

SUMMARY
Epilepsy is a complex neurological disorder characterized by unprovoked seizures. The etiology is heterogeneous with both genetic and environmental causes. Genes that regulate neurotransmitters and ion channels in the central nervous system have been associated with epilepsy. However, a recent screening in human epilepsy patients identified mutations in the PRICKLE1 (PK1) locus, highlighting a potentially novel mechanism underlying seizures. PK1 is a core component of the planar cell polarity network that regulates tissue polarity. Zebrafish studies have shown that Pk1 coordinates cell movement, neuronal migration and axonal outgrowth during embryonic development. Yet how dysfunction of Pk1 relates to epilepsy is unknown. To address the mechanism underlying epileptogenesis, we used zebrafish to characterize Pk1a function and epilepsy-related mutant forms. We show that knockdown of pk1a activity sensitizes zebrafish larva to a convulsant drug. To model defects in the central nervous system, we used the retina and found that pk1a knockdown induces neurite outgrowth defects; yet visual function is maintained. Furthermore, we characterized the functional and biochemical properties of the PK1 mutant forms identified in human patients. Functional analyses demonstrate that the wild-type Pk1a partially suppresses the gene knockdown retinal defects but not the mutant forms. Biochemical analysis reveals increased ubiquitylation of one mutant form and decreased translational efficiency of another mutant form compared with the wild-type Pk1a. Taken together, our results indicate that mutation of human PK1 could lead to defects in neurodevelopment and signal processing, providing insight into seizure predisposition in these patients.
INTRODUCTION
Epilepsy is a chronic condition of unpredictable and recurrent seizures produced by abnormal neuronal activity in the central nervous system (CNS). The etiology of epilepsy is complex, yet has a strong genetic component. A variety of genes, including those that regulate neuronal differentiation, morphology, excitability and signaling, as well as genes of unknown function, have been associated with epilepsy (Wang et al., 2010b). Elucidating the functions of these genes would provide a better understanding of the disease cause and could potentially contribute to clinical therapy.
A genetic screen in epilepsy patients identified mutations in the PRICKLE locus (PK1 and PK2), suggesting an association of PK with epilepsy (Bassuk et al., 2008; Tao et al., 2011). PK is a core component of the planar cell polarity (PCP) pathway that regulates cell movement and tissue polarity. Loss of pk in Drosophila leads to disruption of epithelial planar cell polarity in the wing and ommatidia (Tree et al., 2002; Jenny et al., 2003; Jenny et al., 2005). Pk mutant mice showed decreased seizure threshold following both electrical and convulsant drug induction (Tao et al., 2011). Drosophila pk mutants (spinylegs, sple) likewise display seizure-like behaviors and, interestingly, the seizure-like behaviors can be suppressed by the anti-epileptic drug valproic acid (Tao et al., 2011). The genetic causes of epilepsy have been linked largely to genes encoding for ion channels or neurotransmitter receptors (Mulley et al., 2003; Poduri and Lowenstein, 2011). Understanding how PK dysfunction relates to seizures could elucidate previously unknown mechanisms underlying epilepsy.
In vertebrates and chordates, pk modulates directed cell movement during early embryonic development. At gastrulation stages, sheets of cells undergo convergence extension (CE) movements to form the anterior-posterior axis (A–P axis), and manipulating pk results in CE defects and shorter A–P axis (Carreira-Barbosa et al., 2003; Takeuchi et al., 2003; Veeman et al., 2003; Jiang et al., 2005). Apart from the role in polarity establishment, PCP proteins also regulate axon outgrowth and neuronal migration (Wada et al., 2006; Goodrich, 2008; Zhou et al., 2008; Tissir and Goffinet, 2010; Ng, 2012). Drosophila pk promotes sensory axon outgrowth, suggesting a role as a directional cue for axons (Mrkusich et al., 2011). pk function is required for the migration of facial branchiomotor neurons in zebrafish (Carreira-Barbosa et al., 2003; Bingham et al., 2010; Mapp et al., 2010; Mapp et al., 2011). Although patients with PK mutations present with a seizure disorder, they do not display severe developmental defects and have normal brain MRIs (Bassuk et al., 2008; Tao et al., 2011). These observations suggest that PK1 mutations support general CNS development but could lead to subtle abnormalities in neuronal migration or outgrowth, disrupting signal processing.
Pk1a protein contains PET (Prickle Espinas Testin) and LIM (Lin-1, Isl-1 and Mec-3) protein-protein interaction domains. PK1 mutations identified in human epilepsy patients are point mutations that lead to single amino acid changes (Bassuk et al., 2008; Tao et al., 2011). These changes are found across the entire protein instead of clustering within a specific domain. However, how these mutants differ from the wild type is not fully understood. In previous studies, we have overexpressed mutant Pk1a and Pk2 forms in zebrafish to evaluate their pathogenicity. Pk1 and Pk2 overexpression induces CE defects and intracellular calcium release (Veeman et al., 2003; Tao et al., 2011) and the mutants showed altered activities compared with wild-type Pk (Bassuk et al., 2008; Tao et al., 2011). A similar approach has been used for rare Pk missense mutations associated with neural tube defects (Bosoi et al., 2011). Whereas the overexpression assays have revealed functional differences, biochemical changes in the mutant forms have not been evaluated. Moreover, a partial-knockdown context might better resemble the physiological state in human patients with heterozygous mutations. Thus, we set out to investigate the necessary role of zebrafish Pk1a in neural development and to characterize the biochemical properties of the mutant forms.
TRANSLATIONAL IMPACT.
Clinical issue
Epilepsy is a chronic condition characterized by recurrent, spontaneous seizures. Although epilepsy can be caused by non-genetic factors including stroke, traumatic brain injury and brain infection, genetic causes make a significant contribution to epilepsy development. Mutations in genes that regulate ion channels and neurotransmitters are shown to be frequently involved in epileptogenesis. Understanding the underlying genetics and pathophysiology of epilepsy is necessary to inform therapies that could reduce or offset the incidence of seizures.
Results
Mutations at the human PRICKLE (PK) locus are associated with epilepsy. To investigate the mechanistic role of PK in epilepsy, the authors characterized the functions of wild-type and epilepsy-related mutants in a zebrafish model. Using a zebrafish larva motility assay, the authors showed that the convulsant drug PTZ induces hyperactivity in larva swimming; however, larva with pk1a gene knockdown were found to be more sensitive to PTZ compared with the control. The protein encoded by the PK1a gene, PK1a, is a planar cell polarity protein that regulates tissue polarity and directed cell movements and is involved in neuronal migration and outgrowth. To probe the role of PK1a in neural development, the authors examined neuronal outgrowth in a zebrafish retinal model. Their analysis revealed a necessary role for PK1a in neurite organization in the retina inner plexiform layer. Because the mutations identified in human patients are not clustered in a single domain but are distributed across the protein, the authors tested the different mutant forms for loss of function. Although wild-type PK1a overexpression in the zebrafish embryos was able to partially suppress the neuronal outgrowth defect, mutant forms were found to be less active. In addition, biochemical analysis showed that one of the mutant forms of PK1a, R150H, was more strongly ubiquitylated than the wild-type or Y465H mutant protein.
Implications and future directions
An association of PK with epilepsy has been described in fly and mouse models, but the underlying mechanisms are not well understood. Here, the authors demonstrated the utility of a zebrafish larva epilepsy model and identified novel functions of PK in retinal neurogenesis. Mutations in PK might contribute to epilepsy via its necessary roles in neuronal outgrowth during early development. Patients expressing mutant forms that retain partial function can be predisposed to seizures. Future utilization of the motility assay and retina model could enable epilepsy-drug screening.
