

SUMMARY
The Ras oncogene contributes to ∼30% of human cancers, but alone is not sufficient for tumorigenesis. In a Drosophila screen for oncogenes that cooperate with an activated allele of Ras (RasACT) to promote tissue overgrowth and invasion, we identified the GTP exchange factor RhoGEF2, an activator of Rho-family signalling. Here, we show that RhoGEF2 also cooperates with an activated allele of a downstream effector of Ras, Raf (RafGOF). We dissect the downstream pathways through which RhoGEF2 cooperates with RasACT (and RafGOF), and show that RhoGEF2 requires Rho1, but not Rac, for tumorigenesis. Furthermore, of the Rho1 effectors, we show that RhoGEF2 + Ras (Raf)-mediated tumorigenesis requires the Rho kinase (Rok)–Myosin-II pathway, but not Diaphanous, Lim kinase or protein kinase N. The Rho1–Rok–Myosin-II pathway leads to the activation of Jun kinase (JNK), in cooperation with RasACT. Moreover, we show that activation of Rok or Myosin II, using constitutively active transgenes, is sufficient for cooperative tumorigenesis with RasACT, and together with RasACT leads to strong activation of JNK. Our results show that Rok–Myosin-II activity is necessary and sufficient for Ras-mediated tumorigenesis. Our observation that activation of Myosin II, which regulates Filamentous actin (F-actin) contractility without affecting F-actin levels, cooperates with RasACT to promote JNK activation and tumorigenesis, suggests that increased cell contractility is a key factor in tumorigenesis. Furthermore, we show that signalling via the Tumour necrosis factor (TNF; also known as Egr)-ligand–JNK pathway is most likely the predominant pathway that activates JNK upon Rok activation. Overall, our analysis highlights the need for further analysis of the Rok–Myosin-II pathway in cooperation with Ras in human cancers.
INTRODUCTION
Cancer is a multi-step process involving the acquisition of genetic alterations that deregulate growth and apoptosis pathways, propelling normal cells into a malignant state (Vogelstein and Kinzler, 1993). The following changes in cell physiology arise from such genetic mutations and contribute to malignancy: self-sufficiency in growth signals, insensitivity to anti-growth signals, evasion of apoptosis, sustained angiogenesis, limitless replicative potential, tissue invasion and metastasis (Hanahan and Weinberg, 2000; Hanahan and Weinberg, 2011). Moreover, interactions between the developing tumour and surrounding cells can impact upon tumorigenesis.
Drosophila is an ideal model organism for studying cooperative tumorigenesis, owing to its sophisticated genetics, lower redundancy and fast generation time (Brumby and Richardson, 2005). For example, clonal analysis allows the generation of marked mutant clones with a number of genetic mutations within a wild-type context, enabling the examination of tumour development and the interaction of the tumour with the surrounding wild-type cells. In a two-hit model of tumorigenesis in Drosophila, cooperation was observed between a mutation in the apico-basal cell polarity regulator Scrib, combined with expression of an activated allele of Ras (RasACT) in the developing eye-antennal tissue [eye-antennal discs (EADs)] (Brumby and Richardson, 2003). Although loss of scrib function (scrib1) in clones on its own resulted in hyper-proliferation and altered cell morphology, mutant clones did not over-grow because they were removed by Jun kinase (JNK; also known as Bsk in Drosophila)-mediated apoptosis (Brumby and Richardson, 2003). Expression of RasACT alone in clones in the developing EAD resulted in hyperplasia and ectopic differentiation. In contrast, RasACT expression in scrib mutant EAD clones resulted in massive clonal tissue overgrowth due to increased proliferation, increased survival and loss of differentiation, and was associated with a loss of cell polarity and invasion/metastasis of the mutant tissue, which was not seen with either expression of RasACT alone in clones or in scrib1 mutant clones (Brumby and Richardson, 2003; Pagliarini and Xu, 2003). Furthermore, expressing RasACT in loss-of-function clones of other polarity regulators, such as dlg, lgl, baz, sdt and cdc42, also resulted in invasive overgrowth (Pagliarini and Xu, 2003), suggesting that loss of polarity combined with RasACT is important for cooperative tumorigenesis.
In order to identify other genes that contribute to Ras-mediated tumorigenesis in Drosophila, we carried out a dominant modifier genetic screen to identify genes that, when overexpressed, would act like scrib1 mutants and cooperate with RasACT (Brumby et al., 2011). Among others, this screen identified the guanine nucleotide exchange factor (GEF) RhoGEF2, an activator of Rho-family GTPases (Barrett et al., 1997; Häcker and Perrimon, 1998; Perrimon et al., 1996). Expression of RhoGEF2 enhanced the hyperplastic adult eye phenotype, owing to expression of RasACT in the developing eye under the control of the eyeless-GAL4 driver (ey > RasACT) (Brumby et al., 2011; Karim and Rubin, 1998). In a clonal setting, overexpression of RhoGEF2 with RasACT in EAD clones resulted in clonal tissue overgrowth through an extended larval stage, cell morphology defects and loss of differentiation. Consistent with the importance of RhoGEF2 in Rho-family activation being important for Ras-mediated tumorigenesis, in the genetic screen we also identified Rac1 and an activated allele of Rho1 as cooperating genes with RasACT (Brumby et al., 2011). Cooperation was dependent on activation of the JNK pathway: blocking JNK signalling with bskDN in RhoGEF2 + RasACT-expressing EAD clones suppressed differentiation defects and rescued pupation (Brumby et al., 2011).
The role of RhoGEF2 has been most extensively studied in Drosophila embryos. RhoGEF2 was identified from a screen to uncover genes required for embryo patterning (Perrimon et al., 1996) and also from a screen designed to find binding partners of Rho1 (homologue of mammalian RhoA) in the adult eye (Barrett et al., 1997). The structure of RhoGEF2 is that of a typical GEF, containing a DH domain; in addition RhoGEF2 contains a PDZ binding domain and a PH domain, which might be required for subcellular localisation (Häcker and Perrimon, 1998). RhoGEF2 mutant embryos failed to undergo cell shape changes required for ventral furrow formation during gastrulation (Barrett et al., 1997; Häcker and Perrimon, 1998; Leptin, 1999). This function was linked to Rho1 function, because expression of a dominant negative allele of Rho1 also displayed similar gastrulation defects. This was confirmed by in vitro GDP-GTP exchange assays, which demonstrated that the GEF domain of RhoGEF2 only significantly catalyses release of GDP from Rho1, but not from Rac, RhoL or Cdc42 proteins (Grosshans et al., 2005). However, whether RhoGEF2 also activates Rac1 in vivo in Drosophila – as does Pbl, the related RhoGEF (van Impel et al., 2009) – is not known.
TRANSLATIONAL IMPACT.
Clinical issue
Cancer is a complex disease, involving cooperative interactions between oncogenes and tumour suppressors. A simple model system is needed to dissect the contribution of tumour-promoting mutations to the hallmarks of cancer. The fruit fly, Drosophila, is highly suited to the analysis of tumorigenesis owing to its sophisticated genetics, low molecular redundancy and the evolutionary conservation of signalling pathways. This work focuses on Ras-mediated tumorigenesis. The Ras oncogene contributes to ∼30% of human cancers, but alone is not sufficient for tumorigenesis. Furthermore, Ras-pathway small-molecule inhibitors have proved effective against only a subset of Ras-driven tumours, and resistance often arises. Identifying the factors that cooperate with Ras, and the pathways through which they function in tumorigenesis, is therefore important to improve our understanding of Ras-driven cancers and to reveal new avenues of therapeutic intervention.
Results
In this study, the authors delineated the pathway by which RhoGEF2 cooperates with oncogenic Ras in epithelial tumorigenesis. They provide evidence that RhoGEF2 acts via Rho1, Rok and Myosin II, but does not require Rac1, Limk, Dia or PKN, to upregulate JNK signalling. In addition, Rok–Myosin-II activity was revealed to be necessary and sufficient for Ras-mediated tumorigenesis. The authors observed that activation of Myosin II, which regulates F-actin contractility without affecting F-actin levels, leads to the upregulation of JNK activity and cooperative tumorigenesis with RasACT, suggesting that increased F-actin contractility is a key factor in tumour development. They also show that signalling via the Tumour necrosis factor (TNF; also known in Drosophila as Egr) ligand is the predominant pathway that activates JNK on Rok activation.
Implications and future directions
This study has revealed a role for F-actin contractility and cell tension, in cooperation with oncogenic Ras, in JNK activation and epithelial tumorigenesis. Although the results implicate an extrinsic mechanism involving TNF-induced JNK signalling, further investigation into the mechanism of JNK activation in Drosophila will provide greater mechanistic insights. These findings have the potential to open up new avenues for the development of diagnostics and therapies for Ras-driven human cancers. If it is confirmed that the Rok–myosin-II–JNK pathway is also activated in mammalian Ras-driven tumorigenesis, the phospho-Myosin-II regulatory light-chain antibody would be an ideal biomarker. Moreover, such tumours would be candidates for treatment with Rok or Myosin II small-molecule inhibitors, along with Raf inhibitors.
Determining the downstream effectors of RhoGEF2-Rho1 or Rac that activate JNK is important for understanding RasACT-mediated cooperative tumorigenesis. Extensive analysis in mammalian cells and in Drosophila has revealed that Rho1 signals through the effectors Rho kinase (Rok), Protein kinase N (PKN), Lim kinase (Limk) and Diaphanous (Dia) (reviewed by Amano et al., 2010; Jaffe and Hall, 2005; Johndrow et al., 2004; Settleman, 2001). Rok phosphorylates and activates Myosin II regulatory light chain [MRLC; also known as Spaghetti squash (Sqh) and MLC], which in turn activates Myosin II (Zipper), resulting in actin-myosin contraction. Phosphorylation of MLC-phosphatase by Rok inactivates the phosphatase, resulting in increased phosphorylated MRLC. Dia acts with Profilin to promote actin polymerisation. Rok can also phosphorylate Limk, which phosphorylates and inactivates Cofilin, resulting in actin filament (F-actin) stabilisation. PKN modulates cell shape during Drosophila embryonic dorsal closure (a sheet epithelial migration that occurs during embryogenesis), and regulates F-actin organisation (Lu and Settleman, 1999; Vincent and Settleman, 1997). The effectors of Rac are P21-activated kinase (PAK), which leads to actin filament stabilisation, PKN (Lu and Settleman, 1999) and WAVE/Scar (Wiskott-Aldrich syndrome protein verprolin homologous), which regulates the Arp2/3 complex, resulting in branched-actin polymerisation (reviewed by Derivery and Gautreau, 2010; Szczepanowska, 2009).
In Drosophila, Rho1 can activate JNK via a number of means in different contexts. In the Drosophila epithelium, Rho1 mediates apoptosis via JNK activation by forming a complex with a JNK kinase kinase, Slipper (Neisch et al., 2010; Vidal et al., 2006). Furthermore, Rho1 induces JNK-dependent apoptosis and compensatory proliferation in the developing wing epithelium (Warner et al., 2010). Drosophila Rac1 can also activate JNK in dorsal closure (Glise and Noselli, 1997; Hou et al., 1997; Woolner et al., 2005). However, in the context of Ras-driven tumorigenesis, how JNK is activated downstream of RhoGEF2 is unknown.
In this study, we investigate which effectors of RhoGEF2 signalling are required for cooperation with RasACT in tumorigenesis. We show that Rho1, Rok and Myosin II, but not Rac1, Limk, Dia or PKN, are required for cooperative tumorigenesis with RasACT downstream of RhoGEF2. Furthermore, we show that the RhoGEF2–Rho1–Rok–Myosin-II pathway cooperates with RasACT to activate JNK, and that activated Rok or Myosin II is sufficient for cooperation with RasACT in tumorigenesis.
