

SUMMARY
Apert syndrome is a congenital disorder characterized by severe skull malformations and caused by one of two missense mutations, S252W and P253R, on fibroblast growth factor receptor 2 (FGFR2). The molecular bases underlying differential Apert syndrome phenotypes are still poorly understood and it is unclear why cleft palate is more frequent in patients carrying the S252W mutation. Taking advantage of Apert syndrome mouse models, we performed a novel combination of morphometric, histological and immunohistochemical analyses to precisely quantify distinct palatal phenotypes in Fgfr2+/S252W and Fgfr2+/P253R mice. We localized regions of differentially altered FGF signaling and assessed local cell patterns to establish a baseline for understanding the differential effects of these two Fgfr2 mutations. Palatal suture scoring and comparative 3D shape analysis from high resolution μCT images of 120 newborn mouse skulls showed that Fgfr2+/S252W mice display relatively more severe palate dysmorphologies, with contracted and more separated palatal shelves, a greater tendency to fuse the maxillary-palatine sutures and aberrant development of the inter-premaxillary suture. These palatal defects are associated with suture-specific patterns of abnormal cellular proliferation, differentiation and apoptosis. The posterior region of the developing palate emerges as a potential target for therapeutic strategies in clinical management of cleft palate in Apert syndrome patients.
INTRODUCTION
Apert syndrome [OMIM 101200] is a rare congenital disorder with disease prevalence of 15–16 per million live births. Patients with Apert syndrome are characterized by premature fusion of the coronal suture(s) and severe craniofacial dysmorphology (Cohen and MacLean, 2000), but also exhibit many other developmental defects, including limb abnormalities, heart and lung defects, as well as neural malformations that could lead to cognitive impairment (Cohen and MacLean, 2000). In humans, there are at least 70 nucleotide substitutions that alter the amino acid sequence of fibroblast growth factor receptor (FGFR) genes (Hébert, 2011). Two of these, Ser252Trp (S252W) and Pro253Arg (P253R), occur on neighboring amino acids on the linker region between the second and third extracellular immunoglobulin domain on fibroblast growth factor receptor 2 (FGFR2) and together are responsible for almost 99% of reported cases of Apert syndrome. Approximately 67% of individuals with Apert syndrome have a FGFR2 S252W mutation, while the remaining 33% carry the FGFR2 P253R mutation (Park et al., 1995; Wilkie et al., 1995). The phenotypic outcome of Apert syndrome mutations is generally similar, such that genetic testing is required to identify the causative mutation. Comparative analyses suggest only highly localized phenotypic differences between the two FGFR2 mutations (Slaney et al., 1996). Cleft palate is 3.5 times more frequent in patients carrying the S252W mutation, whereas digit fusion (i.e. syndactyly) is more severe in patients with the P253R mutation (Slaney et al., 1996). These qualitative analyses are, however, based on relatively small samples of patients and details of differential effects of FGFR2 mutations on patients with Apert syndrome remain poorly understood.
Given the relatively low prevalence of Apert syndrome in humans, analysis of appropriate and representative mouse models for Apert syndrome, such as Fgfr2+/S252W and Fgfr2+/P253R mice (Wang et al., 2005; Wang et al., 2010), is crucial for an understanding of the developmental mechanisms underlying the phenotypic differences among patients carrying one FGFR2 mutation or the other. The correspondence between Apert syndrome mouse models and human patients with Apert syndrome has been demonstrated at the morphological, histological and molecular levels (Chen et al., 2003; Wang et al., 2005; Wang et al., 2010; Holmes et al., 2009; Aldridge et al., 2010; Du et al., 2010; Martínez-Abadías et al., 2010; Martínez-Abadías et al., 2011), validating the use of large experimentally controlled samples of mouse models to elucidate the complex etiology of Apert syndrome.
Using a comparative sample of newborn Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mouse models and their unaffected littermates (n=73) we found that murine skull dysmorphologies parallel those described in Apert syndrome patients, and detected significant morphological differences between the two Apert syndrome mouse models (Martínez-Abadías et al., 2010). A particular highly localized difference at the posterior region of the palatine bone prompted us to perform the current detailed analysis of the palate of Apert syndrome mouse models. Here, we analyze with higher precision the palatal dysmorphologies of Fgfr2+/S252W and Fgfr2+/P253R newborn mice (n=120), identifying further shape differences between the two Apert syndrome mouse models, and perform histological and immunohistochemical analyses to identify the cellular and molecular mechanisms affected by the Fgfr2 mutations that contribute to palatal dysmorphology in Apert syndrome.
TRANSLATIONAL IMPACT.
Clinical issue
Apert syndrome is a rare congenital disorder characterized by skull malformations and facial abnormalities. Almost 100% of Apert syndrome cases are caused by one of two amino acid substitutions, S252W or P253R, in a protein involved in FGF/FGFR signaling, fibroblast growth factor receptor 2 (FGFR2). Though the causative FGFR2 mutations have been identified, we still have little understanding of how they contribute to abnormal craniofacial phenotypes. Despite overall similarity between patients carrying one FGFR2 mutation or the other, it has been proposed that patients carrying the S252W mutation present with more severe palatal dysmorphology than patients carrying the P253R mutation. In addition, cleft palate occurs 3.5 times more frequently in the former group. In humans, it is difficult to differentiate the specific effects of each mutation because of the relatively low incidence of the disease and high level of phenotypic variation. For this reason, mouse models have become a valuable tool for determining the molecular mechanisms that alter palate development and lead to cleft palate in Apert syndrome.
Results
To determine how the two major Apert syndrome mutations lead to different palatal dysmorphologies, this study utilizes Fgfr2+/S252W and Fgfr2+/P253R mouse models. The authors report anatomical differences between the two mutant mouse models in terms of size and shape of the palatine bones, and demonstrate that the most striking abnormalities are associated with the Fgfr2 S252W mutation. These include patency of the inter-premaxillary suture, a wider separation of the palatine shelves and fusion of the suture between the palatine bones and maxilla, bones that together define the secondary palate. Furthermore, the authors’ cellular analysis reveals suture-specific aberrant cellular proliferation, differentiation and apoptosis in newborn mice.
Implications and future directions
This work shows that the two Apert syndrome mutations affect the development of the mouse palate in different ways. The results provide further evidence that the S252W mutation causes more severe palatal phenotypes than the P253R mutation. However, in line with previous analyses of craniofacial phenotypes, these results also suggest that ‘severity’ may be a highly localized phenomenon and that the local effects of altered FGF/FGFR signaling should be considered cell- and tissue-specific. Among the localized anatomical sites demonstrating perturbed FGF/FGFR signaling as a result of the S252W mutation is the posterior region of the palate, revealing a potential target for new therapeutic strategies in the clinical management of Apert syndrome.
Palatal malformations occur in 75% of Apert syndrome patients and include cleft soft palate or bifid uvula, as well as highly arched and constricted palates with a median furrow, lateral palatal swellings, relatively shorter hard palate and relatively longer and thicker soft palate (Kreiborg and Cohen, 1992; Cohen and Kreiborg, 1996). Understanding the consequences of altered FGF/FGFR signaling in the palates of Apert syndrome mouse models can help elucidate phenotypic differences among Apert syndrome patients and might also suggest potential mechanisms underlying cleft palate, one of the most common human birth defects (Mossey et al., 2009; Dixon et al., 2011). Palate development is a complex and incompletely understood process that requires a fine spatial and temporal orchestration of molecular and tissue interactions during embryogenesis that enable outgrowth, elevation, reorientation, adhesion and fusion of palatal shelves (Gritli-Linde, 2007; Iwata et al., 2011; Yu and Ornitz, 2011). Failure or change in any of these stages of palatal shelf development, or in any associated tissues of the oropharynx (e.g. tongue), can lead to palatal anomalies including cleft palate. Since FGF/FGFR is one of the interacting signaling pathways involved in palate development (Gritli-Linde, 2007), a detailed morphological examination of palatal morphogenesis using mouse models carrying well-defined mutations in FGF/FGFR signaling will contribute to our growing knowledge of the role of these signaling pathways in palate development.
To further investigate how the two mutations on FGFR2 causing Apert syndrome in humans differently affect palatal morphology in mouse models for Apert syndrome, we combined 3D geometric morphometric methods with histologic and immunohistochemical techniques in a novel characterization of palatal development. We performed geometric morphometric analysis of 3D landmark data registered from microCT (μCT) images of newborn (P0) skulls of Fgfr2+/S252W and Fgfr2+/P253R mouse models and assessed the pattern of fusion of five palatal sutures to precisely define differential palatal traits among Apert syndrome mouse models. Our analyses of large samples of Fgfr2+/S252W and Fgfr2+/P253 Apert syndrome mouse models and unaffected littermates guided detailed histological analysis and immunohistochemical assays that defined the relative roles of cell proliferation, differentiation and apoptosis that lead to palatal dysmorphogenesis.
