gfp mice were transferred with CFSE-labelled OT-II CD4+ T cells and immunized with OVA protein and LPS. Four days later we evaluated the proliferation of transferred cells in the spleen. OT-II CD4+ T cells were activated to the same extent in WT and Rorγtgfp/gfp mice (Fig. 2b), indicating that immune responsiveness is not compromised in the latter. These data support that Rorγtgfp/gfp mice are a reliable model for studying the role of Th17 cells in tumour progression.
Th17 cells do not affect the recruitment or the immunosuppressive phenotype of myeloid-derived suppressor cells and regulatory T cells
Tumour evasion of the immune response is in part mediated through the activation and recruitment of immunosuppressive populations such MDSC and Treg cells to the tumour microenvironment. Inflammatory mediators produced by Th17 cells could participate in the mobilization of immunosuppressive populations present in the tumour.17 To study this possibility, WT and Rorγtgfp/gfp mice were injected with B16.F10 melanoma cells and analysed for the presence of MDSC and Treg cells in spleen and tumour. Our results show that in Rorγtgfp/gfp mice, the proportion of MDSC and Treg cells is not altered in the spleen or tumour compared with WT mice (Fig. 3a,b). To further characterize changes in these populations, we analysed the expression of CD49d, which distinguishes a subpopulation of MDSC that strongly suppresses T-cell proliferation.26 CD49dhigh MDSC were preferentially recruited into the tumour-draining lymph node and tumour, whereas an equal ratio of CD49dint and CD49dhigh MDSC was found in the spleen of tumour-bearing WT mice (see Supplementary material, Fig. S4A). The distribution of CD49dhigh MDSC in Rorγtgfp/gfp mice was similar compared with that in WT mice (Fig. 3c) confirming that MDSC are not preferentially recruited and do not have a higher suppressive activity in Rorγtgfp/gfp mice. Th17 cells are able to recruit neutrophils to inflamed sites through the secretion of IL-17,17 therefore we compared the presence of Gr-1+ CD11b− neutrophils in spleen and tumour. A moderate decrease in the neutrophil population was observed in the spleen of Rorγtgfp/gfp mice compared with WT mice; however, this difference was not observed in the tumour (Fig. 3d), therefore the differences observed in tumour growth are not the result of changes in neutrophil recruitment in the tumour.
Figure 3.
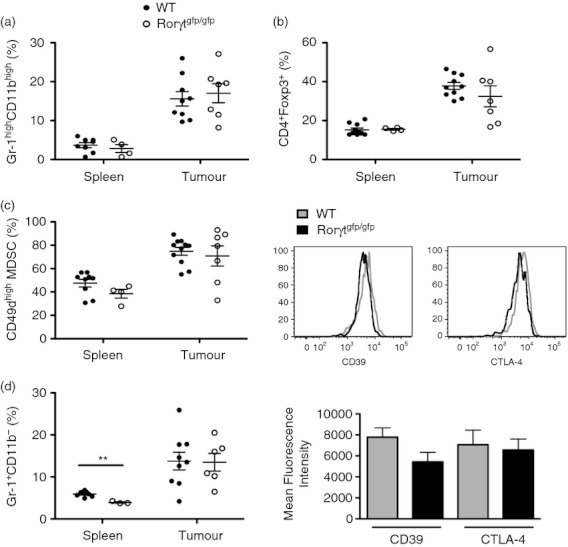
Myeloid-derived suppressor cells (MDSC) and regulatory T (Treg) cells from Rorγtgfp/gfp mice do not present an altered recruitment to the tumour or phenotype. Splenocytes and tumour-infiltrating cells from wild-type (WT) and Rorγtgfp/gfp mice were isolated and analysed by FACS. Frequency of Gr-1highCD11bhigh MDSC in a PI− gate (a). Foxp3+ cells in a CD4+ gate (b). Percentage of CD49dhigh cells in a Gr-1high CD11bhigh gate (c). Frequency of Gr-1high CD11b− neutrophils in a PI− gate (d). CD39 and CTLA-4 mean fluorescence intensity in a CD4+ Foxp3+ gate in cells isolated from WT and Rorγtgfp/gfp tumours (e). Bars show mean ± SEM.
Next, we analysed the suppressive potential of Treg cells and we found that the expression of CD39 and CTLA-4 was up-regulated in tumour-infiltrating Treg cells compared with spleen Treg cells, whereas GARP and PD-L1 had similar expression levels in spleen and tumour Treg cells (see Supplementary material, Fig. S4B). As shown in Figure 3(e), both CD39 and CTLA-4 had similar expression levels in Treg cells from WT and Rorγtgfp/gfp mice.