Zebrafish (Danio rerio) is an emerging model for epilepsy by virtue of its stereotypic behaviors and response to seizure-inducing drugs (Hortopan et al., 2010a; Teng et al., 2010; Teng et al., 2011). Seizure-like behaviors in zebrafish larvae and adults have been characterized by tracking swimming patterns and examining brain electrophysiology (Baraban et al., 2005; Hortopan et al., 2010b). Zebrafish is also amenable to genetic manipulations and to imaging techniques for studying neural development. Here, we use the zebrafish larva as a model and characterize pk1a function in drug-induced seizures. In order to interrogate the role of abnormal pk1a forms in the developing nervous system, we also explore novel aspects of pk1a function in neurite outgrowth in the retina and evaluate biochemical properties of epilepsy-related mutant forms. We confirm the role of pk1a in the sensitivity to drug-induced seizures; show a requirement of pk1a in retina inner plexiform layer (IPL) organization; and find differential processing of wild-type and mutant forms. These results provide insights into the mechanisms by which mutant forms of PK1 might lead to epilepsy.
RESULTS
pk1a knockdown sensitizes zebrafish to PTZ treatment
Pentylenetetrazole (PTZ) is a convulsant drug that induces general motor seizures in human and mouse (Velisek et al., 1992). PTZ also induces seizure-like behaviors, characterized by whirl-like and jerky swimming patterns, as well as electrophysiological changes in adult and larval zebrafish (Baraban et al., 2005; Hortopan et al., 2010b). Knockdown of known epileptogenic genes such as lgi1 (leucine-rich, glioma inactivated 1) in zebrafish leads to sensitized motility in response to PTZ (Teng et al., 2010; Teng et al., 2011). We tracked and quantified total swimming distance in response to exposure to the seizure-inducing drug PTZ (Fig. 1). We monitored the motility of individual larva in a 48-well culture dish by recording 0.5 hours before and 0.5 hours after addition of PTZ to the medium (Fig. 1B). Movement detection software identified ‘small movement’ (0.1–10 cm/second) and ‘large movement’ (above 10cm/second). White areas in Fig 1B are regions of little or no movement (below 0.1 cm/second). The total activity of an individual larva can then be traced within its well. Typically, at 72 hours post fertilization (hpf), wild-type zebrafish displayed small movements around the well (Fig. 1B, top) (supplementary material Movie 1). Upon exposure to PTZ, wild-type zebrafish showed a marked increase in the amount of movement within the well (Fig. 1B, bottom) (supplementary material Movie 2). The total distance travelled during the time course (0.5 hours) can be calculated for each individual larva and then averaged for the entire plate (48 wells). We found increases in total activity associated with increasing doses of PTZ (Fig. 1C).
Fig. 1.

PTZ induces hyperactivity in swimming behavior in a dose-dependent manner. (A) Flow chart of the motility assay and analysis. (B) Representative pictures generated in the Viewpoint Software showing the track of a larva before and after PTZ treatment; total movement over 30 minutes. Red circles depict the shape of the well; green and red lines mark the small and large movements, respectively. (C) Graph of total half-hour activity of larva treated with different doses of PTZ. Data presented as mean + s.e.m.
Pk mutant mice show increased sensitivity to PTZ exposure. To determine whether reduced activity of pk1a sensitizes zebrafish to PTZ treatment, we utilized gene knockdown and PTZ treatment strategies. In order to evaluate the entire range of response, we selected a PTZ dose that induces a mild yet statistically significant increase in activity in wild-type zebrafish (5 mM; Fig. 1C, Fig. 2D). Because pk1a knockdown induces A–P axis defects (Carreira-Barbosa et al., 2003; Veeman et al., 2003), with shorter or curved trunks that would compromise swimming (Fig. 2C), we next identified a sub-phenotypic dose of antisense morpholino-oligonucleotide (MO) directed against pk1a that generates straight-bodied zebrafish to be used for the motility assays (Fig. 2B). We found that the straight-bodied pk1a-MO-injected larvae (morphants) were similar to control morphants in total movement (Fig. 2D, before PTZ). By contrast, after exposure to PTZ, pk1a morphants showed a significantly higher level of activity compared with control morphants (Fig. 2D, after PTZ). To confirm that the sensitivity to PTZ in pk1a morphants is mediated through mechanisms that promote seizures, we tested the ability of the antiepileptic drug valproic acid (VPA) to suppress the increased motility. After pretreatment with VPA for 1 hour before adding PTZ, there was no statistically significant difference between control and pk1a morphants (supplementary material Fig. S1). Taken together, we conclude that pk1a knockdown sensitizes the zebrafish to the seizure-inducing dug PTZ.
Fig. 2.
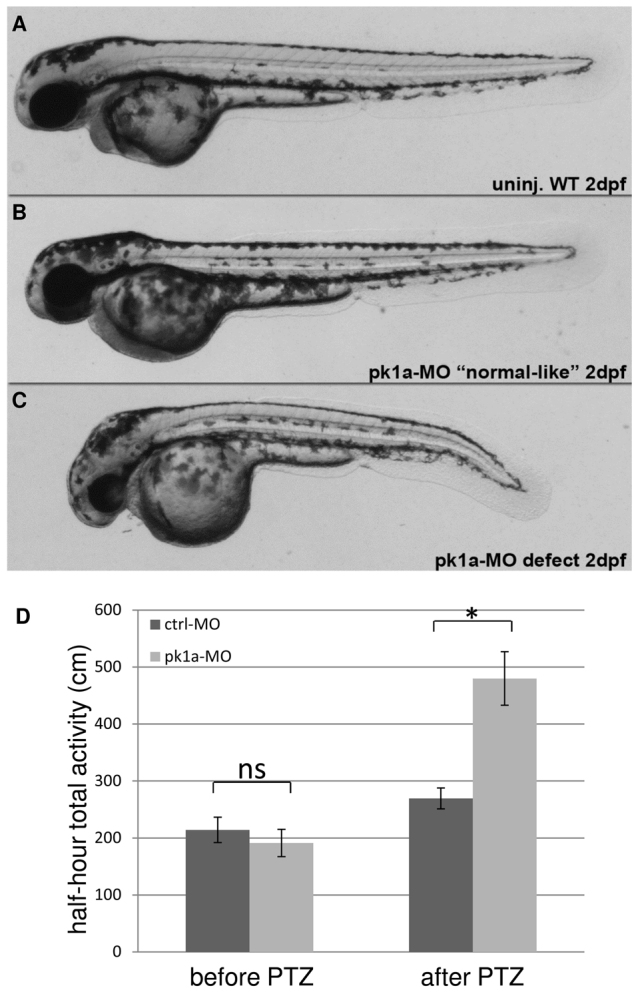
pk1a knockdown sensitizes zebrafish to PTZ treatment. (A–C) Morphology of wild-type and pk1a-MO-injected embryos at 2 dpf: wild-type uninjected embryo (A); pk1a-MO-injected embryos ranging from ‘normal-like’ (B) to curved body axis (C). (D) Graph of total half-hour activity before and after PTZ treatment for larvae injected with control-MO and pk1a-MO. Wilcoxon Rank Sum test found no significant increase (ns) in activity of control-MO and pk1a-MO injected larvae prior to PTZ treatment. There was a significant increase in activity in control after PTZ compared with control before PTZ (P<0.05) and for pk1a morphants after PTZ compared with before PTZ (P<0.05). *P<0.05 is the significant difference between control and pk1a morphant after PTZ treatment. For control-MO, n=48 and for pk1a-MO, n=48. Data presented as mean ± s.e.m.
