

SUMMARY
Amyotrophic lateral sclerosis (ALS) is a fatal disease characterized by complex neuronal and glial phenotypes. Recently, RNA-based mechanisms have been linked to ALS via RNA-binding proteins such as TDP-43, which has been studied in vivo using models ranging from yeast to rodents. We have developed a Drosophila model of ALS based on TDP-43 that recapitulates several aspects of pathology, including motor neuron loss, locomotor dysfunction and reduced survival. Here we report the phenotypic consequences of expressing wild-type and four different ALS-linked TDP-43 mutations in neurons and glia. We show that TDP-43-driven neurodegeneration phenotypes are dose- and age-dependent. In motor neurons, TDP-43 appears restricted to nuclei, which are significantly misshapen due to mutant but not wild-type protein expression. In glia and in the developing neuroepithelium, TDP-43 associates with cytoplasmic puncta. TDP-43-containing RNA granules are motile in cultured motor neurons, although wild-type and mutant variants exhibit different kinetic properties. At the neuromuscular junction, the expression of TDP-43 in motor neurons versus glia leads to seemingly opposite synaptic phenotypes that, surprisingly, translate into comparable locomotor defects. Finally, we explore sleep as a behavioral readout of TDP-43 expression and find evidence of sleep fragmentation consistent with hyperexcitability, a suggested mechanism in ALS. These findings support the notion that although motor neurons and glia are both involved in ALS pathology, at the cellular level they can exhibit different responses to TDP-43. In addition, our data suggest that individual TDP-43 alleles utilize distinct molecular mechanisms, which will be important for developing therapeutic strategies.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder that affects motor neurons and leads to paralysis and respiratory failure, followed by death usually within 2–5 years of diagnosis. Familial ALS (fALS) affects 10% of patients and the remaining 90% of ALS cases are sporadic (sALS) and remain poorly understood. To date, the most common gene linked to ALS is C9ORF72, whose mechanism of action appears to be linked to RNA dysregulation (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Several other loci have been linked to both fALS and sALS and include SOD1, alsin, senataxin, VAMP/synaptobrevin-associated protein B, P150 dynactin, angiogenin, TAR DNA-binding protein (TDP-43), Fused in Sarcoma (FUS) and profilin (Alexander et al., 2002; Beleza-Meireles and Al-Chalabi, 2009; Lagier-Tourenne and Cleveland, 2009; Wu et al., 2012). Based on the known functions of these loci and extensive phenotypic studies in model organisms, ALS appears to be the result of abnormalities in diverse cellular processes ranging from oxidative stress, intracellular transport, RNA metabolism, apoptosis and actin dynamics. Of these, RNA-based mechanisms have recently taken center stage in the ALS field due to findings that the RNA-binding proteins such as TDP-43 and FUS constitute markers of pathology and, in addition, when mutated, neural degeneration occurs in human patients (Colombrita et al., 2011; Kabashi et al., 2008; Kwiatkowski et al., 2009; Sreedharan et al., 2008; Vance et al., 2009). Studies in animal models including worms, flies, zebrafish and rodents support the notion that alterations in these RNA-binding proteins and in RNA metabolism cause motor neuron disease (Couthouis et al., 2011; Estes et al., 2011; Lanson et al., 2011; Li et al., 2010; Liachko et al., 2010; Lu et al., 2009; Wegorzewska et al., 2009). Together with the recent discovery of GGGGCC repeat RNA caused by expansions in C9ORF72, these studies support the hypothesis that RNA dysregulation is a crucial aspect of ALS (DeJesus-Hernandez et al., 2011; Lagier-Tourenne and Cleveland, 2009; Renton et al., 2011).
Of the RNA-binding proteins linked to ALS, TDP-43 has first captured the attention of the field due to pathological studies that identified it as a component of cytoplasmic inclusions in neurons and the surrounding glia (Maekawa et al., 2009; Neumann et al., 2006; Tan et al., 2007). Subsequently, TDP-43 was shown to harbor several missense mutations linked to ALS, some of which are studied here using Drosophila as a model (Kabashi et al., 2008; Rutherford et al., 2008; Sreedharan et al., 2008). Structurally, TDP-43 consists of two RNA recognition motifs (RRM1 and RRM2) and a glycine-rich domain within the C terminus (Johnson et al., 2008). Its known cellular functions include transcriptional repression, splicing, miRNA biogenesis and apoptosis (Banks et al., 2008). In vitro assays and RNA sequencing approaches have shown that TDP-43 binds UG-rich sequences with high affinity and regulates splicing of numerous targets, including its own transcript (Ayala et al., 2011; Buratti and Baralle, 2001; Polymenidou et al., 2011; Sephton et al., 2011; Tollervey et al., 2011). TDP-43 is ubiquitously expressed and associates with other RNA-binding proteins (Freibaum et al., 2010). In cultured neurons, TDP-43 associates with neuronal RNA granules and it colocalizes with RNA-binding proteins such as Fragile X protein (FMRP) and Staufen in an activity-dependent manner, which suggests that TDP-43 regulates the expression of synaptic mRNAs (Fallini et al., 2012; Wang et al., 2008; Yu et al., 2012).
TRANSLATIONAL IMPACT.
Clinical issue
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by rapidly progressive muscle weakness and respiratory difficulties. The underlying mechanisms are poorly understood and there is currently no cure. Studies have shown that RNA dysregulation is a key feature in ALS, and RNA-based mechanisms have recently taken center stage in ALS investigations. Among the proteins that have been linked with ALS, a major player is the RNA-binding protein TDP-43, which is thought to control several aspects of RNA biology. The aim of this study was to determine the phenotypic consequences of mutated and wild-type TDP-43 when expressed in neurons and glia, in an in vivo model of ALS.
Results
The authors developed a Drosophila model of ALS based on the expression of human TDP-43. This model recapitulated several aspects of ALS pathology, including locomotor dysfunction and reduced survival. The team examined the phenotypic effects of several ALS-linked TDP-43 variants when expressed in neurons or glia, compared with wild-type TDP-43. Their investigation revealed that TDP-43 toxicity is age- and dose-dependent. Furthermore, TDP-43 associates with motile neuronal RNA granules in motor neurons. By directly comparing motor neurons versus glia phenotypes they showed that TDP-43 controls the architecture of the neuromuscular junction, its synaptic properties and locomotor behavior. They also provide evidence of sleep fragmentation, which has been documented in patients but remains understudied.
Implications and future directions
These extensive phenotypic analyses reveal that motor neurons and glia elicit differential, variant-dependent responses to TDP-43; however, comparable locomotor defects emerge. These findings suggest distinct molecular mechanisms in motor neurons versus glial cells and underscore the importance of studying individual mutations linked to ALS. Future studies will focus on cell-specific mechanisms and functional studies at the neuromuscular junction synapse, with the aim of identifying new biomarkers and therapeutic candidates for the disease.