RESULTS
RhoGEF2 expression with activated Ras or Raf in clones in the developing EAD results in neoplastic overgrowth
Previously, we have shown that overexpression of RhoGEF2 + RasACT in EAD clones results in clonal tissue overgrowth through an extended larval stage, cell morphology defects and loss of differentiation (Brumby et al., 2011). We had previously found that a gain-of-function allele of the Ras effector protein kinase Raf (RafGOF) in scrib1 clones phenocopied the overgrowth exhibited by scrib1 + RasACT (Brumby and Richardson, 2003); therefore, we tested whether RhoGEF2 could also cooperate with RafGOF. We found that RafGOF expression was able to cooperate with RhoGEF2 in a similar manner to RasACT (Fig. 1; supplementary material Fig. S1) (Brumby et al., 2011). The mosaic larvae exhibited an extended larval period, and the RhoGEF2 + RafGOF tissue [GFP-positive (GFP+)] in the EADs overgrew with time (compare Fig. 1A and 1B–D). RhoGEF2 + RafGOF tissue contained rounded cells with increased F-actin levels (Fig. 1B, arrowheads; quantified in 1E), as revealed by phalloidin staining (Faulstich et al., 1983), similar to RhoGEF2-alone and RhoGEF2 + RasACT-expressing tissues (supplementary material Fig. S1). RhoGEF2 + RafGOF-expressing cells showed reduced photoreceptor cell differentiation (Fig. 2A,B), as revealed by staining with the differentiation marker Elav (Robinow and White, 1991). Large undifferentiated GFP+ clonal masses were observed in the basal sections of the posterior region of the eye disc (arrow, Fig. 2B). This was similar to RhoGEF2 + RasACT tissue (supplementary material Fig. S1D–F), but was in contrast to what occurs with RhoGEF2 alone, where cells still differentiate although many are aberrantly localised basally (supplementary material Fig. S1A–C), and to cells expressing RafGOF (or RasACT) alone, which showed ectopic Elav staining (see below) (Brumby et al., 2011). Thus, RafGOF is sufficient to confer similar tumorigenic effects in cooperation with RhoGEF2, as occurs with RasACT, showing that cooperation involves the MAPK branch of Ras signalling.
Fig. 1.
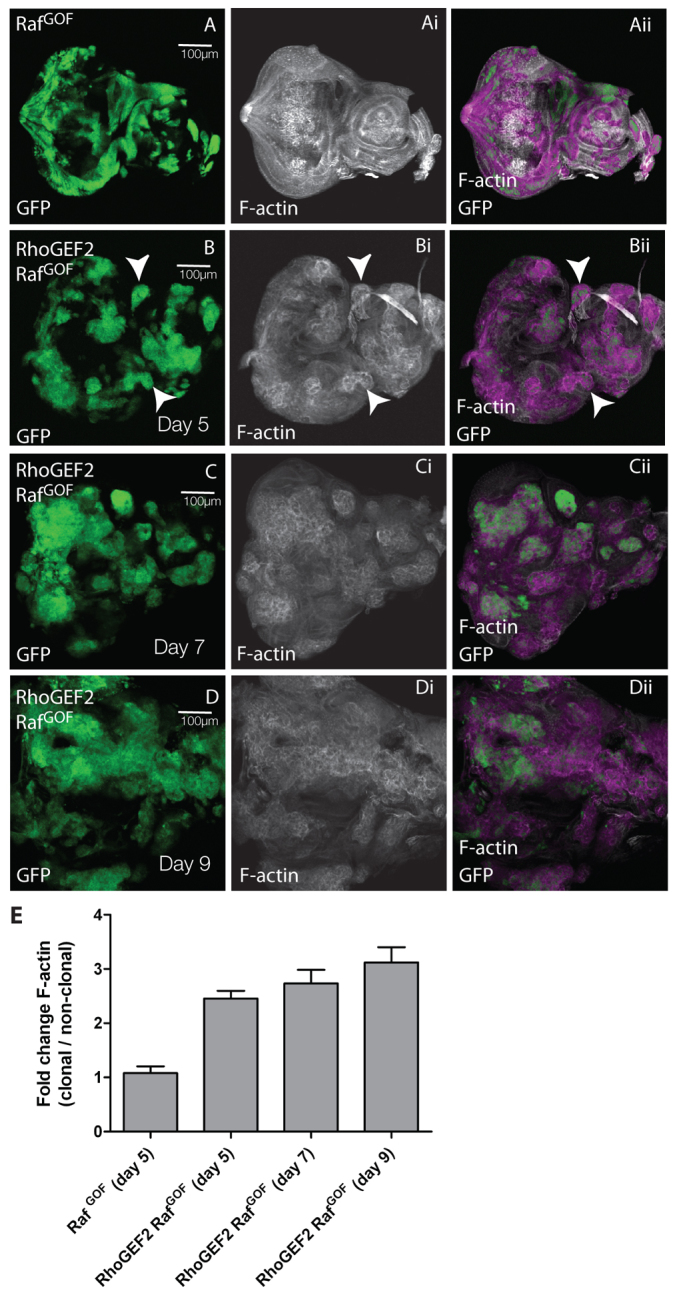
Co-expression of RhoGEF2 + RafGOF in EAD clones results in clonal tissue overgrowth through an extended larval stage. Confocal planar sections through the basal region of the epithelium of third instar larval EADs (A–D). ey-FLP was used to induce clones and the mutant tissue is marked by the expression of GFP in all figures unless otherwise indicated. EADs were dissected from wandering third instar larvae at day 5 (A,B), day 7 (C) and day 9 (D) AEL, and stained with phalloidin-TRITC for F-actin (white). Overlap between white and green appears purple in the merges. Images are orientated with anterior to the right in this and all other figures, unless otherwise stated. (A) RafGOF. (B–D) RhoGEF2 + RafGOF. RafGOF (A) and RhoGEF2 + RafGOF (B) mosaic EADs were similar in size at day 5 and similar to control mosaics and RhoGEF2 mosaic EADs (supplementary material Fig. S1A). RafGOF-expressing mosaic larvae pupated at day 5, but failed to eclose (data not shown). RhoGEF2 + RafGOF EADs increased in size with time (B–D). Both GFP+ clonal and wild-type tissue overgrew, but the proportion of GFP+ tissue to wild-type tissue increased over time (B–D). In B, arrowheads indicate increased F-actin accumulation. (E) Quantification of F-actin levels in RhoGEF2 + RafGOF or RafGOF clones versus wild-type clones. The data was compared using ANOVA analysis; error bars represent s.e.m. and the significance was P<0.05 for RhoGEF2 + RafGOF at day 5, 7 or 9 compared with RafGOF alone.
Fig. 2.

Reducing levels of Rho1 suppressed clonal tissue overgrowth, differentiation, and cell morphology defects, and rescued pupation in RhoGEF2 + RafGOF EADs. Confocal planar sections through the epithelium of third instar larval EADs. Apical (A,C) and basal (B,D) sections are shown. Mutant tissue was marked by the expression of GFP (green). EADs were stained for Elav (white) and with phalloidin-TRITC for F-actin (white). (A,B) RhoGEF2 + RafGOF. (C,D) Rho1RNAi+RhoGEF2 + RafGOF. Many RhoGEF2 + RafGOF-expressing cells in the posterior region do not stain with Elav, especially in the basal sections of the eye disc, where clonal tissue accumulated (arrows, B-Bii). F-actin was enriched in RhoGEF2 + RafGOF-expressing cells, particularly in basal sections (arrowheads, Aii,Aiii,Bii,Biii). Reducing levels of Rho1 in RhoGEF2 + RafGOF-expressing cells reduced the accumulation of undifferentiated clonal tissue (compare B-Bii with D-Dii, arrows) and reduced F-actin levels (arrowheads, Cii,Ciii,Dii,Diii) compared with RhoGEF2 + RafGOF mosaic EADs (Aii,Aiii,Bii,Biii, arrowheads). (E) Depletion of Rho1 in RhoGEF2 + RafGOF mosaic larvae resulted in increased pupation compared with RhoGEF2 + RafGOF mosaic larvae. The data was compared by a t-test and error bars represent s.e.m. The significance was P<0.0001. (F) Quantification of F-actin levels in RhoGEF2 + RafGOF or RhoGEF2 + RafGOF + Rho1RNAi clones versus wild-type clones. The data was compared by a t-test and error bars represent s.e.m. The significance was P<0.0001 for RhoGEF2 + RafGOF + Rho1RNAi versus RhoGEF2 + RafGOF.
Rho1 is required for RhoGEF2 + RafGOF tumorigenesis
We then addressed the issue of which pathways are required downstream of RhoGEF2 for its cooperation with RasACT. Drosophila Rho1, the mammalian homologue of RhoA, was previously shown to act downstream of RhoGEF2 during gastrulation (Barrett et al., 1997; Hacker et al., 1998). However, it is not known whether RhoGEF2 activates Rho1 in RhoGEF2-expressing clones in the developing EAD or whether Rho1 is required for cooperation of RhoGEF2 with RasACT in tumorigenesis.
First, we investigated whether RhoGEF2 activates Rho1 in the eye disc. We did this by using an active Rho1 reporter (Simões et al., 2006), which contains three binding domains of the Rho1 effector PKN fused to GFP and becomes recruited to the apical membrane by active Rho1 (Rho1-GTP). The localisation and intensity of GFP correlates with active Rho1 activity. When we expressed the Rho1 reporter with RhoGEF2 in the posterior compartment of eye discs under the control of GMR-GAL4, RhoGEF2 was localised apically and greater accumulation of GFP was observed in the apical-lateral region of these cells (arrows, supplementary material Fig. S2Bv,Bviii) compared with the control (arrows, supplementary material Fig. S2Aiv,Avi; quantified in S2C). This result indicates that Rho1 is activated by RhoGEF2 in eye disc cells.
To analyse whether Rho1 was functionally required for RhoGEF2 function, we reduced the levels of Rho1 via expression of a previously validated RNAi transgene (Rho1RNAi) (Massarwa et al., 2009; Widmann and Dahmann, 2009; Yan et al., 2009). Expression of Rho1RNAi alone in clones did not substantially affect clone size (supplementary material Fig. S3A,B), but resulted in defects in both differentiation and cell morphology; in the posterior region of the eye disc, cells still differentiated but some Elav-positive nuclei were mislocalised basally (arrows, supplementary material Fig. S3B-Bii) and, consistent with this, the pattern of apical foci of F-actin in the posterior region of the eye disc was altered compared with the surrounding wild-type tissue (supplementary material Fig. S3Aiii,Aiv). Expression of RhoGEF2 + Rho1RNAi in clones rescued the RhoGEF2-clonal phenotype; the small RhoGEF2 clone size was increased (compare supplementary material Fig. S3Ci and Fig. S1Ai), differentiation was increased (compare supplementary material Fig. S3C with Fig. S1A), cell morphology was improved and F-actin levels were reduced (compare supplementary material Fig. S3Ciii,Civ,Diii,Div and Fig. S1Aii,Bii,Cii; quantified in Fig. S3E). In contrast, knocking down Rho1 in RafGOF-expressing clones did not substantially alter the RafGOF phenotype (supplementary material Fig. S4); precocious differentiation was still observed (supplementary material Fig. S4C-Cii compared with S4A-Aii), although some differentiated cells were mislocalised basally (arrows, supplementary material Fig. S4D-Dii), as was also observed with Rho1RNAi alone and RafGOF alone. However, F-actin levels were not significantly changed in any sample relative to surrounding wild-type tissue (supplementary material Fig. S4E). Taken together, these results show that Rho1 acts downstream of RhoGEF2, but not RafGOF, in the developing Drosophila EAD.
We then analysed whether Rho1 was required for RhoGEF2 + RafGOF tumorigenesis, using Rho1RNAi to knock down Rho1 activity in EAD clones. When levels of Rho1 were reduced in RhoGEF2 + RafGOF clones, undifferentiated clonal masses did not accumulate in the basal sections of the eye disc, as was observed for RhoGEF2 + RasACT (arrows, Fig. 2D-Dii, compared with 2B-Bii), differentiation was partially restored as revealed by Elav staining [Fig. 2C-Ciii, 2D-Diii (arrows) compared with 2A-Aiii, 2B-Biii (arrows)] and F-actin levels were significantly reduced (arrowheads, compare Fig. 2Cii,Ciii,Dii,Diii and 2Aii,Aiii,Bii,Biii; quantified in 2F). Importantly, the extended larval period and block to pupation observed for RhoGEF2 + RafGOF was suppressed, with a significantly greater proportion of Rho1RNAi + RhoGEF2 + RafGOF mosaic larvae pupating compared with RhoGEF2 + RafGOF mosaic larvae (Fig. 2E), and no overgrown larvae were observed. Thus, Rho1 is required for the cooperation of RhoGEF2 with RafGOF.