pk1a knockdown induces neurite outgrowth in the retina
pk has been shown to regulate the migration of facial branchiomotor neurons in the hindbrain (Carreira-Barbosa et al., 2003; Mapp et al., 2010; Mapp et al., 2011; Mrkusich et al., 2011; Sanchez-Alvarez et al., 2011), but little is known about the affects of pk1a dysfunction in the central nervous system (CNS). Given the PTZ-sensitivity in pk1a knockdown embryos, we sought a model to explore mechanisms of pk1a function in CNS development. Because the retina originates as an outgrowth of the brain, it serves as a model for the CNS. Whole mount in situ hybridization of the 3 dpf larval brain shows pk1a expression throughout the brain, with enriched expression in the eye (Fig. 3A,B; supplementary material Fig. S2). The retina is organized in layers with stereotypical cell types and dendritic projections similar to but less complicated than the CNS. We further characterized the expression of pk1a transcript within the eye and find it enriched in the retinal ganglion cell layer (RGC), inner nuclear layer (INL) and the lens at 3 dpf (Fig. 3C,D). pk1a is also expressed in adult retina as determined by RT-PCR (data not shown). To determine whether pk1a is necessary for global patterning of the retina, we performed histological analysis of pk1a morphants by hematoxylin and eosin (H&E) staining. At 3 dpf, pk1a morphants have fully laminated retinas similar to wild-type and control morphants (Fig. 3E–G). The photoreceptor outer segments in the central retina revealed no overt differences compared with wild-type and control morphants (Fig. 3E′–G′). However, the inner segments (Fig. 3E″–G″), in particular the IPL, displayed modest disorganization sometimes with nuclei found within the IPL (Fig. 3G″, arrowhead).
Fig. 3.

pk1a is expressed in the brain and retina and is not required for overall retinal lamination. (A,B) Whole-mount in situ hybridization of 3 dpf embryos. Images are from the dorsal view. An antisense RNA riboprobe is used to determine the expression pattern of pk1a, and a riboprobe from the sense strand is used as a control. (C,D) Sections of the eye after whole-mount in situ hybridization at 3 dpf: sense control (C) and pk1a antisense (D). Arrows show different layers in the retina. (E–G″) H&E staining of eye sections at 76–78 hpf. (E–G) Whole retina sections of uninjected (E), control-MO-injected (F) and pk1a-MO-injected larvae (G); magnification 40×. (E′–G″) Enlarged areas of the white boxes in E–G; the arrowhead in G” indicates nuclei disorganization; magnification 63×. PR, photo-receptors; INL, inner nuclear layer; IPL, inner plexiform layer; RGCs, retina ganglion cells; OPL, outer plexiform layer; ONL, outer nuclear layer. Scale bars: 5 μm.
The IPL is a synaptic layer that bears connections between cells in the ganglion cell layer and inner nuclear layer. To more deeply interrogate the subtle anomalies in IPL histology, we used the transgenic line HuC:gfp that expresses GFP in a subset of neuronal cells, the ganglion cells and amacrine cells in the inner nuclear layer (Link et al., 2000; Park et al., 2000). Both cell types extend their projections into the IPL, forming stratified arborizations that present as two continuous tracks in the HuC:gfp line. MO-injected HuC:gfp transgenic embryos were fixed at 3 dpf and transverse sections from the central retina collected for confocal imaging (Fig. 4A–H). In wild-type and control morphants, synaptic connections in the IPL were readily visible as two stratified tracks (Fig. 4A′,B′). In pk1a morphants, we observed defects in the track organization. About 41% of the pk1a morphants displayed a complete loss of parallel organization of the two stratified tracks and were classified as severe (Fig. 4C′). An additional 14% had regions of discontinuous or disorganized parallel tracks, classified as mild (Fig. 4D′, arrowheads). To verify the specificity of the pk1a knockdown defect, we utilized a second MO directed against pk1a with different target sequences. The second pk1a-MO generated an IPL defect with a penetrance of 69% (all defects were severe) (Fig. 4E,E′).
Fig. 4.
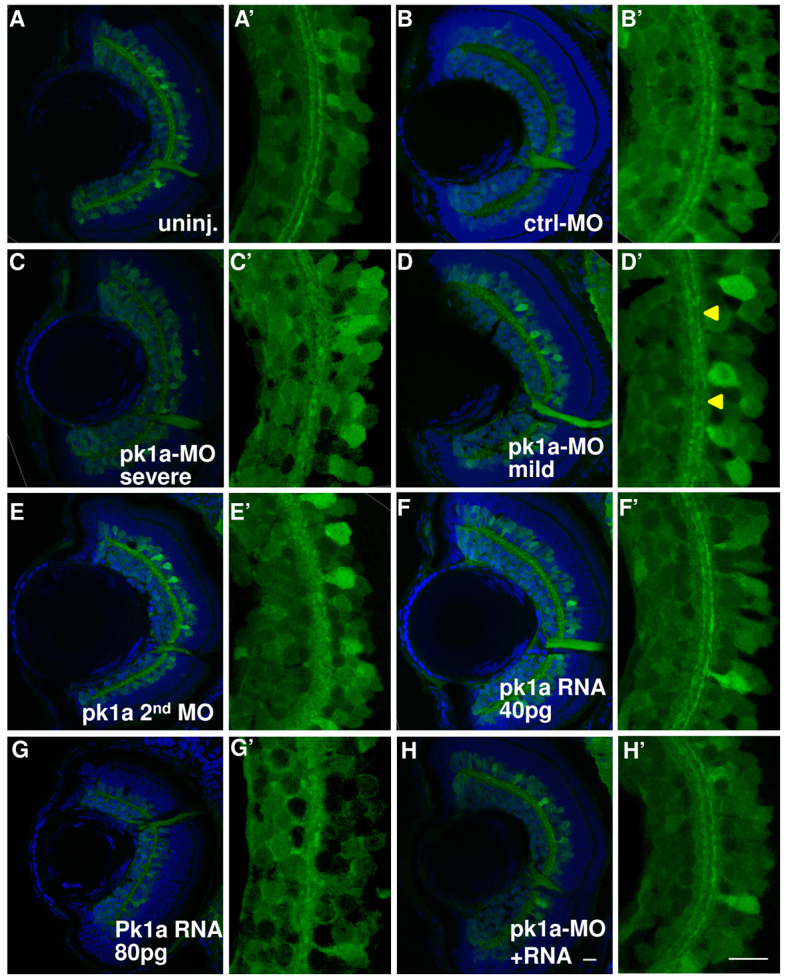
pk1a knockdown induces inner plexiform layer defects. (A–H′) Sections at 76–78 hpf in the HuC:gfp transgenic line. Green channel, GFP; blue channel, To-Pro3 nuclear counter-staining. (A–H) Whole eye visualized at 63×. Representative sections from uninjected embryos (A) and from embryos injected with control-MO (B); pk1a-MO, showing severe IPL defects (C); pk1a-MO, showing mild IPL defects (D); a second pk1a-MO (E); 40 pg pk1a RNA (F); 80 pg pk1a RNA (G); pk1a-MO plus 40 pg pk1a RNA (H). (A′–H′) Digital 3× zoom of the white boxed areas in A–H, showing the green channel only. In the uninjected (A′) and control-MO-injected embryos (B′), there are two clearly stratified tracks. pk1a morphants exhibit either loss of the organization (C′) or discontinuous tracks (arrowheads in D′). A second pk1a-MO also induced similar IPL defects (E′). Although a low dose of pk1a RNA injection does not show a defect (F′), a higher dose does (G′). The defects can be partially suppressed by a low dose pk1a RNA co-expression (H′). Scale bar: 10 μm.
