

Abstract
The success of targeted therapies for cancer is undisputed; strong preclinical evidence has resulted in the approval of several new agents for cancer treatment. The type I insulin-like growth factor receptor (IGF1R) appeared to be one of these promising new targets. Substantial population and preclinical data have all pointed toward this pathway as an important regulator of tumor cell biology. Although early results from clinical trials that targeted the IGF1R showed some evidence of response, larger randomized phase III trials have not shown clear clinical benefit of targeting this pathway in combination with conventional strategies. These disappointing results have resulted in the discontinuation of several anti-IGF1R programs. However, the conduct of these trials has brought to the forefront several important factors that need to be considered in the conduct of future clinical trials. The need to develop biomarkers, a clearer understanding of insulin receptor function, and defining rational combination regimens all require further consideration. In this commentary, the current state of IGF1R inhibitors in cancer therapy is reviewed.
In 2008, Daniel Karp presented data from a phase II trial at the annual meeting of the American Society of Clinical Oncology showing that inhibition of the type I IGF receptor (IGF1R) with a monoclonal antibody (figitumumab) statistically significantly increased the response rate to carboplatin and paclitaxel in small cell lung cancer (1). This exciting result showed a near doubling of the response rate and prolongation of disease-free survival. Particularly striking was the response rate of nearly 80% in squamous cell lung cancer. These findings showed the potential for a targeted therapy in the management of a subset of lung cancer. Based on these findings and substantial preclinical data, numerous anti-IGF1R inhibitors were developed (Table 1).
Table 1.
Anti-insulin-like growth factor-1 receptor (IGF1R) drugs
| Class/agent | Company | Stage of testing |
| Tyrosine kinase inhibitors | ||
| BMS-754807 | Bristol-Myers Squibb | Phase I/II |
| Insm-18 (NDGA) | Insmed | Phase I/II |
| XL-228 | Exelixis | Preclinical |
| OSI-906 (linsitnib) | OSI Pharmaceuticals | Phase I/II |
| GSK 1904529A | Glaxo SmithKline | Preclinical |
| ABDP | AstraZeneca | Preclinical |
| A-928605 | Abbott | Preclinical |
| AXL1717 (PPP) | Alexar | Phase I |
| KW-2450 | Kyowa Kirin | Phase I/II |
| Monoclonal antibodies | ||
| MK 0646 (dalotuzumab) | Merck | Phase III |
| AMG 479 (ganitumumab) | Amgen | Phase III |
| A12 (cixutumumab) | ImClone | Phase III |
| CP 751,871 (figitumumab) | Pfizer | Discontinued |
| AVE1642 | sanofi-aventis | Discontinued |
| Sch717454 (robatumumab) | Schering | Discontinued (Merck) |
| R 1507 | Roche | Discontinued |
| BIIB022 | Biogen Idec | Phase I |
| h10H5 | Genentech | Preclinical |
| Neutralizing antibody to IGF-I and IGF-II | ||
| MEDI-573 | MedImmune | Phase II |
| BI836845 | Boehringer Ingleheim | Phase I |
On December 28, 2009, investigators working with figitumumab received a letter from the drug’s sponsor (Pfizer) stating that the phase III study was being closed “because it has met its predefined boundary for early termination indicating that the addition of figitumumab to paclitaxel plus carboplatin would be unlikely to meet its primary endpoint compared to paclitaxel plus carboplatin alone.” This inability to reproduce the phase II study led to the discontinuation of the entire figitumumab program. Disappointing results were also presented for the combination of Amgen’s monoclonal antibody (ganitumab) and hormonal therapies in the second line treatment of breast cancer. This trial showed no benefit, and a trend toward harm, when ganitumab was combined with either exemestane or fulvestrant (2). Recently published results showed that the Roche IGF1R antibody combined with erlotinib in non-small cell lung cancer provided no benefit over erlotinib alone (3). These negative clinical trials resulted in the discontinuation of many other programs targeted toward this receptor. In a few months, the IGF1R went from the new kid on the block to a has-been.
So what happened? The rationale for targeting IGF signaling as a cancer therapy has been suggested by several observations. IGF-I is produced in the liver in response to pituitary growth hormone release during puberty. Systemic levels of IGF-I are responsible for linear growth of the skeleton and height. Height has been linked to cancer risk (4,5). Early reports showed that higher levels of IGF-I were linked to a higher risk of breast and prostate cancer (6,7). At the opposite end, some humans have very low serum IGF-I levels because they cannot respond to growth hormone due to mutations in the hepatic growth hormone receptor. These populations do not appear to be at risk for developing cancer (8,9). These observations suggest a testable hypothesis; IGF signaling regulates normal cell growth; factors that regulate normal growth might also regulate cancer growth. Certainly, targeting of estrogen receptor α (ER) follows this paradigm, and the IGF system has many analogies to ER.