Our results indicate a differential phenotypic effect of the S252W and P253R FGFR2 mutations in palatal morphogenesis and confirm that, as observed in humans, the FGFR2 S252W mutation is associated with more severe palatal dysmorphology. Histological and immunohistochemical analyses performed at the anatomical sites identified by the morphometric analyses provide suture-specific mechanistic explanations of the cellular and molecular processes leading to the differential palatal traits of Fgfr2+/S252W Apert syndrome mouse models. Translated into the clinical practice, our results could help to improve the management of patients with these craniofacial disorders.
RESULTS
To comparatively assess the effects of FGFR2 mutations on palate morphology, we analyzed patterns of palatal shape variation using geometric morphometric (GM) methods (Dryden and Mardia, 1998; Lele and Richtsmeier, 2001). Three-dimensional (3D) coordinates of a set of ten landmarks located along the medial aspects of the horizontal plate of the right and left palatine bones were collected from the reconstructed isosurfaces of the μCT images of each specimen (Fig. 1, Table 1) using heads of newborn Fgfr2+/S252W (n=24) and Fgfr2+/P253R (n=41) Apert syndrome mice, as well as their unaffected littermates that do not carry the mutation and that we use as controls (Fgfr2+/+ S252W, n=26; Fgfr2+/+ P253R, n=29). We assessed the pattern of fusion of five palatal sutures (inter-premaxillary, inter-maxillary, inter-palatine, right and left maxillary-palatine), qualitatively scoring the sutures as patent, partially fused or completely fused as visualized on μCT (see Fig. 1) (see Materials and Methods for more details).
Fig. 1.

Sutures and landmarks displayed on the palate of a P0 unaffected littermate. The palate is shown from an inferior view of a μCT reconstruction of the mouse skull in which the mandible has been removed. Sutures are shown by colored lines: red, inter-premaxillary suture; purple, inter-maxillary suture; green, right and left maxillary-palatine sutures; yellow, inter-palatine suture. p, premaxilla; m, maxilla; pa, palatine. Landmarks are indicated by red dots; codes and definitions are provided in Table 1.
Table 1.
Anatomical definitions of collected palatal landmarks displayed in Fig. 1

Differential palate dysmorphologies in Fgfr2+/S252W and Fgfr2+/P253 Apert syndrome mice
Palate shape information was extracted from the landmark coordinates using a General Procrustes Analysis (GPA), a procedure that superimposes configurations of landmarks and adjusts for the effects of orientation and scale (Dryden and Mardia, 1998). Allometric shape variation related to size was accounted for statistically by computing a multivariate regression of shape on size (see Materials and Methods for details). We used the allometry-adjusted Procrustes coordinates as input data for canonical variates analysis (CVA), a discriminant analysis used to explore shape variation and to maximize separation between the four pre-defined groups of newborn mice, including: Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mice and their respective unaffected littermates.
The first two canonical variates (CV1 and CV2) explain almost all the morphological variation of the sample (95.92%) and distribute the specimens into three different clusters: a cluster of Fgfr2+/P253R Apert syndrome mice, a cluster of unaffected littermates from both models and a cluster of Fgfr2+/S252W Apert syndrome mice (Fig. 2A). The CVA showed that the main differences in palatal shape occur between Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mice, which are distributed at the positive and negative extremes of the first canonical axis, respectively (Fig. 2A). Unaffected littermates show an intermediation position along CV1 (Fig. 2A).
Fig. 2.

Canonical variates analysis. (A) Scatterplot corresponding to the first two canonical variates (CV1 and CV2). Ellipses account for 90% of within-group variation. Fgfr2+/S252W Apert syndrome mice (extreme positive CV1 values) show a greater range of variation and include those cases representing the most extreme variation of the sample. (B) Shape changes (shown as wireframes) associated with extreme positive and negative values of CV1 and CV2 after adjusting for allometry superimposed on 3D μCT surface reconstructions of the newborn mouse palate. Shape changes associated with CV values are represented by solid red wireframes. Black dotted wireframes represent the palatine morphology of the mean shape. (Ba) Negative values of CV1 (Fgfr2+/P253R); (Bb) positive values of CV1 (Fgfr2+/S252W); (Bc) negative values of CV2 (Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mice); (Bd) positive values of CV2 (Fgfr2+/+ unaffected littermates).
Comparison of the associated shape changes corresponding to the extreme negative and positive CV1 values (Fig. 2B) shows that the palatine bones are contracted along the antero-posterior and medio-lateral axes in Fgfr2+/S252W Apert syndrome mice (Fig. 2Bb). In comparison to the mean shape, the anterior landmarks located along the outline of the palatine bones of Fgfr2+/S252W mice are shifted to a more posterior position (landmarks 1–4), whereas the posterior landmarks are shifted to a more anterior position (landmarks 5–10), and all landmarks of the palatal shelves are displaced laterally (Fig. 2Bb). These shape changes lead to palatine bones that are laterally displaced, with an associated increase in the width of the inter-palatine suture in Fgfr2+/S252W mice (Fig. 2B, top right) relative to Fgfr2+/P253R mutant mice (Fig. 2Ba).
Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mice are separated from their unaffected littermates along CV2 (Fig. 2A), though there is some overlap among groups. Shape differences between mutant and unaffected mice associated with this axis are localized to the posterior aspect of the bony palate (Fig. 2Bc,Bd). In unaffected littermates (positive end of CV2), the posterior aspect of the palatine bones show an arched outline (Fig. 2Bd), that gradually curves laterally and posteriorly away from the midline. This is in marked contrast to the outline of the palatine bones of Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mice that reveal a marked posterior shift of the most posteromedial landmarks (landmarks 5, 6) and an anterior shift of the most posterolateral landmarks (landmarks 9, 10) (Fig. 2Bc).
GDMA confidence interval testing (Lele and Richtsmeier, 1995; Lele and Richtsmeier, 2001) reveals significant differences in the way that the horizontal plates of the palatine bones of Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mice differ from their respective unaffected littermates (α=0.10). These analyses support the results of Procrustes-based analyses and provide statistical evidence of the differences in the localized effects of these two neighboring FGFR2 mutations on palatal development. GDMA confidence intervals demonstrate that the distance between the most anterolateral aspect of the horizontal plate of the palatine bone (where the palatine plates meet and eventually fuse with the maxillary alveolus; between landmarks 1 and 2) is increased relative to unaffected littermates in the Fgfr2+/S252W mutant mice but reduced relative to unaffected littermates in Fgfr2+/P253R mice. Posteriorly (between landmarks 7 and 8), the palate of Fgfr2+/S252W Apert syndrome mice is wider, but there is no difference between Fgfr2+/P253R mice and unaffected littermates. The horizontal plates of the palatine bones are also more profoundly reduced along the anteroposterior axis in Fgfr2+/S252W Apert syndrome mice. Together these observations suggest that the positioning of the maxillary alveolus and deficiency in development of the palatine plate contribute to the differences in the effects of the two FGFR2 mutations.
More severe palate dysmorphologies in Fgfr2+/S252W Apert syndrome mice
To statistically test the degree of differentiation in palatal shape between groups, we computed the Mahalanobis distances between all possible pairs of groups (Klingenberg and Monteiro, 2005). Results indicate that based on palatine morphology, Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mice are significantly different from each other as well as from their unaffected littermates, even after correcting for multiple testing by Bonferroni (Table 2). Unaffected littermates from S252W and P253R models are not significantly different from each other (Table 2). If we consider the Mahalanobis distance from each mutant Apert mouse model to their respective unaffected littermates as a measure of severity of the palatine dysmorphology, our results confirm that the palates of Fgfr2+/S252W Apert syndrome mice are more severely affected than those of Fgfr2+/P253R Apert syndrome mice, because the the Mahalanobis distance between mutant and unaffected littermates is 1.4 times greater in Fgfr2+/S252W mice (Table 2).
Table 2.
Pairwise Mahalanobis distances between Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mice and their unaffected littermate controls

Differential fusion patterns of palatal sutures in Apert syndrome mouse models
Comparison of the qualitative scoring of palatal suture patency showed that the inter-maxillary and the inter-palatine sutures are patent (∼75–100% of cases) or just partially fused (∼0–25%) in all four groups (Fig. 3). Although there are no significant differences between mutant and unaffected littermates in terms of the fusion pattern of these two midline sutures, Fgfr2+/S252W Apert syndrome mice show a greater degree of patency of the inter-palatine suture (Fig. 3).
Fig. 3.