In the early embryo, mis-expression of pk1a generates similar defects to that of knockdown (Carreira-Barbosa et al., 2003; Veeman et al., 2003; Bassuk et al., 2008). We found that mis-expression of pk1a RNA in wild-type embryos also disrupted IPL organization (Fig. 4F,G). In order to test for suppression of the MO-induced IPL defects, we identified the dose of pk1a RNA that does not induce an overexpression defect (Fig. 4F,F′). We used a low dose (40 pg) of pk1a RNA that does not lead to an overexpression defect in the IPL (Fig. 4F,F′). Co-injection of pk1a-MO with this dose of RNA partially suppressed the MO-induced IPL defect (Fig. 4H,H′). We conclude that pk1a function is required for appropriate arborization within the IPL and that this defect is, in part, suppressed by wild-type Pk1a.
Human epilepsy patients with PK1 mutation have normal visual perception (Bassuk et al., 2008; Tao et al., 2011), yet we observe retinal defects in pk1a morphants. We next evaluated visual function of pk1a morphants. The vision startle response in zebrafish is a natural escape response that is elicited when embryos are exposed to rapid changes in light intensity (Baye et al., 2011). Visually responsive 5 dpf embryos changed their swimming behavior when there was a short block in a bright light source (supplementary material Fig. S3A–C). The assay was repeated five times and the average number of responses reported. Wild-type embryos responded an average of 3.6 times. By contrast, the visually impaired cone-rod homeobox (crx) morphants responded only 2.2 times (supplementary material Fig. S3D) (Baye et al., 2011). pk1a morphants with a straight body axis showed a response profile similar to the wild type (supplementary material Fig. S3D). These observations suggest that pk1a knockdown does not compromise basic visual function and indicate that the IPL defect might impact higher order processing. The extent to which similar processing defects, if present in the CNS, relate to epilepsy is unknown.
Pk1a epilepsy mutant forms display distinct functional and biochemical properties compared with the wild-type form
Mutations in PK1 lead to single amino acid changes that locate in different parts of the protein, and PK1 patients have normal stature. Given that the mouse Pk1-null mutant is homozygous lethal and that pk1a complete knockdown in zebrafish leads to trunk defects, we reasoned that each of the mutations found in humans reflects a partially functional product. Moreover, distinct domains harboring mutations might compromise specific aspects of Pk1 function. To test this possibility, we performed functional analyses and investigated the biochemical properties of the mutant forms.
We generated and expressed zebrafish Pk1a carrying analogous mutations. The R150H mutation is an amino acid change of arginine 150 to histidine in the first LIM domain and the Y465H mutation is a change of tyrosine 465 to histidine in the middle of the protein (Fig. 5A) (Tao et al., 2011). In overexpression assays, we monitored IPL formation and found that excess R150H and Y465H pk1a RNA both led to IPL defects (supplementary material Fig. S4). To evaluate activity under more physiologically relevant conditions, we tested the extent to which the mutant RNA can suppress the MO-induced IPL defect. As shown in Fig. 4, pk1a morphants had mild and severe IPL defects and were statistically different to control embryos (Fig. 5B). Co-injection of wild-type pk1a RNA with pk1a-MO not only reduced the severity of the IPL defect but also the embryos were no longer statistically significantly different to uninjected controls (Fig. 5B). By contrast, co-expression of Y465H led to a larger percentage of severe defects (Fig. 5B). Co-expression of R150H with pk1a-MO demonstrated a shift from severe to mild IPL defects but total defects were still significantly different from uninjected control (Fig. 5B). Thus, wild-type pk1a RNA can suppress the MO-induced defect. Although the R150H mutation seemed to attenuate the MO-induced defect, the effect was not statistically significant. The mutant form Y465H, on the other hand, seemed to accentuate the MO-induced IPL defect. We next explored biochemical changes that could account for the different functional activities.
Fig. 5.

pk1a epilepsy mutant forms display reduced ability to suppress the MO-induced IPL defect. (A) Schematic of the zebrafish Pk1a protein. Gray boxes show the PET and LIM protein-protein interaction domains. Arrows denote the position of mutations analogous to those found in human epilepsy patients. (B) Graph showing the range of IPL defects in uninjected controls, pk1a morphants and morphants with mis-expressed mutant Pk1a. Treatment with pk1a-MO caused an increase in mild and severe defects. Wild-type pk1a RNA was able to increase the percentage of normal retinas and retinas with mild defects and decrease the percentage with severe defects; Y465H failed to do so and R150H seemed to increase the percentage of mild defects. However, neither mutant form suppressed the MO-induced IPL defects as efficiently as the wild-type form. Light shading represents normal IPL; medium dark shading represents mild defects, and dark shading represents severe defects. *P<0.05 compared with uninjected controls, Fisher’s exact test; uninjected, n=18; pk1a-MO, n=22; MO+40 pg RNA, n=11; MO+40 pg Y465H, n=14; MO+40 pg R150H, n=10.
We first determined expression of the myc-tagged mutant forms using western blot analysis of embryo lysates. At 80% epiboly (∼8 hpf), we found reduced expression of the R150H form compared with the wild-type Pk1a. By contrast, Y465H was expressed at the same levels as the wild-type protein, if not slightly higher (Fig. 6A). Although wild-type and mutant proteins were not detectable at 76 hpf, the stage at which we evaluate the IPL formation (data not shown), we did find expression of all the forms at 30 hpf when initial retinal ganglion cells start to grow processes (supplementary material Fig. S5A) (Schmitt and Dowling, 1996). The reduced expression of the R150H form of Pk1a could be due to increased turnover or less efficient translation than the wild-type form. To test the possibility of turnover, we expressed myc-tagged R150H Pk1a and treated the embryos with the proteosomal inhibitor MG-132. The expression level of R150H Pk1a was unchanged following drug treatment (supplementary material Fig. S5B), suggesting that the lower R150H expression levels might be a result of low translational efficiency.
Fig. 6.
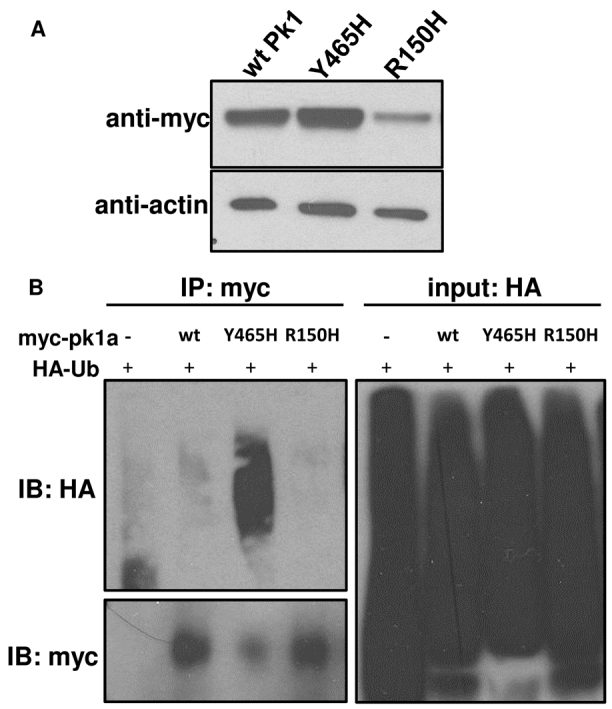
pk1a epilepsy mutants display distinct biochemical properties. (A) Western blot of myc-tagged wild-type (wtPk1) and mutant forms of Pk1a. Protein lysates were made at 80% epiboly stage. β-actin was used as a loading control. (B) Pull-down assays showing the extent to which wild-type (wt) and mutant forms are ubiquitylated. HA-Ub and pk1a RNAs were co-injected into one-cell stage embryos and at 80% epiboly total cell lysates were pulled down by anti-myc antibody beads and blotted for anti-myc or anti-HA. Embryos injected with HA-Ub without pk1a RNA were subjected to anti-myc pull-down as a negative control.