Indeed, this hypothesis was tested over 60 years ago. Before small molecule inhibitors of ER function were developed, surgical removal of the ovaries, adrenals, and pituitary was performed for advanced breast cancer. In this setting, hypophysectomy was performed to remove the pituitary source of ovarian estrogen stimulation. It is notable that hypophysectomy was a useful “second line” surgical therapy in women without an ovarian source of estrogen due to previous oophorectomy (10). We understand now that hypophysectomy reduced the source of growth hormone and, in turn, reduced IGF-I levels. Indeed, administration of growth hormone to patients with advanced breast cancer treated by hypophysectomy resulted in progression of bone metastases as measured by urinary calcium output (11).
In the modern era, the approach to address this hypothesis has been to target the receptors. In support of the human population studies suggesting that reduced IGF-I levels are associated with reduced cancer risk and modulation of cancer growth, IGF1R as a target has been documented through abundant preclinical data. Perhaps the first demonstration that IGF1R antibody targeting might inhibit cancer cell growth came from data obtained more than 20 years ago; use of a monoclonal antibody inhibited growth of breast cancer cells in mouse models with tumor xenografts (12). Small molecule tyrosine kinase inhibitors (TKIs) were also shown to have anticancer activity (13). Like many other growth factor systems, the ligands and receptors that make up the signaling network are complex [reviewed in (14)].
First, there are three ligands for the cell surface receptors: IGF-I, IGF-II, and insulin. Although insulin is not normally thought of as a hormone that regulates tumor cell growth, many studies implicate insulin receptor signaling as an important pathway used by cancer cells (15). As discussed below, the failure of IGF1R antibodies in the clinical trials reported to date may highlight the role for insulin receptor in cancer cells.
Second, in addition to these ligands, there are multiple receptors. The IGF1R is a heterotetramer. The IGF1R gene transcript is translated as a single polypeptide chain and is then processed into an extracellular domain (the α subunit) and a transmembrane or cytoplasmic domain (the β subunit) that contains tyrosine kinase activity. These two subunits are processed and covalently linked to a partner dimer. Thus, the “IGF1R” is a homodimeric structure of two α and two β chains covalently linked in the membrane. This structure dictates the need for ligand binding to activate signaling; the receptor’s tyrosine kinase units are physically constrained from interacting with each other in the absence of ligand binding. Constitutive activation of the receptor is not seen, even in experimental systems, resulting in overexpression of the receptor (16).
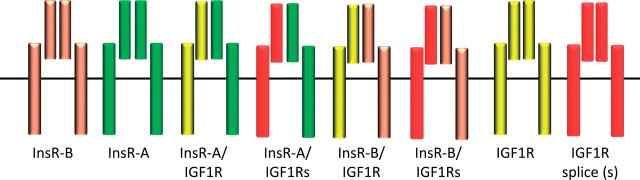
Final assembly of the receptor may also include synthesis of a hybrid receptor composed of linked α and β chains of the IGF1R joined with linked α and β chains of the insulin receptor. Adding complexity to this system, there are two forms of both the insulin receptor and IGF1R proteins that are created by splice variants (17,18). The fetal form of the insulin receptor (insulin receptor A) is of particular note; it can bind IGF-II with high affinity. Thus, if you count all possible homodimer and hybrid receptors, there are potentially eight tyrosine kinase receptors involved in signal transduction (Figure 1).
Figure 1.

Insulin-like growth factor 1 receptor (IGF1R) and insulin receptor (InsR) family members. The IGF1 and insulin receptors are synthesized by similar mechanism. In each case, transcripts from a single gene are translated into a long polypeptide that is cleaved to release the extracellular ligand binding domain (α subunit) and transmembrane tyrosine kinase domain (β subunit). The receptor subunits are covalently linked to form αβ hemireceptors that are further linked to form an α2β2 tetrameric structure. Splicing variants exist for both receptors (InsR-B, InsR-A, IGF1R, and IGF1R splice [shown as IGF1Rs]). The four InsR and IGF1R hemireceptors can homo- and heterodimerize to form multiple receptor subtypes.
Third, there are six high-affinity IGF-binding proteins that complex with the ligand in extracellular fluids. Most circulating IGF-I is complexed to IGFBP-3. In this complex, IGF-I cannot bind to the IGF1R. In times of stress (surgery, burns, pregnancy), IGFBP-3 is proteolytically cleaved and releases IGF-I to its receptor (19). Most IGFBPs have higher affinity for the ligands than for the receptors.
Thus, in the extracellular space, up to 14 interacting proteins compete for the IGF ligands. If IGF ligand interaction with IGF receptors is required for growth stimulation, then what is the best way to inhibit these interactions?
