

Manual evaluation of state-of-the art text mining data reveals promising results for its application in plant network biology. Focusing on Arabidopsis thaliana, an integrated network of text mining and experimental data highlights the complementarity of these resources and the necessity for text mining tools to uncover the latest relevant findings from the literature.
Abstract
Despite the availability of various data repositories for plant research, a wealth of information currently remains hidden within the biomolecular literature. Text mining provides the necessary means to retrieve these data through automated processing of texts. However, only recently has advanced text mining methodology been implemented with sufficient computational power to process texts at a large scale. In this study, we assess the potential of large-scale text mining for plant biology research in general and for network biology in particular using a state-of-the-art text mining system applied to all PubMed abstracts and PubMed Central full texts. We present extensive evaluation of the textual data for Arabidopsis thaliana, assessing the overall accuracy of this new resource for usage in plant network analyses. Furthermore, we combine text mining information with both protein–protein and regulatory interactions from experimental databases. Clusters of tightly connected genes are delineated from the resulting network, illustrating how such an integrative approach is essential to grasp the current knowledge available for Arabidopsis and to uncover gene information through guilt by association. All large-scale data sets, as well as the manually curated textual data, are made publicly available, hereby stimulating the application of text mining data in future plant biology studies.
LARGE-SCALE BIOLOGY ARTICLE
INTRODUCTION
Text mining (i.e., the process of deriving high-quality information from text) has many applications. For instance, text mining can assist in efforts to manually curate biological data, such as the BioCreative initiative, wherein literature-extracted information on protein–protein interactions (PPIs), phenotypes, and gene functions is used as a baseline for manual annotation of these data types (Arighi et al., 2011; Hirschman et al., 2012). In addition, text mining data can be employed in data integration and gene prioritization approaches to construct interaction networks, predict gene functions, identify gene-phenotype associations, rank genes from genome-wide association studies, verify predicted regulators in regulatory network construction, and discover biomarkers (Amoutzias et al., 2007; Tranchevent et al., 2011; Chasman et al., 2012; Faro et al., 2012; Rojas et al., 2012).
Recently, in collaboration with the University of Turku, we developed EVEX, a large-scale text mining resource unprecedented in semantic scope, including information on protein metabolism and protein modifications (e.g., phosphorylation and ubiquitination), fundamental molecular events (e.g., transcription, binding, and localization), regulatory control, specifically negated/speculated statements, and contextual information, such as cellular location (Van Landeghem et al., 2011, 2013). This text mining framework covers all 22 million PubMed abstracts and 460,000 PubMed Central (PMC) Open Access full-text articles. The text mining algorithms were originally developed within the context of the BioNLP Shared Task on Event Extraction of 2009 (Kim et al., 2011). This community-wide evaluation was performed against a data set derived from GENIA (Ohta et al., 2009), a corpus of processed abstracts on blood cell transcription factors in Homo sapiens. The text mining methodology underlying the EVEX framework obtained the highest ranking out of a total of 24 internationa participants (Björne et al., 2009) and obtained top-ranking results on a similar evaluation in 2011 (Björne et al., 2012). However, the specific value of this text mining system for plant biology has not yet been investigated nor has it been used in integration of plant-specific data. Whereas a few frameworks have previously been introduced to allow retrieval of textual data specifically for plants (PLAN2L, Krallinger et al., 2009; Textpresso, Van Auken et al., 2012; Ondex, Köhler et al., 2006), such studies typically build upon labor-intensive manual curation or rely on relatively simple text analytics such as co-occurrence of genes. However, the extraction of complex textual structures or events, as provided by EVEX, is necessary to obtain accurate representations of the complexity of molecular processes.
To investigate the value of text mining in a plant integrative study, we used the publicly available Arabidopsis thaliana CORNET database, which we developed recently to facilitate the integration of coexpression data, PPIs, regulatory interactions, and gene–gene association data (De Bodt et al., 2010, 2012). This database compiles currently available Arabidopsis data, identified through diverse experimental as well as computational approaches. Dealing with various issues, such as heterogeneity and the difference in quality between the various data sources, additional metadata are stored to describe the original databases, identification methods, confidence scores, and literature evidence. Data mining and integration tools such as CORNET are necessary to obtain a comprehensive overview of plant biological data (Brady and Provart, 2009; Bassel et al., 2012).
Construction and analysis of integrated networks is commonly used to exploit the complementary nature of different data sources (Lee et al., 2010; Kourmpetis et al., 2011; Heyndrickx and Vandepoele, 2012). Often, modules of tightly linked components in the network are delineated (Aoki et al., 2007). For instance, modules of coexpressed genes are used for gene function prediction, while modules of interacting proteins can lead to the identification of protein complexes. Furthermore, modules composed of multiple data types reveal the interplay of various types of biological interactions in local network neighborhoods (Zhang et al., 2005; Michoel et al., 2011). Additionally, this integrative approach allows connecting relatively large numbers of genes and/or proteins by assembling paths through different interaction types.