Pk1 was shown to be ubiquitylated in cell culture (Narimatsu et al., 2009). To determine whether ubiquitylation is altered in the mutant forms, we co-injected HA epitope-tagged ubiquitin with myc-tagged Pk1a forms. Denatured embryo lysates were immunoprecipitated with anti-myc antibody-conjugated agarose beads and probed with an antibody specific for the HA epitope by western blot. The epitope-tagged ubiqutin was readily incorporated into zebrafish proteins (Fig. 6B, input: HA). To insure equal loading of Pk1a mutant forms, we adjusted the pull-down samples according to their abundance in myc-tagged proteins (Fig. 6B, IB: myc). We then proportionally loaded pull-down samples for anti-HA immunoblot. Interestingly, we found that the Y465H mutant Pk1a was more strongly ubiquitylated than the wild type or R150H form (Fig. 6B, IB: HA). These results further confirm that the differential expression level of the R150H mutant occurs without detectable ubiquitylation and is probably independent of proteosomal degradation. Moreover, the increased ubiquitylation in Y465H Pk1a appears unrelated to degradation and could impact localization or protein-protein interactions. These data suggest that the distinct biochemical properties displayed by wild-type and mutant forms of Pk1a reflect the functional diversity of the different mutant forms in human patients.
DISCUSSION
Here, we describe the development of a zebrafish model to elucidate the mechanisms by which PK1 dysfunction leads to epilepsy. We first demonstrate that pk1a knockdown sensitizes zebrafish larvae to the convulsant drug PTZ. This finding is consistent with what was reported for Pk mutant mice and flies (Tao et al., 2011). We find broad pk1a expression in the larva brain with enriched expression in the retina. Consistently, pk1a knockdown leads to neurite outgrowth defects in the retina, similar to defects observed in the developing fly nervous system (Tao et al., 2011). These results further highlight the evolutionary role of Pk in regulating seizures and neurodevelopment. Furthermore, we find that mutant forms of Pk1 associated with human epilepsy display distinct functional and biochemical properties.
Human patients heterozygous for the R150H and Y465H mutations of PK1 are otherwise developmentally normal. The fact that Pk1 knockout in mice is lethal suggests that the Pk1 gene is crucial for embryonic development. In the case of human patients with PK1 mutations, it is possible that either PK2 can compensate for PK1 function or these mutations are hypomorphic, maintaining a partially functional PK1 product, or both. Pk is a core PCP protein, and the amount of product as well as the asymmetrical localization of core PCP proteins is necessary for correct cell polarization (Tree et al., 2002; Jenny et al., 2003; Veeman et al., 2003). Therefore, either too much or too little of the protein would lead to similar phenotypes. The extent to which the IPL defect is a PCP phenotype remains to be determined; however, reduced Wnt5b function (a Wnt that generates PCP outputs) leads to defects in IPL formation (Lin et al., 2010). Moreover, we demonstrate that Pk1a overexpression leads to an IPL defect as the mutant forms do, which is consistent with the notion that the mutant forms retain partial or aberrant activity. Although the mutant forms of Pk1a demonstrate overexpression phenotypes, our utilization of the pk1a knockdown approach with expression of the wild-type and mutant forms revealed functional differences in the ability to suppress the IPL defect. This approach might reflect the physiology of the human patients with heterozygous mutations and could provide insights into the development of the CNS and the mechanisms underlying the epilepsy phenotype in PK mutants.
Our biochemical analyses indicate that specific mutations affect distinct aspects of pk1a protein function that, when compromised, display overlapping phenotypes. The reduced expression of R150H Pk1a, even after treatment with the proteosome inhibitor MG-132, suggests that this form is translated less efficiently than wild-type Pk1a. Similar translational efficiency could lead to reduced PK1 protein in human patients and thereby contribute to the epileptic phenotype. The R150H mutant form attenuates the MO-induced defect but not to significant levels. This suggests that the protein, when provided in exogenous levels, can function like the wild type.
The Pk1 protein undergoes post-translational modification. In contrast to previous evidence in cell culture (Narimatsu et al., 2009), the wild-type zebrafish pk1a form did not show extensive ubiquitylation. We reason that in the cell culture studies, addition of E3 ubiquitin ligase as well as other Pk1-interacting factors enhanced this modification whereas, in our studies, the extent of ubiquitylation relies on endogenous ubiquitin ligases and interacting factors expressed in the early embryo. Despite the dependence upon endogenous factors in the embryo, we find robust ubiquitylation of the Y465H mutant form. In addition to proteolytic outcomes, ubiquitylation also regulates protein-protein interactions and protein trafficking in endosomal compartments (Scita and Di Fiore, 2010; Piper and Lehner, 2011; Dupont et al., 2012). Ubiquitylation has also been proposed to act as a general modulator of protein function similar to the role of phosphorylation (Dupont et al., 2012). These alternative functions attributed to ubiquitylation might apply to the Y465H mutant form of Pk1a as it is stably expressed in the zebrafish. The ubiquitylated Y465H form might be sorted through a different compartment than the wild-type or R150H forms of Pk1a, or have different abilities to interact with binding partners (Shimojo and Hersh, 2003; Jenny et al., 2005; Shimojo and Hersh, 2006). Our study revealed that the two mutant forms display distinct biochemical properties that could contribute to epileptogenesis by different mechanisms.
Epileptic seizures are thought to arise from the imbalance of excitatory and inhibitory signaling in the central nervous system. PTZ generates motor seizures in zebrafish by antagonizing the inhibitory γ-aminobutyric acid (GABA)-mediated post-synaptic signaling (Macdonald and Barker, 1977; Baraban et al., 2005; Hortopan et al., 2010b). Although our findings imply that partial loss of pk1a potentiates seizures in response to PTZ, implicating the GABA pathway in pk1 mutant-related epilepsy, the mechanism is unclear. One possibility is that pk1a is necessary for the developmental refinement of axons and dendrites and that subtle deficits in the signal processing within excitatory and inhibitory neurons render the network prone to seizures. Given the IPL organization defects we observe in pk1a knockdown retina, it is possible that the connectivity is affected and thus abnormal signal processing could contribute to the CNS response to stimuli. Similarly, mutations have been shown to link to epilepsy in genes that regulate neuronal proliferation, migration and circuit remodeling (reviewed by Bozzi et al., 2012). For example, these genes include Reln, which codes for Reelin and is responsible for the migration of cortical neurons (D’Arcangelo and Curran, 1998; D’Arcangelo, 2006; Patrylo et al., 2006); Dlx, which codes for transcription factors that are necessary for cortical interneuron differentiation and migration (Anderson et al., 1997; Cobos et al., 2005; Wang et al., 2010a); and Lgi1, lack of which cause defects in pruning and maturation of hippocampal glutamatergic neurons (Kalachikov et al., 2002; Striano et al., 2011). Interestingly, the Dlx5/6+/− mice display electrographic spontaneous seizures without any gross histological abnormalities in the cortex (Wang et al., 2010a), suggesting a connectivity issue. Whether similar types of defects exist in the CNS of pk1a knockdown zebrafish is not known. Moreover, it is not clear whether human PK patients have subtle retinal axonal organization defects.