The monoclonal antibodies directed against IGF1R were developed first. Based on the success of trastuzumab in HER2 amplified breast cancers, it was logical to develop drugs that specifically inhibited a single receptor subtype, despite the known complexity of the IGF receptor family. Although the antibodies described thus far have different Fc domains and are either humanized or fully human, they all have a similar mechanism of action. The antibodies bind to the IGF1R, cause receptor internalization, and thereby prevent binding of ligand to receptor by removing receptors from the cell surface (20). None of the described monoclonal antibodies bind to the insulin receptor. Because the IGF1R is a tyrosine kinase, small-molecule inhibitors designed to disrupt this biochemical activity have also been developed. Unlike the monoclonal antibodies, the small-molecule inhibitors are not specific for the IGF1R; they also maintain activity against the insulin receptor. Finally, neutralizing antibodies for both IGF-I and IGF-II have also entered phase II clinical trials (Table 1).
This is a clearly complex system. Does this complexity explain the failure of the monoclonal antibodies in these early clinical trial reports?
A Need for Biomarkers to Predict an Anti-IGF1R Benefit
Oncologists frequently measure the level of the target to predict benefit from a specific therapy. In breast cancer, proof of the validity of this approach has been most clearly established for the estrogen receptor and HER2. In the absence of evidence of expression, a targeted therapy has no clinical benefit. Measurement of ALK mutation in non-small cell lung cancer identifies the small minority of patients who benefit from crizotinib (21).
Although robust techniques have been developed to measure IGF1R gene expression (quantitative PCR) and protein expression (immunohistochemistry, flow cytometry), these techniques are unable to determine the precise receptor structure. These techniques measure expression of the gene or gene product but cannot distinguish the receptor conformation as shown in Figure 1. For example, suppose that a cancer cell makes 100 molecules of IGF1R mRNA and 100 molecules of insulin receptor mRNA. Because of the multi-subunit structure of the receptor, the distribution of assembled receptors on the cell surface could be 50 homodimers of insulin receptor plus 50 homodimers of IGF1R versus 100 hybrid IGF1R-insulin receptors versus a mix of hybrid and homodimer receptors. If an antibody only interacts with the IGF1R, then a cell with 50 homodimers of insulin receptor still will have a functional signaling pathway that is unaffected by an IGF1R antibody (Figure 2). Furthermore, a cell with 100 hybrid receptors might be extremely sensitive to an IGF1R monoclonal antibody because the entire population of receptor complexes may be internalized by the interaction of antibodies with the IGF1R portion of the hybrid receptor. Of course, cells with mixed numbers of hybrid and holoreceptors are also likely to exist and might demonstrate a partial inhibitory response to an anti-IGF1R antibody.
Figure 2.
Homodimers and heterodimers of insulin receptor (InsR) and insulin-like growth factor-1 receptor (IGF1R) hemireceptors exist in various conformations. The IGF and insulin receptors can homo- and heterodimerize into eight different receptor subtypes that are assumed to take on different conformations and that may appear on the cell in varying proportions. Monoclonal antibodies (moAb) to IGF1R will inactivate and/or decrease the cell surface expression of IGF1R homodimers and InsR-IGF1R heterodimers. However, InsRs are not affected by existing anti-IGF1R monoclonal antibodies. Response to IGF1R monoclonal antibodies might be dependent on receptor subtype distribution and conformation.
These concerns are more than theoretical. We have shown that reduction of IGF1R expression using silencing RNA results in increased sensitivity to insulin (more on that below) (22). In osteosarcoma, there is a clear heterogeneity of receptor conformation. Absolute levels of receptor expression differ among osteoscarcomas with a mixture of homodimers and hybrid receptors (23). Hou et al. have shown that monoclonal Abs to IGF1R actually increase the number of insulin receptors (24). Taken together, these data show that levels of IGF1R protein expression are a weak predictor of benefit (25), but prediction might be enhanced if a more precise method were developed to evaluate receptor conformation. A test, such as the proximity ligation assays used for HER family members, might be a way to solve this question (26). Early classification of gene expression modified by IGF1R activation also shows promise (27). As with other gene expression classifiers, a robust platform validated on clinical specimens needs to be developed.
A Need to Consider IGF1R Inhibitors as Endocrine Disruptors
All hormonal systems are under tight regulation. During puberty, growth hormone released by the pituitary gland interacts with growth hormone receptors in the liver. This interaction increases expression of IGF-I by the liver and stimulates growth in most peripheral tissues. Humans who have mutations in the gene for the growth hormone receptor have low levels of serum IGF-I and are constitutionally short. Interestingly, people with growth hormone receptor deficiency rarely, if ever, develop cancers, thus providing further rationale for targeting this system (8,9).