Rac1 is not required for RhoGEF2 + RasACT tumorigenesis
Because GEFs can activate multiple GTPases (Rossman et al., 2005) and a dominant negative allele of Rac1, Rac1N17 (Rac1DN) (Baek et al., 2010; Luo et al., 1994) showed partial suppression of RhoGEF2 + RasACT cooperative interactions when expressed throughout the tissue with ey > RasACT (Brumby et al., 2011), it was possible that RhoGEF2 also acts through Rac. There are three Drosophila Rac homologues – Rac1, Rac2 and Mig-2-like (Mtl), which can act redundantly (Hakeda-Suzuki et al., 2002). To test whether Rac is also required for RhoGEF2 + RafGOF cooperation, Rac levels were abrogated by a number of methods (supplementary material Fig. S5). First, we expressed Rac1DN, which is thought to block activity of all three Rac homologues (Hakeda-Suzuki et al., 2002), in RhoGEF2 + RafGOF clones. Second, Rac1 levels were decreased by expression of a validated RNAi transgene (Rac1RNAi) (Baek et al., 2010). Third, Rac1, Rac2 and Mtl levels were reduced by halving the dosage of all three of these genes. When Rac activity was reduced in RhoGEF2 + RasACT clones by expressing Rac1DN or levels of Rac1 were reduced in RhoGEF2 + RasACT clones by expressing Rac1RNAi, differentiation in the clones was still significantly reduced at day 5 or 6 after egg laying (AEL) (white arrows, supplementary material Fig. S5B-Bii and 5C-Cii compared with 5A-Aii) in the mutant clones, although there was a small but significant decrease in F-actin in the Rac1RNAi-and Rac1DN-expressing samples (yellow arrows, supplementary material Fig. S5Biv and 5Civ compared with 5Aiv; quantified in supplementary material Fig. S5E). Similar results were obtained when the dosage of Rac was reduced by halving the dosage of Rac1, Rac2 and Mtl in RhoGEF2 + RasACT, except that F-actin levels were not significantly downregulated (supplementary material Fig. S5D,E). Moreover, the block to pupation by RhoGEF2 + RasACT was not rescued by any of these approaches to downregulate Rac activity (data not shown). Thus, these findings show that Rac1 is not required for RhoGEF2 + RafGOF cooperative tumorigenesis in EAD clones, which is in agreement with previous biochemical and genetic data that RhoGEF2 activates Rho1, but not Rac1 (Barrett et al., 1997; Grosshans et al., 2005; Häcker and Perrimon, 1998).
Rho kinase is required for RhoGEF2 + RafGOF tumorigenesis
Because we showed that Rho1 was required for cooperation of RhoGEF2 with RafGOF, we then analysed the downstream effectors Rho1, Rok, Dia, Limk and PKN to determine which of these were required for cooperative tumorigenesis. We knocked down these effectors in RhoGEF2 + RafGOF (or RasACT) clones, using the previously validated RNAi transgenes rokRNAi (Xu et al., 2008), diaRNAi (Widmann and Dahmann, 2009) and PKNRNAi (Warner and Longmore, 2009). For LimkRNAi, we confirmed knock down of the mRNA to 38% by quantitative PCR analysis (data not shown).
Reducing the level of Dia in RhoGEF2 + RasACT clones by the expression of diaRNAi did not restore differentiation in basal sections of the eye disc (arrows, supplementary material Fig. S6B-Bii) or reduce the higher levels of F-actin in RhoGEF2 + RasACT-expressing clones (supplementary material Fig. S6Aiii,Aiv,Biii,Biv; quantified in S6E). Similar effects were observed when PKNRNAi was expressed in RhoGEF2 + RasACT-expressing clones (supplementary material Fig. S6C,D; quantified in S6G), or when LimkRNAi was expressed in RhoGEF2 + RafGOF-expressing clones (supplementary material Fig. S6E,F; quantified in S6G). Moreover, the block to pupation by RhoGEF2 + RasACT was not rescued by reducing dia, Limk or PKN levels (data not shown). Thus, Dia, PKN or Limk individually do not play an important role in RhoGEF2 + RasACT (RafGOF) tumorigenesis.
In contrast, reducing levels of Rok via rokRNAi in RhoGEF2 + RafGOF clones rescued the tumorigenic phenotype (Fig. 3). rokRNAi expression alone did not substantially alter clone size or photoreceptor differentiation (supplementary material Fig. S7A-Aii,B-Bii), although produced minor cell morphology defects (arrows, supplementary material Fig. S7Aiii,Aiv). However, reducing Rok in RhoGEF2 + RafGOF clones partially restored differentiation in the eye disc clones [compare Fig. 3B,Bi (arrows) and Fig. 2B-Bii] and reduced F-actin accumulation compared with RhoGEF2 + RafGOF clones (arrowheads, compare Fig. 3Aii,Aiii,Bii,Biii and Fig. 2Aii,Aiii,Bii,Biii; quantified in Fig. 3F). Furthermore, knockdown of Rok in RhoGEF2 + RafGOF clones rescued the pupation defect of RhoGEF2 + RafGOF (Fig. 3E). Thus, Rok is required for RhoGEF2 + RafGOF tumorigenesis.
Fig. 3.

Reducing levels of Rok in RhoGEF2 + RafGOF co-expressing EAD clones suppressed clonal tissue accumulation, differentiation and cell morphology defects, and rescued pupation. Confocal planar sections through the epithelium of day 5 AEL third instar larval EADs. Apical (A,C) and basal (B,D) sections are shown. Mutant tissue was marked by the expression of GFP (green). EADs were stained for Elav (white) and with phalloidin-TRITC for F-actin (white). (A,B) rokRNAi + RhoGEF2 + RafGOF. (C,D) rokRNAi+ RhoGEF2. Expression of rokRNAi in RhoGEF2 + RafGOF clones resulted in reduced accumulation of undifferentiated clonal tissue compared with RhoGEF2 + RafGOF mosaic eye discs (compare arrows, B-Bii and Fig. 2B-Bii) and reduced the high levels of F-actin in the mutant cells (arrowheads in Aiii,Biii compared with Fig. 2Aii,Aiii,Bii,Biii). rokRNAi in RhoGEF2 clones rescued the reduced clone size, F-actin accumulation and morphology defects of RhoGEF2-alone clones (arrowheads in Cii,Ciii,Dii,Diii compared with supplementary material Fig. S1Aii,Aiii,Bii,Biii). Note that the high levels of F-actin observed in Biii are due to the axonal projections that are present in this basal section. (E) Expression of rokRNAi in RhoGEF2 + RafGOF-expressing cells resulted in an increase in pupation compared with RhoGEF2 + RafGOF expression alone. The data was compared by a t-test and error bars represent s.e.m. The significance was P<0.0001. (F) Quantification of F-actin levels in RhoGEF2 + RafGOF + rokRNAi or RhoGEF2 + rokRNAi clones versus wild-type clones. The data was compared by a t-test and error bars represent s.e.m. The significance was P<0.0004 for RhoGEF2 + rokRNAi versus RhoGEF2 and P<0.0001 for RhoGEF2 + RafGOF + Rho1RNAi versus RhoGEF2 + RafGOF.
To explore how Rok contributed to RhoGEF2 + RafGOF tumorigenesis, we examined whether rokRNAi affected RafGOF as well as RhoGEF2 phenotypes. As expected, expression of rokRNAi with RhoGEF2 in clones rescued the small clone size (compare Fig. 3C,D and supplementary material Fig. S1Ai,Bi), the differentiation defects (compare Fig. 3C-Cii,D-Dii and supplementary material Fig. S1A,Ai,B,Bi,Ci,Cii), the cell morphology defects and the elevated F-actin levels of RhoGEF2 (compare Fig. 3Ciii,Diii and supplementary material Fig. S1Aii,Aiii,Bii,Biii,Cii,Ciii; quantified in Fig. 3F). However, expression of rokRNAi in RafGOF clones did not substantially alter the RafGOF phenotype; precocious Elav-positive cells were still observed anterior to the morphogenetic furrow (MF; arrows, supplementary material Fig. S7C-Cii) and some differentiated cells were present in the basal sections of the eye disc (arrows, supplementary material Fig. S7D-Dii), but F-actin levels were not affected compared with the surrounding wild-type tissue (supplementary material Fig. S7Ciii,Civ,Diii,Div,E). Thus, Rok is required for the RhoGEF2, but not the RafGOF, phenotypes.
Myosin II is required for RhoGEF2 + RafGOF tumorigenesis
Given the finding that Rok suppressed RhoGEF2 + RafGOF cooperation, we then sought to examine which effectors downstream of Rok are required. Mammalian Rok can act via activation of Limk and MRLC (Amano et al., 2010). In Drosophila, Rok has not been shown to regulate Limk, but phosphorylates the Drosophila MRLC (Winter et al., 2001), encoded by sqh (Jordan and Karess, 1997), which in turn activates Myosin II heavy chain, encoded by zipper (zip) (Young et al., 1993), to regulate cell contractility. Furthermore, RhoGEF2 regulates actin-myosin contractility during gastrulation through Rho1, Rok and Zip (Barrett et al., 1997; Dawes-Hoang et al., 2005; Hacker et al., 1998), and RhoGEF2 and zip genetically interact in leg imaginal disc morphogenesis (Halsell et al., 2000).
Because we have already ruled out the involvement of Limk for RhoGEF2 + RafGOF cooperation (see supplementary material Fig. S6E,F), we examined whether Myosin II was required. We knocked down Myosin II activity with a previously validated zipRNAi transgene (Kwon et al., 2010) in RhoGEF2 + RafGOF clones and assessed the effect on differentiation and cell morphology. Expression of zipRNAi alone resulted in a slight reduction in clone size relative to wild-type clones, and in some defects in differentiation and cell morphology (supplementary material Fig. S8A-Aii and arrows in S8B-Bii). However, Myosin II depletion in RhoGEF2 + RafGOF clones partially restored differentiation compared with RhoGEF2 + RafGOF-expressing mosaic eye discs (compare Fig. 4B-Bii and Fig. 2B-Bii), and suppressed the cell morphology and elevated F-actin defects of the mutant tissue (compare Fig. 4Aii,Aiii,Bii,Biii and Fig. 2Aii,Aiii,Bii,Biii; quantified in Fig. 4F). Moreover, reducing Myosin II levels rescued the pupation defect of RhoGEF2 + RafGOF larvae (Fig. 4E). Thus, Myosin II is required for RhoGEF2 + RafGOF tumorigenesis.
Fig. 4.

Reducing levels of Myosin II (Zip) in RhoGEF2 + RafGOF-expressing EAD clones suppressed clonal tissue accumulation, differentiation, and cell morphology defects, and restored pupation. Confocal planar sections through the epithelium of day 5 AEL third instar larval EADs. Apical (A,C) and basal (B,D) sections are shown. Mutant tissue was marked by the expression of GFP (green). EADs were stained for Elav (white) and with phalloidin-TRITC for F-actin (white). (A,B) zipRNAi + RhoGEF2 + RafGOF. (C,D) zipRNAi + RhoGEF2. Expression of zipRNAi in RhoGEF2 + RafGOF co-expressing cells resulted in reduced accumulation of undifferentiated clonal tissue basally compared with RhoGEF2 + RafGOF mosaic eye discs (compare arrows in B-Bii and Fig. 2B-Bii) and reduced F-actin accumulation in the mutant cells (compare arrowheads in Aii,Aiii,Bii,Biii and Fig. 2Aii,Aiii,Bii,Biii). (C,D) zipRNAi in RhoGEF2 clones rescued the reduced clone size, F-actin accumulation and morphology defects of RhoGEF2-alone clones (arrowheads in Dii,Diii compared with supplementary material Fig. S1Bii,Biii). (E) Expression of zipRNAi in RhoGEF2 + RafGOF-expressing cells resulted in an increase in pupation compared with RhoGEF2 + RafGOF expression alone. The data was compared by a t-test and error bars represent s.e.m. The significance was P<0.0001. (F) Quantification of F-actin levels in RhoGEF2 + RafGOF + zipRNAi or RhoGEF2 + zipRNAi clones versus wild-type clones. The data was compared by a t-test and error bars represent s.e.m. The significance was P<0.0001 for RhoGEF2 + zipRNAi compared with RhoGEF2 and for RhoGEF2 + RafGOF + zipRNAi compared with RhoGEF2 + RafGOF.
To determine how Myosin II knockdown was affecting RhoGEF2 + RafGOF tumorigenesis, we examined its affect on the RhoGEF2 or RafGOF phenotypes alone. As expected, knockdown of Myosin II in RhoGEF2-expressing clones rescued the clone size relative to RhoGEF2 alone (compare Fig. 4Ci,Di and supplementary material Fig. S1Ai,Bi). The differentiation defects were also partially rescued (compare Fig. 4C and supplementary material Fig. S1A,B), as was the cell morphology defects and F-actin accumulation (compare Fig. 4Ciii,Diii and supplementary material Fig. S1Aii,Bii,Cii; quantified in Fig. 4F). In contrast, knockdown of Myosin II did not alter the RafGOF phenotype; ectopic differentiation was still observed anterior to the MF (arrows, supplementary material Fig. S8C-Cii) and some photoreceptor nuclei were basally localised (arrows, supplementary material Fig. S8D-Dii), but F-actin levels were not affected compared with the surrounding wild-type tissue (supplementary material Fig. S8E). Thus, Myosin II is required for the RhoGEF2, but not the RafGOF, phenotypes.