Here, we aim to present a critical assessment of the use of complex event extraction from literature, in combination with a comprehensive integrative network approach for plants. First, we performed a systematic manual evaluation of the textual data, using predictions derived from 1176 Arabidopsis articles. Next, the added value of incorporating computationally derived text mining data from all PubMed abstracts and all PMC Open Access full-text articles was evaluated through the construction and analysis of integrated networks for Arabidopsis. We demonstrate that the combination of text mining and experimentally derived interaction data greatly increases the density and connectivity of biological networks.
RESULTS AND DISCUSSION
A Systematic Evaluation of the Text Mining Events
Text mining can extract a wealth of information on various types of interactions or events, including binding and regulation (Table 1). Regulatory events may further involve specific physical events, such as phosphorylation or gene expression, and longer chains of regulatory control can include a variety of such different event types. Additionally, the polarity of regulatory events is marked as positive (upregulation), negative (downregulation), or unspecified/unknown. Figure 1 illustrates a representative example of event extraction.
Table 1. Examples of Different Event Types, with the Association Term and the Relevant Gene(s)/Protein(s) Marked in Bold.
| Example | Event Type | Sentence | PubMed ID |
|---|---|---|---|
| 1 | Localization | Arabidopsis RNA binding protein UBA2a relocalizes into nuclear speckles in response to abscisic acid. | 16828085 |
| 2 | Transcription | A phytochrome-mediated signaling pathway(s) activates the transcription of APRR9. | 14634162 |
| 3 | Gene expression | Truncated AtGCP2- and AtGCP3-green fluorescent protein fusion proteins were expressed in BY-2 cells. | 17714428 |
| 4 | Single-argument binding | A transcription factor, Auxin Response Factor1 (ARF1), that binds to the sequence TGTCTC. | 9188533 |
| 5 | Double-argument binding | Cat1 can bind with the PTS1 receptor (Pex5p). | 12943550 |
| 6 | Double-argument binding | The PH domain alone binds equally well to both PtdIns-3-P and phosphatidylinositol 4-phosphate. | 12105222 |
| 7 | Phosphorylation | BRs induce dephosphorylation and accumulation of BZR1 protein. | 12114546 |
| 8 | Regulation (unspecified) | CK2 can modulate CCA1 activity. | 9724822 |
| 9 | Positive regulation | Arabidopsis LIP5 acts as a positive regulator of SKD1. | 17468262 |
| 10 | Negative regulation | 2b specifically inhibits AGO1 cleavage activity. | 17158744 |
Figure 1.

Example of Event Extraction.
Biomolecular interactions and regulations are automatically extracted from text as directed graphs with genes and proteins as leaves (circles) and event types as the intermediate nodes (rectangular boxes). The “Theme” denotes the subject of the association (e.g., what is being regulated), while the “Cause” denotes the object (e.g., the regulator). In a subsequent step, pairwise relations are extracted, directly linking two genes in a format compatible with systems biology studies.
[See online article for color version of this figure.]
The text mining algorithms underlying the EVEX resource have been proven to achieve state-of-the-art results on a small-scale text corpus on human biology, concerning transcription factors specific to blood cells (Björne et al., 2009; Kim et al., 2011). However, it remained to be seen whether such promising results also could be obtained for more general applications in plant biology. To investigate this issue, we collected an unbiased set of 1176 PubMed articles on Arabidopsis. In total, the abstracts of these articles contain 7691 automatically predicted events, of which 1787 were randomly selected for manual evaluation (see Methods). The evaluation data set, containing all correct events and all wrongly predicted events, as well as an indication of the type of error, is available as the PLEV corpus (short for plant evaluation) (see Supplemental Data Set 1 online).
Global Results
Table 2 summarizes the results of the manual evaluation effort. The precision rate of all events in the plant data set (58.6%) corresponds well to the 58.5% precision rate previously obtained on the human data set (Björne et al., 2009), warranting the application of the textual data to plant integrative studies. In the next sections, we summarize additional findings arising from this manual evaluation effort.
Table 2. Systematic Evaluation of Text Mining Events from 1176 PubMed Abstracts Related to Arabidopsis.
| Event Type | No. of Evaluated Events | Correct Event Type |
Correct Event Type and Arguments |
||
|---|---|---|---|---|---|
| Single-argument binding | 259 | 22 | 8% | 19 | 7% |
| Double-argument binding | 431 | 402 | 93% | 317 | 74% |
| Phosphorylation | 314 | 307 | 98% | 204 | 65% |
| Transcription | 89 | 53 | 60% | 41 | 46% |
| Gene expression | 53 | 49 | 92% | 43 | 81% |
| Regulation (unspecified) | 385 | 360 | 94% | 270 | 70% |
| Positive regulation | 208 | 166 | 80% | 119 | 57% |
| Negative regulation | 48 | 44 | 92% | 35 | 73% |
| All | 1787 | 1403 | 79% | 1048 | 59% |
This PLEV corpus is available as Supplemental Data Set 1 online. For every event type, the evaluated events were randomly picked. All regulatory events assigned to a certain polarity (positive/negative) were also evaluated as unspecified regulation.