Comparison of palatal suture patency. (A–D) Stacked columns represent the percentage of specimens with complete patent (green), partially fused (yellow) or completely fused (blue) palatal sutures for (A) Fgfr2+/S252W Apert syndrome mice, (B) Fgfr2+/+ unaffected littermates of the S252W model, (C) Fgfr2+/P253R Apert syndrome mice and (D) Fgfr2+/+ unaffected littermates of the P253R model.
Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mice show a higher tendency towards patency (∼60%) or partial fusion (∼20–40%) of the inter-premaxillary suture relative to their unaffected littermates, in which the inter-premaxillary suture is usually fused (∼40–95%) or partially fused (∼60–5%) at P0 (Fig. 3). Apparent differences in the pattern of fusion of the inter-premaxillary suture between the two groups of unaffected littermates are not relevant. Complete inter-premaxillary suture fusion (FFF) was observed in 22 out of 23 unaffected littermates of the S252W model, whereas 26 out of 30 unaffected littermates of the P253R model have the inter-premaxillary suture completely (FFF) or almost completely fused (PFF). The palates of unaffected littermates from both Apert syndrome mouse models are thus morphologically similar and comparable, as confirmed by the rest of the morphometric analyses (Fig. 2; Table 2).
Finally, we identified a difference in fusion patterns of the maxillary-palatine sutures between Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mice. In unaffected littermates of both models and in Fgfr2+/P253R Apert syndrome mice, the right and left maxillary-palatine sutures are patent (∼70–95%) or just partially fused (∼5–30%) (Fig. 3B–D), whereas these sutures are either partially (∼35%) or completely fused (∼25%) in Fgfr2+/S252W Apert syndrome mice at P0 (Fig. 3A).
Abnormal palate development in Fgfr2+/S252W Apert syndrome mice
Patterns of cell proliferation, differentiation and apoptosis were assessed to establish a baseline for understanding the effects of the FGFR2 mutations on localized cellular behaviors that underlie Apert syndrome. We focused our comparative histological analysis and immunohistochemical assays on the most significant differential effects detected on the palate morphology of Fgfr2+/S252W Apert syndrome mice at P0: the greater degree of fusion of the maxillary-palatine sutures, the greater separation of palatine bones at the midline and the higher tendency towards inter-premaxillary suture patency. Similar analyses of Fgfr2+/P253R Apert syndrome mice were not performed because the fusion pattern of the right/left maxillary-palatine in Fgfr2+/P253R mutant mice mimics the pattern of unaffected littermates (Fig. 3) and palatal shape changes in Fgfr2+/P253R mice are relatively mild, with morphologies that overlap with those of unaffected littermates (Fig. 2A).
Histological analyses confirmed that the maxillary-palatine sutures in Fgfr2+/S252W Apert syndrome mice commonly presented with localized partial premature fusion, showing variably fused osteogenic fronts with deposition of osteoid as compared with the distinct osteogenic fronts separated by undifferentiated mesenchymal cells in the unaffected littermates (Fig. 4A,B). In Fig. 4, a maxillary palatine suture of a Fgfr2+/S252W Apert syndrome mouse displays various aspects of early fusion. The opposing osteogenic fronts meet initially at a center of fusion midway between the dorsal and ventral aspects of the suture (Fig. 4B). In regions of the suture that remain patent, the mutant palatine and maxillary bones show the overlap typical of the unaffected littermates, although the mesenchymal interface between the two bones is often less regular in mutants (Fig. 4A,B and not shown). Alkaline phosphatase (ALP) activity, an early marker of osteogenesis, is low or absent in normal suture mesenchyme (Fig. 4C). In the mutant suture, ALP is similarly absent from half the suture mesenchyme dorsal to the central locus of fusion, but appears in the half ventral to the point of fusion (Fig. 4D). Cell proliferation assessed by Ki67 staining showed similar numbers of positive cells in the unfused mutant suture mesenchyme and adjacent osteogenic faces as compared with sutures of unaffected littermates. Proliferation was maintained even as ALP activity rose within the mesenchyme of fusing sutures (Fig. 4E,F,O). Runx2 expression, an additional marker of osteoblast differentiation, was similar in mutants and unaffected littermates, being higher in osteoblasts closer to bone and lower in the suture mesenchyme (Fig. 4G,H). As revealed by TUNEL staining, there was almost no apoptosis in the suture mesenchyme of unaffected littermates, but a low frequency of apoptosis was evident within the palatine and maxillary bones (Fig. 4I). In mutant sutures, the distribution and frequency of apoptotic cells was similar to sutures of unaffected specimens, even in the area of increased ALP activity. However, focal areas of intense apoptosis occurred in fusing sutures at the point where osteoid deposition bridges the overlapping bones in Fgfr2+/S252W mice (Fig. 4J). Antibody detection of the phosphorylated forms of the MAPK p38 and ERK1/2, major downstream effectors of FGFR signaling, showed no appreciable differences between mutant and unaffected littermates, although signals were decreased in the area of fusion dominated by apoptotic cells (Fig. 4K–N).
Fig. 4.
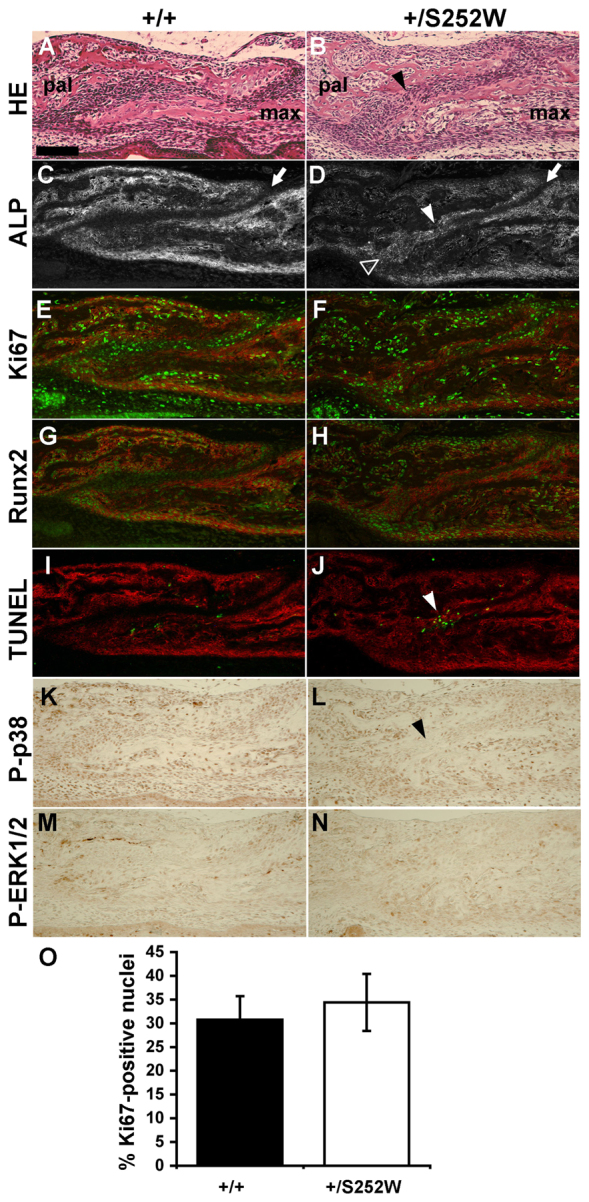
Maxillary-palatine suture at P0. (A,B) H&E staining shows a similar overlap of maxillary (max) and palatal (pal) bones in unaffected littermates (Fgfr2+/+) and Fgfr2+/S252W sutures, but secreted osteoid bridges the two bones (arrowhead) in a mutant suture beginning to fuse. (C,D) Osteoblast-specific ALP activity is absent from normal suture mesenchyme (C, arrow). In the early stages of fusion, ALP is still absent in mutant mesenchyme (D, arrow) dorsal to the point of fusion (white arrowhead), but has risen throughout mesenchyme ventral (black arrowhead) to this point (D). ALP activity is imaged fluorescently and shown in gray channel. (E,F) Immunofluorescent staining for Ki67 (green) shows a similar frequency of positive cells in the suture mesenchyme and adjacent osteogenic surfaces (stained for ALP activity; red) of the maxillary and palatal bones between unaffected littermates and mutant mice. (G,H) Immunofluorescent staining for Runx2 (green) shows similar expression levels between unaffected littermates and mutant mice, with expression being generally lower in the suture mesenchyme than in osteoblasts within osteogenic fronts (stained for ALP activity; red). (I,J) TUNEL staining (green) shows that apoptosis was negligible within the suture mesenchyme and infrequent in the bones of wild-type sutures. In mutant sutures, apoptosis was intense specifically in areas undergoing fusion (arrowhead). (K,L) Phospho-p38 levels within bone and suture mesenchyme were similar in mutant and unaffected littermate sutures, although expression was decreased in the area of mutant suture dominated by apoptotic cells (arrowhead). (M,N) Phospho-ERK 1/2 expression was concentrated around bone at similar levels in mutant and unaffected littermate sutures. (O) The percentage of suture mesenchyme cells expressing Ki67 was not significantly different between mutant and unaffected littermates. All images are from near-adjacent parasagittal sections from the same littermate pair. Rostral direction is to the right. Scale bar: 100 μm.