In summary, we describe the use of zebrafish larvae as a model to study epilepsy mechanisms. We provide evidence that reduced pk1a function leads to increased sensitivity to PTZ-induced seizure behaviors. We also demonstrate the role of pk1a in the developing retina, potentially also in CNS development, which suggests that synaptic connectivity might affect signal processing. Biochemical characterization of mutant forms could inform treatment paradigms in human patients. Further studies to dissect other epilepsy mechanisms with the zebrafish model will be useful for illuminating potential treatment paradigms for human patients harboring specific mutations.
MATERIALS AND METHODS
Ethical statement
All the work with zebrafish was approved by University Animal Care and Use Committee of the University of Iowa.
Zebrafish maintenance
Zebrafish adults were maintained under standard conditions and eggs collected from natural spawnings (Westerfield, 1993). Staging of the embryos was performed according to Kimmel et al. (Kimmel et al., 1995).
Morpholinos, expression constructs and primers
Control MO, 5′-CCTCTTACCTCAGTTACAATTTATA-3′; pk1a-MO, 5′-CAGCTCCATCACTAACACCCCCTCA-3′ and pk1a second MO, 5′-GCCCACCGTGATTCTCCAGCTCCAT-3′ were purchased from Gene Tools. The second pk1a-MO completely overlaps with pk1a RNA sequence so was not used for rescue studies. HA-Ub expression construct was a gift from Douglas Houston (University of Iowa, Iowa City, IA). The myc-pk1a wild-type form was cloned from 8–12 somite stage cDNA library with GeneAmp highfidelity polymerase (Applied Biosystems, Life Technologies, Carlsbad, CA). PCR primers for cloning pk1a were: (forward) 5′-ATGGAGCTGGAGAATCACGGTGG-3′ and (reverse) 5′-AATTCCCTCTCAAAGTGGGC-3′. PCR products were cloned into PCR8/GW/TOPO entry vector and recombined with the destination vector with an N-terminal 6× myc (from Nathan Lawson’s Lab at University of Massachusetts Medical School, Worcester, MA). TA cloning and LR recombination kits were purchased from Life Technologies (Carlsbad, CA). The myc-pk1a mutant forms were generated by site-directed mutagenesis as described (Tao et al., 2011). In vitro transcription was performed to make synthetic RNAs using the mMessage mMachine in vitro transcription kit (Ambion, Life Technologies, Carlsbad, CA). Microinjections were done at the 1–2 cell stage by injecting into the yolk. Needles for injections were calibrated after each injection set. The injection volume varied between 2.0 nl/pump and 3.0 nl/pump. For the motility assay and the retina axon outgrowth analyses, 1.0 ng and 2.0–3.0 ng pk1a-MO was injected into the embryos, respectively. For the vision assay, 1.0 ng crx-MO was used. RNA injection doses were 40 pg and 80 pg wild-type pk1a for rescue experiments and 250 pg wild-type and mutant forms for biochemical analyses.
Drug treatment and motility assay
Age-matched zebrafish larvae were incubated at 28.5°C. Larvae at 60 hpf were transferred individually into a 48-well culture dish and incubated in the Zebrabox overnight. Each well contained 1.2 ml egg water. Larvae at 3 dpf were monitored for swimming behavior by the Zebrabox and the Viewpoint Software (Viewpoint Life Sciences, Lyon, France). The behavior was recorded once every second for 0.5 hours before and after PTZ treatment. Detection/movement threshold was set at 0.1/10. Activities below 0.1, from 0.1 to 10, and above 10 were designated by the software as ‘freezing’ (white areas in Fig. 1A), ‘small movement’ (green lines in Fig. 1A) and ‘large movement’ (red lines in Fig. 1A), respectively. For quantification, total swimming distances across the whole 0.5 hours, including small and large movements, were calculated and averaged by larvae number. Given that the total activity by individual larvae was not normally distributed (by the Shapiro-Wilk normality test), the Wilcoxon Rank Sum Test was used.
Powdered PTZ (Sigma-Aldrich, St Louis, MO) was dissolved in egg water to make fresh working stock before each experiment. For dose-dependent response experiments, PTZ working stock was added to each well to achieve the desired final concentration in a final volume of 1.6 ml. For tracking the MO-injected larvae, 0.4 ml of 20 mM PTZ was added to each well to make a final 5 mM concentration. To minimize the disturbance to the larvae during the addition of PTZ, the room was kept dark and quiet.
Powdered VPA (Sigma-Aldrich) was dissolved in egg water to make a working stock of 3 mM. Larva were incubated in 0.8 ml egg water overnight and tracked for 1 hour before addition of 0.4 ml VPA working stock, making the final VPA concentration 1 mM. Then 0.4 ml of 20 mM PTZ was added to make a final 5 mM final concentration and the larva were tracked for 1 hour.
Larvae of 3 dpf in egg water or 15 mM PTZ were placed in an Eppendorf tube cap and video-taped at about 7 frames/second. Extracted image sequences from the video were imported into Quicktime and made into a movie at 10 frames/second.
In situ hybridization
Whole mount in situ hybridization was performed according to previously described protocols (Thisse et al., 1993). Embryos were fixed at 76–78 hpf in 4% paraformaldehyde in 1× PBS. Degoxigenin-UTP labeled RNA riboprobes were made from linearized constructs using the MAXIscript in vitro transcription kit (Ambion, Life Technologies, Carlsbad, CA). Brain sections (10 μm) were collected from post-fixed embryos by cryosectioning as described below. Sections were mounted in 1:1 sterile glycerol and 1× PBS and imaged at 20× magnification.
Retina histology and fluorescence microscopy
MO-injected embryos were fixed at 76–78 hpf for 24 hours and infiltrated in 15% sucrose and 30% sucrose and in OCT (optimal cutting temperature medium, Sakura, Alphen aan den Rijn, The Netherlands) overnight at 4°C. Then, embryos were aligned and embedded in OCT, frozen and sectioned at −21°C. Sections (12 μm) were mounted on glass slides and left to dry overnight. H&E staining was performed according to standard protocols. For fluorescence microscopy, HuC:gfp transgenic embryos were used. Dried slides were rehydrated with 1× PBS and stained with To-Pro3 (Molecular Probes, Life Technologies, Carlsbad, CA) diluted 1:1000 in PBS. Sections (12 μm) were mounted with Vectorshield mounting medium (Vector Laboratories, Burlingame, CA), coverslipped and imaged with Leica TCS SP2 confocal microscope at 63× magnification with a 3× zoom. Zoomed images were maximum projections of four consecutive scans in a series.