Event Ranking
All text mining predictions were automatically assigned a certain confidence score (see Methods). To evaluate whether these scores can be used for a meaningful ranking of the text mining results, we plotted them against the precision rate, obtained through the manual evaluation effort. Supplemental Figure 1A online depicts the precision rate of the set of regulatory events, plotted as a function of the confidence value threshold, which defines the minimal value for events to be included in the output. The complete set of regulatory events within the PLEV data set had an average precision rate of around 68%. However, when applying a threshold of −0.65, discarding all events with a lower confidence value, we obtained a precision rate of around 75%. Applying a more stringent cutoff of −0.20 would retain only the most confident ones (90% precision). Supplemental Figure 1B online depicts a similar graph for binding events. The general trend of obtaining higher precision with more stringent cutoffs was preserved. However, the precision rate did not reach 100% for the most stringent criteria, and in fact dropped to 50%. This artifact at the end of the graph is caused by a few sentences that highly resemble true statements of PPIs and were consequently assigned high confidence scores, even though they were in fact false-positive predictions. Examples of such cases are sentences containing negation information like failed to bind, or the presence of highly ambiguous words, such as associated. In conclusion, the confidence values can be used for ranking the textual information roughly from less to more reliable. To increase precision even further, manual curation can be applied to this ranked list.
Event Type–Specific Evaluation
While in general the text mining data have been shown to be of high quality, manual evaluation has also revealed a few important differences between interaction types, which are summarized here. These findings can serve as a reference point in future studies that apply large-scale text mining information within data integration efforts.
Within the original event extraction challenge of the BioNLP Shared Task 2009 (Kim et al., 2011), a text mining algorithm had to differentiate between a variety of event types, including localization, transcription, and gene expression (Table 1, examples 1 to 3). However, the difference in text between these event types is not always clear or even relevant. In particular, we noticed that a substantial part of the predicted positive regulation events were in fact unspecified (e.g., effects) or even negative regulations (e.g., inhibits), resulting in a relatively low precision rate of 57%. However, since it is often already informative or even sufficient to know that a particular protein has an effect on the expression of a gene, we suggest grouping all regulatory interactions together for large-scale text mining analyses and relying on additional external data or manual curation to define the final effect of the regulation. Within the PLEV data set, this approach yielded a precision rate of 70% (Table 2).
Two additional event types in the text mining data are single-argument and double-argument binding events. Cases of single-argument binding events involve those sentences where it is difficult to identify the second argument of the interaction, such as protein-DNA binding (Table 1, example 4). By contrast, when two proteins are said to interact, a double-argument binding event should be produced (Table 1, example 5). Through manual evaluation, we established a remarkable difference in performance between these two different event types: While only 7% of the single-argument binding events were correct, the precision rate of the double-argument events was 74% (Table 2). Excluding the single-argument binding events from further analysis, we note that double-argument binding events are extracted with high precision and can be incorporated into network studies as such.
To conclude, we have shown that the state-of-the-art event extraction mechanism, underlying our text mining framework EVEX, identifies highly precise plant-specific biological data. Our manual evaluation confirmed that the results of a previous small-scale evaluation on articles about human blood cell transcription factors can be transferred also to other domains. Remarkably, the limited data in the original human training set did not lead to a bias of the text mining algorithms toward a specific topic, author, or grammatical structure in those data.
As we identified a few remaining challenges regarding the identification of the specificities of regulatory interactions, we recommend grouping all regulatory interactions together as unspecified. Similarly, we advise excluding the error-prone single-theme binding events. These simple preprocessing rules can be straightforwardly applied to the textual data, allowing easy integration of text mining data within any application. Within the following sections, we thus focus specifically on binding interactions (74% precision) and regulatory associations (70% precision) from the Arabidopsis literature. We also include indirect regulatory events, which refer to text mining events where a gene has an indirect effect on a protein, for example, by interacting with a direct regulator.
Integration of Text Mining and Biological Interaction Data
To evaluate the potential of information retrieved through text mining, we compared the textual data to experimental results recorded in authoritative databases. To this end, we used CORNET, a comprehensive plant database for coexpression, PPI, regulatory interaction, and gene–gene association data (De Bodt et al., 2010, 2012). The text mining data was derived by selecting the subset of Arabidopsis interactions within the EVEX resource, which covers all available PubMed abstracts and PMC Open Access full-text articles (Van Landeghem et al., 2011, 2013). The algorithms and data sets used by these resources are detailed in Methods.