Our results indicate that mice with decreased endogenous Th17 cells are more vulnerable against tumour development but they present no differences in the infiltration and immunosuppressive function of MDSC or Treg cells in the tumours.
Effect of Th17 cells in the recruitment of effector populations to the tumour
To understand the mechanisms underlying the enhanced tumour growth observed in Rorγtgfp/gfp mice, we compared the presence of effector populations such as natural killer cells, CD8+ T cells and CD4+ T cells in the tumour of Rorγtgfp/gfp and WT mice. Although we observed no differences in the percentage of natural killer cells in WT versus Rorγtgfp/gfp mice (Fig. 4a), we observed a significant increase in the percentage of tumour-infiltrating IFN-γ-producing CD8+ T cells in Rorγtgfp/gfp mice compared with WT mice (Fig. 4b,c).
Figure 4.
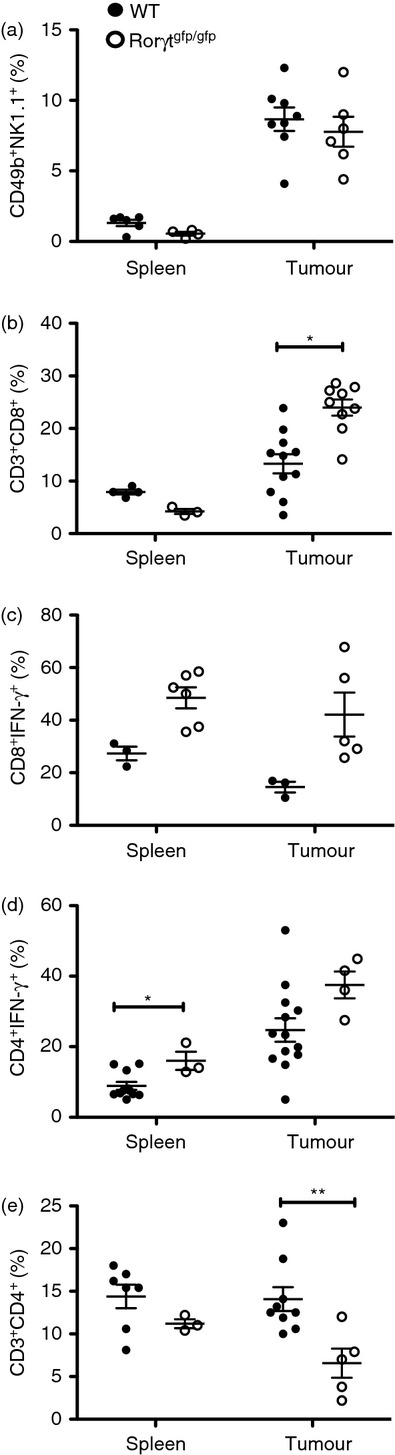
Altered recruitment of CD4+ and CD8+ T-cell effector populations in Rorγtgfp/gfp mice. Cells from spleen and tumour of wild-type (WT) and Rorγtgfp/gfp mice were isolated and cell populations were analysed by FACS. Percentage of CD49b+ NK1.1+ natural killer cells in a PI− gate (a).Total and interferon-γ+ (IFN-γ+) CD8+ T cells (b and c). Total and IFN-γ+ CD4+ T cells (d and e). Bars show mean ± SEM. **P < 0·001.
We also observed a higher proportion of Th1 cells in the CD4+ population in the spleen of Rorγtgfp/gfp mice compared with WT mice (Fig. 4e); however, we observed no difference in the levels of secreted IFN-γ either in spleen or tumour between WT and Rorγtgfp/gfp mice (Fig. 1c). Importantly, our data also showed a significant reduction of total CD4+ T cells in the tumour while maintaining similar levels in the spleen, compared with WT mice (Fig. 4d), suggesting that in Rorγtgfp/gfp mice there is an overall decrease in Th1 cells in the tumour. These results suggest that enhanced tumour growth observed in Rorγtgfp/gfp mice could be the outcome, at least in part, of a decrease of Th1 effector cells in the tumour, despite the observed increase in CD8+ T cells.
To confirm that tumour progression is mediated by the decline in the percentage of effector rather than by the accumulation of immunosuppressive populations within the tumour, we studied the percentage of MDSC, Treg cells and Th1 cells in tumours of different size from WT mice. We observed a reduction in Th1 cells but no evident accumulation of MDSC or Treg cells in advanced tumours (Fig. 5). This suggests that the degree of tumour severity is not correlated with the expansion of immunosuppressive populations but rather with a decline in effector cells.