The Rho1–Rok–Myosin-II pathway contributes to the upregulation of JNK in RhoGEF2-expressing clones
We have previously shown that JNK is upregulated and required for RhoGEF2 + RasACT tumorigenesis (Brumby et al., 2011). Because our results here have shown that cooperation of RhoGEF2 with RasACT depends on Rho1, Rok and Myosin II, we then investigated whether this pathway also contributes to JNK upregulation in RhoGEF2-expressing clones (Fig. 5). Expression of RhoGEF2 resulted in upregulation of the JNK reporter msn-lacZ (Mattila et al., 2005) in some cells, which was most obvious in clones in the anterior-lateral regions of the eye disc (arrows, Fig. 5A-Aii). When levels of Rho1 or Zip were reduced in RhoGEF2-expressing clones, msn-lacZ expression was still detectable in the lateral region clones, but was significantly reduced relative to RhoGEF2 expression alone, reflecting a decrease in either the number of expressing cells or in the level of expression (arrows, Fig. 5B-Bii,C-Cii; quantified in 5F). However, msn-lacZ expression was upregulated when Rho1RNAi or zipRNAi were expressed alone in clones (arrows, Fig. 5D-Dii,E-Eii), perhaps owing to a disruption of cell morphology. This suggests that the JNK activation still observed in RhoGEF2-expressing clones upon Rho1RNAi or zipRNAi expression might be due to the depletion of Rho1 or Myosin II to below normal levels. Taken together, these data show that activation of Rho1 and Myosin II contributes to JNK pathway activation in RhoGEF2-expressing EAD clones.
Fig. 5.

Knockdown of Rho1 or Myosin II (Zipper) in RhoGEF2 EAD clones resulted in partial suppression of the JNK pathway activation. Confocal planar sections through the epithelium of third instar larval EADs. Mutant tissue was marked by the presence of GFP (green). JNK activity was detected by msn-lacZ enhancer trap expression, marked by β-galactosidase (β-gal) staining (white). (A) RhoGEF2 msn-lacZ. (B) Rho1RNAi+ RhoGEF2 msn-lacZ. (C) zipRNAi+ RhoGEF2 msn-lacZ. (D) Rho1RNAi msn-lacZ. (E) zipRNAi msn-lacZ. Compared with the control, msn-lacZ was upregulated in some RhoGEF2-expressing cells, particularly in the lateral regions of the EAD, indicating JNK pathway activation (arrows, A-Aii). Reducing levels of Rho1 or Zip by expressing Rho1RNAi or zipRNAi in RhoGEF2-expressing cells partially suppressed either the intensity of, or number of cells with, msn-lacZ expression (arrows, B-Bii,C-Cii). Expression of Rho1RNAi or zipRNAi in clones resulted in upregulation of msn-lacZ in the lateral regions of the eye disc (arrows, D-Dii,E-Eii). Note that, in E, the eye disc is folded and the arrow is pointing to the lateral edge. (F) Quantification of the level of msn-lacZ staining in RhoGEF2-, Rho1RNAi + RhoGEF2- or zipRNAi + RhoGEF2-expressing clones in the lateral regions of the eye disc relative to adjacent wild-type clones. This reflects the intensity of staining as well as the number of cells that are positive for msn-lacZ. The data was compared using ANOVA analysis; error bars represent s.e.m. and the significance was P<0.05 for Rho1RNAi + RhoGEF2 compared with RhoGEF2 and for zipRNAi + RhoGEF2 compared with RhoGEF2.
Activation of Rok or Myosin II is sufficient for RasACT-mediated cooperative tumorigenesis
Having shown the importance of Rok and Myosin II downstream of RhoGEF2 in cooperative tumorigenesis with RasACT, we then sought to investigate whether upregulation of Rok or Myosin II was sufficient for tumorigenesis. Therefore, we expressed constitutively active versions of rok [rokCAT (Verdier et al., 2006)] or MRLC [sqhEE (Wang and Riechmann, 2007)] in clones alone or with RasACT (Figs 6, 7).
Fig. 6.

Expression of activated rok with RasACT leads to clonal tissue overgrowth and reduced pupation. Apical or basal confocal planar sections through the epithelium of third instar larval EADs (A–D). Mutant tissue was marked by the expression of GFP (green). EADs were stained for Elav (white) and with phalloidin-TRITC for F-actin (white). (A,B) rokCAT. (C,D) rokCAT + RasACT. Expression of rokCAT resulted in disruption to the pattern of differentiation (arrows, A-Aii). Some rokCAT-expressing Elav+ cells were basally localised (arrows, B-Bii). However, unlike RhoGEF2-expressing clones, F-actin was not upregulated in rokCAT-expressing clones (arrowheads, Biii and Biv) compared with adjacent wild-type tissue. Expression of rokCAT + RasACT in clones resulted in large masses of undifferentiated clonal tissue (arrows, D-Dii) particularly in basal sections of EADs and upregulation of F-actin in the clones (Ciii,Civ and arrowheads in Diii-Div), similar to RhoGEF2 + RasACT expression (supplementary material Fig. S1Dii,Diii,Eii,Eiii). (E) Expression of rokCAT + RasACT resulted in decreased pupation compared with rokCAT expression alone. The data was compared by a t-test and error bars represent s.e.m. The significance was P<0.0001. (F) Quantification of F-actin levels in rokCAT + RasACT or rokCAT clones versus wild-type clones. The data was compared using ANOVA analysis and error bars represent s.e.m. The significance was P<0.05 for rokCAT + RasACT versus rokCAT and for rokCAT + RasACT versus RasACT.
Fig. 7.

Expression of activated Myosin II regulatory light chain (Sqh) with Ras ACT leads to clonal tissue overgrowth and decreased pupation. Apical or basal confocal planar sections through the epithelium of third instar larval EADs (A–D). Mutant tissue was marked by the expression of GFP (green). EADs were stained for Elav (white) and with phalloidin-TRITC for F-actin (white). (A,B) sqhEE. (C,D) sqhEE + RasACT. Expression of sqhEE did not noticeably affect differentiation or cell morphology (A,B). When sqhEE was expressed with RasACT in clones (C,D), large masses of undifferentiated clonal tissue were observed in basal sections of eye discs (arrows, D-Dii), although F-actin levels were not substantially upregulated (Ciii,Civ and arrowheads in Diii-Div). (E) Expression of sqhEE + RasACT resulted in decreased pupation compared with sqhEE expression alone. The data was compared by a t-test and error bars represent s.e.m. The significance was P<0.05. (F) Quantification of F-actin levels in sqhEE + RasACT or sqhEE clones versus wild-type clones. The data was compared using ANOVA analysis and error bars represent s.e.m. The significance was P<0.05 for sqhEE + RasACT versus RasACT.
Expression of rokCAT alone in clones did not substantially affect clone size compared with wild type, but showed mild disruption to differentiation, with Elav-positive cells detected in basal sections (arrows, Fig. 6A-Aii,B-Bii). Morphological defects were also apparent, although F-actin was not significantly upregulated (Fig. 6Aiii,Aiv and arrowheads in Biii,Biv; quantified in 6F). When rokCAT was expressed with RasACT, cooperative tumorigenesis occurred, leading to a blockage to pupation (reduced to ∼30%; Fig. 6E) similar to that caused by RhoGEF2 + RafGOF (or RasACT) (Fig. 2E; and data not shown). Massive tumours developed over an extended larval stage, similar to RhoGEF2 + RasACT (or RafGOF) (supplementary material Fig. S9; and data not shown). Analysis of the EADs revealed an accumulation of mutant tissue basally, reduced differentiation (Fig. 6Ci,Cii and arrows in 6Di,Dii) and morphological defects with accumulation of F-actin (Fig. 6Ciii,Civ and arrowheads in 6Diii,Div; quantified in 6F). Thus, rokCAT cooperates with RasACT similarly to RhoGEF2 with RasACT.
When we expressed the activated version of sqh alone in clones, no substantial effect on clone size, differentiation or cell morphology was observed (Fig. 7A,B). However, when sqhEE was expressed with RasACT in clones, cooperative tumorigenesis was observed, with large masses of undifferentiated clonal tissue present in basal sections of eye discs and reduced differentiation (arrowheads, Fig. 7D-Dii). However, in contrast to rokCAT + RasACT or RhoGEF2 + RasACT, F-actin levels were not significantly upregulated in day 5/6 larval EAD clones (compare Fig. 7Ciii,Civ,Diii,Div and Fig. 6Ciii,Civ,Diii,Div and supplementary material Fig. S1Dii,Diii,Eii,Eiii,Fii,Fiii; quantified in Fig. 7F). Despite not showing a significant effect on F-actin upregulation, sqhEE showed robust cooperation with RasACT, resulting in massive tumour overgrowth over an extended larval stage (supplementary material Fig. S10), and reduced pupation (reduced to ∼60%; Fig. 7E), which, although not as strongly reduced as with RhoGEF2 + RafGOF (or RasACT) (Fig. 2E; and data not shown), was still significantly decreased. Thus, activation of Myosin II is sufficient for cooperation with RasACT. Furthermore, because F-actin was not upregulated in sqhEE + RasACT tissue at least at the early stages of tumorigenesis, this suggests that cooperative tumorigenesis initiates independently of increased F-actin levels.
Activated Rok and Myosin II cooperate with RasACT to activate JNK
We then tested the effect of activated Rok and Myosin II on JNK pathway activation, using the expression of a JNK target, Mmp1 (Uhlirova and Bohmann, 2006). Expression of rokCAT did not upregulate Mmp1 in the majority of EADs, although it was weakly upregulated in some clones (Fig. 8A; quantified in 8F). However, Mmp1 was strongly upregulated in rokCAT + RasACT clones (Fig. 8C; quantified in 8F), whereas RasACT clones alone did not substantially upregulate Mmp1 in the majority of EADs (Fig. 8B). Similarly, sqhEE expression alone did not upregulate Mmp1 (Fig. 8D; quantified in 8F); however, Mmp1 was robustly upregulated in some sqhEE + RasACT clones (Fig. 8E,F). Taken together, these results show that activation of Rok or Myosin II cooperates with RasACT to induce JNK activation. Although we did not detect significant effects on Mmp1 upregulation in rokCAT or sqhEE clones alone, since Mmp1 is a weaker reporter of JNK pathway activation compared with msn-lacZ (data not shown), JNK activity might be induced at low levels by Rok or Myosin II activation.
Fig. 8.

Activation of Rok and Myosin II lead to increased expression of the JNK target Mmp1 in EAD clones. Confocal planar apical sections through the epithelium of third instar larval EADs. Mutant clones are marked by the presence of GFP (green). EADs were stained for Mmp1 to reveal JNK activity (white) and with phalloidin-TRITC to detect F-actin (white). (A) rokCAT. (B) RasACT. (C) rokCAT + RasACT. (D) sqhEE. (E) sqhEE + RasACT. Third instar EADs from rokCAT mosaic larvae did not show upregulation of Mmp1 in the majority of clones, although expression was mildly induced in some clones (not shown). Expression of RasACT alone did not upregulate Mmp1 in the majority of clones. Mmp1 was strongly upregulated in most rokCAT + RasACT clones (arrows, C-Cii) compared with adjacent wild-type tissue. Expression of sqhEE did not lead to Mmp1 upregulation in the clones (D), whereas expression of sqhEE + RasACT resulted in induction of Mmp1 in many but not all clones (arrows, E-Eii). (F) Quantification of Mmp1 levels in sqhEE, sqhEE + RasACT, rokCAT or rokCAT + RasACT clones versus wild-type clones. The data was compared by a t-test and error bars represent s.e.m. The significance was P<0.0001 for rokCAT + RasACT compared with rokCAT and P<0.08 for sqhEE + RasACT compared with sqhEE.
How is JNK upregulated by the RhoGEF2–Rho1–Rok–Myosin-II pathway in cooperation with RasACT?
Recent studies have revealed an extrinsic mechanism by which JNK is activated during tumorigenesis in the developing Drosophila wing or EAD epithelia, involving exogenous activation of JNK via the tumour necrosis factor (TNF) homologue Eiger (Egr) (Cordero et al., 2010; Igaki et al., 2009; Ohsawa et al., 2011). JNK activation and induction of apoptosis in scrib mutant clones is dependent on Egr, which is produced by circulating haemocytes (macrophage-like cells) to induce JNK upregulation in the scrib mutant cells, leading to their elimination through apoptosis (Cordero et al., 2010; Igaki et al., 2009; Lolo et al., 2012; Ohsawa et al., 2011). How the scrib mutant cells are recognised is currently unknown, but might involve changes in cell morphology. Similarly, the change in cell morphology in rokCAT clones might trigger such a pathway. scrib mutant clones in the EAD are smaller than wild-type clones due to JNK-mediated cell death (Brumby and Richardson, 2003), but in a homozygous egr mutant background the small clone size is rescued owing to blockage of JNK-mediated apoptosis (Cordero et al., 2010; Igaki et al., 2009). rokCAT clones are not significantly smaller than wild-type clones (Fig. 9A,B,E); however, they also show increased cell death, as revealed by TUNEL staining (Fig. 9B compared with the control in 9A). Most apoptosis was cell autonomous; however, some apoptotic cells were also observed in the surrounding wild-type cells. To investigate whether the increased cell death in the rokCAT mosaic EADs was due to Egr signalling, we examined whether global removal of egr reduced apoptosis in rokCAT clones (Fig. 9C). In rokCAT clones in a homozygous egr mutant background, apoptosis was strongly reduced throughout the EAD, but not eliminated, and the rokCAT clones did not overgrow (Fig. 9C compared with 9A,B; quantified in 9D). This was in contrast to the effect of the egr mutation on blocking apoptosis in scrib mutant tissue, which eliminated apoptotic cells and resulted in overgrowth of the scrib mutant clones (Cordero et al., 2010; Igaki et al., 2009) (Fig. 9E). These results suggest that an extrinsic mechanism involving TNF (Egr)-induced JNK activation plays an important role in the increased apoptosis that occurs in rokCAT clones. However, because apoptosis was not completely eliminated in rokCAT clones in an egr mutant background, this suggests that another mechanism also contributes to JNK activation and the increased cell death in rokCAT mosaic EADs (see Discussion).