This endocrine system is under negative feedback control. Disruption of the brain’s ability to sense IGF-I levels results in compensatory increases in growth hormone and IGF-I production by the liver. This phenomenon has been well documented in the phase I clinical trials of IGF1R monoclonal antibodies (28–30), with elevation of serum growth hormone and IGF-I above baseline levels. This finding might not be of clinical relevance if there were only one receptor and if the drug were potent enough to block this receptor as is the case with tamoxifen in premenopausal women. Administration of tamoxifen to women with functioning ovaries results in supraphysiologic levels of estradiol, yet tamoxifen is still effective in treating breast cancer. By contrast, there is concern that supraphysiologic levels of IGF-I might activate insulin and/or IGF-1 receptors not inhibited by the therapeutic anti-IGF1R antibody, thus promoting tumor growth. Furthermore, some cancer cells express growth hormone receptor making it possible that elevated growth hormone levels could drive tumor cell biology (31).
In addition, elevation of growth hormone levels results in insulin resistance. This phenomenon is well known by endocrinologists who treat patients with growth hormone excess (acromegaly), and it is likely to be due to increased lipolysis and free fatty acid production by the liver (32). Thus, the GH-IGF feedback system allows serum insulin levels to rise. Patients may become hyperglycemic on figitumumab with elevation of insulin levels (28). Elevated insulin levels, coupled with the inability of anti-IGF1R monoclonal antibodies to block insulin receptors, could lead to harm. Indeed, analysis of the effects of figitumumab in non-small cell lung cancer trials suggested increased toxicity if patients had evidence of insulin resistance as measured by hemoglobin A1c (S. Meech, personal communication). This concern is especially important because the steroids commonly used in antiemetic regimens and as premedications for taxane administration can augment insulin resistance.
It is notable that the effects of monoclonal antibodies on the endocrine systems of rodents differ substantially from those of humans. Most monoclonal antibodies are specific for human IGF1R binding; thus, disruption of the feedback loop and subsequent elevation of growth hormone, IGF-I, and insulin levels are not seen in mouse models of cancer. Moreover, postnatally, mice have low circulating levels of IGF-II, whereas humans have high levels of this hormone (33). Despite the preclinical data showing that blocking the IGF1R results in tumor inhibition in mice, it must be recognized that mice remain an imperfect model system to study drugs with endocrine targets. Rodents cannot model the ability of IGF-II to interact with the insulin receptor. Again, if the monoclonal antibodies directed against the IGF1R result in enhanced insulin receptor signaling, then there is great potential to do harm. Because of the species specificity of the antibodies, this endocrine effect would never be seen in mice.
A Need to Define Optimal Combination Therapies
As with any signaling system, there are multiple linked networks that might be exploited by inhibiting multiple targets. IGF1R signaling can also link to key biological pathways relevant to tumor biology. Thus, it would be ideal to link pathway inhibition to observable clinical outcomes. Although this is a simple idea, in the IGF system it is not always so simple to execute.
First, it is clear that IGF1R activation can lead to multiple phenotypes including cell proliferation, inhibition of apoptosis, and stimulation of cell motility and metastasis. It is also evident that some cells may not display all of those phenotypes when the IGF1R is stimulated. For example, we have shown that the IGF1R plays an important role in cancer cell motility and metastasis, but it may not be linked to proliferation (34). It has been suggested that these differences in cancer cell phenotypes are regulated not by the receptor but by the adaptor protein(s) utilized by the receptor (35-36). Because inhibition of metastasis is not necessarily linked to tumor growth, inhibition of an activated IGF1R may not be linked to an objective response or clinical benefit as defined in most phase II clinical trials.
Second, downstream pathways identified in preclinical model systems may not be clearly modeled in patients enrolled on clinical trials. For example, although multiple preclinical studies have defined a link between IGF1R and estrogen receptor function in breast cancer (37-38), patients enrolled in clinical trials rarely have an untreated tumor. The importance of modeling becomes apparent because patients with tamoxifen-resistant tumors have reduced IGF1R expression compared with the expression levels before tamoxifen exposure (39). Thus, an IGF1R monoclonal antibody might be expected to fail in a hormone refractory subset of breast cancer patients, as was the case with ganitumab in endocrine- resistant tumors (2). Similar results might be expected from the combination of an epidermal growth factor receptor (EGFR) TKI and an anti-IGF1R monoclonal antibody. Preclinical data have modeled the utility of blocking IGF1R function in cells that have become resistant to an EGFR TKI (40). However, a clinical trial that examined erlotinib with an IGF1R monoclonal antibody in patients with non-small cell lung cancer excluded patients who had previously been treated with an EGFR TKI (3).