Figure 5.
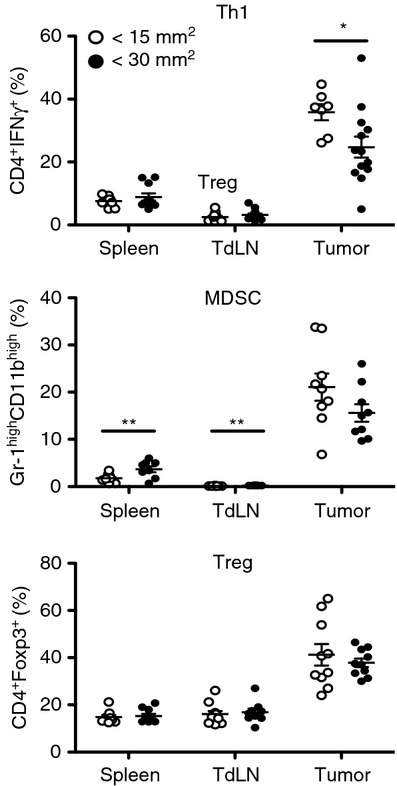
Infiltration of effector T helper type 1 (Th1) cells takes place during initial tumour development and declines with tumour growth. Wild-type mice injected with B16.F10 melanoma cells were divided in two groups that were euthanized at two different points of tumour development, before tumour size had reached 15 mm2 and when the tumour had grown over 30 mm2. Th1, myeloid-derived suppressor cell (MDSC) and regulatory T (Treg) cell frequencies in spleen, tumour-draining lymph node (TdLN) and tumour were analysed by FACS. Data are presented as mean ± SEM.*P < 0·05.
Discussion
Although Th17 cells have been found in higher frequency in patients with cancer and are particularly enriched in human and mouse tumours, the function of Th17 cells in tumour immunity remains controversial, because they have been described as presenting both tumour-promoting and anti-tumour properties. The main pro-tumour role of Th17 cells is based on their production of IL-17 and its pro-angiogenic effect over surrounding vascular endothelial cells and fibroblasts. It has been demonstrated that by acting on stromal cells and fibroblasts, IL-17 induces a wide range of angiogenic mediators,27,28 including vascular endothelial growth factor, which promotes tumour angiogenesis.29 Additional reports have analysed the role of Th17 function by deleting IL-17 or the IL-17 receptor in mice, while overlooking other cytokines and factors secreted by Th17 cells.5–8 The group of Yu has reported that IL-17-deficient mice present a reduction in tumour growth and progression. They propose that IL-17 could contribute to tumour growth by stimulation of tumour cell proliferation through the induction of IL-6 in the tumour environment and activation of STAT3 in tumour cells.9 In the same line of evidence, using IL-17R-deficient mice, another group has reported that IL-17 promotes the tumour growth through the recruitment of MDSC to the tumour microenvironment.22
Although IL-17 is the signature cytokine produced by Th17 cells, the production of this cytokine is not the only function of Th17 cells, so the biological activity of Th17 cells should not be equated to biological activity of IL-17. In our model, RORγt-deficient mice lack Th17 cells and all the biological functions related to these cells. This includes not only IL-17 production but other cytokines and chemokines produced by Th17 cells capable of altering the tumour microenvironment, such as CCL20, IFN-γ and granulocyte–macrophage colony-stimulating factor, the latter being recently proposed as a novel Th17s ‘weapon’.30–32 This important difference in the models used could explain why in the absence of Th17 cells we observe an increase in tumour burden whereas other studies using IL-17 and IL-17R-deficient mice have reported that IL-17 promotes tumour growth.
In this work, a new approach was taken to study the function of endogenous Th17 cells in tumour development. Here we analyse how the endogenous development of Th17 cells influences tumour growth in a model of intradermal melanoma. In agreement with previous evidence from other groups,8,10 our results show that Th17 cells confer protection against tumour development. Moreover, we did not find evidence to associate Th17-mediated inflammation with recruitment of immunosuppressive populations in the tumour. Instead, we show that Th17 cells promote antitumour responses by enhancing the recruitment of CD4+ T cells to the tumour.