Fig. 9.

TNF (Eiger) is required for apoptosis in rokCAT clones. Confocal planar compiled sections through the epithelium of third instar larval EADs. Mutant clones are marked by the presence of GFP (white, and green in the merge) and TUNEL (white, and red in the merge). (A) FRT82B control. (B) rokCAT. (C) egr1/3rokCAT. Third instar EADs from rokCAT mosaic larvae show higher levels of apoptotic cells, as revealed by TUNEL, both cell autonomously (arrows, Bi,Bii) and non-cell autonomously (arrowheads, Bi,Bii). Removing egr reduced, but did not eliminate, apoptotic cells in the rokCAT EAD in both rokCAT (arrows, Ci,Cii) and wild-type clones (arrowheads, Ci,Cii). (D) Quantification of the number of TUNEL-positive cells in EADs of each genotype indicated. The data was compared by a t-test and error bars represent s.e.m. The significance was P<0.05 for rokCAT compared with the FRT control, and for rokCAT compared with egr1/3rokCAT. (E) Quantification of the percentage of mutant clonal tissue relative to the total EAD size for each genotype indicated. The data was compared by a t-test and error bars represent s.e.m. The significance was P<0.05 for scrib1 compared with egr1/3 scrib1, for scrib1 compared with FRT, and for egr1/3scrib1 compared with FRT.
DISCUSSION
In this study, we have dissected the contribution of the RhoGEF2 pathway in Ras-mediated tumorigenesis in Drosophila epithelial tissues. We show here that the cooperation of RhoGEF2 with activated Raf phenocopies that observed with RasACT. Moreover, our analysis revealed that Rho1, Rok and Myosin II, but not Rac, Dia, Limk or PKN, are important downstream of RhoGEF2 for cooperation with RasACT (or RafGOF). We also demonstrated that the RhoGEF2–Rho1–Rok–Myosin-II pathway contributes to JNK activation in cooperative tumorigenesis with RasACT (or RafGOF). Indeed, activation of Rok or Myosin II alone is sufficient for cooperative tumorigenesis with RasACT and induces JNK activity in association with RasACT. Cooperative tumorigenesis of RhoGEF2, Rho1 and Rok with RasACT is correlated with increased F-actin; however, activation of Myosin II with RasACT is not, suggesting that actin-myosin contractility might be an important factor in cooperative tumorigenesis with RasACT. Moreover, we show that TNF (Egr)-JNK activation plays an important role in RokCAT-induced cell death; however, because apoptosis was still observed in an egr mutant background, a more direct effect of Rok–Myosin-II on JNK activation might also be involved (see below). These findings are likely to have important implications in the understanding of human Ras-driven cancers.
Contribution of Rho1 effectors to F-actin accumulation in RhoGEF2 clones
The Rho1 effectors Dia, Rok, Limk and PKN have all been shown previously to induce actin polymerisation (Grosshans et al., 2005; Maekawa et al., 1999; Ohashi et al., 2000a; Ohashi et al., 2000b; Vincent and Settleman, 1997). Our results here have revealed that the accumulation of F-actin in RhoGEF2-expressing clones depends on Rho1 and Rok as expected; however, knocking down Limk, Dia or PKN did not significantly reduce F-actin levels in RhoGEF2 + RasACT-expressing clones. It is possible that these Rho1 effectors could play more minor roles or act redundantly, such that double or triple knockdowns are required to reduce F-actin levels in RhoGEF2-expressing cells. Interestingly, we found that reducing Myosin II heavy chain levels (using zipRNAi) also led to reduced F-actin levels in RhoGEF2 + RasACT-expressing clones. This was unexpected given the role of Myosin II in actin-myosin contractility, but not in actin polymerisation. However, expression of activated Myosin II light chain (SqhEE) did not lead to upregulation of F-actin, suggesting that it is more likely that Myosin II heavy chain depletion might have secondary effects on the regulation of F-actin polymerisation, perhaps due to reduced levels of F-actin–Myosin-II fibres upon the depletion of Myosin II protein, or to feedback mechanisms that affect the activity of actin polymerisation regulators. Nonetheless, the cooperation of SqhEE with RasACT in tumorigenesis without effects on F-actin suggests that actin-myosin contractility or other events triggered by activated Myosin II light chain are crucial for cooperative tumorigenesis.
The Rho1–Rok–Myosin-II pathway in JNK activation and RasACT-mediated tumorigenesis
From data presented here, as well as in our previous study (Brumby et al., 2011), we have shown that Rho1, Rok and Myosin II activity are necessary and sufficient for cooperative tumorigenesis with RasACT (Fig. 10). This pathway leads to the activation of JNK (Brumby et al., 2011), which is important for invasion, and the blockage of differentiation and pupation, enabling the tumour to overgrow through an extended larval phase (Brumby et al., 2011; Leong et al., 2009). Blocking JNK activity (using a dominant negative transgene) in RhoGEF2 + RasACT tumours results in increased differentiation and pupation (Brumby et al., 2011). In tumorigenesis with RasACT, JNK has been shown to be activated by a cell extrinsic mechanism in scrib mutant tissue, via TNF (Egr) supplied by the haemocytes (Cordero et al., 2010; Igaki et al., 2009; Lolo et al., 2012; Ohsawa et al., 2011). We have shown that the TNF pathway is partially responsible for the increased apoptosis in RokCAT EADs; however, unlike scrib mutant tissue (Cordero et al., 2010; Igaki et al., 2009), cell death was not completely eliminated in an egr mutant background and the clones did not overgrow. Therefore, it is likely that a novel mechanism also exists for JNK activation and/or triggering apoptosis by RokCAT.
Fig. 10.
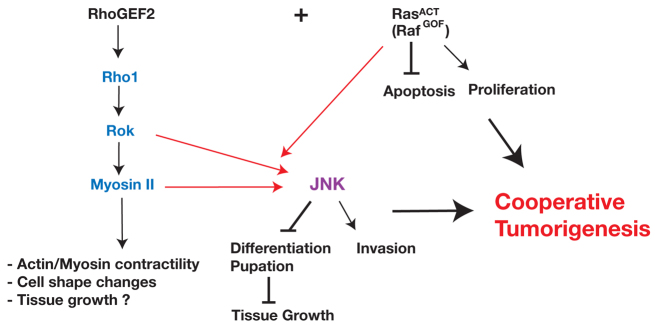
Model of the RhoGEF2 effector pathway required for cooperation with RasACT in tumorigenesis. Expression of RasACT (or RafGOF) alone promotes proliferation and blocks apoptosis, which contributes to the tumour growth. Our epistasis experiments show that RhoGEF2 acts via Rho1–Rok–Myosin-II activity in its cooperation with activated Ras signalling in tumorigenesis. Rho1, Rok and Myosin II are required downstream of RhoGEF2 for JNK activation. Activation of Rok (RokCAT) or Myosin II activity (SqhEE) only robustly promotes JNK activation in cooperation with RasACT. JNK promotes tumour invasion, and blocks differentiation and pupation, which contributes to the tumour growth during the extended larval stage. Myosin II activity has been documented by others to result in increased F-actin contractility and cell shape changes, but might also contribute to tumour growth in combination with RasACT.
Previous studies have provided evidence that the Rho1–Rok–Myosin-II pathway activates JNK, although the precise mechanism is not clear. One study showed that loss of the cell polarity regulators aPKC, Cdc42 or Par6 results in Rho1–Rok–Myosin-II pathway activation of JNK-dependent apoptosis and compensatory proliferation (Warner et al., 2010). Interestingly, this study showed that JNK activation was independent of Rho1-Rok pathway effects on F-actin polymerisation, because inhibiting Cofilin (by reducing levels of Slingshot, a Cofilin phosphatase, which activates Cofilin) and thereby promoting F-actin stabilisation, did not result in JNK activation or hyperproliferation when apoptosis was blocked. This result is consistent with our finding that activation of Myosin II activity (via sqhEE expression) results in cooperative tumorigenesis with RasACT without upregulation of F-actin. Another study revealed that overexpression of wild-type Rho1 in the anterior-posterior boundary of the developing wing epithelium resulted in JNK-mediated apoptosis (Vidal et al., 2006). Myosin II activity has also been linked to JNK activation in a study of the Drosophila non-muscle myosin phosphatase PP1β (Flapwing), which negatively regulates JNK activity through the inhibition of Myosin II activity in the developing wing (Kirchner et al., 2007). Furthermore, a recent study has shown that depletion of the Sds22/PP1 phosphatase can cooperate with RasACT, via upregulation of Myosin II activity and JNK activation (Jiang et al., 2011). However, in all of these studies, cell morphology changes occur and might induce recruitment of haemocytes and the activation of JNK via the extrinsic TNF (Egr) pathway. There are, however, examples where JNK is induced without cell morphology changes, such as in response to cellular damage due to irradiation, which is due to a feedback loop involving caspases and p53 (Shlevkov and Morata, 2012), and by oxidative stress, which induces reactive oxygen species (ROS) that activate JNK (Ohsawa et al., 2012; Wang et al., 2003). However, in both these examples, there is evidence suggesting that Egr could also be involved in JNK activation (Kanda et al., 2011; Shlevkov and Morata, 2012). Although we have shown that RokCAT-induced apoptosis shows a strong dependence on Egr, the fact that there was still some remaining apoptotic cells might suggest that an Egr-independent mechanism is also induced. Whether this mechanism involves p53, Ros or more direct effects of Myosin II on JNK activity remains to be determined. It is possible that activated Myosin II activates JNK directly, because Sqh and Bsk were shown to form a complex in vivo in the large-scale proteomics project carried out in Drosophila cultured cells (Guruharsha et al., 2011). Furthermore, it is possible that Flapwing or Sds22 phosphatases are inhibited by RhoGEF2-Rho1 signalling and are also involved in Myosin II and JNK activation.
In RasACT-driven cooperative tumorigenesis with RhoGEF2 overexpression or Rho1 activation, the activation of JNK is clearly involved in invasion and the block to differentiation and pupation (Brumby et al., 2011). Although we have not yet tested the role of JNK in rokCAT or sqhEE cooperative tumorigenesis with RasACT, it most likely plays a similar role. Moreover, JNK might also contribute to cell morphology changes in RasACT-driven cooperative tumorigenesis, because blocking JNK activity in wing disc cells depleted for the apico-basal cell polarity regulator Lgl can rescue the cell morphology defects and rescue the overgrowth phenotype (Zhu et al., 2010). JNK might also contribute to the tumour overgrowth: a recent study showed that, in lgl-depleted wing disc tissue, JNK can inactivate the Hippo tissue growth control pathway, thereby promoting tissue overgrowth (Sun and Irvine, 2011). However, in lgl mutant clones in the EAD, blocking JNK enhances the clonal overgrowth and leads to loss of polarity and an invasive phenotype (Grzeschik et al., 2010), and blocking JNK in scrib mutant EAD clones does not prevent, but rather enhances, the impairment of the Hippo pathway (Doggett et al., 2011). Interestingly, a recent study has revealed that, in scrib− + RasACT tumours, JNK activation upregulates expression of an F-actin cross-linking protein, Filamin (Cher), which acts to inhibit the Hippo pathway to promote overgrowth (Külshammer and Uhlirova, 2012). Therefore, although RhoGEF2-Rho1 + RasACT-mediated tumorigenesis requires JNK activation for the block to differentiation and pupation, and to promote invasion, whether JNK can also have a role in cell morphology changes or promoting proliferation via inhibition of the Hippo pathway in these tumours requires further investigation.