Finally, the combination of IGF1R inhibitors and cytotoxic chemotherapy requires some consideration of the sequence in which the drugs are delivered. The activation of IGF1R signaling clearly causes cells to progress through the cell cycle. In addition, IGF1R signaling activates prosurvival signaling. Both of these pathways may affect a cell’s response to cytotoxic chemotherapy. If cell cycle progression is inhibited, then cell cycle–specific agents might be less effective. By contrast, if survival pathways are disrupted, then a cell’s response to chemotherapy could be enhanced. Both situations can be observed in breast cancer cells. If conventional chemotherapy (doxorubicin, gemcitabine) is administered before IGF1R inhibition, then growth inhibition is improved. By contrast, the opposite sequence results in no further benefit (41,42). This attention to detail is important in understanding the results of the published clinical trials. In the positive trial that combined paclitaxel and carboplatin with an anti-IGF1R antibody to treat non-small cell lung cancer, the chemotherapy was administered before the therapeutic antibody (1). However, because the antibody had a long half-life, this “chemo first” regimen effectively occurred only during the first cycle of treatment. In this regard, TKIs, which have a relatively short half-life, might be easier to combine with cytotoxic chemotherapy.
Is There a Baby in There Somewhere?
Despite these initial discouraging results in large randomized clinical trials, still there is some hope that IGF1R inhibitors could be useful in the treatment of cancer. Several trials have shown the activity of monoclonal antibodies to the IGF1R in the treatment of uncommon diseases including Ewing’s sarcoma and adrenocortical carcinoma (43–45). Unfortunately, in these examples, the development of the antibody has been discontinued by the manufacturer. There are also several large ongoing trials testing anti-IGF1R monoclonal antibodies in combination with chemotherapy in patients with pancreatic and ovarian cancer. In the case of pancreatic cancer, preliminary data were reported suggesting the activity of ganitumab with gemcitabine (46). Cixutumumab is being tested in many disease states including prostate, colorectal, mesothelioma, head and neck, and breast cancer. Many of these studies use antibody alone, antibody in combination with cytotoxic chemotherapy, and antibody combined with other signaling disruptors such as cetuximab, temsirolimus, or lapatinib. Dalotuzumab is undergoing similar development plans, in which the antibody will be combined with Akt, Notch, or mTORC1 inhibition.
These ongoing clinical trials will test the efficacy of IGF1R inhibitors in combination with cytotoxic chemotherapy and other targeted therapies. The lessons of the previous trials are well-known, and ongoing analysis of insulin resistance should help define the ability of these drugs to augment conventional therapy. Recently, a clinical trial reported a trend toward benefit in combining an IGF1R antibody (figitumumab) with exemestane as first-line treatment for advanced estrogen receptor-positive breast cancer, but only in patients with normal hemoglobin A1C levels at the time of enrollment (47). Thus, patients with preexisting metabolic syndrome (hyperinsulinemia) did not benefit from blocking the IGF1R. As stated earlier, these patients might actually be harmed by further worsening of their hyperinsulinemia.
If the insulin receptor plays an important role in tumor biology, then there are several ways by which this could be addressed. First, inhibition of both IGF1R and insulin receptor tyrosine kinase activity could be useful. Two drugs (OSI-906 and BMS 754807) are undergoing clinical testing in a variety of different settings. It is notable that the BMS compound is being tested in a patient population in which ganitumab (2) failed. This trial will help directly address the necessity to inhibit both receptors.
It may also be possible to control insulin receptor sensitivity or activation of downstream signaling pathways. The I-SPY2 trial is now testing new therapies in the neoadjuvant setting (48). This trial will evaluate ganitumab in combination with metformin as a way to manage insulin sensitivity. Metformin has many potential mechanisms of action in breast cancer (49), but the purpose of the use of metformin in I-SPY2 is to control the growth-hormone induced hyperinsulinemia stimulated by the anti-IGF1R antibody.
Hyperinsulinemia, by itself, has been shown to accelerate breast tumor growth in a rodent model of type 2 diabetes. Interestingly, inhibition of mTOR results in worsened hyperglycemia but is also associated with better tumor control (50). mTOR might be a critical downstream signaling pathway required for insulin receptor stimulation of tumor growth. Although there are many clinical trials examining mTOR inhibition in cancer, preliminary reports suggest that this combination may have activity in estrogen receptor expressing breast cancer (51). While mTOR inhibition could have many potential mechanisms of action, including disruption of intracellular feedback mechanisms (52), it might blunt the effects of hyperinsulinemia induced by the IGF1R monoclonal antibody. Early reports suggest that this combination of IGF1R and mTOR inhibition has clinical benefits in Ewing’s sarcoma (53).
In summary, the reported clinical trials have raised serious concerns about the ability of IGF1R inhibition to serve as an effective cancer treatment. In some ways, this concern is not completely fair; meaningful single-agent long-term responses have been documented (29,45) in subsets of patients treated in early phase trials. Unfortunately, these tumors, mostly sarcomas, are relatively uncommon, and anti-IGF1R inhibition likely only benefits a subset of these uncommon tumors. Thus, development of anti-IGF1R drugs as single agents desperately needs predictive biomarker analysis to improve patient selection. At a minimum, a means to clearly identify the relative proportions of IGF1R-related receptor subtypes and their conformations in tumors is necessary. Osteosarcomas have a mixture of homodimer and hybrid insulin and IGF-1 receptors (23), and the relative proportions of these receptors and their hybrids might be a simple way to predict responses to a targeted anti-IGF1R monoclonal antibody.