Western blot and pull-down assay
RNA-injected embryos were lysed at 80% epiboly stage and protein samples analyzed with mouse anti-myc antibody (9E10; Santa Cruz Biotechnology, Santa Cruz, CA) in 1:10,000 dilution. Rabbit anti-β-actin antibody (Sigma-Aldrich) in 1:2000 dilution was used as loading control. For MG-132 treatment, RNA-injected embryos were dechorionated at the ∼1000-cell stage. DMSO or 10 mM MG-132 (EMD Millipore, Billerica, MA) was added to the egg water at a dilution of 1:1000. Embryo lysate was collected at 80% epiboly stage. For pull-down assays, myc-tagged proteins were immunoprecipitated with anti-myc antibody diluted 1:400 using protein A/G agarose beads (Fisher Scientific, Loughborough, UK). The beads were then washed three times with buffer (20 mM Tris, 100 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 2% SDS, 100 μM PMSF and 5 μg/ml leupeptin) and boiled at 100°C for 5 minutes.
Vision startle response assay
The vision startle response assay was as previously described (Nishimura et al., 2010; Pretorius et al., 2010; Baye et al., 2011) and was modified from Easter and Nicola (Easter and Nicola, 1996). Zebrafish larvae at 5 dpf were light-adapted for at least 1 hour. A visual stimulus was applied to the larvae by rapidly closing and opening the shutter, allowing for ∼1 second of darkness. A positive response would be seen as an abrupt direction change of the larva during the 1 second (either at the immediate onset of darkness or at the onset of light). Five trials were performed on the same fish spaced 30 seconds apart. A blunt needle probe was used to test the embryo for touch-responsiveness. Larvae with no touch-response were excluded from the data analysis. Data were plotted as the average number out of five trials that a larva was scored as positive.
Supplementary Material
Acknowledgments
We thank Trudi Westfall and Hilary Griesbach for technical expertise, Lisa Baye and Pamela Pretorius for providing reagents and feedback to the manuscript, and Robert A. Cornell and Amanda R. Decker for their help with the behavior assay and use of the Zebrabox.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
D.C.S. and A.G.B. conceived and designed the experiments. X.M. performed the experiments. S.W. contributed to the reagents. D.C.S. and X.M. analyzed the data. X.M. and D.C.S. wrote the paper.
FUNDING
This work is supported by the National Institutes of Health [grant number R01NS064159 to A.G.B. and D.C.S.].
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.010793/-/DC1
REFERENCES
- Anderson S. A., Eisenstat D. D., Shi L., Rubenstein J. L. (1997). Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278, 474–476 [DOI] [PubMed] [Google Scholar]
- Baraban S. C., Taylor M. R., Castro P. A., Baier H. (2005). Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience 131, 759–768 [DOI] [PubMed] [Google Scholar]
- Bassuk A. G., Wallace R. H., Buhr A., Buller A. R., Afawi Z., Shimojo M., Miyata S., Chen S., Gonzalez-Alegre P., Griesbach H. L., et al. (2008). A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am. J. Hum. Genet. 83, 572–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baye L. M., Patrinostro X., Swaminathan S., Beck J. S., Zhang Y., Stone E. M., Sheffield V. C., Slusarski D. C. (2011). The N-terminal region of centrosomal protein 290 (CEP290) restores vision in a zebrafish model of human blindness. Hum. Mol. Genet. 20, 1467–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham S. M., Sittaramane V., Mapp O., Patil S., Prince V. E., Chandrasekhar A. (2010). Multiple mechanisms mediate motor neuron migration in the zebrafish hindbrain. Dev. Neurobiol. 70, 87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosoi C. M., Capra V., Allache R., Trinh V. Q.-H., De Marco P., Merello E., Drapeau P., Bassuk A. G., Kibar Z. (2011). Identification and characterization of novel rare mutations in the planar cell polarity gene PRICKLE1 in human neural tube defects. Hum. Mutat. 32, 1371–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzi Y., Casarosa S., Caleo M. (2012). Epilepsy as a neurodevelopmental disorder. Front. Psychiatry 3, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira-Barbosa F., Concha M. L., Takeuchi M., Ueno N., Wilson S. W., Tada M. (2003). Prickle 1 regulates cell movements during gastrulation and neuronal migration in zebrafish. Development 130, 4037–4046 [DOI] [PubMed] [Google Scholar]
- Cobos I., Calcagnotto M. E., Vilaythong A. J., Thwin M. T., Noebels J. L., Baraban S. C., Rubenstein J. L. (2005). Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat. Neurosci. 8, 1059–1068 [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G. (2006). Reelin mouse mutants as models of cortical development disorders. Epilepsy Behav. 8, 81–90 [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G., Curran T. (1998). Reeler: new tales on an old mutant mouse. BioEssays 20, 235–244 [DOI] [PubMed] [Google Scholar]
- Dupont S., Inui M., Newfeld S. J. (2012). Regulation of TGF-β signal transduction by mono- and deubiquitylation of Smads. FEBS Lett. 586, 1913–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easter S. S., Jr, Nicola G. N. (1996). The development of vision in the zebrafish (Danio rerio). Dev. Biol. 180, 646–663 [DOI] [PubMed] [Google Scholar]
- Goodrich L. V. (2008). The plane facts of PCP in the CNS. Neuron 60, 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortopan G. A., Dinday M. T., Baraban S. C. (2010a). Spontaneous seizures and altered gene expression in GABA signaling pathways in a mind bomb mutant zebrafish. J. Neurosci. 30, 13718–13728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortopan G. A., Dinday M. T., Baraban S. C. (2010b). Zebrafish as a model for studying genetic aspects of epilepsy. Dis. Model. Mech. 3, 144–148 [DOI] [PubMed] [Google Scholar]
- Jenny A., Darken R. S., Wilson P. A., Mlodzik M. (2003). Prickle and Strabismus form a functional complex to generate a correct axis during planar cell polarity signaling. EMBO J. 22, 4409–4420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenny A., Reynolds-Kenneally J., Das G., Burnett M., Mlodzik M. (2005). Diego and Prickle regulate Frizzled planar cell polarity signalling by competing for Dishevelled binding. Nat. Cell Biol. 7, 691–697 [DOI] [PubMed] [Google Scholar]
- Jiang D., Munro E. M., Smith W. C. (2005). Ascidian prickle regulates both mediolateral and anterior-posterior cell polarity of notochord cells. Curr. Biol. 15, 79–85 [DOI] [PubMed] [Google Scholar]
- Kalachikov S., Evgrafov O., Ross B., Winawer M., Barker-Cummings C., Martinelli Boneschi F., Choi C., Morozov P., Das K., Teplitskaya E., et al. (2002). Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat. Genet. 30, 335–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 [DOI] [PubMed] [Google Scholar]
- Lin S., Baye L. M., Westfall T. A., Slusarski D. C. (2010). Wnt5b-Ryk pathway provides directional signals to regulate gastrulation movement. J. Cell Biol. 190, 263–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link B. A., Fadool J. M., Malicki J., Dowling J. E. (2000). The zebrafish young mutation acts non-cell-autonomously to uncouple differentiation from specification for all retinal cells. Development 127, 2177–2188 [DOI] [PubMed] [Google Scholar]