Statistics on the Arabidopsis text mining data set are summarized in Table 3. Almost 24,400 articles were found to discuss ∼2500 distinct Arabidopsis genes or proteins. While there are fewer than 10,000 associations derived from text mining in total, the experimental data set contained more than one million associations covering almost 10 times as many genes (Table 4). Additionally, functional annotation from Gene Ontology (GO) and MapMan is available for over 20,000 Arabidopsis genes and was used to evaluate the functional relevance of the text mining data (Berardini et al., 2004; Usadel et al., 2005).
Table 3. Statistics on EVEX Text Mining Data Relevant to Arabidopsis, Extracted from 24,391 PubMed Abstracts and PMC Full-Text Articles Related to Arabidopsis.
| Event Type | No. of Genes | No. of Events | No. of Nonredundant Events |
|---|---|---|---|
| Binding | 1844 | 4440 | 3098 |
| Regulation | 1568 | 3960 | 2986 |
| Indirect regulation | 295 | 317 | 298 |
| All | 2461 | 8718 | 6382 |
The nonredundant events represent the number of unique events between two Arabidopsis Genome Initiative identifiers, independent of the number of sentences and articles in which the event was detected.
Table 4. Statistics on Arabidopsis CORNET Data.
| CORNET Type | No. of Genes | No. of Interactions |
|---|---|---|
| Experimental PPI | 7,994 | 34,519 interactions |
| Regulatory interactions (AGRIS) | 9,440 | 13,038 interactions |
| Regulatory interactions (Microarray) | 17,481 | 156,563 interactions |
| Gene–gene associations (AraNet) | 19,647 | 1,062,222 interactions |
| All experimental relations (CORNET) | 24,279 | 1,245,692 interactions |
| GO | 23,046 | na |
| MapMan | 33,265 | na |
For GO and MapMan, genes annotated only with root or general terms are not considered (see Methods). na, not applicable.
In general, text mining events supported by other resources tended to have higher confidence values (Figure 2; see Methods). A detailed comparison of the resources further showed the complementary nature of textual data and experimental interactions (Table 5). A considerable number of experimentally derived associations could not be identified through automated text mining. To a large extent, this observation can be attributed to the mid- and large-scale protein interactome mapping studies that have been performed recently and for which the individual interactions are not described in literature (see Supplemental Table 1 online). In total, 28,382 of 33,863 PPIs (or 84%) that are unique to the CORNET database were identified in large-scale studies (see Supplemental Table 1 online). Such data, often published in (supplemental) tables or external databases, fall out of the scope of the text mining algorithm. Conversely, only 35% of the text mining binding events could be found in the public PPI databases or the AraNet database (Table 5). However, the majority of these interactions, uniquely found by text mining, were supported by common functional annotation between the interacting proteins (Table 5; see Methods).
Figure 2.

Distribution of Confidence of Textual Binding Data.
(A) Confidence of all textual binding events.
(B) Confidence of textual binding events supported by experimental data.
Table 5. Summary of the Comparison of EVEX Text Mining Data to CORNET Experimental Data and Functional Annotation Data.
| Reference Data Source | EVEX Binding |
EVEX Regulation |
EVEX Indirect Regulation |
|||
|---|---|---|---|---|---|---|
| CORNET PPI | 760 | 25% | 467 | 16% | 41 | 14% |
| AGRIS | 160 | 5% | 146 | 5% | 9 | 3% |
| AraNet | 533 | 17% | 327 | 11% | 20 | 7% |
| CORNET PPI, AGRIS, or AraNet | 1071 | 35% | 723 | 24% | 56 | 19% |
| GO, at least 1 | 3060 | 99% | 2956 | 97% | 293 | 98% |
| GO, at least 4 | 3002 | 97% | 2901 | 97% | 293 | 98% |
| MapMan | 1596 | 19% | 1408 | 47% | 170 | 57% |
Percentages are calculated relative to the total number of EVEX events. The support through functional annotation is calculated based on the occurrence of GO categories common to both genes taking part in an EVEX interaction (see Methods).
The overlap between the EVEX regulatory events (24% for direct interactions; 19% for indirect interactions) and CORNET data was proportionally even smaller than the PPI overlap (35%) (Table 5). The low support of regulatory interactions is probably due to the limited number of experimental studies currently represented in the public databases.
The Integrated Arabidopsis Text and Interaction Network
To further investigate the value of text mining in data integration, a network of all EVEX binding events and all direct and indirect regulatory associations, consisting of 2461 genes (nodes) and 6382 connections (edges), was constructed. Integrating this information with the CORNET PPI data, a network of 8900 genes and 43,237 connections was generated. Next, the integrated network was clustered using the ClusterOne method (Methods), allowing the detection of partially overlapping connectivity-based clusters (Nepusz et al., 2012). Subsequent to the clustering, we incorporated experimentally derived and microarray-inferred regulatory interactions into the generated clusters for visualization and analysis. These regulatory interactions were not integrated prior to the clustering step, as their relatively large contribution would result in many clusters containing only this type of association and no text mining data.