The majority of TDP-43 mutations found in ALS patients represent amino acid substitutions that are thought to increase TDP-43 phosphorylation and target it for degradation (Kabashi et al., 2008; Rutherford et al., 2008; Sreedharan et al., 2008). Work from several laboratories including ours has provided evidence that these missense mutations mimic a loss of function for TDP-43 and that the RNA-binding domain is required to mediate neurotoxicity (Estes et al., 2011; Feiguin et al., 2009; Voigt et al., 2010). Recently, different TDP-43 mutations have been shown to differentially regulate stress granule formation in cell lines subjected to environmental stress (Dewey et al., 2011; Liu-Yesucevitz et al., 2010; McDonald et al., 2011). Using a battery of phenotypic assays, we have also shown previously that wild-type and mutant A315T TDP-43 exhibit differential levels of neurotoxicity in a Drosophila model of ALS (Estes et al., 2011). Taken together, these findings suggest that different TDP-43 alleles utilize distinct mechanisms in motor neuron disease and underscore the importance of studying the effects of both wild-type and individual TDP-43 mutations linked to ALS. Here we address this important issue by comparing the phenotypic consequences of expressing wild-type TDP-43 and four different variants (D169G, G298S, A315T and N345K) in motor neurons versus glia, in vivo, using Drosophila as a model. The D169G mutation lies within the first RNA-binding domain of TDP-43 (RRM1) and has been linked to sporadic ALS, whereas G298S, A315T and N345K reside within the C-terminus domain of TDP-43 and have been linked to familial ALS (Neumann, 2009). Here, using the bipartite Gal4-UAS expression system in Drosophila, we first show that the expression of D169G, G298S and N345K mutations in the eye neuroepithelium leads to a dose- and age-dependent neurodegeneration, which is consistent with our previous work on wild-type and A315T TDP-43 (Estes et al., 2011). We confirm the presence of TDP-43 puncta in photoreceptor axons and, using a cellular fractionation approach, show that TDP-43 is detectable in insoluble aggregates. Next, we compare the effects of TDP-43 expression in motor neurons versus glia and find that these cells, which play a crucial role in the disease pathology, handle TDP-43 expression differently, both in a cell type- and a variant-dependent manner. We find that TDP-43 is primarily restricted to nuclei in motor neurons in vivo, whereas in glial cells, TDP-43 puncta are detected in the cytosol including the glial cytoplasmic extensions (i.e. glial ‘feet’) that envelope the neuromuscular junction synapse (Fuentes-Medel et al., 2009). Interestingly, we find that the motor neuron nuclei are grossly misshapen whereas glial nuclei exhibit milder shape abnormalities. These data suggest that TDP-43 elicits different responses in glial cells than in motor neurons. Upon culturing primary motor neurons from the ventral ganglia, we were able to detect cytoplasmic TDP-43-positive puncta, consistent with the notion that these structures represent stress granules, which have been suggested to have a protective role (Dormann and Haass, 2011). These TDP-43 cytoplasmic puncta are motile and exhibit dynamic features typical of neuronal RNA transport granules. Interestingly, fluorescence recovery after photobleaching (FRAP) experiments show that wild-type TDP-43 exhibits different kinetic properties to the mutant TDP-43 alleles used in this study. We also compared the consequences of expressing TDP-43 in motor neurons versus glia on the architecture as well as the synaptic marker distribution at the larval neuromuscular junction. These experiments indicate profound phenotypic differences, including opposite effects on the number of active zones and glutamate receptor distribution. Interestingly, larval locomotor activity, a measure of synaptic function, appears to be similarly affected by TDP-43 expression in both motor neurons and glia. Finally, we examine adult activity and sleep patterns and find evidence of locomotor dysfunction and sleep fragmentation. Our experiments provide the first in vivo comparison of several neuroanatomical, cellular and functional phenotypes due to motor neuron versus glial expression of multiple TDP-43 variants linked to ALS in humans. We conclude that although the observable functional phenotypes such as locomotor function and sleep are comparable, the mechanisms utilized by different TDP-43 variants in different tissues (e.g. motor neurons versus glia) might be divergent at the cellular level. These studies are crucial to our understanding of the molecular mechanisms underlying ALS in humans and the future development of therapies targeting individual mutations in TDP-43.
RESULTS
Overexpression of several mutant forms of TDP-43 leads to age- and dose-dependent neurodegeneration in the eye
We have previously shown that overexpression of human wild-type TDP-43 or the ALS-linked variant A315T TDP-43 in the developing neuroepithelium results in neurodegeneration (Estes et al., 2011). Here we set out to determine the phenotypic consequences of expressing additional missense mutations linked to ALS, namely D169G, G298S and N345K in the retina. With the exception of D169G, which lies within the first RNA-binding domain RRM1 and has been linked to sporadic ALS, the remainder of the mutations used in this study have been linked to familial ALS and are found within the C-terminus domain of TDP-43 (Kabashi et al., 2008). For each variant, we surveyed several transgenic lines and selected to work with those that exhibited similar levels of expression (for a representative set, see Fig. 1Y). Note that although some mutant variant lines displayed clearly visible truncated C-terminal fragments (∼25 kDa) that have been previously shown to be the result of caspase activation (reviewed by Baloh, 2011), some lines only showed a modest or barely detectable truncation (Fig. 1Y). Using the bipartite Gal4-UAS system we found that overexpression of all TDP-43 variants in the developing retina resulted in progressive neurodegeneration (as indicated by depigmentation, Fig. 1D–L) compared with GMR Gal4 controls (Fig. 1A–C). Notably, despite being expressed at comparable levels to other TDP-43 variants used in this study, the D169G mutant exhibited a milder phenotype (especially at day 15, compare Fig. 1D–F with 1G–L). These phenotypic differences appeared less pronounced when TDP-43 levels were increased by raising the flies at a higher temperature (29°C versus the standard 25°C), which led to an enhanced neurodegeneration in the retina (compare Fig. 1A–L with 1M–X). This phenotypic enhancement can be explained by higher levels of TDP-43, which are due to an increase in Gal4 activity at higher temperatures. These results demonstrate that the D169G, G298S and N345K variants are toxic in vivo and are consistent with previous reports that TDP-43 expression in the retina leads to an age- and dose-dependent neurodegeneration phenotype (Estes et al., 2011; Li et al., 2010). In addition, our data suggest that the D169G mutation in the RRM1 domain exhibits a lower level of toxicity than other variants, at least in the retina (see Table 1 for a summary of variant-specific phenotypes).
Fig. 1.
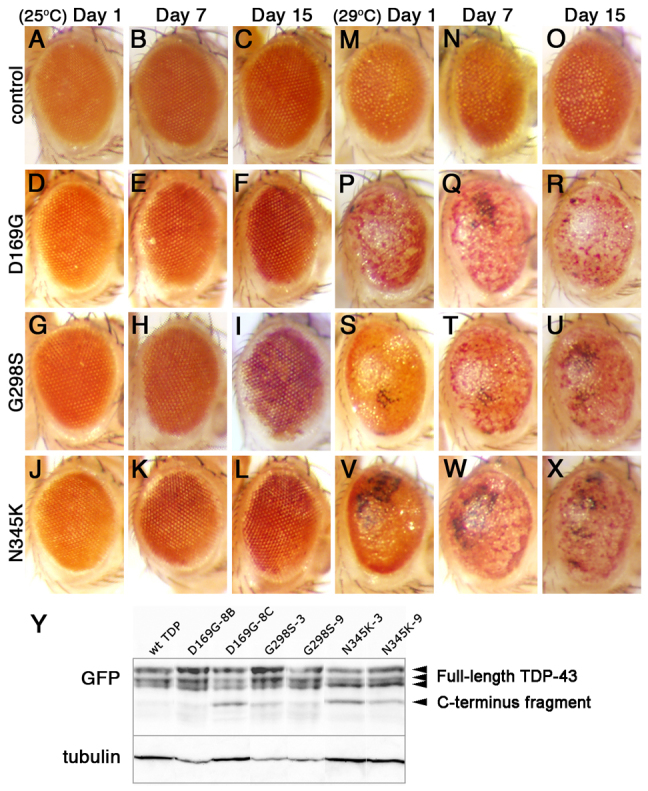
Overexpression of mutant TDP-43 leads to age- and dose-dependent neurodegeneration in the adult retina. (A–L) GMR Gal4 expression of TDP-43 D169G (D–F), G298S (G–I) and N345K (J–L) variants results in age-dependent loss of pigment compared with GMR Gal4 driver controls (A–C) at 25°C. Note that D169G depigmentation is milder than in G298S and N345K variants. (M–X) TDP-43 variants D169G TDP-43 (P–R), G298S TDP-43 (S–U) and N345K TDP-43(V–X) exhibit stronger surface phenotypes, including necrosis, compared with controls (M–O) when expressed at higher levels (29°C). Genotypes and ages are indicated. Anterior right, dorsal up. (Y) Western blot analyses showing TDP-43 expression in several transgenic lines. For each mutant variant we characterized multiple lines, two of which are shown. Genotypes are indicated on top; blotting antibodies are indicated on the left. Arrowheads on the right indicate full-length and C-terminus truncation of TDP-43. Tubulin was used as a loading control.