The reason positive clinical trial results in non-small cell lung cancer (1) could not be reproduced is uncertain. As mentioned, careful attention to preexisting metabolic syndrome, insulin levels after figitumumab administration, and the sequence of antibody and chemotherapy administration might affect outcomes. Future trials should collect data to evaluate these important regulators of IGF action. These concerns are not limited to anti-IGF1R therapies alone; any of the promising new drugs targeting the PI3K-Akt-mTOR pathway could result in the disruption of glucose homeostasis.
Finally, TKIs directed against IGF1R and insulin receptors could address the concern about insulin receptor serving as a bypass pathway. As shown in animal models, this type of receptor can be effective at controlling tumor growth while at the same time making glucose control worse (54). Preclinical data suggest that some TKIs have a differential distribution to insulin target organs, with less distribution to muscle (55). These pharmacologic differences could play a key role in defining a therapeutic window for these TKIs that would at first glance have substantial host toxicity.
While we have thought of IGF1R disruption as a relatively new targeted therapy, it must be remembered that IGF-I ligand-lowering strategies—via hypophysectomy—were successfully employed in hormone-responsive breast cancer (10). Although these clinical benefits cannot be unequivocally associated with reduced IGF receptor signaling, these clinical data are consistent with a role for IGF signaling in cancer. Like all important advances in cancer therapy, inhibition of IGF1R is travelling the bench-to-bedside-to-bench pathway. Hopefully, the information we have learned in the initial clinical development of these agents will guide future clinical trials. By analogy to another successful targeted therapy, it took almost 100 years to determine the mechanism of oophorectomy in breast cancer and to develop medical therapies to accomplish the same goals. Let us not take that long this time!
Funding
This work was supported by NCI grants R01 CA074285, P50CA116201, P30 CA077598 and Komen for the Cure SAC110039.
Notes
The author declares no conflict of interests.
References
- 1. Karp DD, Paz-Ares LG, Novello S, et al. Phase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancer J Clin Oncol. 2009;27 15):2516 2522 [DOI] [PubMed] [Google Scholar]
- 2. Kaufman PA, Ferrero JM, Bourgeois H, et al. A randomized, double-blind, placebo-controlled, phase 2 study of AMG 479 with exemestane (E) or fulvestrant (F) in postmenopausal women with hormone-receptor positive (HR+) metastatic (M) or locally advanced (LA) breast cancer (BC) Cancer Res. 2010;70suppl 24):76s [Google Scholar]
- 3. Ramalingam SS, Spigel DR, Chen D, et al. Randomized phase II study of erlotinib in combination with placebo or R1507, a monoclonal antibody to insulin-like growth factor-1 receptor, for advanced-stage non-small-cell lung cancer J Clin Oncol. 2011;29 34):4574 4580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ahlgren M, Melbye M, Wohlfahrt J, Sorensen TI. Growth patterns and the risk of breast cancer in women N Engl J Med. 2004;351 16):16191626 [DOI] [PubMed] [Google Scholar]
- 5. Green J, Cairns BJ, Casabonne D, Wright FL, Reeves G, Beral V. Height and cancer incidence in the Million Women Study: prospective cohort, and meta-analysis of prospective studies of height and total cancer risk Lancet Oncol. 2011;12 8):785 794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hankinson SE, Willett WC, Colditz GA, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer Lancet. 1998;351:1373 1375 [DOI] [PubMed] [Google Scholar]
- 7. Chan JM, Stampfer MJ, Giovannucci E, et al. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study Science. 1998;279 5350):563 565 [DOI] [PubMed] [Google Scholar]
- 8. Shevah O, Laron Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: a preliminary report Growth Horm IGF Res. 2007;17 1):54 57 [DOI] [PubMed] [Google Scholar]
- 9. Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans Sci Transl Med. 2011;3 70):70ra13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ray BS, Pearson OH. Hypophysectomy in treatment of disseminated breast cancer Surg Clin North Am. 1962;42:419 433 [DOI] [PubMed] [Google Scholar]
- 11. Pearson OH, West CD, Li MC, Maclean JP, Treves N. Endocrine therapy of metastatic breast cancer AMA Arch Intern Med. 1955;95 2):357 364 [DOI] [PubMed] [Google Scholar]