Within the integrated network, 701 clusters containing 2513 genes were delineated. Functional enrichment analysis based on GO and MapMan annotations was used to investigate the functional relevance of the identified clusters. Overall, 86.0% of the clusters were enriched for at least one GO category, and 70.8% of all clusters showed significant enrichment for at least one MapMan category (Table 6). After removal of clusters that contained only CORNET interactions, 427 clusters containing 1919 genes remained, of which 87.6% of all clusters showed enrichment for at least one GO category, and 76.8% for at least one MapMan category. Clusters containing both text-based and experimentally derived associations thus showed a tendency to reinforce each other resulting in higher functional enrichment. We examined several clusters in detail and manually inspected the literature in which the genes and relationships between genes in these subnetworks are described to verify the automatically retrieved text mining data.
Table 6. Number of Clusters in the Integrated Networks and Their Functional Enrichment.
| Event Type | All |
GO Enriched |
MapMan Enriched |
||
|---|---|---|---|---|---|
| Whole network | 701 | 603 | 86.0% | 496 | 70.8% |
| Whole network, without CORNET-only clusters | 427 | 374 | 87.6% | 328 | 76.8% |
As a first example of the added value of integrating text mining data, Figure 3 displays an integrated subnetwork for genes involved in trichome differentiation and response to jasmonic acid stimulus (see Supplemental Table 2 online for GO enrichment). Overall, 13 out of the 35 genes in this cluster were, according to the GO analysis, involved in trichome differentiation. Furthermore, this network contained a high number of PPIs (62 edges, red) and regulatory interactions (19 edges, yellow) from authoritative databases as well as binding events (84 edges, green) and regulatory associations (74 edges, blue) from text mining. However, only 27 out of the 167 edges were supported by at least two data sources, showing the complementary nature of the resources. Specifically, a number of microRNA (miRNA) genes were also part of the network, illustrating the strength of the text mining algorithm to uncover regulatory and binding events of miRNA genes and between miRNA genes and protein-coding genes.
Figure 3.

Integrated Network for Epidermal Cell Differentiation, Response to Jasmonic Acid, and Trichome Differentiation.
(A) The network of experimental interactions, excluding text mining data.
(B) The full network, including also text mining interactions. Experimental PPIs are depicted in red, experimental regulatory interactions in yellow, textual binding events in green, textual regulatory events in dark blue, and textual indirect regulatory events in light blue. When the polarity of the regulation is negative, an inhibitory edge is drawn. When the polarity of the regulation is positive or uncertain, an edge with an arrowhead is drawn.
To illustrate the type of information underlying the text mining edges, we randomly inspected a number of sentences from which the textual associations, displayed in Figure 3, were extracted (see Supplemental Table 3 online). Figure 4 depicts one of the inspected sentences in detail. In this example, regulatory events were correctly extracted, recognizing two negative regulatory events (loss) of TRANSPARENT TESTA GLABRA1 (TTG1) and GLABRA1 (GL1) and an additional regulatory event (affects) involving GLABRA3 (GL3). However, the text mining algorithm was not able to combine these statements into causal relationships. Nevertheless, in some cases, complex nested structures could be correctly identified from text (see Supplemental Table 3 online). Another specific challenge for text mining is the often hypothetical, speculative, or negating nature of text. These examples demonstrate the challenges, as well as the strengths, of text mining in general.
Figure 4.

Example of Three Complex Regulatory Events Expressed in Text.
In this particular case, the text mining algorithm did not correctly combine the regulatory events into causal relations, but the individual pieces of information were correctly extracted.
[See online article for color version of this figure.]
Figure 5 displays a second cluster of pollen developmental genes (based on GO enrichment). Three members of the MIR159 family were connected to three MYB genes and the germ line–specific transcription factor DUO POLLEN1. Although The Arabidopsis Information Resource (TAIR) phenotype data describe that the miR159a miR159b double mutant shows curled leaves and reduced stature, a role in pollen development cannot be deduced based on these data. However, through text mining, a link between miR159 and the MYB genes involved in pollen development could be inferred. Recent experimental studies indeed show that miR159 and MYB33 are cotranscribed in aleurone and embryo during seed germination, where miR159 tunes MYB33 expression (Alonso-Peral et al., 2012). These results highlight the potential of text mining information to retrieve the latest experimental findings that might not (yet) be included in external knowledge bases.
Figure 5.

Text-Based Network of Pollen Developmental Genes, Including Many Regulatory Events between miRNAs and MYB Genes.
When the polarity of the regulation is negative, an inhibitory edge is drawn. When the polarity of the regulation is positive or uncertain, an edge with an arrowhead is drawn. No experimental interactions were available between these genes.
[See online article for color version of this figure.]