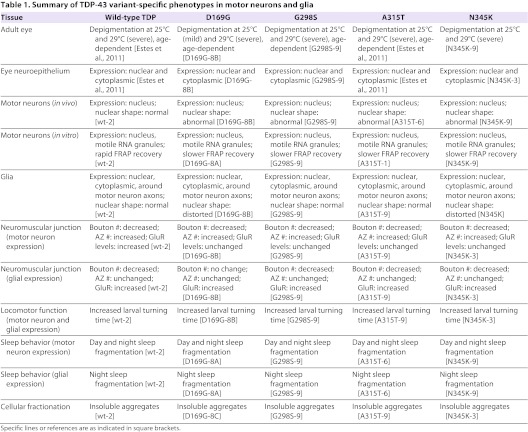
Table 1.
Summary of TDP-43 variant-specific phenotypes in motor neurons and glia
TDP-43 localizes to axonal aggregates in the developing neuroepithelium
A hallmark of ALS pathology is the presence of cytoplasmic aggregates containing TDP-43 (Neumann et al., 2006). Although the role of these protein structures in the pathology of disease and their relationship with RNA stress granules remains a matter of much debate (Baloh, 2011; Parker et al., 2012), we set out to evaluate the distribution of TDP-43 in the eye neuroepithelium cytoplasm. Consistent with our previous report showing that wild-type and A315T TDP-43 localized to axonal puncta (Estes et al., 2011), we found that TDP-43-positive structures were visible in axons for all variants tested here (Fig. 2A–H, note high magnification insets in E–H). To determine whether these correspond to insoluble aggregates found in patient samples, we took a cellular fractionation approach and found that all the TDP variants were largely found in low salt (LS) or detergent-soluble fractions (Triton X-100 and Sarkosyl fractions in Fig. 2I). In our hands, a relatively small, albeit clearly visible, amount of TDP-43 was found in detergent-insoluble aggregates (urea fraction in Fig. 2I). Notably, Liachko and colleagues found that most of the phosphorylated mutant TDP-43 was found in the detergent-soluble (e.g. Triton X-100 and Sarkosyl) and insoluble (i.e. urea) fractions, which correlates with more misfolded protein (Liachko et al., 2010). Taken together, these data indicate a positive correlation between photoreceptor neurodegeneration and the accumulation of TDP-43 in cytoplasmic puncta, some of which are insoluble aggregates.
Fig. 2.

TDP-43 forms axonal aggregates in developing eyes. (A–H) Confocal images (single slices, 1 μm each or 2–3 slice projections) showing GFP NLS (A,E) or TDP-43 variant (B–D,F–H) localization when expressed in the developing eye neuroepithelium with GMR-Gal4. TDP-43 visualized via individual fluorescent YFP tags (indicated as GFP here). Filamentous actin labeled with phalloidin, DNA stained with Hoechst 33342 as indicated. Note TDP-43 puncta in axons (arrowheads in B–D,F–H and insets); compare with GFP NLS controls (arrowheads in A,E and inset). (I) Cellular fractionation of adult head tissue shows TDP-43 distribution complexes ranging from highly soluble (LS, low salt) to insoluble (U, urea). Triton X-100 (TX) and sarkosyl (SK) correspond to non-ionic and ionic detergent soluble fractions, respectively. TDP-43 was detected using anti-GFP antibodies. Tubulin was used as a loading control. Scale bar: 75 μm.
TDP-43 overexpression in motor neurons in vivo does not result in detectable cytoplasmic puncta but affects nuclear morphology
Next, we examined the localization of TDP-43 variants when expressed in motor neurons using the D42 Gal4 driver (Fig. 3A–F). Consistent with our previous observations, TDP-43 appeared mostly restricted to the nucleus compared with controls (compare Fig. 3B–F with 3A). Upon close examination, we discovered that motor neuron nuclei expressing TDP-43 were misshapen, a feature associated with changes in gene expression and apoptosis (Fig. 3A–F, insets). Using Cell Profiler software we quantified eccentricity, form factor and compactness, all of which measure shape, of nuclei expressing TDP-43 in comparison to control nuclei, which appear round and smooth. As shown in Fig. 3G–I, all mutant TDP variants used in our study (D169G, G298S, A315T and N345K) exhibited a significantly higher eccentricity and compactness compared with D42 driver controls. In keeping with these findings, TDP-43 mutations led to a reduction in form factor, which is consistent with nuclear malformation from a sphere to a random three-dimensional shape (Fig. 3H). Taken together, these results indicate that the overexpression of TDP-43 mutations in motor neurons results in shape abnormalities and distorted nuclei, which is consistent with previous reports in HeLa cells (Ayala et al., 2008). Interestingly, all mutant variants exhibited stronger phenotypes than wild-type TDP-43, suggesting that this assay might be useful in distinguishing variant-specific effects of TDP-43 in motor neurons in vivo.
Fig. 3.

TDP-43 expression in motor neurons or glia leads to cytoplasmic aggregates and nuclear morphology defects. (A–F) TDP-43 expressed with D42 Gal4 was visualized using anti-GFP antibodies (B–F) and compared with D42 Gal4 >GFP NLS controls (A). Motor neurons within the ventral ganglia were labeled by GFP; DNA was stained with Hoechst 33342. (G–I) Mutant variants but not wild-type TDP-43 alter nuclear shape by increasing eccentricity (G), decreasing the form factor (H) and increasing compactness (I) compared with D42 Gal4 driver controls. (J–P) TDP-43 expressed in glia with repo Gal4 was visualized using anti-GFP antibodies (K–P) and compared with repo Gal4 >GFP NLS controls stained with Hoechst 33342 to visualize DNA (J). Note the presence of TDP-43 puncta in the cytoplasm (K–P). (Q–V) TDP-43 is expressed in puncta (arrows) within glial cells enveloping the neuromuscular junction synapse labeled by CSP and HRP. (W–Y) Glial expression causes relatively mild changes in nuclear shape, primarily in D169G and N345K compared with repo Gal4 driver controls. Eccentricity, form factor and compactness are shown as means ± s.e.m.; ***P<0.001. Scale bars: 20 μm.
TDP-43 forms cytoplasmic puncta in glia but has little or no effect on nuclear shape
Recent evidence suggests that glial cell toxicity has a contribution to ALS pathology (Ince et al., 2011). To determine whether TDP-43-mediated pathology has a glial component, we expressed wild-type and mutant TDP-43 in glial cells using the panglial driver repo Gal4 (Xiong et al., 1994). In contrast to motor neurons, in glial cells, we found that TDP-43 exits the nucleus and localizes to cytoplasmic puncta (Fig. 3J–P) that can be observed as far from the nucleus as the glial ‘feet’ surrounding the neuromuscular junction synapse (Fuentes-Medel et al., 2009) (Fig. 3Q–V, arrows). Upon examining the glial nuclei we found that only D169G and N345K TDP had a significant effect on nuclear shape (see insets in Fig. 3J–P and see 3W–Y for quantification). These data indicate an inverse correlation between nuclear restriction or cytoplasmic localization and nuclear shape distortion due to TDP-43 expression in glia and provide insights into phenotypic differences between TDP-43 variants.
TDP-43 forms dynamic puncta in motor neurons in culture
Next, we addressed the issue of the subcellular localization of TDP-43 using a primary culture approach. Although we could not detect TDP-43 in cytoplasmic puncta in motor neurons in vivo, it is possible that TDP-43 assembles in small RNA granules that are below the limit of resolution for the confocal microscope. To further probe the distribution of TDP-43 in motor neurons, we turned to primary cultures derived from the larval ventral ganglia and found that TDP-43 formed well-defined puncta distributed throughout the soma and neurites (Fig. 4B–F, arrows). Interestingly, D169G-containing puncta appeared to be less defined (compare Fig. 4C with 4B,D–F). Because the D169G mutation lies within the RRM1 domain, it is tempting to speculate that this variant’s ability to associate with neuronal RNA granules might be impaired. Although at this time we do not know whether D169G affects the function of the RRM1 domain, it is interesting to note that previous studies have linked the RNA-binding activity of RRM1 to neurotoxicity in vivo (Voigt et al., 2010).