Given that Th17 cells represent a minor percentage of immune cells in the tumour, it is notable that a reduction in this population can produce such profound effects. Despite expressing phenotypic markers of terminally differentiated effector cells, it has been demonstrated that human and mouse primary Th17 cells also present stem cell-like properties. For instance, it has been described that Th17 cells have a high proliferation potential, self renewal, multipotency and resistance to apoptosis compared with Th1 cells.33,34 Interestingly, Th17 cells are significantly more effective than Th1 cells at mediating tumour rejection; however, this strong effect is dependent on the acquisition of Th1-like features, including IFN-γ production.10,34 This strongly suggests that Th17 cells are polyfunctional and may represent a critical component for a long-term anti-tumour response as a persistent source of Th1-like effector cells.
In our model, adoptively transferred Th17 cells presented features of multipotency because upon transfer into Rorγtgfp/gfp mice, a percentage of these cells are converted into IFN-γ-producing cells within the tumour. This observation confirms that in vitro-generated Th17 cells present stem cell-like features and may require conversion to Th1 cells to exert their anti-tumour function in our model as well.
Immunosuppression within the tumour is the major obstacle for the antitumour response and represents a critical barrier for the development of successful cancer immunotherapy. Two central components that contribute to immunosuppression in the tumour are MDSC and Treg cells. We observed that MDSC and Treg cells are dramatically expanded in tumour compared with spleen and tumour-draining lymph nodes (Fig. 5). It was recently reported that IL-17 is required for the infiltration and tumour-promoting activity of MDSC in a subcutaneous lymphoma model.22 In contrast, we provide evidence that a decrease in endogenous Th17 cells does not affect the percentages of MDSC and Treg cell populations in intradermal melanoma (Fig. 3), indicating that the enhanced tumour growth rate observed in Rorγtgfp/gfp mice is not associated with an increased percentage of these populations in the tumour. MDSC-mediated suppressive mechanisms have been extensively described but until recently, no surface markers had been associated with the suppressive properties of MDSC. Recently, CD49d was reported to be expressed exclusively on monocytic MDSC, which are more efficient at suppressing T-cell proliferation compared with CD49d− MDSC.26 Analysis of the expression of CD49d in WT mice showed that CD49dhigh MDSC are predominantly accumulated in the tumour-draining lymph node and tumour (see Supplementary material, Fig. S4A). However, we observed no change in the proportion of the CD49high subpopulation in the spleen or within tumours of Rorγtgfp/gfp mice compared with WT mice, suggesting that the suppressive potential of MDSC present in the tumour of Rorγtgfp/gfp mice is similar compared with WT mice. In contrast with MDSC, several surface proteins have been described to mediate immunosuppressive mechanisms by Treg. CD39 and CTLA-4 suppress T-cell activation,35 PD-L1 expression induces T-cell death,36 while GARP participates in the release of surface-bound transforming growth factor-β37. Our analysis showed an increase in the expression levels of CD39 and CTLA-4 in Treg cells isolated from the tumour compared with spleen Treg cells (see Supplementary material, Fig. S4B), suggesting that these mechanisms play an important role in the regulation of T-cell priming and expansion in the tumour. However, we found no difference in the expression of CD39 or CTLA-4 in Treg cells from Rorγtgfp/gfp mice, indicating that the degree of immunosuppression mediated by Treg cells in these mice is comparable with that seen in WT mice. Altogether, we show that a decrease of Th17 cells does not influence the recruitment or phenotype of immunosuppressive populations in melanoma tumours.
By comparing immunosuppressive populations (MDSC and Treg cells) and effector Th1 cells during initial and advanced stages of tumour development in WT mice, we observed that immunosuppressive and effector cells infiltrate the tumour from early development. Whereas the percentage of MDSC and Treg cells in the tumour is sustained, there is a significant drop in the percentage of Th1 cells (Fig. 5). This suggested that the tumour severity is not correlated with the expansion of immunosuppressive populations but rather with a decline in effector cells. The percentage of Th1 cells was moderately increased in the spleen and tumour of Rorγtgfp/gfp mice compared with WT mice (Fig. 4e); however, tumours of Rorγtgfp/gfp mice were significantly less infiltrated by total CD4+ T cells compared with WT mice. This finding is consistent with decreased infiltration of CD4+ T cells in tumour-burdened lungs of IL-17 knockout mice8 and increased CD4+ T-cell infiltration in a model that enhances Th17 differentiation in the tumour microenvironment.11
Our results support the concept that Th17 cells promote signalling in the tumour environment that specifically favours the trafficking of effector CD4+ T cells. In human ovarian cancer ascites, the levels of IL-17 are positively correlated with that of CXCL9 and CXCL10, two chemokines that are associated with a Th1 response.2 Additionally, the group of Dutton demonstrated that OVA-specific Tc17 cells, IL-17-producing CD8+ T cells, adoptively transferred into tumour-bearing mice control tumour growth in a model of melanoma. They observed that these Tc17 cells are a source for various cytokines and chemokines that elicit further chemokines involved in the attraction of inflammatory cells to the tumour, including CXCR3+ effector cells and neutrophils.38 Additional evidence of the recruitment of Th1 effector cells by Th17 cells came from a study in mice showing the protective role of Th17 cells in response to vaccination during Mycobacterium tuberculosis challenge. The authors proposed that vaccination induces IL-17-producing CD4+ T cells that populate the lung and, after challenge, trigger the production of chemokines such as CXCL9, CXCL10 and CXCL11 that recruit IFN-γ-producing CD4+ T cells, which ultimately restrict bacterial growth.39 It remains to be studied whether in our model, Th17 cells in the tumour can influence chemokine expression in melanoma tumour and stromal cells as a mechanism to coordinate the recruitment of CXCR3+ effector Th1 cells to the tumour thus favouring anti-tumour responses.