Histological analyses of the inter-palatine suture revealed palatine bones of Fgfr2+/S252W mice to be thicker and more robust relative to unaffected littermates (Fig. 5A,B). Both the osteogenic mesenchyme of the suture and the non-osteogenic mesenchyme ventral to the palatal bones were also thicker. Patency of the midline inter-palatine suture in both mutant and unaffected littermates at P0 (Fig. 5A,B), as well as the relatively wider separation of the palatine shelves in mutants detected by the CVA analysis was evident histologically (Fig. 5C,D). Ki67 staining showed no proliferation differences in either the osteogenic fronts of the palatine shelves or the intervening suture mesenchyme (Fig. 5E,F,O). Runx2 expression levels and distribution were similar in mutants and unaffected littermates, although the increased thickness of the mutant osteogenic mesenchyme is evidenced by the wider domain of Runx2 expression (Fig. 5G,H). Apoptotic cells, indicated by TUNEL staining, were infrequent among mesenchymal cells or within osteogenic fronts of both Fgfr2+/S252W Apert syndrome mice and unaffected littermates (Fig. 5I,J). Immunohistochemical staining for phosphorylated p38 and ERK1/2 showed no difference between mutants and unaffected littermates (Fig. 5K–N).
Fig. 5.
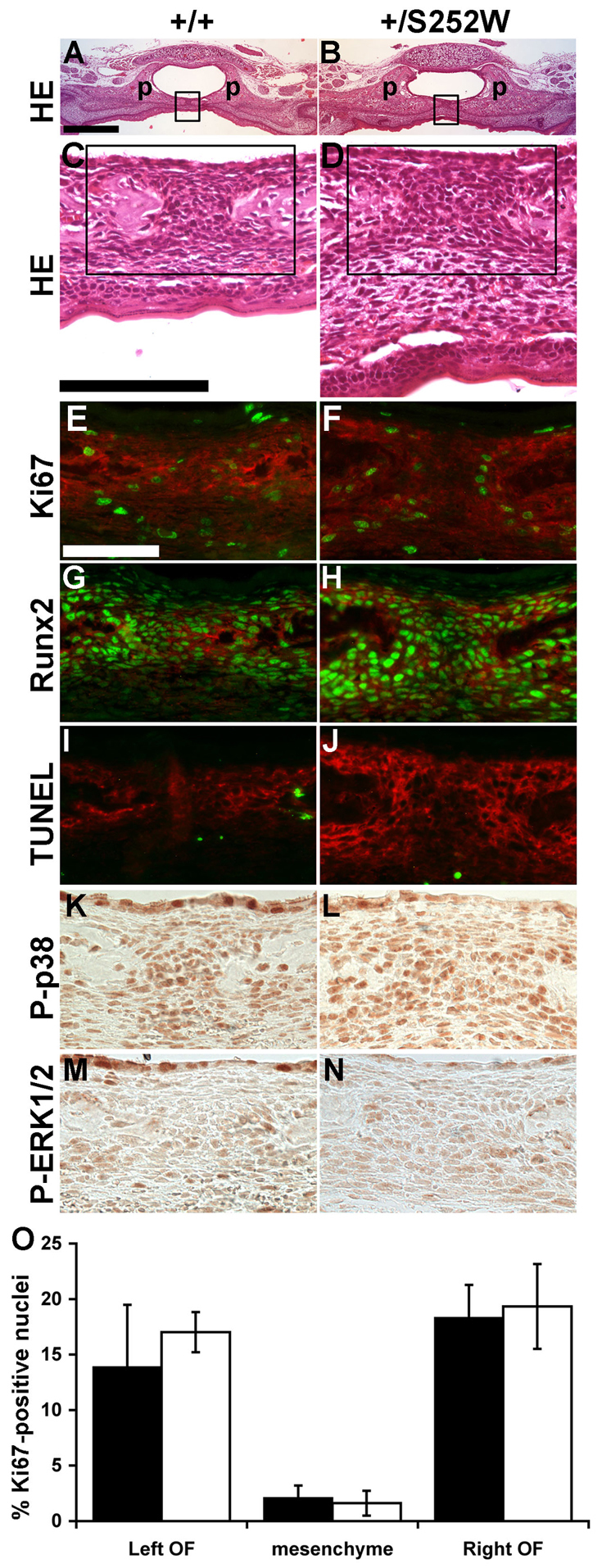
Inter-palatine suture at P0. (A,B) H&E staining shows that mutant palatal (p) bones are more robust than in unaffected littermates. Boxed areas are shown in C and D. (C,D) Within the suture, mutant palatal bones are separated further apart and both the osteogenic mesenchyme and underlying connective mesenchyme are thicker. Boxed areas are shown in E–N. (E,F) Immunofluorescent staining for Ki67 (green) shows a similar frequency of proliferating cells concentrated in the osteogenic fronts (stained for ALP activity; red) and lower in the intervening suture mesenchyme between unaffected littermates and mutant mice. (G,H) Immunofluorescent staining for Runx2 (green) shows similar expression levels between unaffected littermates and mutant mice (ALP activity; red). (I,J) TUNEL staining (green) was negligible within the suture mesenchyme of unaffected littermates and mutant mice. (K,L) Phospho-p38 levels and (M,N) phospho-ERK 1/2 levels within osteogenic fronts and suture mesenchyme were similar in mutant and unaffected littermate sutures. (O) The percentage of proliferating (Ki67-positive) cells in the left and right osteogenic fronts (OF) and intervening mesenchyme was not significantly different between mutant and unaffected littermates. Images in A–J are from near-adjacent sections from the same littermate pair. Images in K–N are from other littermate pairs. Scale bars: (A,B) 500 μm; (C,D) 100 μm; (E–N) 50 μm.
Histological analyses of the inter-premaxillary suture revealed striking differences between unaffected littermates and Fgfr2+/S252W mutants. The paired premaxillary bones in unaffected littermates are broad, consisting of trabecular bone ventrally, and are separated at the midline by a thick mesenchyme. Each premaxilla extends vertically as a thin compact sheet of bone curving around the overlying vomeronasal cartilage to end adjacent to the cartilage of the nasal septum. The mesenchyme separating these vertical sheets is sparse and flattened osteoblasts line their surface, separated from the vomeronasal cartilage on the opposite face by a mesenchyme a few cells in thickness (Fig. 6A). In contrast to unaffected littermates, the mutant premaxillary bones are thicker ventrally and extend vertically as broad trabecular wedges tapering to points adjacent to the cartilage of the nasal septum. The thin compact sheets of bone seen in unaffected littermates seem to be retained in these wedges, forming the lateral side curving along the vomeronasal cartilages, from which trabecular bone extends medially. The mesenchyme of Fgfr2+/S252W mice separating these bones remains thick as it extends dorsally, but typically thins below the nasal septum. The septal cartilage is often misaligned with the inter-premaxillary suture mesenchyme in the mutant suture. Only a single cell layer typically separates the premaxillary bone from the vomeronasal cartilage. The vertical thickening of the inter-premaxillary bones and intervening mesenchyme greatly widens the distance between the paired vomeronasal organs in mutants compared with unaffected littermates (Fig. 6B).
Fig. 6.

Inter-premaxillary suture at P0. (A,B) H&E staining shows that mutant sutures present with aberrant trabecular overgrowth of the vertical extensions of the premaxillary bones (pm), thickening of the intervening suture mesenchyme and misalignment of the suture with the cartilage of the nasal septum (s). Widening of the mutant suture is evident in the greater separation of the cartilages (c) encapsulating the vomeronasal organs. (C,D,M) Immunofluorescent staining for Ki67 (gray channel shown) shows increased proliferation in mutant suture mesenchyme compared with unaffected littermates. (E,F) Immunofluorescent staining for Runx2 (gray channel shown) shows similar expression levels between unaffected littermates and mutant mice, although mutant suture mesenchyme is more abundant and highly Runx2-positive cells are more numerous along the bone-mesenchyme interface. (G,H,N) TUNEL staining (green) shows minimal apoptosis within the suture mesenchyme of unaffected littermates and mutant mice but a high frequency in the adjacent osteogenic domains (stained for ALP activity; red). In these regions, apoptosis is significantly higher in mutants compared with unaffected littermates. (I–L) Phospho-p38 and phospho-ERK 1/2 levels are similar in mutant and unaffected littermate sutures. (M) Proliferation is significantly increased in mutant suture mesenchyme relative to unaffected littermates; *P=0.035. (N) Apoptosis is significantly increased in bone bordering the suture mesenchyme in mutants relative to unaffected littermates; *P=0.026. Images from A–L are from near-adjacent sections from the same littermate pair. Scale bar: 100 μm.