Fig. 4.

TDP-43 is expressed in dynamic cytoplasmic puncta in primary motor neurons. (A–F) TDP-43 expressed using D42 Gal4, visualized with anti-GFP antibody, localizes to distinct puncta within neurites (arrows). Neuronal membranes were labeled with anti-HRP antibodies. (G) Velocity quantification of TDP-43-containing puncta (mean, maximum and minimum velocities). (H) Quantification of total and net distances for TDP-43 puncta. (I) FRAP indicates that TDP-43 is mobile within neurites. Note that wild-type kinetics are significantly different to those of mutant TDP-43 (see text for discussion). Values show means ± s.e.m. Scale bar: 15 μm.
It has been previously shown that TDP-43 associates with RNA stress granules and, in motor neurons, TDP-43-containing granules undergo axonal transport (Fallini et al., 2012; Liu-Yesucevitz et al., 2010; McDonald et al., 2011). Upon stimulation with brain-derived neurotrophic factor, TDP-43 mutants mobilized to axons in greater amounts than wild-type TDP-43, which suggests an axonal transport-dependent mechanism of disease (Fallini et al., 2012). Using live imaging in primary motor neurons we compared the dynamic properties of several TDP-43 variants and found that TDP-43 puncta undergo transport at rates comparable to neuronal RNA granules containing Fragile X protein (FMRP) (Estes et al., 2008) (Fig. 4G). In this assay, we could not detect any significant differences between wild-type and mutant TDP-43, with granule speeds that ranged between 0.01 and 0.1 μm/second, consistent with microtubule-based transport. Similarly, total and net distances traveled within neurites were comparable among the TDP-43 variants used in our study (4.1–5.4 μm for total and 0.4–0.8 μm for net distances; Fig. 4H). To further characterize the kinetics of TDP-43 in neurites, we used FRAP, which determines the ability of TDP-43 to shuttle in and out of stationary RNA granules (i.e. mobility). These experiments indicate that TDP-43 is highly mobile within neurites, as indicated by a 55–64% recovery of the initial fluorescence intensity (Fig. 4I). Notably, wild-type TDP-43 exhibited significantly different dynamics compared to mutant TDP-43, with a recovery half-time of 9.7 seconds compared with 51.2 seconds for D169G, 56.8 seconds for G298S, 76.1 seconds for A315T and 62.0 for N345K TDP-43. These results are consistent with our experimental observations that wild-type TDP-43 granules are difficult to bleach down to 20% or less fluorescence (which was our set threshold for bleaching RNA granules with the laser; see Materials and Methods) due to their fast recovery. Furthermore, our data suggest that although both wild-type and mutant TDP-43 overexpression result in ALS-like phenotypes, the mechanisms by which mutant TDP-43 is neurotoxic is likely to be different and underscore the need to focus our attention on mutant alleles linked to disease (see also Estes et al., 2011).
TDP-43 overexpression in motor neurons or glia affects axonal growth and locomotor function at the neuromuscular junction
We and others have previously shown that TDP-43 is required for the architecture of the larval neuromuscular junction synapse (NMJ) (Estes et al., 2011; Feiguin et al., 2009; Li et al., 2010). Each NMJ contains several varicosities referred to as synaptic boutons, which form as motor neuron axons innervate the larval body wall musculature. We examined the synaptic vesicle marker cysteine string protein (CSP) (Bronk et al., 2005) and the neuronal membrane marker horseradish peroxidase (HRP) and focused our analyses on the well-characterized synapses at muscles 6/7 in abdominal segment 3 (Koh et al., 2000). Here we compared the effects of overexpressing wild-type as well as D169G, G298S, A315T and N345K TDP-43 in motor neurons (Fig. 5A–F) or glia (Fig. 5I–N) on the morphology of the NMJ. When expressed in motor neurons, all TDP-43 variants tested resulted in smaller synapses, as indicated by a reduced number of synaptic boutons (Fig. 5G). This anatomical phenotype was accompanied by an impairment in locomotor function, as indicated by a significant increase in larval turning time compared with controls (Fig. 5H).
Fig. 5.

TDP-43 variants affect the growth and function of the neuromuscular junction synapse. (A–F) Neuromuscular junctions in larvae expressing TDP-43 driven by D42 Gal4 were labeled by CSP (synaptic vesicle marker) and HRP (neuronal membrane marker). (G) Quantification of synaptic boutons indicates a reduction in synaptic size due to TDP-43 overexpression. (H) Larvae expressing TDP-43 in motor neurons are impaired in larval turning behavior. (I–N) Neuromuscular junctions in larvae expressing TDP-43 driven by repo Gal4 were labeled by CSP (synaptic vesicle marker) and HRP (neuronal membrane marker). (O) Quantification of synaptic boutons indicates a decrease in synaptic size due to TDP-43 overexpression. (P) Larvae expressing TDP-43 in glia are impaired in larval turning behavior. Values show means ± s.e.m. All comparisons were performed using D42 Gal4 driver controls and Student’s t-test was used to calculate significance; *P<0.5, **P<0.01, ***P<0.001. Scale bars: 30 μm.
Similarly, when expressed in glial cells, all variants except D169G TDP-43 resulted in significantly smaller NMJs, with a decreased number of synaptic boutons (Fig. 5O). Interestingly, when tested for their ability to turn, larvae expressing TDP-43 in glia also exhibited a significant impairment in locomotor function (Fig. 5P). These data show that, when expressed in either motor neurons or glia, the phenotypic consequences of TDP-43 on synaptic architecture and locomotor function are comparable.
Synaptic markers are differentially affected by TDP-43 expression in motor neurons versus glia
Our findings that TDP-43 modulates the architecture of the NMJ suggest potential effects on synaptic function. In order to test this possibility and to further investigate the molecular mechanisms underlying TDP-43 neurotoxicity in motor neurons versus glia, we examined the distribution of various synaptic markers including Bruchpilot (a core component of active zones recognized by the NC82 antibody) (Kittel et al., 2006; Wagh et al., 2006) and the glutamate receptor GluRIIC (Marrus and DiAntonio, 2004). In wild-type larvae there is generally a 1:1 correspondence of pre-synaptic active zones to post-synaptic glutamate receptor densities, which is thought to contribute to the efficient relay of synaptic signals from motor neurons to muscles. As shown in Fig. 6, we found that TDP-43 overexpression in motor neurons led to a significant increase in active zones (areas of neurotransmitter release) compared with controls (see NC82 staining in Fig. 6A–F and quantification in 6G). In contrast, there was no significant effect on glutamate receptor levels (as measured by quantitative confocal fluorescence) when TDP-43 mutations were expressed in motor neurons (Fig. 6H). We found an increase in glutamate receptor levels due to wild-type TDP-43 expression, which provides further support for the notion that the TDP-43 mutants utilize distinct cellular mechanisms. These data suggest an impairment in synaptic function at the NMJ due to overexpression of TDP-43 in motor neurons and provide an explanation for the larval turning defect shown in Fig. 5H.
Fig. 6.

Active zones and postsynaptic glutamate receptor distribution are differentially affected at the neuromuscular junction by TDP-43 expression in motor neurons versus glia. (A–F) The presynaptic marker Bruchpilot (visualized with NC82 antibodies) and postsynaptic marker glutamate receptor (GluR) were used to label neuromuscular junctions in larvae expressing TDP-43 in motor neurons (staining and genotype as indicated). (G,H) Quantification of NC82 puncta indicates a significant increase in the number of presynaptic active zones per bouton (G), which is not accompanied by a similar increase in postsynaptic GluR (except for wild-type TDP, see H). (I–N) The presynaptic marker Bruchpilot (visualized with NC82 antibodies) and postsynaptic marker GluR label neuromuscular junctions in larvae expressing TDP-43 in glia (staining and genotype as indicated). (O,P) Quantification of NC82 puncta shows no effect on the number of presynaptic active zones per bouton (O), but a significant increase in postsynaptic GluR expression (P). Values show means ± s.e.m. All comparisons were performed using Gal4 driver controls and Student’s t-test was used to calculate significance; *P<0.5, **P<0.01, ***P<0.001. Scale bars: 5 μm.