It has been proposed that a shift in the ratio of the Th17/Treg numbers leads to reduced tumour growth and improved survival in a model of pancreatic cancer.11 Although Rorγtgfp/gfp mice have a higher proportion of Th1 cells, it is possible that the absolute number of cells in the tumour is reduced compared with that in WT mice, because of a significant decrease in total CD4+ T cells. It is possible that changes in the ratio of effector Th1 and Th17 cells versus immunosuppressive populations also have a contributory effect on tumour progression.
A surprising difference observed in the tumour environment of Rorγtgfp/gfp mice was a higher frequency of CD8+ T cells. If such an increase were accompanied by a stronger tumour-specific cytotoxic response, the expected outcome would be a reduction in tumour size. As this is not the case, the enhanced tumour growth that is seen in these mice suggests that a better assessment of cytotoxic functions of CD8+ T cells is needed to fully comprehend how impaired Th17 development affects the anti-tumour response. It is important to note that tumour rejection following adoptive transfer of tumour-specific CD8+ T cells can be greatly enhanced when co-transferred with Th17 cells,33 therefore it is possible that disruption of the collaborative response between Th17 and CD8+ T cells also contributes to enhanced tumour growth in Rorγtgfp/gfp mice.
In conclusion, our data show that Th17 cells do not affect the recruitment of immunosuppressive populations such as Treg cells or MDSC to the tumour, but favour the recruitment of effector Th1 cells. This evidence suggests that Th17 cells orchestrate the recruitment of Th1 effector cells to the tumour, and therefore strongly argues in favour of a protective role of Th17 cells during anti-tumour responses.
Acknowledgments
This work was supported by Grants 3100077, 1100557 and 1100448 from Fondo Nacional de Desarrollo Cientifico y Tecnologico. G.T. and P.R. hold fellowships from Comision Nacional de Investigacion Cientifica y Tecnologica.
We thank Dr Randolph Noelle for providing B16.OVA cells and helpful guidance.
Glossary
- IFN
interferon
- IL-17
interleukin-17
- MDSC
myeloid-derived suppressor cell
- Rorγt
retinoic acid-like orphan receptor γt
- Th
T helper
- Treg
regulatory T
- WT
wild-type
Disclosures
The authors declare no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Rorγtgfp/gfp mice present enhanced tumour growth.
Figure S2. T helper type 17 cells produce interferon-γ upon adoptive transfer to wild-type or Rorγt gfp/gfp mice.
Figure S3. Impaired T helper type 17 polarization in Rorγtgfp/gfp mice.
Figure S4. Myeloid-derived suppressor cell and regulatory T cell surface markers related to a suppressive phenotype are up-regulated in the tumour microenvironment.