Activation of the Rho1–Rok–Myosin-II pathway might also contribute to tumour growth with RasACT, independently of JNK (Fig. 10), because our previous analysis has shown that JNK activation alone with RasACT does not result in as potent cooperative tumorigenesis as occurs with RhoGEF2 (or Rho1ACT, rokCAT or sqhEE) + RasACT (Brumby et al., 2011) (and this study). It is possible that F-actin polymerisation, actin-myosin contractility or cell morphology changes contribute to tumour overgrowth. Recently, increased F-actin polymerisation, due to expression of activated Dia or loss-of-function mutants in the actin capping proteins Cpa and Cpb, was shown to inactivate the Hippo tissue growth control pathway and drive tissue growth (Fernández et al., 2011; Richardson, 2011; Sansores-Garcia et al., 2011). However, as discussed above, RhoGEF2 expression alone did not result in increased cell proliferation in clones, suggesting that a specific form of F-actin is required to regulate the Hippo pathway, which is not generated by the RhoGEF2-Rho1 pathway, or that RhoGEF2 also activates another tissue growth inhibitory pathway. Furthermore, increased F-actin polymerisation seems not to be necessary, because activated Myosin-II regulatory light chain (SqhEE) induced cooperative tumorigenesis with RasACT without affecting F-actin levels.
It is possible that, owing to activation of Myosin II, actin-myosin contractility, cellular tension and cell morphology changes might contribute to tissue growth in RhoGEF2 + RasACT tumours. In mammalian cells, RhoA–Rok–Myosin-II pathway-mediated cell contractility regulates cell proliferation; microinjection of activated forms of RhoA, Rac and Cdc42 in Swiss 3T3 fibroblasts promotes S-phase entry, whereas reducing RhoA, Rac or Cdc42 activity blocked DNA replication (Olson et al., 1995). Actin-myosin contractility can also increase cellular tension, and cellular tension can induce cell proliferation (reviewed by Assoian and Klein, 2008; Mammoto and Ingber, 2009; Yu et al., 2011). Indeed, in cultured monolayers of cells in an elastomeric force sensor array, inhibition of Rok or Myosin II suppressed proliferation, suggesting that contractile force and tension resulting from the RhoA–Rok–Myosin-II pathway regulates cell proliferation (Nelson et al., 2005). More recently, a role for cellular tension in cell proliferation via activation of the Hippo tissue growth control pathway transcriptional co-activator Yap/Taz (homologues of Drosophila Yorkie) has been revealed in mammalian cells (Dupont et al., 2011; Wada et al., 2011). The precise mechanism by which this occurs is unclear; however, it has been recently discovered that Scrib can bind to Taz, thereby tethering Taz to the cell cortex (Cordenonsi et al., 2011). Thus, any alteration of cell morphology or cellular tension that leads to mis-localisation of Scrib would release Taz, thereby enabling it to enter the nucleus and upregulate cell survival and proliferation genes. Interestingly, the mechanism was shown in one study to require Rho-GTPase activity (Dupont et al., 2011). Furthermore in a mouse model, activated Rock2 (homologue of Drosophila Rok) induced tissue stiffness and activation of the transcription factor β-catenin to promote epidermal hyperplasia (Samuel et al., 2011; Samuel and Olson, 2011). Whether these mechanisms contribute to the tumour growth of RhoGEF2–Rho1–Rok–Myosin-II with activated Ras in Drosophila epithelial tissues will require further analysis. Interestingly, a recent study revealed that Drosophila Filamin (Cher) binds to Myosin II and is important for activation of Myosin II activity in scrib− + RasACT tumours, and also that Myosin II activity is important for tumour overgrowth, via Hippo pathway inactivation, as well as invasion (Külshammer and Uhlirova, 2012). Thus, the cooperation of cell polarity mutants with oncogenic Ras might also require the RhoGEF2–Rho1–Rok–Myosin-II pathway. Indeed, our research here together with this previous study suggests that a positive feedback loop between JNK activation and Myosin II activity might be critical downstream of cell morphology or polarity disruption for cooperative tumorigenesis with oncogenic Ras.
The involvement of RhoGEF2, Rho1, Rok and Myosin II in cooperative tumorigenesis with oncogenic Ras in human cancer
Activation of the Ras signalling pathway occurs in about 30% of all human cancers; however, Ras activation is not sufficient for tumorigenesis because of the induction of cellular senescence (Kern et al., 2011; Serrano et al., 1997). We have identified here that the RhoGEF2–Rho1–Rok–Myosin-II pathway cooperates with oncogenic Ras in Drosophila epithelial tissues, raising the question of whether cooperation occurs between these genes in mammalian cancer. There is strong evidence that overexpression of some mammalian RhoGEFs such as Vav1 (Katzav, 2007) and Ect2 (Fields and Justilien, 2010) contribute to human cancers. Furthermore, the mammalian RhoGEF2 homologues PDZ-RhoGEF, p115-RhoGEF and Leukemia associated RhoGEF (LARG) can transform Swiss NIH 3T3 cells (Fukuhara et al., 2001), and LARG fused to the mixed lineage leukemia (MLL) gene results in acute myeloid leukemia (Kourlas et al., 2000). However, whether Ras is also activated in these cells and contributes to tumorigenesis with the RhoGEFs is not known.
RhoA and Rock (Rok) have been found to be upregulated in many human cancers (reviewed by Karlsson et al., 2009; Narumiya et al., 2009), and upregulation of Rock has been shown to be crucial for RhoA-mediated tumorigenesis (Sahai et al., 1999). Because of this, Rho and Rock have been considered to be good drug targets for cancer therapy (Fritz and Kaina, 2006; Lu et al., 2009; Maruta et al., 2003; Olson, 2008; Rath and Olson, 2012). Whether Ras signalling is also upregulated in cancers in which RhoA or Rock is activated remains to be thoroughly examined. However, in vitro studies have revealed that Rho and Rock cooperate with the Ras-Raf pathway in cellular transformation to promote cell proliferation and motility (Coleman et al., 2004; Fleming et al., 2009; Olson et al., 1998; Sahai et al., 1999; Sahai et al., 2001). Furthermore, activated Rock2 induced epidermal hyperplasia in mice, and after treatment with carcinogens promoted the formation of papillomas and progression to carcinomas (Samuel et al., 2011). The progression of these papillomas to carcinomas is associated with activation of the Ras signalling pathway (Quintanilla et al., 1986), and therefore it is likely that, in this system, the activation of Rock cooperates with activated Ras signalling in tumour progression. Importantly, and consistent with our studies, Samuel et al. showed that the hyperplasia induced by activation of Rock was dependent on Myosin II activity (Samuel et al., 2011). Interestingly, another study reported that actin-myosin contractility can promote K-Ras pathway flux, leading to phosphorylation of Erk; however, this was dependent on Myosin-II light chain kinase, but not Rock (Helfman and Pawlak, 2005). In mammalian cells, activation of Myosin II is not only associated with proliferation (see above), but also cell migration (Clark et al., 2007; Ng et al., 2012). Thus, Myosin II has potentially potent tumorigenic properties that might contribute to Ras-induced mammalian cancer. Whether activation of Myosin II induces JNK to promote these properties in mammalian cells and in human cancer remains to be determined. Thus, further analysis of the mechanism of cooperation between Rock–Myosin-II, JNK activity and oncogenic Ras in mammalian cell models and human cancers is clearly warranted.
MATERIALS AND METHODS
Fly stocks, conditions of culture, overexpression and clonal analysis
Fly stocks used in this study were: UAS-RhoGEF2 (Mulinari et al., 2008); UAS-RasACT (Halfar et al., 2001); UAS-RafGOF (Brand and Perrimon, 1994); UAS-rokCAT [Bloomington #6669 (Verdier et al., 2006)]; UAS-sqhEE [sqhE20E21 (Wang and Riechmann, 2007)]; UAS-zip (Franke et al., 2005); msn-lacZ [msn06946 (Mattila et al., 2005)]; UAS-RhoRNAi (#12734 – VDRC); UAS-Rac1RNAi (#17411 – VDRC); UAS-Rac1N17 (Luo et al., 1994); Rac1, Rac2, mtl (Hakeda-Suzuki et al., 2002); UAS-diaRNAi (#20518 – VDRC); UAS-PKNRNAi (#42927 – VDRC); UAS-LimkRNAi (#25344 – VDRC); UAS-rokRNAi (#3793 – VDRC); UAS-zipRNAi (#7819 – VDRC); UAS-sqhAA (Jordan and Karess, 1997); the Rho sensor was UAS-PKN58A-eGFP (Simões et al., 2006), scrib1 (Bilder and Perrimon, 2000), and the egr homozygous viable alleles, egr1 and egr3 (Igaki et al., 2002; Igaki et al., 2009).
The MARCM (mosaic analysis with repressible cell marker) system (Lee and Luo, 2001) with FRT82B, ey-FLP and UAS-GFP (eyFLP1, UASmCD8-GFP ;; Tub-GAL4 FRT82B Tub-GAL80/TM6B) or with FRT40A, ey-FLP and UAS-GFP (eyFLP1, UASmCD8-GFP; Tub-GAL80 FRT40A/CyO; Tub-GAL4/TM6B) were used to induce GFP-positively marked clones. Stocks containing the relevant UAS-transgenes and either FRT82B or FRT40A were generated for clonal analysis using the MARCM system.
Staged lays were carried out by allowing the females in the cross to lay eggs for 8–12 hours before removing the flies. Vials were aged for 5, 7 or 9 days before larvae were collected for dissection. All flies were raised on a standard semolina agar food at 25°C.
Pupation rate
To determine pupation rate, flies were allowed to lay for 12 hours on apple juice agar plates and larvae were left to develop until early third instar at 25°C. Five replicates of 20 larvae of the relevant genotype were placed into fresh food vials. After 9 days AEL, the number of pupae was counted and used to calculate pupation percentage. Data was analysed using GraphPad Prism 5, using a two-tailed t-test. Error bars represent standard error of the mean (s.e.m.) and the significance was set at P<0.05.
Immunocytochemistry for analysis of Drosophila tissues
For analysis of third instar larval EADs, the discs were dissected in PBS, fixed in 4% PFA, washed in PBT (0.1% Triton X-100) and blocked in PBT + 2% normal goat serum. Antibodies used were: mouse Elav [Developmental Studies Hybridoma Bank (DSHB); at 1:20], mouse Mmp1 [DSHB (mouse 5H7B11, 3D8D12, 316B4; at 1/20)], rabbit anti-RhoGEF2 [(Rogers et al., 2004); at 1:200] and mouse β-galactosidase (Rockland; at 1:500). Secondary antibodies were: anti-mouse Alexa-Fluor-647 (Invitrogen; 1:400) or anti-mouse Alexa-Fluor-488 (Invitrogen; 1:400). F-actin was detected with phalloidin-tetramethylrhodamine isothiocyanate (Rhodamine; Sigma; 0.3 mM).
Quantification of F-actin, Mmp1 and Rho1 activity levels
Images of EADs were captured on the Olympus FV Laser Scanning confocal microscope. Multiple EADs were imaged for every genotype. Three clones per EAD were randomly selected and the average pixel intensity in the F-actin, Mmp1 or Rho1 channel was measured using Adobe Photoshop CS3 in two 50×−50 pixel areas, one on either side of the clonal boundary. The average pixel intensity within each clone was normalised using the average pixel intensity outside of that clone. At least three EADs were analysed for each sample. For F-actin measurements, regions of mutant tissue containing cysts, with very high F-actin levels and different morphology compared with the surrounding wild-type tissue, were not used for the measurements. These data for the various samples were then compared using GraphPad Prism 5, using t-tests or ANOVA as indicated. Error bars represent s.e.m. and the significance was set at P<0.05.
Quantification of β-gal intensity in clones
β-gal intensity of mutant clones was measured by MetaMorph Version 7.7.2.0 after images were captured on the Olympus FV laser scanning confocal microscope. Measurements of β-gal intensity in mutant clones were taken from a ventral or dorsal area of 50×−50 pixels for each EAD section, for at least nine EADs per sample. Only average intensity values over a base line threshold of arbitrary units were recorded. Each measurement was normalised to adjacent wild-type tissue of the same pixel area and expressed as a ratio. The average intensity of β-gal for each sample was compared with others using GraphPad Prism 5, using a two-tailed t-test. Error bars represent s.e.m. and the significance was set at P<0.05.
TUNEL staining and quantification, and clonal size measurements
TUNEL staining was carried out as described in the manufacturer’s protocol (Roche Applied Science). Maximum projections were generated using Fluoview 1.7 Software. Quantification of TUNEL staining was measured by Adobe Photoshop CS5. The number of red pixels was divided by the total area of the EAD as counted in pixels and expressed as a percentage. For determining the clonal size, the area of GFP-marked clones relative to the total area of the EAD was calculated for each sample, averaged for each genotype and expressed as a percentage. At least five EADs were analysed for each sample. These data for the various samples were then compared using GraphPad Prism 5. Error bars represent s.e.m. and the significance was set at P<0.05.