Next, we investigated the synaptic phenotypes due to TDP-43 overexpression in glia and found that although there were no obvious changes in the number and distribution of pre-synaptic active zones (Fig. 6I–N,O), the levels of post-synaptic glutamate receptor were significantly increased (Fig. 6I–N,P). Taken together these data suggest a non-autonomous role for TDP-43 in glia, in modulating synaptic properties and are consistent with the locomotor function defects observed when TDP-43 is overexpressed in glia (Fig. 5P). Furthermore, these findings show that when expressed in either motor neurons or glia, TDP-43 leads to a mismatch, albeit in opposite direction between pre-synaptic active zones and post-synaptic glutamate receptor densities, which in turn, translates into synaptic and locomotor dysfunction.
Sleep and locomotor activity in adult flies are impaired due to TDP-43 expression
In addition to impaired motor function, ALS patients exhibit sleep disturbances (Lo Coco et al., 2011). To determine whether sleep patterns are also affected in our fly model we used Drosophila Activity Monitors (DAMs) and quantified both locomotor and sleep activity in adult flies expressing TDP-43. DAMs are kept at 25°C in an incubator equipped with 12-hour alternating dark-light cycles and can hold several long and narrow glass tubes filled with food on one end, plugged with cotton at the other end and housing one fly each. As a fly walks back and forth within its tube, it interrupts an infrared beam that crosses the tube at its midpoint and this interruption, detected by the onboard electronics, is added to the tube’s activity count as a measure of fly activity. This daily record can be simultaneously acquired from several flies and provides a good measure of both the intensity of locomotor activity and the relative periods of rest. A period of rest of 5 minutes or more is defined as sleep (Sehgal and Mignot, 2011). Locomotor activity was evaluated for populations of flies with the same genotype as an average activity over time using Pysolo, a multiplatform software for analyzing sleep and locomotor patterns in Drosophila (Gilestro and Cirelli, 2009). As seen in Fig. 7, these experiments showed that TDP-43 expression in motor neurons led to a significant reduction in locomotor activity (Fig. 7A). In addition, total sleep was altered, with wild-type TDP-43-expressing flies sleeping more that those expressing mutant TDP-43, which exhibited a reduction in total sleep (Fig. 7B). Interestingly, when the sleep data was analyzed in more detail, we found that although adults expressing TDP-43 variants exhibited, on average, shorter sleep episodes both during day and night (Fig. 7C,E), overall they exhibit significantly more sleep episodes (Fig. 7D,F). These findings are consistent with the notion that TDP-43 leads to sleep fragmentation, possibly due to hyperexcitability.
Fig. 7.

TDP-43 overexpression affects sleep and locomotor activity in adult flies. (A–F) TDP-43 variants were expressed in motor neurons using D42 Gal4. Measurements using DAMs show that locomotor activity is significantly reduced overall (A) and total sleep is significantly altered, with wild-type TDP-43 leading to longer overall sleep than the mutant variants (B). During the day (C,D) as well as during the night (E,F), TDP-43 expression results in significantly more sleep episodes (D,F) that last shorter intervals of time (C,E). (G–L) TDP-43 variants were expressed in glial cells using repo Gal4. Measurements using DAMs show variable effects on locomotor activity (G) and a significant reduction in total sleep (H). During the day (I,J) there are variable effects on sleep. During the night (K,L), TDP-43 expression in glia results in significantly more sleep episodes (L) that last shorter amounts of time (K). Values show means ± s.e.m. All comparisons were performed using D42 Gal4 driver controls and significance was determined using a non-parametric Wilcox test calculated in R; *P<0.5, **P<0.01, ***P<0.001.
When expressed in glia, TDP-43 also led to alterations in locomotor activity, although only D169G and N345K were significantly different from controls (Fig. 7G). With regard to sleep activity, total sleep was significantly reduced for all variants used in this study (Fig. 7H). During the day, we found alterations in both the number of sleep episodes and their duration; however we could not consistently detect statistically significant differences (Fig. 7I,J). In contrast, during the night we found significantly more (Fig. 7L), albeit shorter, sleep episodes (Fig. 7K) for all TDP-43-expressing flies compared with controls. Our results indicate that both motor neuron and glial expression of TDP-43 led to alterations in adult locomotion and sleep patterns, although glial expression was mildly less toxic, especially during the day. These data are consistent with sleep fragmentation and parallel the disturbances in sleep patterns reported in patients (Lo Coco et al., 2011), suggesting that the fly model could provide useful insights into this elusive aspect of ALS pathology.
DISCUSSION
In human patients, ALS is primarily diagnosed by eliminating other known motor neuron or neurodegenerative conditions. This is due in part to a lack of biomarkers and in part due to the presence of complex and variable phenotypes, an issue further compounded by genetic background effects in sporadic ALS, which represents the majority of cases. To address these significant issues, efforts are being made to identify disease biomarkers and to characterize disease phenotypes by modeling ALS in various animal models. We have developed a Drosophila model of ALS based on TDP-43, an RNA-binding protein that has emerged as a common denominator for most ALS cases known to date, due to its presence in cytoplasmic inclusions as well as the discovery of point mutations in ALS patients (Baloh, 2011; Colombrita et al., 2011; Fiesel and Kahle, 2011; Neumann, 2009).
Consistent with our previous observations on wild-type and A315T TDP-43 (Estes et al., 2011), the overexpression of the D169G, G298S and N345K variants in the developing neuroepithelium leads to a progressive and dose-dependent neurodegeneration. Interestingly, the D169G mutation, which lies in the RRM1 domain, appears less toxic in the eye than the other alleles, consistent with previous reports that RNA-binding mediated by the RRM1 domain is crucial for mediating neurotoxicity (Voigt et al., 2010). Notably, in mammalian cells, the D169G mutation has no effect on the formation of stress granules in response to oxidative stress, as opposed to a mutation lying within the C-terminal domain, namely R361S (McDonald et al., 2011). We also found that D169G behaves differently to the other TDP-43 variants in multiple phenotypic assays. For example, there appear to be less defined D169G puncta in primary motor neurons compared with other variants, suggesting that the association with neuronal RNA granules is dependent on a fully functional RRM1 domain. Conversely, when TDP-43 variants were expressed in glia, the D169G mutant was one of two (the other was N345K) that showed significant effects on nuclear shape and adult locomotor activity (see Table 1 for a summary of phenotypes). Furthermore, D169G expression in glia had no effect on synaptic size, as indicated by the number of synaptic boutons that were similar to controls.
In other assays, similar phenotypic effects were generated by the different TDP-43 mutations, including the presence of cytoplasmic puncta in photoreceptor axons, a detectable truncated C-terminal fragment, nuclear shape defects and transport kinetics when expressed in motor neurons. Larval locomotor function and the distribution of synaptic markers such as presynaptic active zones and postsynaptic glutamate receptors were also similarly affected by the different TDP-43 variants used in our study. Notably, wild-type TDP-43 showed a different effect to that of the mutant variants on nuclear shape, transport kinetics and glutamate receptor distribution when expressed in motor neurons. These data provide a much needed map of the phenotypic complexities due to overexpression of various TDP-43 alleles, including wild-type and several mutants linked to ALS. Our findings support the assertion that various TDP-43 alleles are divergent and underscore the importance of determining their in vivo phenotypes.