References
- 1.Kryczek I, Wei S, Zou L, Altuwaijri S, Szeliga W, Kolls J, Chang A, Zou W. Cutting edge: Th17 and regulatory T cell dynamics and the regulation by IL-2 in the tumor microenvironment. J Immunol. 2007;178:6730–3. doi: 10.4049/jimmunol.178.11.6730. [DOI] [PubMed] [Google Scholar]
- 2.Kryczek I, Banerjee M, Cheng P, et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood. 2009;114:1141–9. doi: 10.1182/blood-2009-03-208249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miyahara Y, Odunsi K, Chen W, Peng G, Matsuzaki J, Wang RF. Generation and regulation of human CD4+ IL-17-producing T cells in ovarian cancer. Proc Natl Acad Sci USA. 2008;105:15505–10. doi: 10.1073/pnas.0710686105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, Peng G. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol. 2010;184:1630–41. doi: 10.4049/jimmunol.0902813. [DOI] [PubMed] [Google Scholar]
- 5.Benchetrit F. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood. 2002;99:2114–21. doi: 10.1182/blood.v99.6.2114. [DOI] [PubMed] [Google Scholar]
- 6.Numasaki M, Fukushi J, Ono M, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101:2620–7. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
- 7.Kryczek I, Wei S, Szeliga W, Vatan L, Zou W. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood. 2009;114:357–9. doi: 10.1182/blood-2008-09-177360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin-Orozco N, Muranski P, Chung Y, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787–98. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206:1457–64. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muranski P, Boni A, Antony PA, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–73. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gnerlich JL, Mitchem JB, Weir JS, et al. Induction of Th17 cells in the tumor microenvironment improves survival in a murine model of pancreatic cancer. J Immunol. 2010;185:4063–71. doi: 10.4049/jimmunol.0902609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176:284–90. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 15.Cheng P, Corzo CA, Luetteke N, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–49. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghannam S, Pene J, Torcy-Moquet G, Jorgensen C, Yssel H. Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J Immunol. 2010;185:302–12. doi: 10.4049/jimmunol.0902007. [DOI] [PubMed] [Google Scholar]
- 17.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–67. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67:4507–13. doi: 10.1158/0008-5472.CAN-06-4174. [DOI] [PubMed] [Google Scholar]
- 19.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–75. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 21.Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+ CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 22.He D, Li H, Yusuf N, Elmets CA, Li J, Mountz JD, Xu H. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J Immunol. 2010;184:2281–8. doi: 10.4049/jimmunol.0902574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Z, Zhang B, Li D, Lv M, Huang C, Shen GX, Huang B. Mast cells mobilize myeloid-derived suppressor cells and Treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PLoS ONE. 2010;5:e8922. doi: 10.1371/journal.pone.0008922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 25.Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORγ(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 26.Haile LA, Gamrekelashvili J, Manns MP, Korangy F, Greten TF. CD49d is a new marker for distinct myeloid-derived suppressor cell subpopulations in mice. J Immunol. 2010;185:203–10. doi: 10.4049/jimmunol.0903573. [DOI] [PubMed] [Google Scholar]
- 27.Numasaki M, Lotze MT, Sasaki H. Interleukin-17 augments tumor necrosis factor-α-induced elaboration of proangiogenic factors from fibroblasts. Immunol Lett. 2004;93:39–43. doi: 10.1016/j.imlet.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi H, Numasaki M, Lotze MT, Sasaki H. Interleukin-17 enhances bFGF-, HGF- and VEGF-induced growth of vascular endothelial cells. Immunol Lett. 2005;98:189–93. doi: 10.1016/j.imlet.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 29.Honorati MC, Neri S, Cattini L, Facchini A. Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthritis Cartilage. 2006;14:345–52. doi: 10.1016/j.joca.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–7. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 31.El-Behi M, Ciric B, Dai H, et al. The encephalitogenicity of TH17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–75. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGeachy MJ. GM-CSF: the secret weapon in the TH17 arsenal. Nat Immunol. 2011;12:521–2. doi: 10.1038/ni.2044. [DOI] [PubMed] [Google Scholar]
- 33.Kryczek I, Zhao E, Liu Y, et al. Human TH17 cells are long-lived effector memory cells. Sci Transl Med. 2011;3:104ra0. doi: 10.1126/scitranslmed.3002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muranski P, Borman ZA, Kerkar SP, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. 2011;35:972–85. doi: 10.1016/j.immuni.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–32. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Q, Munger ME, Highfill SL, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010;116:2484–93. doi: 10.1182/blood-2010-03-275446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Battaglia M, Roncarolo MG. The Tregs' world according to GARP. Eur J Immunol. 2009;39:3296–300. doi: 10.1002/eji.200940117. [DOI] [PubMed] [Google Scholar]
- 38.Garcia-Hernandez Mde L, Hamada H, Reome JB, Misra SK, Tighe MP, Dutton RW. Adoptive transfer of tumor-specific Tc17 effector T cells controls the growth of B16 melanoma in mice. J Immunol. 2010;184:4215–27. doi: 10.4049/jimmunol.0902995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khader SA, Bell GK, Pearl JE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–77. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.