Ki67 staining showed that the proportion of proliferating cells was significantly increased within the midline mesenchyme separating premaxillary bones in Fgfr2+/S252W mice compared with unaffected littermates. This difference was especially notable comparing the dorsal portion of the sutures (Fig. 6C,D,M). The expression levels of Runx2 were similar in Fgfr2+/S252W mice and unaffected littermates, with high expression in osteoblasts and low expression in the intervening mesenchyme, but this mesenchyme was more extensive in the mutant suture. The difference in the cellularity of the medial and lateral faces of the premaxillary bones is also readily appreciated with Runx2 imaging, with more Runx2-expressing osteoblasts lining the medial surfaces and fewer lining the lateral surfaces of mutant premaxillary bones compared with unaffected littermates (Fig. 6E,F). ALP activity levels were also similar in Fgfr2+/S252W mice and unaffected littermates, but decreased strongly in the sparse mesenchyme separating the vertical bone extensions in unaffected littermates (Fig. 6G,H). Apoptotic cells detected by TUNEL staining were infrequent in the suture mesenchyme but were common in the adjacent trabecular bone of both mutants and unaffected littermates and occurred with significantly increased frequency in the Fgfr2+/S252W mice (Fig. 6G,H,N). The levels of phosphorylated p38 (Fig. 6I,J) and phosphorylated ERK1/2 (Fig. 6K,L) were similar in mutant sutures and unaffected littermates.
DISCUSSION
Our results demonstrate the differential effects of the S252W and P253R FGFR2 mutations on palatal morphogenesis and reveal the aberrant cellular behaviors associated with the phenotypic differences between Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mice. The morphometric analyses and the detailed scoring of palatal suture patency detected significant palatal shape differences between Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mice and underscored what has been reported in humans: individuals carrying the S252W mutation show more severe palatal dysmorphologies. The histological and immunohistochemical analyses highlighted that these palatal defects are highly localized and suture-specific and can be associated with dynamic combinations of abnormal patterns of cellular proliferation, differentiation and apoptosis. The extent of these changes at P0 suggests their beginnings earlier in embryogenesis.
From bone palatal defects to cellular and molecular alterations in Apert syndrome mouse models
Differential palatal defects in Fgfr2+/S252W and Fgfr2+/P253R mutant mice involve gross shape changes of the palatine bones (Fig. 2) and differences in fusion patterns of palatal sutures (Fig. 3). The most striking palatal defects of Fgfr2+/S252W mice include palatal shelves that were contracted and reduced in length with an enlarged inter-palatine distance (Fig. 2B), a greater tendency to fuse the right and left maxillary-palatine sutures and a tendency for the inter-premaxillary suture to remain patent (Fig. 3). Analysis of several litters and specimens per genotype was crucial in revealing highly localized differences in dynamic and variable developmental processes such as suture closure.
Our data confirm that premature fusion of the maxillary-palatine suture is a feature of palatal dysplasia in Apert syndrome (Purushothaman et al., 2011; Holmes and Basilico, 2012) and provide a snapshot of the process of closure of this suture (Fig. 4B,D). Apoptosis in the mutant maxillary-palatine suture is specifically increased in the areas of bone deposition that bridge the two bones, but not in areas of the suture with increased ALP activity that presumably precede fusion, or along bone surfaces in other regions of the maxillary and palatine bones (Fig. 4I,J). Increased apoptosis appears to be a later event in maxillary-palatine suture fusion, similar to its late occurrence in the fusing coronal suture in this Apert mouse model (Holmes et al., 2009).
Regarding the inter-palatine suture, no significant changes in cell proliferation (Fig. 5E,F), differentiation (Fig. 5G,H) or death (Fig. 5I,J) were found. However, the histological analysis revealed that the suture mesenchyme and the palatal bones were thicker in Fgfr2+/S252W mice relative to unaffected littermates. The greater midline separation of the right and left palatine bones observed in Fgfr2+/S252W mice (Fig. 2B; Fig. 5A,B) might be the result of changes occurring earlier in embryogenesis and undetectable at P0 and/or of other cellular processes and mechanisms not analyzed here. Considering that Fgfr2 is expressed in the outer layer of immature osteoblasts of the palatine bones during development (Rice et al., 2003), a possible explanation that requires further testing is that altered growth kinetics bestowed by a mutant Fgfr2 might affect bone shape throughout development, causing the localized thickening of palatine bones and the wider separation in the midline suture.
Our results reveal significantly abnormal cell patterns of proliferation, differentiation and apoptosis local to the inter-premaxillary suture, causing a lack of fusion and maldevelopment of the anterior palate in Fgfr2+/S252W mice. In contrast to what was observed at the maxillary-palatine suture, a high incidence of apoptosis was common to both the unaffected and mutant inter-premaxillary suture, revealing a distribution largely restricted to the bone-forming surfaces adjacent to the suture mesenchyme (Fig. 6G,H,N), occurring in the absence of suture fusion. Although the incidence was significantly higher in the mutant suture, this would appear to be a normal developmental process at this location, accentuated by aberrant FGF/FGFR signaling, and perhaps related to the differentiation of osteoblasts from mesenchyme with the function of limiting osteoblast numbers. Coupled with the increased proliferation seen in mutant mesenchyme, the inter-premaxillary suture could provide an in vivo example of the finding in vitro that chronic FGF signaling or the expression of FGFR2 craniosynostosis mutant proteins induces proliferation in immature osteoblasts but apoptosis in mature osteoblasts (Mansukhani et al., 2000). The little-known roles of apoptosis in normal and pathological facial suture formation appears to be suture-specific and requires further study.
Finally, immunohistochemical staining suggested that altered FGF/FGFR signaling in palatal sutures did not trigger significant differences in activation of MAPK, p38 and ERK1/2 in any of the sutures analyzed, at least at P0. However, p38 and ERK1/2 are major effectors of the FGF signaling pathway and it has been shown that inhibition of ERK1/2 in mice carrying the S252W mutation significantly improves facial development, suggesting the importance at least of ERK1/2 activity in the Apert facial phenotype (Shukla et al., 2007). Although we did not see significant differences in FGFR2 signaling within sutures as measured by detection of phosphorylated p38 and phosphorylated ERK1/2, such changes might be subtle in vivo, generating aberrant phenotypes incrementally over time.
Palate is a preferential target of the FGFR2 S252W Apert syndrome mutation
Our results suggest that the S252W mutation leads to more severe palate dysmorphologies than the P253R mutation, confirming the pattern already described in patients with Apert syndrome (Slaney et al., 1996). Larger Mahalanobis distances (Table 2) and greater ranges of palatal shape variation (Fig. 2A) show that palates of Fgfr2+/S252W mutant mice are more severely affected. The palatal phenotype of Fgfr2+/P253R mice was not unaffected, however. Rather, the palates of the two Apert syndrome mouse models were affected differently (Fig. 2), resulting in varying palatal dysmorphologies that were more intense in Fgfr2+/S252W mutant mice. Fgfr2+/P253R mice might show more severe malformations at other anatomical locations or have other tissues or developmental systems more severely affected. In fact, patients carrying the P253R mutation present with more severe syndactyly (Slaney et al., 1996).
Perturbed FGF/FGFR signaling leads to palate dysmorphology in Apert syndrome
Compelling evidence suggests that FGF/FGFR signaling plays an essential role in palatal development mediating cell communication and interaction (Wilke et al., 1997). Misregulation of FGF/FGFR signaling can result in severe dysmorphogenesis and disease, including malformations of the palate that occur as a part of a syndrome or contribute to the occurrence of isolated cleft palate (Dixon et al., 2011). For instance, 3–5% of human cases of nonsyndromic cleft lip and palate are associated with mutations and single nucleotide polymorphisms affecting genes directly involved in the FGF/FGFR pathway (Riley et al., 2007). Mouse models carrying mutations on Fgf10, Fgf18, Fgf8 and Fgfr1, Fgfr2 display abnormal or cleft palate providing further support of the involvement of FGF/FGFR signaling in the pathogenesis of cleft palate (De Moerlooze et al., 2000; Abu-Issa et al., 2002; Liu et al., 2002; Ohbayashi et al., 2002; Trokovic et al., 2003; Rice et al., 2004; Wang et al., 2005; Martínez-Abadías et al., 2010; Snyder-Warwick et al., 2010; Wang et al., 2010; Purushothaman et al., 2011).