- Macdonald R. L., Barker J. L. (1977). Pentylenetetrazol and penicillin are selective antagonists of GABA-mediated post-synaptic inhibition in cultured mammalian neurones. Nature 267, 720–721 [DOI] [PubMed] [Google Scholar]
- Mapp O. M., Wanner S. J., Rohrschneider M. R., Prince V. E. (2010). Prickle1b mediates interpretation of migratory cues during zebrafish facial branchiomotor neuron migration. Dev. Dyn. 239, 1596–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapp O. M., Walsh G. S., Moens C. B., Tada M., Prince V. E. (2011). Zebrafish Prickle1b mediates facial branchiomotor neuron migration via a farnesylation-dependent nuclear activity. Development 138, 2121–2132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrkusich E. M., Flanagan D. J., Whitington P. M. (2011). The core planar cell polarity gene prickle interacts with flamingo to promote sensory axon advance in the Drosophila embryo. Dev. Biol. 358, 224–230 [DOI] [PubMed] [Google Scholar]
- Mulley J. C., Scheffer I. E., Petrou S., Berkovic S. F. (2003). Channelopathies as a genetic cause of epilepsy. Curr. Opin. Neurol. 16, 171–176 [DOI] [PubMed] [Google Scholar]
- Narimatsu M., Bose R., Pye M., Zhang L., Miller B., Ching P., Sakuma R., Luga V., Roncari L., Attisano L., et al. (2009). Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell 137, 295–307 [DOI] [PubMed] [Google Scholar]
- Ng J. (2012). Wnt/PCP proteins regulate stereotyped axon branch extension in Drosophila. Development 139, 165–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura D. Y., Baye L. M., Perveen R., Searby C. C., Avila-Fernandez A., Pereiro I., Ayuso C., Valverde D., Bishop P. N., Manson F. D., et al. (2010). Discovery and functional analysis of a retinitis pigmentosa gene, C2ORF71. Am. J. Hum. Genet. 86, 686–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H. C., Kim C. H., Bae Y. K., Yeo S. Y., Kim S. H., Hong S. K., Shin J., Yoo K. W., Hibi M., Hirano T., et al. (2000). Analysis of upstream elements in the HuC promoter leads to the establishment of transgenic zebrafish with fluorescent neurons. Dev. Biol. 227, 279–293 [DOI] [PubMed] [Google Scholar]
- Patrylo P. R., Browning R. A., Cranick S. (2006). Reeler homozygous mice exhibit enhanced susceptibility to epileptiform activity. Epilepsia 47, 257–266 [DOI] [PubMed] [Google Scholar]
- Piper R. C., Lehner P. J. (2011). Endosomal transport via ubiquitination. Trends Cell Biol. 21, 647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduri A., Lowenstein D. (2011). Epilepsy genetics; past, present, and future. Curr. Opin. Genet. Dev. 21, 325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretorius P. R., Baye L. M., Nishimura D. Y., Searby C. C., Bugge K., Yang B., Mullins R. F., Stone E. M., Sheffield V. C., Slusarski D. C. (2010). Identification and functional analysis of the vision-specific BBS3 (ARL6) long isoform. PLoS Genet. 6, e1000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Alvarez L., Visanuvimol J., McEwan A., Su A., Imai J. H., Colavita A. (2011). VANG-1 and PRKL-1 cooperate to negatively regulate neurite formation in Caenorhabditis elegans. PLoS Genet. 7, e1002257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt E. A., Dowling J. E. (1996). Comparison of topographical patterns of ganglion and photoreceptor cell differentiation in the retina of the zebrafish, Danio rerio. J. Comp. Neurol. 371, 222–234 [DOI] [PubMed] [Google Scholar]
- Scita G., Di Fiore P. P. (2010). The endocytic matrix. Nature 463, 464–473 [DOI] [PubMed] [Google Scholar]
- Shimojo M., Hersh L. B. (2003). REST/NRSF-interacting LIM domain protein, a putative nuclear translocation receptor. Mol. Cell Biol. 23, 9025–9031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojo M., Hersh L. B. (2006). Characterization of the REST/NRSF-interacting LIM domain protein (RILP): localization and interaction with REST/NRSF. J. Neurochem. 96, 1130–1138 [DOI] [PubMed] [Google Scholar]
- Striano P., Busolin G., Santulli L., Leonardi E., Coppola A., Vitiello L., Rigon L., Michelucci R., Tosatto S. C., Striano S., et al. (2011). Familial temporal lobe epilepsy with psychic auras associated with a novel LGI1 mutation. Neurology 76, 1173–1176 [DOI] [PubMed] [Google Scholar]
- Takeuchi M., Nakabayashi J., Sakaguchi T., Yamamoto T. S., Takahashi H., Takeda H., Ueno N. (2003). The prickle-related gene in vertebrates is essential for gastrulation cell movements. Curr. Biol. 13, 674–679 [DOI] [PubMed] [Google Scholar]
- Tao H., Manak J. R., Sowers L., Mei X., Kiyonari H., Abe T., Dahdaleh N. S., Yang T., Wu S., Chen S., et al. (2011). Mutations in prickle orthologs cause seizures in flies, mice, and humans. Am. J. Hum. Genet. 88, 138–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y., Xie X., Walker S., Rempala G., Kozlowski D. J., Mumm J. S., Cowell J. K. (2010). Knockdown of zebrafish Lgi1a results in abnormal development, brain defects and a seizure-like behavioral phenotype. Hum. Mol. Genet. 19, 4409–4420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y., Xie X., Walker S., Saxena M., Kozlowski D. J., Mumm J. S., Cowell J. K. (2011). Loss of zebrafish lgi1b leads to hydrocephalus and sensitization to pentylenetetrazol induced seizure-like behavior. PLoS ONE 6, e24596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse C., Thisse B., Schilling T. F., Postlethwait J. H. (1993). Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development 119, 1203–1215 [DOI] [PubMed] [Google Scholar]
- Tissir F., Goffinet A. M. (2010). Planar cell polarity signaling in neural development. Curr. Opin. Neurobiol. 20, 572–577 [DOI] [PubMed] [Google Scholar]
- Tree D. R. P., Shulman J. M., Rousset R., Scott M. P., Gubb D., Axelrod J. D. (2002). Prickle mediates feedback amplification to generate asymmetric planar cell polarity signaling. Cell 109, 371–381 [DOI] [PubMed] [Google Scholar]
- Veeman M. T., Slusarski D. C., Kaykas A., Louie S. H., Moon R. T. (2003). Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr. Biol. 13, 680–685 [DOI] [PubMed] [Google Scholar]
- Velisek L., Kubova H., Pohl M., Stankova L., Mareš P., Schickerova R. (1992). Pentylenetetrazol-induced seizures in rats: an ontogenetic study. Naunyn Schmiedebergs Arch. Pharmacol. 346, 588–591 [DOI] [PubMed] [Google Scholar]
- Wada H., Tanaka H., Nakayama S., Iwasaki M., Okamoto H. (2006). Frizzled3a and Celsr2 function in the neuroepithelium to regulate migration of facial motor neurons in the developing zebrafish hindbrain. Development 133, 4749–4759 [DOI] [PubMed] [Google Scholar]
- Wang Y., Dye C. A., Sohal V., Long J. E., Estrada R. C., Roztocil T., Lufkin T., Deisseroth K., Baraban S. C., Rubenstein J. L. (2010a). Dlx5 and Dlx6 regulate the development of parvalbumin-expressing cortical interneurons. J. Neurosci. 30, 5334–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. Y., Smith P., Murphy M., Cook M. (2010b). Global expression profiling in epileptogenesis: does it add to the confusion? Brain Pathol. 20, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. (1993). The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Brachydanio rerio). Eugene, OR: M. Westerfield [Google Scholar]
- Zhou L., Bar I., Achouri Y., Campbell K., De Backer O., Hebert J. M., Jones K., Kessaris N., de Rouvroit C. L., O’Leary D., et al. (2008). Early forebrain wiring: genetic dissection using conditional Celsr3 mutant mice. Science 320, 946–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.