- 12. Arteaga CL, Kitten LJ, Coronado EB, et al. Blockade of the type I somatomedin receptor inhibits growth of human breast cancer cells in athymic mice J Clin Invest. 1989;84:1418 1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garcia-Echeverria C, Pearson MA, Marti A, et al. In vivo antitumor activity of NVP-AEW541: a novel, potent, and selective inhibitor of the IGF-IR kinase Cancer Cell. 2004;5 3):231 239 [DOI] [PubMed] [Google Scholar]
- 14. Sachdev D, Yee D. Disrupting insulin-like growth factor signaling as a potential cancer therapy Mol Cancer Ther. 2007;6 1):1 12 [DOI] [PubMed] [Google Scholar]
- 15. Belfiore A, Roberta M. The insulin receptor and cancer Endocr Relat Cancer. 2011;18 4):R125 R147 [DOI] [PubMed] [Google Scholar]
- 16. Kaleko M, Rutter WJ, Miller AD. Overexpression of the human insulin-like growth factor I receptor promotes ligand-dependent neoplastic transformation Mol Cell Biol. 1990;10 2):464 473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frasca F, Pandini G, Scalia P, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin- like growth factor II receptor in fetal and cancer cells Mol Cell Biol. 1999;19 5):3278 3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tobin G, Yee D, Brunner N, Rotwein P. A novel human insulin-like growth factor I messenger RNA is expressed in normal and tumor cells Mol Endocrinol. 1990;4 12):1914 1920 [DOI] [PubMed] [Google Scholar]
- 19. Jogie-Brahim S, Feldman D, Oh Y. Unraveling insulin-like growth factor binding protein-3 actions in human disease Endocr Rev. 2009;30 5):417 437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sachdev D, Li SL, Hartell JS, Fujita-Yamaguchi Y, Miller JS, Yee D. A chimeric humanized single-chain antibody against the type I insulin-like growth factor (IGF) receptor renders breast cancer cells refractory to the mitogenic effects of IGF-I. Cancer Res. 2003;63 3):627 635 [PubMed] [Google Scholar]
- 21. Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer N Engl J Med. 2010;363 18):1693 1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang H, Pelzer AM, Kiang DT, Yee D. Down-regulation of type I insulin-like growth factor receptor increases sensitivity of breast cancer cells to insulin Cancer Res. 2007;67 1):391 397 [DOI] [PubMed] [Google Scholar]
- 23. Avnet S, Sciacca L, Salerno M, et al. Insulin receptor isoform A and insulin-like growth factor II as additional treatment targets in human osteosarcoma Cancer Res. 2009;69 6):2443 2452 [DOI] [PubMed] [Google Scholar]
- 24. Hou X, Harrington SC, Macedo LF, Weroha SJ, Brodie A, Haluska P. Hormonal therapies differentially enhance insulin receptor isoform A and erbB receptor up-regulation in response to IGF-1R inhibitor MK-0646 in vivo [abstract] Proc Am Assoc Cancer Res. 2009;Abs 2812 [Google Scholar]
- 25. Gualberto A, Dolled-Filhart M, Gustavson M, et al. Molecular analysis of non-small cell lung cancer identifies subsets with different sensitivity to insulin-like growth factor I receptor inhibition Clin Cancer Res. 2010;16 18):46544665 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26. Spears M, Taylor KJ, Munro AF, et al. In situ detection of HER2:HER2 and HER2:HER3 protein-protein interactions demonstrates prognostic significance in early breast cancer Breast Cancer Res Treat. 2012;132 2):463 470 [DOI] [PubMed] [Google Scholar]
- 27. Litzenburger BC, Creighton CJ, Tsimelzon A, et al. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy Clin Cancer Res. 2011;17 8):2314 2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haluska P, Shaw HM, Batzel GN, et al. Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors Clin Cancer Res. 2007;13 19):5834 5840 [DOI] [PubMed] [Google Scholar]
- 29. Tolcher AW, Sarantopoulos J, Patnaik A, et al. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1 J Clin Oncol. 2009;27 34):5800 5807 [DOI] [PubMed] [Google Scholar]
- 30. Atzori F, Tabernero J, Cervantes A, et al. A phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors Clin Cancer Res. 2011;17 19):6304 6312 [DOI] [PubMed] [Google Scholar]
- 31. van Garderen E, Schalken JA. Morphogenic and tumorigenic potentials of the mammary growth hormone/growth hormone receptor system Mol Cell Endocrinol. 2002;197 1–2):153 165 [DOI] [PubMed] [Google Scholar]
- 32. Moller N, Jorgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects Endocr Rev. 2009;30 2):152 177 [DOI] [PubMed] [Google Scholar]
- 33. DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting Nature. 1990;345 6270):78 80 [DOI] [PubMed] [Google Scholar]
- 34. Sachdev D, Hartell JS, Lee AV, Zhang X, Yee D. A dominant negative type I insulin-like growth factor receptor inhibits metastasis of human cancer cells J Biol Chem. 2004;279 6):5017 5024 [DOI] [PubMed] [Google Scholar]