Several mechanisms involving alternative splicing of the FGFR2 molecules and abnormal interaction of mutant receptors with inappropriate ligands have been proposed to explain the pathogenesis of cleft palate in Apert syndrome. Ibrahimi and colleagues suggested that the differential increase in FGFR2-IIIc affinity for a specific ligand such as FGF2, which is expressed in both facial ectoderm and mesenchyme, could explain the higher frequency of cleft palate in Apert syndrome patients with the FGFR2 S252W mutation (Ibrahimi et al., 2001). Britto and colleagues proposed that the incomplete expressivity of cleft palate in humans with Apert syndrome is consistent with a dose-dependent effect of FGFR2 mutations on palatal shelf fusion via the FGFR2-IIIb isoform (Britto et al., 2002). In this case, the Apert mutations might confer a dominant-negative functional effect on FGFR2-IIIb in human palatal epithelia, resulting in failure of fusion of the palatal shelves (Britto et al., 2002). Hajihosseini and colleagues hypothesized that cleft palate in Apert syndrome is the result of loss-of-FGFR2-IIIb function in epithelial cells, secondary to alleles that cause a gain-of-FGFR2 function in mesenchymal cells (Hajihosseini et al., 2009).
These varying lines of evidence highlight altered cross-talk between epithelial and mesenchymal cells, mediated by FGF/FGFR signaling and causing palate malformations. Our findings in Apert syndrome mouse models suggest that cellular miscommunication, evidenced by local variation in patterns of cell proliferation, differentiation and apoptosis at P0, is associated with highly localized palate dysmorphologies that might be associated with aberrant signaling specific to early undifferentiated cell populations, formative sutures and/or bony precursors.
Insights into altered palate development in Apert syndrome
Patterns of palatal suture fusion scored in unaffected littermates suggest that in normal palate development the fusion progresses from anterior to posterior, with anterior palatal sutures fusing first before birth (i.e. inter-premaxillary suture) and posterior palatal sutures fusing postnatally (i.e. inter-maxillary, inter-palatine and maxillary-palatine sutures) (Fig. 3). This gradient is reminiscent of that described for normal development of the palate and tongue during elevation of the palatal shelves (Yu and Ornitz, 2011), suggesting a continuation of gradients of morphogenetic processes during early and late palatal development. Here we show that palate development in Fgfr2+/S252W Apert syndrome mice is altered by aberrant cellular behaviors that cause a premature fusion of anterior transverse palatal sutures (e.g. right and left maxillary-palatine sutures) (Fig. 4) as well as failure of closure of palatal midline sutures, such as the inter-palatine (Fig. 5) and the inter-premaxillary sutures (Fig. 6), which are patent or show osteogenic fronts that are more separated in Fgfr2+/S252W mutant mice. Interestingly, Apert syndrome FGFR2 mutations also cause premature fusion of other non-midline sutures of the craniofacial complex, such as the coronal and the premaxillary-maxillary sutures (Wang et al., 2005; Wang et al., 2010; Martínez-Abadías et al., 2010). This abnormal pattern of suture patency and closure is associated with the typical wide and short head shape (brachycephaly) and midfacial retrusion of Apert syndrome patients.
Differences in fusion patterns of palatal sutures could stem from structural and/or suture-specific differences in localized FGF/FGFR signaling environments. Overall, the three sutures examined histologically at P0 have strikingly different fates and structural characteristics. The maxillary-palatine suture contains broadly overlapping bone edges separated by a reasonably narrow mesenchyme, similar to the coronal suture, where the degree of overlap is much shorter but which consistently fuses in Apert syndrome mice (Holmes et al., 2009; Wang et al., 2005). The inter-palatine suture, on the other hand, has a butted structure similar to the sagittal suture, which fuses infrequently if at all in Apert syndrome mice. The inter-premaxillary suture is unique, having a butted structure but also an extensive region of proximity between the two premaxillary bones, and it is the suture that in unaffected littermates more readily fuses. Although the basis for suture-specific differences in FGF signaling are unknown, in the mutant inter-premaxillary suture the result is a more extensive and proliferative suture mesenchyme relative to unaffected littermates, which might prevent close contact and fusion between the paired premaxillary bones at P0.
Considering the potential differential sensitivity of mesenchymal cells occupying specific sutures to the presence of mutant receptors and the selective distribution or concentration of FGF ligands (Hajihosseini et al., 2004), it is not surprising that the same FGFR2 mutation appears to have different or even opposite effects on the patency of varying cranial sutures. FGF/FGFR signaling is known to be complex, and tissue-specific responses are widespread and variable (Wilke et al., 1997; Ornitz and Marie, 2002; Dorey and Amaya, 2010; Hébert, 2011; Martínez-Abadías et al., 2011; Martínez-Abadías et al., 2013). The identity of FGFs expressed during early craniofacial morphogenesis and palatal formation have been extensively characterized (Britto et al., 2002; Bachler and Neubüser, 2001; Welsh et al., 2007), but the complement of FGFs expressed in the facial sutures during late embryological development is undefined. A broad range of FGFs is identifiable in the coronal suture during late gestation (Hajihosseini and Heath, 2002), so the repertoire of FGF signaling within the various facial sutures, modified by their particular affinity for the FGFR2 S252W and P253R mutant receptors, could be similarly complex and lead to such diverse phenotypic outcomes.
Our results add to the accumulating evidence that although FGF/FGFR signaling contributes to the development of many cranial tissues, the effects can be highly localized, varying according to the tissue considered and the time of development. Our analysis reveals several localized sites of altered palatal development and confirms that the posterior palate is one of the palatal regions most affected by perturbed FGF/FGFR signaling caused by the FGFR2 S252W mutation. These anatomical sites might serve as a potential target for therapeutic strategies in clinical management of cleft palate in Apert syndrome patients.
MATERIALS AND METHODS
Mouse models
Fgfr2+/S252W and Fgfr2+/P253R Apert syndrome mouse models were bred on C57BL/6J genetic background for 20 generations to minimize phenotypic variation and were generated, euthanized, fixed and imaged in compliance with animal welfare guidelines approved by the Johns Hopkins University, the Mount Sinai School of Medicine and the Pennsylvania State University Animal Care and Use Committees. Further details on generation of targeting construct can be found elsewhere (Wang et al., 2005; Wang et al., 2010).
Skull imaging
High resolution micro-computed tomography (μCT) images of the mouse heads were acquired by the Center for Quantitative Imaging at the Pennsylvania State University (www.cqi.psu.edu) using the HD-600 OMNI-X high-resolution X-ray computed tomography system (Bio-Imaging Research Inc., Lincolnshire, IL). Pixel sizes ranged from 0.015 to 0.020 mm, and slice thickness from 0.016 to 0.025 mm. Image data were reconstructed on a 1024×1024 pixel grid as a 16-bit TIFF but reduced to 8 bit for image analysis. Based on hydroxyapatite phantoms imaged with the specimens, the minimum thresholds used to create isosurfaces from these 8-bit images ranged from 70 to 100 mg/cm3 partial density of hydroxyapatite. To visualize skull morphology, isosurfaces were reconstructed from the μCT images using the software package Avizo 6.0 (Visualization Sciences Group). A set of 10 palatal landmarks (Fig. 1, Table 1) were collected twice by the same observer to minimize measurement error; deviations between the two trials were restricted to 0.05 mm.
Scoring patterns of suture fusion
We assessed the pattern of fusion of five palatal sutures (inter-premaxillary, inter-maxillary, inter-palatine, right and left maxillary-palatine), qualitatively scoring the sutures as patent, partially fused or completely fused (Fig. 1). For the inter-premaxillary suture, we divided the whole length of the suture into three different segments (anterior, middle, posterior) and scored the degree of fusion at each segment as open (O), partial (P) or fused (F) independently. Once a code was obtained for each of the three sections of the inter-premaxillary suture, the three codes were assembled into a pattern of suture fusion. For example, individuals presenting with a completely open inter-premaxillary suture were coded as ‘OOO’ and were included in the analysis as ‘patent’, whereas individuals with a totally fused inter-premaxillary suture were coded as ‘FFF’ and were included in the analysis as ‘completely fused’. All those cases in which at least one segment was partially fused (P) were pooled into the ‘partially fused’ category.
Geometric morphometric analyses
General Procrustes Analysis (GPA), a procedure that superimposes configurations of landmarks by shifting them to a common position, rotating and scaling them to a standard size until a best fit of corresponding landmarks is achieved, was used to extract shape information (Dryden and Mardia, 1998). GPA, however, does not eliminate the allometric shape variation that is related to size. We adjusted for the effect of allometry (Drake and Klingenberg, 2008) by computing a multivariate regression of shape on centroid size, measured as the square root of the summed distances between each landmark coordinate and the centroid of the landmark configuration (Dryden and Mardia, 1998).