Additionally, a cluster of organ polarity genes was delineated (Figure 6). ASYMMETRIC LEAVES1 (AS1) and ASYMMETRIC LEAVES2 (AS2), both part of this subnetwork, are known to be involved in the specification of the leaf proximo-distal axis, playing a role in lateral organ growth together with BREVIPEDICELLUS (BP) or KNOTTED-like from Arabidopsis thaliana1 (KNAT1) (Sun et al., 2002; Kojima et al., 2011). Although text mining evidence suggests a link between KNAT1 and several members of the subnetwork shown in Figure 6 (Hay et al., 2006; Larue et al., 2009), KNAT1 is not included in this specific cluster due to the lower connectivity of KNAT1 to the other members. However, KNAT2, a homolog of BP, is linked to AS1 and AS2 (Ikezaki et al., 2010) and present in the cluster. Other genes that are part of the subnetwork are KANADI1 (KAN1), required for abaxial identity in leaves and carpels; PETAL LOSS (PTL), involved in limiting lateral growth of organs; and homolog of histone chaperone HIRA, CUP-SHAPED COTYLEDON1 (CUC1), and CUC3, which are all involved in shoot apical meristem formation and auxin-mediated lateral root formation (Phelps-Durr et al., 2005; Wu et al., 2008; Hasson et al., 2011; Lampugnani et al., 2012). RS2-interacting KH protein (RIK), a predicted histone chaperone interacting with AS1, and ASYMMETRIC LEAVES 2-like protein1 (ASL1) are also linked to the cluster based on experimental PPI data (Chalfun-Junior et al., 2005; Phelps-Durr et al., 2005).
Figure 6.

Integrated Network of Organ Polarity Genes.
This cluster contains a number of edges that were identified with a confidence level lower than −0.7 (dashed edges), which are discussed in the text. Visual properties of the network are as in Figure 3.
Supplemental Table 4 online presents an overview of the findings for eight low-confidence text mining events in the network displayed in Figure 6, with scores ranging from −0.86 to −1.43. Overall, the majority of the low-confidence events (75%) were true or at least stated in a speculative context (e.g., sentence 3). In only two cases, binding events were falsely predicted: CUC1-CUC3 binding (sentence 8) and, to a lesser extent, AS1-general transcription factor group E6 (GTE6) binding (sentence 5). In the first case, “homologous” was wrongly interpreted as a binding interaction word. In the second case, the first sentence describes the binding of GTE6 to the promoter of AS1, detected through chromatin immunoprecipitation, while the second sentence does not describe a binding event but rather the regulation of AS1 by GTE6. It is important to note that even low-confidence text mining events often still refer to biologically meaningful associations.
Finally, we applied our integrative approach to a case study of the Arabidopsis cell cycle (Inzé and De Veylder, 2006). To this end, a network analysis on 61 previously documented core cell cycle (CC) genes (Vandepoele et al., 2002) was performed. In the core network, 508 associations were found between the 61 core CC genes, most of which were PPIs (446 from databases and 41 from text mining). Next, we selected all direct associations to these 61 genes, resulting in a set of 507 candidate genes and a network of 1685 associations (Figure 7). Out of these associations, there were 1120 PPIs (1045 from databases and 75 from text mining) and 565 regulatory interactions (248 from AGRIS, 39 from text mining, and 278 inferred from microarray data). A large fraction of the experimentally identified PPIs were identified through tandem affinity purification performed on all core CC genes (Van Leene et al., 2010). Nevertheless, a substantial number of additional interactions that were not identified in the latter study were found from the literature.
Figure 7.

Integrated Network of the 61 Core CC Genes (Depicted in Purple) and Their Direct Associations.
Visual properties of the network are as in Figure 3. Self-regulation and homodimerization were removed from the network figure, as we aimed to elucidate the interplay between the different CC genes and putative interactors, regulators, or targets.
Inspecting one of the core CC genes in detail (Figure 8), we found many associations for RBR1 (AT3G12280). RBR1 encodes a RETINOBLASTOMA-RELATED protein (RBR or RBR1). RBR1 is involved in the determination of the G1-to-S transition of the cell cycle (Zhao et al., 2012) and in the regulation of imprinted genes (Johnston and Gruissem, 2009). Most connections to RBR1 were only supported by one data type (Figure 8). However, some RBR1 targets were supported by text mining as well as experimental validation, such as GL1 (AT3G27920) and GL3 (AT5G41315). GL1 and GL3 have both been determined to be involved in trichome differentiation (Larkin et al., 1994; Luo and Oppenheimer, 1999). The text mining evidence for the links between RBR1 and GL1/GL3 was as follows: “Interestingly and highlighting the false negative discovery rate of ChIP-chip experiments, SIM, RBR1, CPL3, and FDH, which were only found in the ChIP-chip experiments with GL3-YFP, showed reproducible tethering of both GL3 and GL1 to the corresponding promoters in ChIP assays, suggesting that they should be added to the list of shared direct targets of GL3 and GL1” (Morohashi and Grotewold, 2009). That study specifically investigated the regulatory events associated with the differentiation of Arabidopsis epidermal cells into trichomes, using expression data and genome-wide binding studies (chromatin immunoprecipitation-chip) for GL1 and GL3. This example illustrates the potential of text mining information to rapidly provide additional background information on the data recorded in public databases.