RNA extraction and quantitative real-time PCR
Total RNA was harvested from third instar Drosophila larvae by homogenising cells with TRIzol reagent according to the manufacturer’s instructions (Invitrogen). cDNA was created from 2 μg total RNA using random primers and M-MLV reverse transcriptase RNase H (Promega), and amplified by PCR in triplicate with SYBR Green® dye detection method (Agilent Technologies) as per the manufacturer’s specifications. PCR product detection, using the Limk primers listed below, was performed on the ABI PRISM 7700 system (Applied Biosystems), under standard conditions and normalised with a GFP control.
Primers used were: Limk forward 5′-TCGGACTTCAGTCTCAATCAG-3′; Limk reverse 5′-GGGATTAAGATCACAGCACAC-3′.
Supplementary Material
Acknowledgments
We are grateful to Linda Parsons for critical review of the manuscript, and to all members of the Richardson lab for helpful discussions during the course of this work. We thank Steven Rogers, Udo Hacker, Dan Kiehart, Barry Dickson, the Bloomington Stock Center and the Developmental Studies Hybridoma Bank for fly stocks and antibodies. We acknowledge the Flybase website resource, which greatly facilitated this study.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
H.E.R. and A.M.B. conceived the study and designed the experiments. P.K. and K.A. carried out all experiments and A.M.B. and H.E.R. assisted with stock generation. P.K., K.A., H.E.R. and A.M.B. interpreted the data. L.W. carried out pixel intensity measurements, comparisons and statistical analysis. H.E.R. and P.K. wrote the paper, K.A. and L.W. contributing to the method descriptions, P.K., K.A., L.W. and H.E.R. prepared the figures, and A.M.B. contributed editorial guidance. All authors read and approved the final manuscript.
FUNDING
This work was supported by the National Health & Medical Research (NHMRC) grants 350369 and 400211, The Oncology Childhood Foundation (OCF), and funds from the Peter MacCallum Cancer Centre (PMCC) to H.E.R. P.K. was supported by an Australian Postgraduate Award, K.A. from the OCF, L.W. from the PMCC and A.M.B. from NHMRC 350396 and 509051 grants, and H.E.R. by a NHMRC Senior Research Fellowship.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.010066/-/DC1
REFERENCES
- Amano M., Nakayama M., Kaibuchi K. (2010). Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 67, 545–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assoian R. K., Klein E. A. (2008). Growth control by intracellular tension and extracellular stiffness. Trends Cell Biol. 18, 347–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S. H., Kwon Y.-C., Lee H., Choe K.-M. (2010). Rho-family small GTPases are required for cell polarization and directional sensing in Drosophila wound healing. Biochem. Biophys. Res. Commun. 394, 488–492 [DOI] [PubMed] [Google Scholar]
- Barrett K., Leptin M., Settleman J. (1997). The Rho GTPase and a putative RhoGEF mediate a signaling pathway for the cell shape changes in Drosophila gastrulation. Cell 91, 905–915 [DOI] [PubMed] [Google Scholar]
- Bilder D., Perrimon N. (2000). Localization of apical epithelial determinants by the basolateral PDZ protein Scribble. Nature 403, 676–680 [DOI] [PubMed] [Google Scholar]
- Brand A. H., Perrimon N. (1994). Raf acts downstream of the EGF receptor to determine dorsoventral polarity during Drosophila oogenesis. Genes Dev. 8, 629–639 [DOI] [PubMed] [Google Scholar]
- Brumby A. M., Richardson H. E. (2003). scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 22, 5769–5779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumby A. M., Richardson H. E. (2005). Using Drosophila melanogaster to map human cancer pathways. Nat. Rev. Cancer 5, 626–639 [DOI] [PubMed] [Google Scholar]
- Brumby A. M., Goulding K. R., Schlosser T., Loi S., Galea R., Khoo P., Bolden J. E., Aigaki T., Humbert P. O., Richardson H. E. (2011). Identification of novel Ras-cooperating oncogenes in Drosophila melanogaster: a RhoGEF/Rho-family/JNK pathway is a central driver of tumorigenesis. Genetics 188, 105–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K., Langeslag M., Figdor C. G., van Leeuwen F. N. (2007). Myosin II and mechanotransduction: a balancing act. Trends Cell Biol. 17, 178–186 [DOI] [PubMed] [Google Scholar]
- Coleman M. L., Marshall C. J., Olson M. F. (2004). RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat. Rev. Mol. Cell Biol. 5, 355–366 [DOI] [PubMed] [Google Scholar]
- Cordenonsi M., Zanconato F., Azzolin L., Forcato M., Rosato A., Frasson C., Inui M., Montagner M., Parenti A. R., Poletti A., et al. (2011). The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 147, 759–772 [DOI] [PubMed] [Google Scholar]
- Cordero J. B., Macagno J. P., Stefanatos R. K., Strathdee K. E., Cagan R. L., Vidal M. (2010). Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev. Cell 18, 999–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawes-Hoang R. E., Parmar K. M., Christiansen A. E., Phelps C. B., Brand A. H., Wieschaus E. F. (2005). folded gastrulation, cell shape change and the control of myosin localization. Development 132, 4165–4178 [DOI] [PubMed] [Google Scholar]
- Derivery E., Gautreau A. (2010). Generation of branched actin networks: assembly and regulation of the N-WASP and WAVE molecular machines. Bioessays 32, 119–131 [DOI] [PubMed] [Google Scholar]
- Doggett K., Grusche F. A., Richardson H. E., Brumby A. M. (2011). Loss of the Drosophila cell polarity regulator Scribbled promotes epithelial tissue overgrowth and cooperation with oncogenic Ras-Raf through impaired Hippo pathway signaling. BMC Dev. Biol. 11, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S., et al. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 [DOI] [PubMed] [Google Scholar]
- Faulstich H., Trischmann H., Mayer D. (1983). Preparation of tetramethylrhodaminyl-phalloidin and uptake of the toxin into short-term cultured hepatocytes by endocytosis. Exp. Cell Res. 144, 73–82 [DOI] [PubMed] [Google Scholar]
- Fernández B. G., Gaspar P., Brás-Pereira C., Jezowska B., Rebelo S. R., Janody F. (2011). Actin-Capping Protein and the Hippo pathway regulate F-actin and tissue growth in Drosophila. Development 138, 2337–2346 [DOI] [PubMed] [Google Scholar]
- Fields A. P., Justilien V. (2010). The guanine nucleotide exchange factor (GEF) Ect2 is an oncogene in human cancer. Adv. Enzyme Regul. 50, 190–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming Y. M., Ferguson G. J., Spender L. C., Larsson J., Karlsson S., Ozanne B. W., Grosse R., Inman G. J. (2009). TGF-beta-mediated activation of RhoA signalling is required for efficient (V12)HaRas and (V600E)BRAF transformation. Oncogene 28, 983–993 [DOI] [PubMed] [Google Scholar]
- Franke J. D., Montague R. A., Kiehart D. P. (2005). Nonmuscle myosin II generates forces that transmit tension and drive contraction in multiple tissues during dorsal closure. Curr. Biol. 15, 2208–2221 [DOI] [PubMed] [Google Scholar]
- Fritz G., Kaina B. (2006). Rho GTPases: promising cellular targets for novel anticancer drugs. Curr. Cancer Drug Targets 6, 1–14 [PubMed] [Google Scholar]
- Fukuhara S., Chikumi H., Gutkind J. S. (2001). RGS-containing RhoGEFs: the missing link between transforming G proteins and Rho? Oncogene 20, 1661–1668 [DOI] [PubMed] [Google Scholar]
- Glise B., Noselli S. (1997). Coupling of Jun amino-terminal kinase and Decapentaplegic signaling pathways in Drosophila morphogenesis. Genes Dev. 11, 1738–1747 [DOI] [PubMed] [Google Scholar]
- Grosshans J., Wenzl C., Herz H.-M., Bartoszewski S., Schnorrer F., Vogt N., Schwarz H., Müller H.-A. (2005). RhoGEF2 and the formin Dia control the formation of the furrow canal by directed actin assembly during Drosophila cellularisation. Development 132, 1009–1020 [DOI] [PubMed] [Google Scholar]
- Grzeschik N. A., Parsons L. M., Richardson H. E. (2010). Lgl, the SWH pathway and tumorigenesis: It’s a matter of context and competition! Cell Cycle 9, 3202–3212 [DOI] [PubMed] [Google Scholar]
- Guruharsha K. G., Rual J. F., Zhai B., Mintseris J., Vaidya P., Vaidya N., Beekman C., Wong C., Rhee D. Y., Cenaj O., et al. (2011). A protein complex network of Drosophila melanogaster. Cell 147, 690–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häcker U., Perrimon N. (1998). DRhoGEF2 encodes a member of the Dbl family of oncogenes and controls cell shape changes during gastrulation in Drosophila. Genes Dev. 12, 274–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker B. M., Tomlinson J. E., Wayman G. A., Sultana R., Chan G., Villacres E., Disteche C., Storm D. R. (1998). Cloning, chromosomal mapping, and regulatory properties of the human type 9 adenylyl cyclase (ADCY9). Genomics 50, 97–104 [DOI] [PubMed] [Google Scholar]
- Hakeda-Suzuki S., Ng J., Tzu J., Dietzl G., Sun Y., Harms M., Nardine T., Luo L., Dickson B. J. (2002). Rac function and regulation during Drosophila development. Nature 416, 438–442 [DOI] [PubMed] [Google Scholar]
- Halfar K., Rommel C., Stocker H., Hafen E. (2001). Ras controls growth, survival and differentiation in the Drosophila eye by different thresholds of MAP kinase activity. Development 128, 1687–1696 [DOI] [PubMed] [Google Scholar]
- Halsell S. R., Chu B. I., Kiehart D. P. (2000). Genetic analysis demonstrates a direct link between rho signaling and nonmuscle myosin function during Drosophila morphogenesis. Genetics 156, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R. A. (2000). The hallmarks of cancer. Cell 100, 57–70 [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- Helfman D. M., Pawlak G. (2005). Myosin light chain kinase and acto-myosin contractility modulate activation of the ERK cascade downstream of oncogenic Ras. J. Cell. Biochem. 95, 1069–1080 [DOI] [PubMed] [Google Scholar]
- Hou X. S., Goldstein E. S., Perrimon N. (1997). Drosophila Jun relays the Jun amino-terminal kinase signal transduction pathway to the Decapentaplegic signal transduction pathway in regulating epithelial cell sheet movement. Genes Dev. 11, 1728–1737 [DOI] [PubMed] [Google Scholar]
- Igaki T., Kanda H., Yamamoto-Goto Y., Kanuka H., Kuranaga E., Aigaki T., Miura M. (2002). Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 21, 3009–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T., Pastor-Pareja J. C., Aonuma H., Miura M., Xu T. (2009). Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev. Cell 16, 458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe A. B., Hall A. (2005). Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- Jiang Y., Scott K. L., Kwak S. J., Chen R., Mardon G. (2011). Sds22/PP1 links epithelial integrity and tumor suppression via regulation of myosin II and JNK signaling. Oncogene 30, 3248–3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johndrow J. E., Magie C. R., Parkhurst S. M. (2004). Rho GTPase function in flies: insights from a developmental and organismal perspective. Biochem. Cell Biol. 82, 643–657 [DOI] [PubMed] [Google Scholar]
- Jordan P., Karess R. (1997). Myosin light chain-activating phosphorylation sites are required for oogenesis in Drosophila. J. Cell Biol. 139, 1805–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda H., Igaki T., Okano H., Miura M. (2011). Conserved metabolic energy production pathways govern Eiger/TNF-induced nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 108, 18977–18982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F. D., Rubin G. M. (1998). Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development 125, 1–9 [DOI] [PubMed] [Google Scholar]
- Karlsson R., Pedersen E. D., Wang Z., Brakebusch C. (2009). Rho GTPase function in tumorigenesis. Biochim. Biophys. Acta 1796, 91–98 [DOI] [PubMed] [Google Scholar]
- Katzav S. (2007). Flesh and blood: the story of Vav1, a gene that signals in hematopoietic cells but can be transforming in human malignancies. Cancer Lett. 255, 241–254 [DOI] [PubMed] [Google Scholar]
- Kern F., Niault T., Baccarini M. (2011). Ras and Raf pathways in epidermis development and carcinogenesis. Br. J. Cancer 104, 229–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner J., Gross S., Bennett D., Alphey L. (2007). The nonmuscle myosin phosphatase PP1beta (flapwing) negatively regulates Jun N-terminal kinase in wing imaginal discs of Drosophila. Genetics 175, 1741–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourlas P. J., Strout M. P., Becknell B., Veronese M. L., Croce C. M., Theil K. S., Krahe R., Ruutu T., Knuutila S., Bloomfield C. D., et al. (2000). Identification of a gene at 11q23 encoding a guanine nucleotide exchange factor: evidence for its fusion with MLL in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 97, 2145–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Külshammer E., Uhlirova M. (2012). The actin cross-linker Filamin/Cheerio mediates tumor malignancy downstream of JNK signaling. J. Cell Sci. [Epub ahead of print] doi: 10.1242/jcs.114462 [DOI] [PubMed] [Google Scholar]