Canonical variates analysis (CVA) was performed to explore shape variation and to maximize discrimination between Apert syndrome mouse models and their unaffected littermates. CVA derives discriminant functions called canonical variates (CVs) that result from an optimal combination of variables (the Procrustes coordinates), so that the first one provides the most overall discrimination between groups; the second provides the second-most, and so on (Manly, 2004). The discriminant functions correspond to shape features that are independent or orthogonal, that is, their contributions to the discrimination between groups does not overlap.
Mahalanobis distances, a distance measure used in cluster analysis and classification techniques, were computed from the pooled within-group covariance matrix for all groups after adjusting for allometry to assess the degree of differentiation between all possible pairs of groups of Apert syndrome mouse models and their unaffected littermates (Klingenberg and Monteiro, 2005). Statistical significance for all pairwise comparisons was tested using permutation tests based on 10,000 iterations under the null hypothesis of complete dissimilarity. The P values obtained were adjusted for the lack of independence using the Bonferroni method: the total comparisons were c=k(k–1)/2 (where k is the number of populations) and the significance level 0.05 was divided by c to guarantee real significance. All geometric morphometric methods based on Procrustes analysis were performed using MorphoJ (Klingenberg, 2011).
To statistically determine differences in the way the two mutant phenotypes differed from their respective unaffected littermates, we used a confidence interval procedure developed within EDMA, Euclidean Distance Matrix Analysis (Lele and Richtsmeier, 1995; Lele and Richtsmeier, 2001). EDMA converts 3D landmark data into a matrix of all possible linear distances between unique landmark pairs and tests for statistical significance of differences between shapes using non-parametric confidence intervals (Lele and Richtsmeier, 1995; Lele and Richtsmeier, 2001). An average form is estimated using the linear distance data and differences in 3D size and shape are statistically compared as a matrix of ratios of all like linear distances in the two samples. We tested for morphological differences in each mutant group as compared with its unaffected littermates, and for differences in the mutant/unaffected contrasts between the two mouse models for Apert syndrome. The null hypothesis for each comparison is that there is no difference in shape between groups (or no difference in shape contrasts). Statistical tests for differences in specific linear distances were evaluated by a non-parametric bootstrapping procedure (Lele and Richtsmeier, 1995). For each linear distance, a ratio between the average values of that distance for each group was computed and confidence intervals for the null hypothesis of similarity in shape were estimated from 100,000 pseudo-samples generated from the data using a non-parametric bootstrapping algorithm. For each linear distance, the null hypothesis was rejected if the 90% confidence interval produced from the bootstrapping method did not include 1.0. Rejection of the null hypothesis enables localization of differences to specific landmarks and linear distances (Lele and Richtsmeier, 1995; Lele and Richtsmeier, 2001). EDMA analyses were performed using WinEDMA (Cole, 2002).
Histological and immunohistochemical analyses
Histological sections (5 μm) were prepared from selected tissues that had been fixed in 4% paraformaldehyde for 24 hours, demineralized in 10% EDTA in PBS for 5 days, and embedded in paraffin. We studied the maxillary-palatine sutures by sectioning the head in the sagittal plane using the anterior-posterior axis from the nose to vertebra; whereas the inter-palatine and inter-premaxillary sutures were analyzed by sectioning the head in the coronal plane. Sections were stained with haematoxylin and eosin (H&E) for histological analysis. Alkaline phosphatase (ALP) staining was performed as described (Miao and Scutt, 2002). Antibodies used were rabbit anti-Ki67 (1:200; Vectorlabs, VP-RM04), rabbit anti-Runx2 (1:200; Sigma, HPA022040), rabbit anti-phospho-MAPK ERK1/2 (1:200; Cell Signaling, 4376) and rabbit anti-phospho-p38 MAPK (1:100; Cell Signaling, 4631). Antibodies were detected by the sequential application of biotinylated anti-rabbit IgG (1:200; Vectorlabs, BA-1000) and streptavidin-Alexa Fluor 488 conjugate (1:200 of a 1 mg/ml stock; Invitrogen, S-11223) for immunofluorescence or by the SignalStain Boost IHC Detection Reagent and Signalstain DAB Substrate (Cell Signaling, 8115 and 8059) for MAPK immunohistochemistry. Nuclei were stained with Hoechst 33258 (1:20,000 of a 100 mg/ml stock; Invitrogen, H1398). The TUNEL assay was carried out using the In Situ Cell Death Detection Kit, POD (Roche Applied Science, 11684817910) to detect apoptotic cells. For each staining, at least two litters of mice were examined. Brightfield and fluorescent photomicrographs were taken on a Nikon Eclipse 80i microscope with a Q Imaging Retiga 4000R camera using QCapture software. Images were processed in Adobe Photoshop. For statistical analysis, at least three animals per genotype were used, and data was analyzed using the unpaired, two-tailed Student’s t-test. Differences with a P value below 0.05 were considered significant.
Acknowledgments
We gratefully acknowledge Colin Shaw, Tim Ryan and Tim Stecko from CQI (Pennsylvania State University) for their excellent technical skill and support in microCT scanning. We thank Yann Heuzé for fruitful discussions of earlier versions of this manuscript, as well as editorial suggestions. We thank the editor and two anonymous reviewers whose comments and suggestions greatly improved the overall quality of the manuscript.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
N.M.-A., J.T.R., Y.W. and E.W.J. conceived and designed the experiments. N.M.-A., G.H., T.P., X.Z. and Y.W. collected data and performed the experiments. N.M.-A. and J.T.R. analyzed the data. G.H., Y.W. and E.W.J. contributed reagents and materials. N.M.-A. and J.T.R. wrote the paper. T.P., G.H., Y.W., E.W.J. and J.T.R. provided critical revision and final approval of the manuscript.
FUNDING
This work was funded by the National Institutes of Craniofacial and Dental Research, National Institutes of Health and the American Recovery and Reinvestment Act [grant numbers R01 DE018500, 3R01 DE018500-02S1, 5R01 DE022988]; and Comissionat per a Universitats i Recerca, Generalitat de Catalunya (Spain) [grant number 2008 BP A 00170].
REFERENCES
- Abu-Issa R., Smyth G., Smoak I., Yamamura K., Meyers E. N. (2002). Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development 129, 4613–4625 [DOI] [PubMed] [Google Scholar]
- Aldridge K., Hill C. A., Austin J. R., Percival C., Martínez-Abadias N., Neuberger T., Wang Y., Jabs E. W., Richtsmeier J. T. (2010). Brain phenotypes in two FGFR2 mouse models for Apert syndrome. Dev. Dyn. 239, 987–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachler M., Neubüser A. (2001). Expression of members of the Fgf family and their receptors during midfacial development. Mech. Dev. 100, 313–316 [DOI] [PubMed] [Google Scholar]
- Britto J. A., Evans R. D., Hayward R. D., Jones B. M. (2002). Toward pathogenesis of Apert cleft palate: FGF, FGFR, and TGF beta genes are differentially expressed in sequential stages of human palatal shelf fusion. Cleft Palate Craniofac. J. 39, 332–340 [DOI] [PubMed] [Google Scholar]
- Chen L., Li D., Li C., Engel A., Deng C. X. (2003). A Ser252Trp [corrected] substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone 33, 169–178 [DOI] [PubMed] [Google Scholar]
- Cohen M. M., Jr, Kreiborg S. (1996). A clinical study of the craniofacial features in Apert syndrome. Int. J. Oral Maxillofac. Surg. 25, 45–53 [DOI] [PubMed] [Google Scholar]
- Cohen M. J., MacLean R. (2000). Craniosynostosis: diagnosis, evaluation, and management. J. Med. Genet. 37, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole T., III (2002). WinEDMA Version 1.0.1 beta. Windows-Based Software for Euclidean distance Matrix Analysis [Google Scholar]
- De Moerlooze L., Spencer-Dene B., Revest J. M., Hajihosseini M., Rosewell I., Dickson C. (2000). An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development 127, 483–492 [DOI] [PubMed] [Google Scholar]
- Dixon M. J., Marazita M. L., Beaty T. H., Murray J. C. (2011). Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet. 12, 167–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorey K., Amaya E. (2010). FGF signalling: diverse roles during early vertebrate embryogenesis. Development 137, 3731–3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake A. G., Klingenberg C. P. (2008). The pace of morphological change: historical transformation of skull shape in St Bernard dogs. Proc. Biol. Sci. 275, 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryden I., Mardia K. (1998). Statistical Shape Analysis. Chichester, UK: John Wiley [Google Scholar]