Figure 8.

Cell Cycle Subnetwork of RBR1 Interactors.
Visual properties of the network are as in Figure 3. Core CC genes are depicted in purple. Self-regulation and homodimerization were removed from the network figure.
We further investigated how many of the direct neighbors of the 61 core CC genes were included in the network based solely on text mining. Out of 26 such cases, we were able to select a number of valid candidates through limited manual curation effort (see Supplemental Table 5 online). For example, both FERTILIZATION-INDEPENDENT SEED 2 (FIS2) (AT2G35670 or FERTILIZATION-INDEPENDENT ENDOSPERM 2) and FLOWERING WAGENINGEN (FWA) (AT4G25530), involved in imprinting, could be located in a subnetwork involving RBR1, where only limited experimental data were available. Figure 9 displays the network of direct interactions partners of FWA in the cell cycle network. In this network, only one experimental PPI and two regulatory interactions inferred from microarray data were available, in addition to five binding and 13 regulatory events from text mining. The textual evidence for the links between RBR1 and FIS2/FWA is detailed in Supplemental Table 5 online (numbers 5 and 6, respectively). It is interesting to note that the text mining data may occasionally be derived from author statements that do not express direct physical interactions, such as the association between RBR1 and FWA: “Our results suggest that MSI1 and RBR1 antagonize the repressive action of MET1, which regulates the expression of FIS2 and FWA. … Hence, both MSI1 and RBR1 are required for FWA and FIS2 expression” (Jullien et al., 2008). Supplemental Table 5 online lists a few additional candidate genes found to associate with core CC genes only through text mining, such as the transcription factor SHORT ROOT (AT4G37650).
Figure 9.

Cell Cycle Subnetwork of FWA Interactors.
Visual properties of the network are as in Figure 3. Core CC genes are depicted in purple. Self-regulation and homodimerization were removed from the network figure.
Conclusions
We performed an extensive manual evaluation of text mining data for the model plant Arabidopsis and illustrated that the text mining performance is of high quality. Comparison of text mining data, such as those derived by the EVEX framework, and PPIs, regulatory interactions, gene–gene associations, and functional annotation data, such as compiled in CORNET, were found to be highly complementary. Network-based data integration allowed mining of the information hidden in both data sources, and highly connected subnetworks composed of both types of data were observed to be more biologically relevant in terms of GO enrichment. The richness of the integrated networks allows the discovery and better understanding of the functions of genes and molecular mechanisms active in particular biological processes. Through this study, we have shown that text mining has matured substantially, and we believe that text-based data will become indispensable in future large-scale biological studies. We foresee that current and future text mining will be increasingly used to assist manual curation efforts of biological data, as well as in data integration and network-based biomarker discovery.
Interesting future avenues for improving the text mining data include the design and implementation of a set of postprocessing rules that capture common mistakes such as those identified in our study. In addition to the binding and regulation events that were the focus of this study, the EVEX resource can also be employed to specifically analyze modifications, such as phosphorylation and methylation. Moreover, EVEX will be extended with other data types, such as mutations, and research into table mining will be considered to capture meaningful relations from published (supplemental) tables. Finally, a plant-specific extension will focus on the extraction of phenotypic information from literature, including data on growth processes and plant properties.
METHODS
Text Mining Methodology
The text mining methodology employed in this study covers the extraction of detailed molecular events from text, using advanced natural language processing techniques. Figure 1 depicts an illustrative example of such an event extracted from text. Several steps are involved in this pipeline (Van Landeghem et al., 2013). First, gene and protein names are recognized in text with the widely used BANNER system (Leaman and Gonzalez, 2008). Next, gene name normalization assigns a unique gene identifier for ambiguous gene mentions in text. This step is performed with GenNorm (Wei et al., 2012), which achieved state-of-the-art performance in the BioCreative III challenge (Arighi et al., 2011). Finally, relations or events are extracted between these genes and proteins, detecting a variety of different event types ranging from phosphorylation and ubiquitination to PPIs and regulatory associations. Event extraction is performed with the Turku Event Extraction System (Björne et al., 2009), a machine learning system based on support vector machines, which achieved top-ranking performance in both the BioNLP Shared Task of 2009 and 2011: 46 to 49% recall, 57 to 58% precision, and 52 to 53% F-score (definitions are given in the next section) (Kim et al., 2011; Björne et al., 2012).