- 35. Nagle JA, Ma Z, Byrne MA, White MF, Shaw LM. Involvement of insulin receptor substrate 2 in mammary tumor metastasis Mol Cell Biol. 2004;24 22):9726 9735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Byron SA, Horwitz KB, Richer JK, Lange CA, Zhang X, Yee D. Insulin receptor substrates mediate distinct biological responses to insulin-like growth factor receptor activation in breast cancer cells Br J Cancer. 2006;95 9):1220 1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cho HS, Aronica SM, Katzenellenbogen BS. Regulation of progesterone receptor gene expression in MCF-7 breast cancer cells—a comparison of the effects of cyclic adenosine 3′,5′-monophosphate, estradiol, insulin-like growth factor-I, and serum factors Endocrinology. 1994;134 2):658 664 [DOI] [PubMed] [Google Scholar]
- 38. Becker MA, Ibrahim YH, Cui X, Lee AV, Yee D. The IGF pathway regulates ERalpha through a S6K1-dependent mechanism in breast cancer cells Mol Endocrinol. 2011;25 3):516 528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Drury SC, Detre S, Leary A, et al. Changes in breast cancer biomarkers in the IGF1R/PI3K pathway in recurrent breast cancer after tamoxifen treatment Endocr Relat Cancer. 2011;18 5):565 577 [DOI] [PubMed] [Google Scholar]
- 40. Morgillo F, Kim WY, Kim ES, Ciardiello F, Hong WK, Lee HY. Implication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib Clin Cancer Res. 2007;13 9):2795 2803 [DOI] [PubMed] [Google Scholar]
- 41. Zeng X, Zhang H, Oh A, Zhang Y, Yee D. Enhancement of doxorubicin cytotoxicity of human cancer cells by tyrosine kinase inhibition of insulin receptor and type I IGF receptor Breast Cancer Res Treat. 2012;133 1):117 126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khatri A, Brundage RC, Hull JM, Williams BW D Yee, Kirstein MN. Pharmacodynamic modeling of sequence-dependent antitumor activity of insulin-like growth factor blockade and gemcitabine AAPS J. 2012;14 1):1 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Olmos D, Postel-Vinay S, Molife LR, et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study Lancet Oncol. 2010;11 2):129 135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haluska P, Worden F, Olmos D, et al. Safety, tolerability, and pharmacokinetics of the anti-IGF-1R monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma Cancer Chemother Pharmacol. 2010;65 4):765 773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pappo AS, Patel SR, Crowley J, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration Study J Clin Oncol. 2011;29 34):4541 4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu J, Deng H, Tang RP, et al. Exposure-response (E-R) analysis to facilitate phase III (P3) dose selection for ganitumab (GAN, AMG 479) in combination with gemcitabine (G) to treat metastatic pancreatic cancer (mPC) J Clin Oncol. 2011;29:abstr 4049 [Google Scholar]
- 47. Ryan PD, Neven P, Blackwell KL, et al. Figitumumab plus exemestane versus exemestane as first-line treatment of postmenopausal hormone receptor-positive advanced breast cancer: a randomized, open-label phase II trial Cancer Res. 2011;71(suppl 24):239sAbs nr P1-17-01. [Google Scholar]
- 48. Barker AD, Sigman CC, Kelloff GJ, Hylton NM, Berry DA, Esserman LJ. I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy Clin Pharmacol Ther. 2009;86 1):97 100 [DOI] [PubMed] [Google Scholar]
- 49. Dowling RJ, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment BMC Med. 2011;9:33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fierz Y, Novosyadlyy R, Vijayakumar A, Yakar S, LeRoith D. Mammalian target of rapamycin inhibition abrogates insulin-mediated mammary tumor progression in type 2 diabetes Endocr Relat Cancer. 2010;17 4):941 951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Di Cosimo S, Bendell JC, Cervantes-Ruiperez A, et al. A phase I study of the oral mTOR inhibitor ridaforolimus (RIDA) in combination with the IGF-1R antibody dalotozumab (DALO) in patients (pts) with advanced solid tumors J Clin Oncol. 2010;28 15):abstr 3008 [Google Scholar]
- 52. O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt Cancer Res. 2006;66 3):1500 1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Naing A, Lorusso P, Fu S, et al. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing’s sarcoma family tumors Clin Cancer Res. 2012;18 9):2625 2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Novosyadlyy R, Lann DE, Vijayakumar A, et al. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes Cancer Res. 2010;70 2):741 751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dool C, Mashhedi H, Zakikhani M, et al. IGF-1/insulin receptor kinase inhibition by BMS-536924 is better tolerated than alloxan-induced hypoinsulinemia and more effective than metformin in the treatment of experimental insulin responsive breast cancer Endocr Relat Cancer. 2011;18 6):699 709 [DOI] [PubMed] [Google Scholar]