- Du X., Weng T., Sun Q., Su N., Chen Z., Qi H., Jin M., Yin L., He Q., Chen L. (2010). Dynamic morphological changes in the skulls of mice mimicking human Apert syndrome resulting from gain-of-function mutation of FGFR2 (P253R). J. Anat. 217, 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritli-Linde A. (2007). Molecular control of secondary palate development. Dev. Biol. 301, 309–326 [DOI] [PubMed] [Google Scholar]
- Hajihosseini M. K., Heath J. K. (2002). Expression patterns of fibroblast growth factors-18 and −20 in mouse embryos is suggestive of novel roles in calvarial and limb development. Mech. Dev. 113, 79–83 [DOI] [PubMed] [Google Scholar]
- Hajihosseini M. K., Lalioti M. D., Arthaud S., Burgar H. R., Brown J. M., Twigg S. R. F., Wilkie A. O. M., Heath J. K. (2004). Skeletal development is regulated by fibroblast growth factor receptor 1 signalling dynamics. Development 131, 325–335 [DOI] [PubMed] [Google Scholar]
- Hajihosseini M. K., Duarte R., Pegrum J., Donjacour A., Lana-Elola E., Rice D. P., Sharpe J., Dickson C. (2009). Evidence that Fgf10 contributes to the skeletal and visceral defects of an Apert syndrome mouse model. Dev. Dyn. 238, 376–385 [DOI] [PubMed] [Google Scholar]
- Hébert J. M. (2011). FGFs: neurodevelopment’s Jack-of-all-trades – how do they do it? Front. Neurosci. 5, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes G., Basilico C. (2012). Mesodermal expression of Fgfr2S252W is necessary and sufficient to induce craniosynostosis in a mouse model of Apert syndrome. Dev. Biol. 368, 283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes G., Rothschild G., Roy U. B., Deng C. X., Mansukhani A., Basilico C. (2009). Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology. Dev. Biol. 328, 273–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahimi O. A., Eliseenkova A. V., Plotnikov A. N., Yu K., Ornitz D. M., Mohammadi M. (2001). Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc. Natl. Acad. Sci. USA 98, 7182–7187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata J., Parada C., Chai Y. (2011). The mechanism of TGF-β signaling during palate development. Oral Dis. 17, 733–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg C. P. (2011). MorphoJ: an integrated software package for geometric morphometrics. Mol. Ecol. Resour. 11, 353–357 [DOI] [PubMed] [Google Scholar]
- Klingenberg C. P., Monteiro L. R. (2005). Distances and directions in multidimensional shape spaces: implications for morphometric applications. Syst. Biol. 54, 678–688 [DOI] [PubMed] [Google Scholar]
- Kreiborg S., Cohen M. M., Jr (1992). The oral manifestations of Apert syndrome. J. Craniofac. Genet. Dev. Biol. 12, 41–48 [PubMed] [Google Scholar]
- Lele S., Richtsmeier J. T. (1995). Euclidean distance matrix analysis: confidence intervals for form and growth differences. Am. J. Phys. Anthropol. 98, 73–86 [DOI] [PubMed] [Google Scholar]
- Lele S., Richtsmeier J. T. (2001). An Invariant Approach to the Statistical Analysis of Shapes. Boca Raton, FL: Chapman and Hall/CRC Press [Google Scholar]
- Liu Z., Xu J., Colvin J. S., Ornitz D. M. (2002). Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 16, 859–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manly B. F. J. (2004). Multivariate Statistical Methods: A Primer, Third Edition Boca Raton, FL: Chapman and Hall/CRC [Google Scholar]
- Mansukhani A., Bellosta P., Sahni M., Basilico C. (2000). Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J. Cell Biol. 149, 1297–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Abadías N., Percival C., Aldridge K., Hill C. A., Ryan T., Sirivunnabood S., Wang Y., Jabs E. W., Richtsmeier J. T. (2010). Beyond the closed suture in apert syndrome mouse models: evidence of primary effects of FGFR2 signaling on facial shape at birth. Dev. Dyn. 239, 3058–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Abadías N., Heuzé Y., Wang Y., Jabs E. W., Aldridge K., Richtsmeier J. T. (2011). FGF/FGFR signaling coordinates skull development by modulating magnitude of morphological integration: evidence from Apert syndrome mouse models. PLoS ONE 6, e26425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Abadías N., Motch S. M., Pankratz T. L., Wang Y., Aldridge K., Jabs E. W., Richtsmeier J. T. (2013). Tissue-specific responses to aberrant FGF signaling in complex head phenotypes. Dev. Dyn. 242, 80–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao D., Scutt A. (2002). Histochemical localization of alkaline phosphatase activity in decalcified bone and cartilage. J. Histochem. Cytochem. 50, 333–340 [DOI] [PubMed] [Google Scholar]
- Mossey P. A., Little J., Munger R. G., Dixon M. J., Shaw W. C. (2009). Cleft lip and palate. Lancet 374, 1773–1785 [DOI] [PubMed] [Google Scholar]
- Ohbayashi N., Shibayama M., Kurotaki Y., Imanishi M., Fujimori T., Itoh N., Takada S. (2002). FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 16, 870–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz D. M., Marie P. J. (2002). FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 16, 1446–1465 [DOI] [PubMed] [Google Scholar]
- Park W. J., Theda C., Maestri N. E., Meyers G. A., Fryburg J. S., Dufresne C., Cohen M. M., Jr, Jabs E. W. (1995). Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am. J. Hum. Genet. 57, 321–328 [PMC free article] [PubMed] [Google Scholar]
- Purushothaman R., Cox T. C., Muga A. M., Cunningham M. L. (2011). Facial suture synostosis of newborn Fgfr1(P250R/+) and Fgfr2(S252W/+) mouse models of Pfeiffer and Apert syndromes. Birth Defects Res. A Clin. Mol. Teratol. 91, 603–609 [DOI] [PubMed] [Google Scholar]
- Rice D. P., Rice R., Thesleff I. (2003). Fgfr mRNA isoforms in craniofacial bone development. Bone 33, 14–27 [DOI] [PubMed] [Google Scholar]
- Rice R., Spencer-Dene B., Connor E. C., Gritli-Linde A., McMahon A. P., Dickson C., Thesleff I., Rice D. P. (2004). Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. Invest. 113, 1692–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley B. M., Mansilla M. A., Ma J., Daack-Hirsch S., Maher B. S., Raffensperger L. M., Russo E. T., Vieira A. R., Dodé C., Mohammadi M., et al. (2007). Impaired FGF signaling contributes to cleft lip and palate. Proc. Natl. Acad. Sci. USA 104, 4512–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla V., Coumoul X., Wang R.-H., Kim H.-S., Deng C.-X. (2007). RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat. Genet. 39, 1145–1150 [DOI] [PubMed] [Google Scholar]
- Slaney S. F., Oldridge M., Hurst J. A., Moriss-Kay G. M., Hall C. M., Poole M. D., Wilkie A. O. (1996). Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am. J. Hum. Genet. 58, 923–932 [PMC free article] [PubMed] [Google Scholar]
- Snyder-Warwick A. K., Perlyn C. A., Pan J., Yu K., Zhang L., Ornitz D. M. (2010). Analysis of a gain-of-function FGFR2 Crouzon mutation provides evidence of loss of function activity in the etiology of cleft palate. Proc. Natl. Acad. Sci. USA 107, 2515–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trokovic N., Trokovic R., Mai P., Partanen J. (2003). Fgfr1 regulates patterning of the pharyngeal region. Genes Dev. 17, 141–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Xiao R., Yang F., Karim B. O., Iacovelli A. J., Cai J., Lerner C. P., Richtsmeier J. T., Leszl J. M., Hill C. A., et al. (2005). Abnormalities in cartilage and bone development in the Apert syndrome FGFR2(+/S252W) mouse. Development 132, 3537–3548 [DOI] [PubMed] [Google Scholar]
- Wang Y., Sun M., Uhlhorn V. L., Zhou X., Peter I., Martínez-Abadias N., Hill C. A., Percival C. J., Richtsmeier J. T., Huso D. L., et al. (2010). Activation of p38 MAPK pathway in the skull abnormalities of Apert syndrome Fgfr2(+P253R) mice. BMC Dev. Biol. 10, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh I. C., Hagge-Greenberg A., O’Brien T. P. (2007). A dosage-dependent role for Spry2 in growth and patterning during palate development. Mech. Dev. 124, 746–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke T. A., Gubbels S., Schwartz J., Richman J. M. (1997). Expression of fibroblast growth factor receptors (FGFR1, FGFR2, FGFR3) in the developing head and face. Dev. Dyn. 210, 41–52 [DOI] [PubMed] [Google Scholar]
- Wilkie A. O., Slaney S. F., Oldridge M., Poole M. D., Ashworth G. J., Hockley A. D., Hayward R. D., David D. J., Pulleyn L. J., Rutland P., et al. (1995). Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat. Genet. 9, 165–172 [DOI] [PubMed] [Google Scholar]
- Yu K., Ornitz D. M. (2011). Histomorphological study of palatal shelf elevation during murine secondary palate formation. Dev. Dyn. 240, 1737–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]