- Kwon Y.-C., Baek S. H., Lee H., Choe K.-M. (2010). Nonmuscle myosin II localization is regulated by JNK during Drosophila larval wound healing. Biochem. Biophys. Res. Commun. 393, 656–661 [DOI] [PubMed] [Google Scholar]
- Lee T., Luo L. (2001). Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 24, 251–254 [DOI] [PubMed] [Google Scholar]
- Leong G. R., Goulding K. R., Amin N., Richardson H. E., Brumby A. M. (2009). Scribble mutants promote aPKC and JNK-dependent epithelial neoplasia independently of Crumbs. BMC Biol. 7, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leptin M. (1999). Gastrulation in Drosophila: the logic and the cellular mechanisms. EMBO J. 18, 3187–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolo F. N., Casas-Tintó S., Moreno E. (2012). Cell competition time line: winners kill losers, which are extruded and engulfed by hemocytes. Cell Rep. 2, 526–539 [DOI] [PubMed] [Google Scholar]
- Lu Y., Settleman J. (1999). The Drosophila Pkn protein kinase is a Rho/Rac effector target required for dorsal closure during embryogenesis. Genes Dev. 13, 1168–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q., Longo F. M., Zhou H., Massa S. M., Chen Y. H. (2009). Signaling through Rho GTPase pathway as viable drug target. Curr. Med. Chem. 16, 1355–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L., Liao Y. J., Jan L. Y., Jan Y. N. (1994). Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev. 8, 1787–1802 [DOI] [PubMed] [Google Scholar]
- Maekawa M., Ishizaki T., Boku S., Watanabe N., Fujita A., Iwamatsu A., Obinata T., Ohashi K., Mizuno K., Narumiya S. (1999). Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285, 895–898 [DOI] [PubMed] [Google Scholar]
- Mammoto A., Ingber D. E. (2009). Cytoskeletal control of growth and cell fate switching. Curr. Opin. Cell Biol. 21, 864–870 [DOI] [PubMed] [Google Scholar]
- Maruta H., Nheu T. V., He H., Hirokawa Y. (2003). Rho family-associated kinases PAK1 and rock. Prog. Cell Cycle Res. 5, 203–210 [PubMed] [Google Scholar]
- Massarwa R., Schejter E. D., Shilo B.-Z. (2009). Apical secretion in epithelial tubes of the Drosophila embryo is directed by the Formin-family protein Diaphanous. Dev. Cell 16, 877–888 [DOI] [PubMed] [Google Scholar]
- Mattila J., Omelyanchuk L., Kyttälä S., Turunen H., Nokkala S. (2005). Role of Jun N-terminal Kinase (JNK) signaling in the wound healing and regeneration of a Drosophila melanogaster wing imaginal disc. Int. J. Dev. Biol. 49, 391–399 [DOI] [PubMed] [Google Scholar]
- Mulinari S., Barmchi M. P., Häcker U. (2008). DRhoGEF2 and diaphanous regulate contractile force during segmental groove morphogenesis in the Drosophila embryo. Mol. Biol. Cell 19, 1883–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumiya S., Tanji M., Ishizaki T. (2009). Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 28, 65–76 [DOI] [PubMed] [Google Scholar]
- Neisch A. L., Speck O., Stronach B., Fehon R. G. (2010). Rho1 regulates apoptosis via activation of the JNK signaling pathway at the plasma membrane. J. Cell Biol. 189, 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C. M., Jean R. P., Tan J. L., Liu W. F., Sniadecki N. J., Spector A. A., Chen C. S. (2005). Emergent patterns of growth controlled by multicellular form and mechanics. Proc. Natl. Acad. Sci. USA 102, 11594–11599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M. R., Besser A., Danuser G., Brugge J. S. (2012). Substrate stiffness regulates cadherin-dependent collective migration through myosin-II contractility. J. Cell Biol. 199, 545–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K., Hosoya T., Takahashi K., Hing H., Mizuno K. (2000a). A Drosophila homolog of LIM-kinase phosphorylates cofilin and induces actin cytoskeletal reorganization. Biochem. Biophys. Res. Commun. 276, 1178–1185 [DOI] [PubMed] [Google Scholar]
- Ohashi K., Nagata K., Maekawa M., Ishizaki T., Narumiya S., Mizuno K. (2000b). Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J. Biol. Chem. 275, 3577–3582 [DOI] [PubMed] [Google Scholar]
- Ohsawa S., Sugimura K., Takino K., Xu T., Miyawaki A., Igaki T. (2011). Elimination of oncogenic neighbors by JNK-mediated engulfment in Drosophila. Dev. Cell 20, 315–328 [DOI] [PubMed] [Google Scholar]
- Ohsawa S., Sato Y., Enomoto M., Nakamura M., Betsumiya A., Igaki T. (2012). Mitochondrial defect drives non-autonomous tumour progression through Hippo signalling in Drosophila. Nature 490, 547–551 [DOI] [PubMed] [Google Scholar]
- Olson M. F. (2008). Applications for ROCK kinase inhibition. Curr. Opin. Cell Biol. 20, 242–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson M. F., Ashworth A., Hall A. (1995). An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 269, 1270–1272 [DOI] [PubMed] [Google Scholar]
- Olson M. F., Paterson H. F., Marshall C. J. (1998). Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature 394, 295–299 [DOI] [PubMed] [Google Scholar]
- Pagliarini R. A., Xu T. (2003). A genetic screen in Drosophila for metastatic behavior. Science 302, 1227–1231 [DOI] [PubMed] [Google Scholar]
- Perrimon N., Lanjuin A., Arnold C., Noll E. (1996). Zygotic lethal mutations with maternal effect phenotypes in Drosophila melanogaster. II. Loci on the second and third chromosomes identified by P-element-induced mutations. Genetics 144, 1681–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla M., Brown K., Ramsden M., Balmain A. (1986). Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 322, 78–80 [DOI] [PubMed] [Google Scholar]
- Rath N., Olson M. F. (2012). Rho-associated kinases in tumorigenesis: reconsidering ROCK inhibition for cancer therapy. EMBO Rep. 13, 900–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson H. E. (2011). Actin up for Hippo. EMBO J. 30, 2307–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinow S., White K. (1991). Characterization and spatial distribution of the ELAV protein during Drosophila melanogaster development. J. Neurobiol. 22, 443–461 [DOI] [PubMed] [Google Scholar]
- Rogers S. L., Wiedemann U., Häcker U., Turck C., Vale R. D. (2004). Drosophila RhoGEF2 associates with microtubule plus ends in an EB1-dependent manner. Curr. Biol. 14, 1827–1833 [DOI] [PubMed] [Google Scholar]
- Rossman K. L., Der C. J., Sondek J. (2005). GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- Sahai E., Ishizaki T., Narumiya S., Treisman R. (1999). Transformation mediated by RhoA requires activity of ROCK kinases. Curr. Biol. 9, 136–145 [DOI] [PubMed] [Google Scholar]
- Sahai E., Olson M. F., Marshall C. J. (2001). Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO J. 20, 755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel M. S., Olson M. F. (2011). Actomyosin contractililty: force power drives tumor growth. Cell Cycle 10, 3409–3410 [DOI] [PubMed] [Google Scholar]
- Samuel M. S., Lopez J. I., McGhee E. J., Croft D. R., Strachan D., Timpson P., Munro J., Schröder E., Zhou J., Brunton V. G., et al. (2011). Actomyosin-mediated cellular tension drives increased tissue stiffness and β-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 19, 776–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansores-Garcia L., Bossuyt W., Wada K., Yonemura S., Tao C., Sasaki H., Halder G. (2011). Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J. 30, 2325–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M., Lin A. W., McCurrach M. E., Beach D., Lowe S. W. (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 [DOI] [PubMed] [Google Scholar]
- Settleman J. (2001). Rac ’n Rho: the music that shapes a developing embryo. Dev. Cell 1, 321–331 [DOI] [PubMed] [Google Scholar]
- Shlevkov E., Morata G. (2012). A dp53/JNK-dependant feedback amplification loop is essential for the apoptotic response to stress in Drosophila. Cell Death Differ. 19, 451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões S., Denholm B., Azevedo D., Sotillos S., Martin P., Skaer H., Hombría J. C.-G., Jacinto A. (2006). Compartmentalisation of Rho regulators directs cell invagination during tissue morphogenesis. Development 133, 4257–4267 [DOI] [PubMed] [Google Scholar]
- Sun G., Irvine K. D. (2011). Regulation of Hippo signaling by Jun kinase signaling during compensatory cell proliferation and regeneration, and in neoplastic tumors. Dev. Biol. 350, 139–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanowska J. (2009). Involvement of Rac/Cdc42/PAK pathway in cytoskeletal rearrangements. Acta Biochim. Pol. 56, 225–234 [PubMed] [Google Scholar]
- Uhlirova M., Bohmann D. (2006). JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 25, 5294–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Impel A., Schumacher S., Draga M., Herz H.-M., Grosshans J., Müller H. A. J. (2009). Regulation of the Rac GTPase pathway by the multifunctional Rho GEF Pebble is essential for mesoderm migration in the Drosophila gastrula. Development 136, 813–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdier V., Guang-Chao-Chen, Settleman J. (2006). Rho-kinase regulates tissue morphogenesis via non-muscle myosin and LIM-kinase during Drosophila development. BMC Dev. Biol. 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M., Larson D. E., Cagan R. L. (2006). Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev. Cell 10, 33–44 [DOI] [PubMed] [Google Scholar]
- Vincent S., Settleman J. (1997). The PRK2 kinase is a potential effector target of both Rho and Rac GTPases and regulates actin cytoskeletal organization. Mol. Cell. Biol. 17, 2247–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B., Kinzler K. W. (1993). The multistep nature of cancer. Trends Genet. 9, 138–141 [DOI] [PubMed] [Google Scholar]
- Wada K., Itoga K., Okano T., Yonemura S., Sasaki H. (2011). Hippo pathway regulation by cell morphology and stress fibers. Development 138, 3907–3914 [DOI] [PubMed] [Google Scholar]
- Wang Y., Riechmann V. (2007). The role of the actomyosin cytoskeleton in coordination of tissue growth during Drosophila oogenesis. Curr. Biol. 17, 1349–1355 [DOI] [PubMed] [Google Scholar]
- Wang M. C., Bohmann D., Jasper H. (2003). JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev. Cell 5, 811–816 [DOI] [PubMed] [Google Scholar]
- Warner S. J., Longmore G. D. (2009). Distinct functions for Rho1 in maintaining adherens junctions and apical tension in remodeling epithelia. J. Cell Biol. 185, 1111–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner S. J., Yashiro H., Longmore G. D. (2010). The Cdc42/Par6/aPKC polarity complex regulates poptosis-induced compensatory proliferation in epithelia. Curr. Biol. 20, 677–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann T. J., Dahmann C. (2009). Dpp signaling promotes the cuboidal-to-columnar shape transition of Drosophila wing disc epithelia by regulating Rho1. J. Cell Sci. 122, 1362–1373 [DOI] [PubMed] [Google Scholar]
- Winter C. G., Wang B., Ballew A., Royou A., Karess R., Axelrod J. D., Luo L. (2001). Drosophila Rho-associated kinase (Drok) links Frizzled-mediated planar cell polarity signaling to the actin cytoskeleton. Cell 105, 81–91 [DOI] [PubMed] [Google Scholar]
- Woolner S., Jacinto A., Martin P. (2005). The small GTPase Rac plays multiple roles in epithelial sheet fusion – dynamic studies of Drosophila dorsal closure. Dev. Biol. 282, 163–173 [DOI] [PubMed] [Google Scholar]
- Xu N., Keung B., Myat M. M. (2008). Rho GTPase controls invagination and cohesive migration of the Drosophila salivary gland through Crumbs and Rho-kinase. Dev. Biol. 321, 88–100 [DOI] [PubMed] [Google Scholar]
- Yan J., Lu Q., Fang X., Adler P. N. (2009). Rho1 has multiple functions in Drosophila wing planar polarity. Dev. Biol. 333, 186–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young P. E., Richman A. M., Ketchum A. S., Kiehart D. P. (1993). Morphogenesis in Drosophila requires nonmuscle myosin heavy chain function. Genes Dev. 7, 29–41 [DOI] [PubMed] [Google Scholar]
- Yu H., Mouw J. K., Weaver V. M. (2011). Forcing form and function: biomechanical regulation of tumor evolution. Trends Cell Biol. 21, 47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M., Xin T., Weng S., Gao Y., Zhang Y., Li Q., Li M. (2010). Activation of JNK signaling links lgl mutations to disruption of the cell polarity and epithelial organization in Drosophila imaginal discs. Cell Res. 20, 242–245 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.