A major finding resulting from our direct comparison of the effects of TDP-43 in motor neurons and glia is that these different cell types exhibit a differential response to TDP-43 expression. This might simply reflect the different functions of these individual cell types in the nervous system and/or be a consequence of TDP-43 regulating motor neurons versus glial-specific RNA targets. Notably, TDP-43 appears restricted to the nucleus in motor neurons but forms cytoplasmic puncta in glial cells, in vivo. These differences in subcellular localization translate into differential effects on nuclear shape, which is often an indicator of cellular health (Dahl et al., 2008). Furthermore, motor neurons and glia appear to handle TDP-43 expression by differentially modulating the distribution of synaptic proteins including Bruchpilot, a presynaptic active zone marker, and postsynaptic glutamate receptors. The significant increase in active zones due to TDP-43 overexpression in motor neurons is not matched by a similar increase in glutamate receptors, suggesting that excess glutamate is released in the synaptic cleft. This is consistent with increased excitability, one of the proposed mechanisms underlying ALS (Vucic et al., 2008). It will be interesting to see whether manipulating the levels of the glutamate transporter EAAT in glial cells can modulate the functional defects due to TDP-43 expression in motor neurons. Conversely, when TDP-43 is expressed in glia, there is an increase in postsynaptic glutamate receptor levels but not in presynaptic active zones. This mismatch between the pre- and postsynaptic sides of the neuromuscular junction is consistent with the functional defects identified using larval turning assays. Taken together, these findings demonstrate that TDP-43 expression in glia is also toxic with respect to locomotor function, although its precise molecular mechanism remains to be elucidated. Future experiments will be aimed at investigating the effect of TDP-43 expression on synaptic physiology at the neuromuscular junction. It is interesting to note that glial cells are clearly involved in ALS and related neurological disorders (Ince et al., 2011) and that our fly model recapitulates this less-understood aspect of pathology. Given the availability of Gal4 drivers specific to subsets of glia, including astrocytes and peripheral glia that surround motor neuron axons, it will be possible in the near future to investigate the contribution of these different glial subtypes to ALS pathology.
Our side-by-side comparison of motor neuron and glial expression showed that adult locomotor activity is also impaired in both conditions, although TDP-43 toxicity in glia appears to be slightly milder. This could be attributed in part to differences in levels of expression between the D42 and repo Gal4 lines that were used to drive expression in motor neurons and glia, respectively. It is interesting to note, however, that total sleep time was similarly affected by TDP-43 expression in both motor neurons and glia, suggesting that the sleep phenotypes are less sensitive to differences between Gal4 driver levels of expression. Although the sleep phenotypes we detected could be due to restlessness caused by impaired motor function rather than being a circadian rhythm defect per se, our studies uncovered a sleep fragmentation phenotype similar to that reported in ALS patients. This is a novel phenotypic aspect of TDP-43 and can provide a useful assay for testing candidate interacting genes and small molecules with therapeutic potential for ALS.
In summary, we provide further support for the fly model as an excellent experimental system for studying ALS in vivo. We provide evidence that motor neurons and glia are both susceptible to TDP-43 toxicity; however, it is important to note that these distinct cell types appear to mount different responses to TDP-43 expression. TDP-43 is required autonomously in motor neurons and non-autonomously in glia to regulate various morphological and functional aspects of the neuromuscular junction. The presence of TDP-43 cytoplasmic puncta in glia, but not in motor neurons, in vivo is intriguing. Together with the fact that both motor neuron and glial expression leads to functional defects, our data suggest that although the end point effect is the same, i.e. locomotor dysfunction, the cellular pathways employed by different cell types might be different. Elucidation of these differential responses of neuronal versus glial cells together with the phenotypic consequences of different TDP-43 variants is needed to further our understanding of the disease mechanisms involved in ALS.
MATERIALS AND METHODS
Drosophila genetics
All Drosophila stocks and crosses were kept on standard yeast/cornmeal/molasses food at 25°C unless otherwise noted. Human TDP-43, wild-type and D169G, G298S, A315T, and N345K mutants with YFP C-terminal tags (originally in pRS416 yeast expression vector, from Aaron Gitler, Stanford University, CA) were cloned into the pUAST germline transformation vector and transgenic lines were made as previously described (Estes et al., 2011). Gal4 drivers used in this study included the eye-specific GMR Gal4 R13, the motor neuron driver D42 Gal4 (Gustafson and Boulianne, 1996) and the glial-specific driver repo (Xiong et al., 1994). Several insertions were surveyed for each TDP-43 variant to ensure that the phenotypes observed in the eye are caused by TDP-43 and not due to positional effects. One or two lines for each variant were chosen for further study in individual assays (see Table 1). For controls, w1118 or UAS GFP NLS was crossed with the appropriate Gal4 drivers. In our hands, the expression of GFP NLS resulted in mild phenotypes, presumably due to the accumulation of high levels of GFP in the nucleus compared with TDP-43, which is expressed at lower levels than GFP NLS and also shuttles in and out of the nucleus (data not shown). Furthermore, because untagged and tagged wild-type TDP-43 produced comparable eye neurodegeneration and larval turning defects (data not shown), for quantification purposes we used Gal4 driver crossed to w1118 as controls. GFP NLS was used to mark the cells where individual Gal4 lines are driving the expression of TDP-43.
Western blots
Western blots were performed as described (Estes et al., 2011). TDP-43 transgenes were detected using rabbit polyclonal anti-GFP (Invitrogen) at 1:3000. Tubulin was used as a loading control and was detected using a mouse monoclonal anti-tubulin antibody (Millipore) at 1:1000. Secondary antibodies were HRP-conjugated goat anti-mouse or goat anti-rabbit, as appropriate, at 1:1000 (Thermo Scientific).
Immunohistochemistry and imaging
Adult fly eyes were imaged with a Leica MZ6 microscope equipped with an Olympus DP71 camera and controlled by Olympus DP Controller and Olympus DP Manager software. Individual images were processed using Adobe Photoshop CS2 (Adobe). Larval neuromuscular junction preparations have been described previously (Estes et al., 2011). Briefly, wandering third instar larvae were filleted in HL-3 saline, pinned out on Sylgard dishes and fixed in 3.5% formaldehyde in PBS, pH 7.2 for 20 minutes, then permeabilized with 0.1% Triton X-100. The blocking agent consisted of 2% BSA and 5% normal goat serum. The following antibodies were used: anti-DCSP2 at 1:300 (DSHB), anti-HRP-FITC at 1:50 (Sigma), anti Dlg polyclonal at 1:900 (gift from Peter Bryant, University of California, CA), anti-GluRC polyclonal at 1:2000 (gift from Aaron DiAntonio, Washington University, MO) and anti-Bruchpilot, NC82 at 1/50 (DSHB). Secondary antibodies were used at 1:1000 (Molecular Probes). Larval muscles 6–7 were imaged in abdominal segment A3 on a Nikon PCM 2000 or Zeiss Meta 510 confocal microscope and were displayed as projections of 1-μm serial sections. Boutons were manually counted and total CSP, glutamate receptor and HRP total pixel intensities were determined using Metamorph Image Analysis software (Universal Imaging). NC82 spot counts were performed using the Cell Counting Macro in the Metamorph software. All measurements were divided by total muscle area, and in some cases bouton number, to take into account variations in the size of the individual larvae or synapse size.
Eye discs and larval ventral ganglia were dissected and prepared for immunohistochemistry as described above. Eye discs were stained with anti-GFP-FITC (Rockland) at 1:200, rhodamine phalloidin at 1:300 (Molecular Probes), as well as Hoechst 33342 (Invitrogen) at 1:10,000. Ventral ganglia were stained as above except that instead of phalloidin, anti-HRP-TRITC (Sigma) was used at 1:50. Samples were imaged on a Zeiss Meta 510 confocal microscope and displayed as projections of 1-μm serial sections.
Nuclear shape features were measured by CellProfiler 2.0, subversion 11710 (Carpenter et al., 2006) (pipeline provided upon request). Shape measurements of nuclei were quantified in CellProfiler as follows: nuclei stained with Hoechst 33342 were identified using the Otsu global threshold algorithm and filtered to represent those along the midline of the ventral ganglia. Shapes were then measured by their individual descriptions. Compactness is measured as the variance of the radial distance of the nuclei’s perimeter from the centroid divided by the area. Form factor is measured as 4π × area/perimeter2. Eccentricity is measured as the ratio of the distance between the foci of a fitted ellipse and its major axis length. Measurements were first checked for normality using the Anderson-Darling test and then for significant differences using t-tests for normal data and Wilcox tests for non-normal data. Data processing, statistical tests and box plots were computed by the statistical software package R, version 2.15.0 (R Core Development Team).
Behavioral assays
Larval turning assays
Assays were performed as previously described (Estes et al., 2011). Briefly, wandering third instar larvae were placed on a grape juice plate at room temperature. After becoming acclimated, crawling larvae were gently turned onto their backs (ventral side up) and monitored until they were able to turn back (dorsal side up) and continue their forward movement. The amount of time that it took each larva to complete this task was recorded. Twenty to thirty larvae were used per genotype. Student’s t-test was performed to assess statistical significance.
Sleep assays
Adult flies of 1–3 days old were monitored individually in autoclavable plastic tubes (5 mm in diameter and 95 mm in length) on the same fly food described above under 12-hour light-dark cycles at 22°C with 45–60% humidity. Preparation of individual fly vials was performed prior to the experiment by melting pre-made fly food to liquidity and ¾ filling plastic vial end-caps with the mixture. Vials were then inserted into the caps and the food was allowed to set for ∼30 minutes prior to use. Sleep and locomotor activity data were collected using the Drosophila Activity Monitoring (DAM) system (Trikinetics, Waltham, MA) and analyzed using Pysolo, a multiplatform software for analyzing sleep and locomotor activity in Drosophila (Gilestro and Cirelli, 2009). Flies were given 32 hours to habituate to the experimental conditions prior to data collection. Approximately 32 flies were gathered and placed in each monitor (maximum capacity 32) for each genotype for each experimental replicate. Sleep and locomotor activity data were collected at 1-minute intervals for 5 days. Three experimental replicates were performed and data from each replicate was compiled. Pre-processing of the Pysolo data was performed in Microsoft Excel. Normality of the data was tested using the Anderson-Darling test using statistical software package R. Based on the fact that at least one group per parameter was not normally distributed compared with the control, a non-parametric Wilcox test was used in R (R Core Development Team) to calculate all P values.
Primary neuron cultures and trafficking measurements
Larval motor neurons were cultured as described (Estes et al., 2008) with the following exceptions: cells were isolated from wandering third instar larval ventral ganglia and imaged 5–6 days after plating at 25°C. Measurements of FRAP and speed were done as previously described (Estes et al., 2008).
Cellular fractionations
Cellular fractionation was carried out as described in (Liachko et al., 2010) using as a starting material adult heads snap-frozen in liquid nitrogen and ground to a fine powder.
Acknowledgments
We are indebted to the Himelic family and friends for their generous and continued support. We thank Dr Aaron Gitler (Stanford University) for the human TDP-43 constructs, Dr Carol Barnes (The McKnight Institute at UA) for support and discussions, Bloomington Stock Center for reagents, Iowa Developmental Studies Hybridoma Bank for antibodies, Genetics Services (Cambridge, MA) for generating transgenic flies and Dr Gio Bosco (UA, MCB) for access to the Olympus DP71 camera. We also acknowledge the support of Dr Carl Boswell and Patty Jansma (the MCB Imaging Facility), Anna Burns and Dr Lynne Oland (Department of Neuroscience Tissue Culture Facility), Donovan Lockwood and Briana Buscemi for technical assistance, and members of the Zarnescu laboratory for comments on the manuscript.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
P.S.E., S.G.D., A.P.M. and D.C.Z. designed the experiments and analyzed the data. P.S.E., S.G.D., A.P.M., A.V.B., A.S. and R.Z. performed the experiments. P.S.E., S.G.D. and D.C.Z. wrote the manuscript.
FUNDING
This work was supported by the Jim Himelic Foundation, the Muscular Dystrophy Association (in part through support from Mr and Mrs Dunham in memory of their daughter Susan Hope Nearing Dunham) [grant number MDA173230 to D.C.Z.] and the National Institutes of Health [grant number 1R21NS078429-01A1 to D.C.Z.]. Partial support was also provided by the McKnight Brain Research Foundation and the Undergraduate Biology Research Program funded by the Howard Hughes Medical Institute [grant number HHMI 52006942 to A.S.S.].
REFERENCES
- Alexander M. D., Traynor B. J., Miller N., Corr B., Frost E., McQuaid S., Brett F. M., Green A., Hardiman O. (2002). “True” sporadic ALS associated with a novel SOD-1 mutation. Ann. Neurol. 52, 680–683 [DOI] [PubMed] [Google Scholar]
- Ayala Y. M., Misteli T., Baralle F. E. (2008). TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc. Natl. Acad. Sci. USA 105, 3785–3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala Y. M., De Conti L., Avendaño-Vázquez S. E., Dhir A., Romano M., D’Ambrogio A., Tollervey J., Ule J., Baralle M., Buratti E., et al. (2011). TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 30, 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloh R. H. (2011). TDP-43: the relationship between protein aggregation and neurodegeneration in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. FEBS J. 278, 3539–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks G. T., Kuta A., Isaacs A. M., Fisher E. M. (2008). TDP-43 is a culprit in human neurodegeneration, and not just an innocent bystander. Mamm. Genome 19, 299–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beleza-Meireles A., Al-Chalabi A. (2009). Genetic studies of amyotrophic lateral sclerosis: controversies and perspectives. Amyotroph. Lateral Scler. 10, 1–14 [DOI] [PubMed] [Google Scholar]
- Bronk P., Nie Z., Klose M. K., Dawson-Scully K., Zhang J., Robertson R. M., Atwood H. L., Zinsmaier K. E. (2005). The multiple functions of cysteine-string protein analyzed at Drosophila nerve terminals. J. Neurosci. 25, 2204–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E., Baralle F. E. (2001). Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 276, 36337–36343 [DOI] [PubMed] [Google Scholar]
- Carpenter A. E., Jones T. R., Lamprecht M. R., Clarke C., Kang I. H., Friman O., Guertin D. A., Chang J. H., Lindquist R. A., Moffat J., et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrita C., Onesto E., Tiloca C., Ticozzi N., Silani V., Ratti A. (2011). RNA-binding proteins and RNA metabolism: a new scenario in the pathogenesis of Amyotrophic lateral sclerosis. Arch. Ital. Biol. 149, 83–99 [DOI] [PubMed] [Google Scholar]
- Couthouis J., Hart M. P., Shorter J., DeJesus-Hernandez M., Erion R., Oristano R., Liu A. X., Ramos D., Jethava N., Hosangadi D., et al. (2011). A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. USA 108, 20881–20890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl K. N., Ribeiro A. J., Lammerding J. (2008). Nuclear shape, mechanics, and mechanotransduction. Circ. Res. 102, 1307–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., Nicholson A. M., Finch N. A., Flynn H., Adamson J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey C. M., Cenik B., Sephton C. F., Dries D. R., Mayer P., 3rd, Good S. K., Johnson B. A., Herz J., Yu G. (2011). TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol. 31, 1098–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D., Haass C. (2011). TDP-43 and FUS: a nuclear affair. Trends Neurosci. 34, 339–348 [DOI] [PubMed] [Google Scholar]
- Estes P. S., O’Shea M., Clasen S., Zarnescu D. C. (2008). Fragile X protein controls the efficacy of mRNA transport in Drosophila neurons. Mol. Cell. Neurosci. 39, 170–179 [DOI] [PubMed] [Google Scholar]
- Estes P. S., Boehringer A., Zwick R., Tang J. E., Grigsby B., Zarnescu D. C. (2011). Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet. 20, 2308–2321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C., Bassell G. J., Rossoll W. (2012). The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 21, 3703–3718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiguin F., Godena V. K., Romano G., D’Ambrogio A., Klima R., Baralle F. E. (2009). Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583, 1586–1592 [DOI] [PubMed] [Google Scholar]
- Fiesel F. C., Kahle P. J. (2011). TDP-43 and FUS/TLS: cellular functions and implications for neurodegeneration. FEBS J. 278, 3550–3568 [DOI] [PubMed] [Google Scholar]
- Freibaum B. D., Chitta R. K., High A. A., Taylor J. P. (2010). Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 9, 1104–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Medel Y., Logan M. A., Ashley J., Ataman B., Budnik V., Freeman M. R. (2009). Glia and muscle sculpt neuromuscular arbors by engulfing destabilized synaptic boutons and shed presynaptic debris. PLoS Biol. 7, e1000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilestro G. F., Cirelli C. (2009). pySolo: a complete suite for sleep analysis in Drosophila. Bioinformatics 25, 1466–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson K., Boulianne G. L. (1996). Distinct expression patterns detected within individual tissues by the GAL4 enhancer trap technique. Genome 39, 174–182 [DOI] [PubMed] [Google Scholar]
- Ince P. G., Highley J. R., Kirby J., Wharton S. B., Takahashi H., Strong M. J., Shaw P. J. (2011). Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol. 122, 657–671 [DOI] [PubMed] [Google Scholar]
- Johnson B. S., McCaffery J. M., Lindquist S., Gitler A. D. (2008). A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 105, 6439–6444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E., Valdmanis P. N., Dion P., Spiegelman D., McConkey B. J., Vande Velde C., Bouchard J. P., Lacomblez L., Pochigaeva K., Salachas F., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574 [DOI] [PubMed] [Google Scholar]
- Kittel R. J., Wichmann C., Rasse T. M., Fouquet W., Schmidt M., Schmid A., Wagh D. A., Pawlu C., Kellner R. R., Willig K. I., et al. (2006). Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science 312, 1051–1054 [DOI] [PubMed] [Google Scholar]
- Koh Y. H., Gramates L. S., Budnik V. (2000). Drosophila larval neuromuscular junction: molecular components and mechanisms underlying synaptic plasticity. Microsc. Res. Tech. 49, 14–25 [DOI] [PubMed] [Google Scholar]
- Kwiatkowski T. J., Jr, Bosco D. A., Leclerc A. L., Tamrazian E., Vanderburg C. R., Russ C., Davis A., Gilchrist J., Kasarskis E. J., Munsat T., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208 [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C., Cleveland D. W. (2009). Rethinking ALS: the FUS about TDP-43. Cell 136, 1001–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanson N. A., Jr, Maltare A., King H., Smith R., Kim J. H., Taylor J. P., Lloyd T. E., Pandey U. B. (2011). A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet. 20, 2510–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Ray P., Rao E. J., Shi C., Guo W., Chen X., Woodruff E. A., 3rd, Fushimi K., Wu J. Y. (2010). A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 107, 3169–3174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachko N. F., Guthrie C. R., Kraemer B. C. (2010). Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci. 30, 16208–16219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L., Bilgutay A., Zhang Y. J., Vanderweyde T., Citro A., Mehta T., Zaarur N., McKee A., Bowser R., Sherman M., et al. (2010). Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS ONE 5, e13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Coco D., Mattaliano P., Spataro R., Mattaliano A., La Bella V. (2011). Sleep-wake disturbances in patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 82, 839–842 [DOI] [PubMed] [Google Scholar]
- Lu Y., Ferris J., Gao F. B. (2009). Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Mol. Brain 2, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa S., Leigh P. N., King A., Jones E., Steele J. C., Bodi I., Shaw C. E., Hortobagyi T., Al-Sarraj S. (2009). TDP-43 is consistently co-localized with ubiquitinated inclusions in sporadic and Guam amyotrophic lateral sclerosis but not in familial amyotrophic lateral sclerosis with and without SOD1 mutations. Neuropathology 29, 672–683 [DOI] [PubMed] [Google Scholar]
- Marrus S. B., DiAntonio A. (2004). Preferential localization of glutamate receptors opposite sites of high presynaptic release. Curr. Biol. 14, 924–931 [DOI] [PubMed] [Google Scholar]
- McDonald K. K., Aulas A., Destroismaisons L., Pickles S., Beleac E., Camu W., Rouleau G. A., Vande Velde C. (2011). TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 20, 1400–1410 [DOI] [PubMed] [Google Scholar]
- Neumann M. (2009). Molecular neuropathology of TDP-43 proteinopathies. Int. J. Mol. Sci. 10, 232–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- Parker S. J., Meyerowitz J., James J. L., Liddell J. R., Crouch P. J., Kanninen K. M., White A. R. (2012). Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochem. Int. 60, 415–424 [DOI] [PubMed] [Google Scholar]
- Polymenidou M., Lagier-Tourenne C., Hutt K. R., Huelga S. C., Moran J., Liang T. Y., Ling S. C., Sun E., Wancewicz E., Mazur C., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A. E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J. R., Schymick J. C., Laaksovirta H., van Swieten J. C., Myllykangas L., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford N. J., Zhang Y. J., Baker M., Gass J. M., Finch N. A., Xu Y. F., Stewart H., Kelley B. J., Kuntz K., Crook R. J., et al. (2008). Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 4, e1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal A., Mignot E. (2011). Genetics of sleep and sleep disorders. Cell 146, 194–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton C. F., Cenik C., Kucukural A., Dammer E. B., Cenik B., Han Y., Dewey C. M., Roth F. P., Herz J., Peng J., et al. (2011). Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 286, 1204–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J., Blair I. P., Tripathi V. B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J. C., Williams K. L., Buratti E., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C. F., Eguchi H., Tagawa A., Onodera O., Iwasaki T., Tsujino A., Nishizawa M., Kakita A., Takahashi H. (2007). TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol. 113, 535–542 [DOI] [PubMed] [Google Scholar]
- Tollervey J. R., Curk T., Rogelj B., Briese M., Cereda M., Kayikci M., König J., Hortobágyi T., Nishimura A. L., Zupunski V., et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C., Rogelj B., Hortobágyi T., De Vos K. J., Nishimura A. L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt A., Herholz D., Fiesel F. C., Kaur K., Müller D., Karsten P., Weber S. S., Kahle P. J., Marquardt T., Schulz J. B. (2010). TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS ONE 5, e12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic S., Nicholson G. A., Kiernan M. C. (2008). Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131, 1540–1550 [DOI] [PubMed] [Google Scholar]
- Wagh D. A., Rasse T. M., Asan E., Hofbauer A., Schwenkert I., Dürrbeck H., Buchner S., Dabauvalle M. C., Schmidt M., Qin G., et al. (2006). Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron 49, 833–844 [DOI] [PubMed] [Google Scholar]
- Wang I. F., Wu L. S., Chang H. Y., Shen C. K. (2008). TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J. Neurochem. 105, 797–806 [DOI] [PubMed] [Google Scholar]
- Wegorzewska I., Bell S., Cairns N. J., Miller T. M., Baloh R. H. (2009). TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 106, 18809–18814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. H., Fallini C., Ticozzi N., Keagle P. J., Sapp P. C., Piotrowska K., Lowe P., Koppers M., McKenna-Yasek D., Baron D. M., et al. (2012). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W. C., Okano H., Patel N. H., Blendy J. A., Montell C. (1994). repo encodes a glial-specific homeo domain protein required in the Drosophila nervous system. Genes Dev. 8, 981–994 [DOI] [PubMed] [Google Scholar]
- Yu Z., Fan D., Gui B., Shi L., Xuan C., Shan L., Wang Q., Shang Y., Wang Y. (2012). Neurodegeneration-associated TDP-43 interacts with fragile X mental retardation protein (FMRP)/Staufen (STAU1) and regulates SIRT1 expression in neuronal cells. J. Biol. Chem. 287, 22560–22572 [DOI] [PMC free article] [PubMed] [Google Scholar]