The EVEX framework contains the results of applying this text mining pipeline to all available PubMed abstracts and PMC Open Access full-text articles. All textual results are aggregated and assigned a certain confidence score, derived automatically from the output of the text mining classifiers by taking into account the distance to the decision hyperplane of the linear classifier, with higher scores associated with more confident decisions (Van Landeghem et al., 2012). Negative values are generated by normalizing the confidence scores to zero mean and do not have any other special meaning. Five confidence categories are delineated: very high, high, average, low, and very low.
To allow straightforward usage of complex event structures within network analyses, the EVEX resource additionally provides a pairwise view for each event extracted from text (Figure 1). This pairwise view ensures compatibility with the common practice of analyzing relations between exactly two arguments in systems biology studies, labeling the relationship between the two genes/proteins with a concise description of the event structure.
Evaluation of the Text Mining Data
The F-score is a criterion widely used to measure the performance of text mining systems. It is calculated as the harmonic mean between precision (p) and recall (r). Precision and recall can be expressed as a function of the number of true positives (tp; correct predictions), false positives (fp; incorrect predictions), and false negatives (fn; missing predictions):
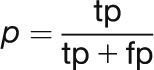 |
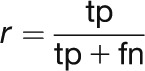 |
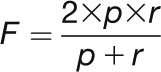 |
In general, measuring recall is difficult, as the set of all true biological events is unknown. Additionally, within our small-scale manual evaluation of 1176 Arabidopsis articles, measuring recall would require the full annotation of these abstracts, an extremely time-consuming task. For these reasons, we restricted our analysis to the measurement of precision, which is by itself a useful indicator of the amount of noise that is to be expected when integrating text mining with other data types. Finally, considering the fact that the precision of our plant evaluation highly resembles that of the previous evaluation on the human blood cell transcription factor data, we can postulate that the recall will be comparable as well.
To facilitate the evaluation of the textual data, an in-house framework was developed for displaying the sentence and metadata together with link-out functionality to the original PubMed article. For each event, a manual curator recorded whether the sentence was annotated with the correct event type and whether all assigned gene and protein symbols were indeed involved in the interaction. Note that text mining does not unveil the difference between genes or proteins, as text is often ambiguous in this respect. For example, in the sentence “The GRXS13 gene plays a role in protection against photooxidative stress,” a colloquial shortcut is used by referring to the gene rather than the gene product. Consequently, the difference between genes and proteins cannot be found directly in text mining data but can be deduced by inspecting the various event types in which they are involved (e.g., gene expression and protein catabolism).
CORNET 2.0 Data
Experimental PPIs of CORNET consist of data retrieved from BIND (Bader et al., 2003), IntAct (Hermjakob et al., 2004), BioGRID (Stark et al., 2006), DIP (Salwinski et al., 2004), MINT (Chatr-aryamontri et al., 2007), TAIR (Rhee et al., 2003), Arabidopsis Interactome Mapping (Arabidopsis Interactome Mapping Consortium, 2011), MIND 0.5 (Lalonde et al., 2010), and the G-protein interactome (Klopffleisch et al., 2011). Predicted PPIs were not considered in this study.
AraNet consists of a probabilistic functional gene network that represents gene–gene associations inferred through the integration of diverse functional genomics, proteomics, and comparative genomics data sets (Lee et al., 2010). The data sets include mRNA coexpression patterns measured from DNA microarray experiments, known Arabidopsis PPIs, protein sequence features including sharing of protein domains, similarity of phylogenetic profiles, or genomic context of bacterial or archaebacterial homologs, and diverse gene–gene associations transferred from yeast, fly, worm, and human genes based on orthology.
Regulatory interactions consist of experimentally identified interactions retrieved from AGRIS and computationally inferred interactions based on microarray data of genetically modified plants (De Bodt et al., 2010, 2012; Yilmaz et al., 2011).
Integration of EVEX and CORNET Data
To facilitate the construction of integrated networks containing text mining data, we added the EVEX binding events, as well as EVEX regulatory information to the CORNET database (http://bioinformatics.psb.ugent.be/cornet) (De Bodt et al., 2010, 2012). The binding data can be explored using the CORNET PPI tool, while the regulatory associations can be searched using the CORNET TF tool. These two tools can be combined as a pipeline to construct integrated networks of PPIs and regulatory interactions. Furthermore, EVEX metadata are included in the CORNET database, allowing the user to inspect text mining confidence values and the detailed regulation type (e.g., positive regulation of expression) as Cytoscape attributes and to link to the original EVEX database (http://www.evexdb.org) through unique event IDs. This link can be reached by right clicking on the event ID and selecting “Search on Web > Plants_Arabidopsis > EVEX_eventid”. When an interaction was extracted from text multiple times, all event IDs and according metadata are shown in Cytoscape. In addition to the network visualization with Cytoscape, database search results of CORNET can also be viewed and exported in text format. The EVEX data in CORNET will be updated for each major new release to include newly published articles.
Clustering
The command-line version of the ClusterOne (clustering with overlapping neighborhood expansion) algorithm was applied to cluster the integrated networks based on graph connectivity (Nepusz et al., 2012). This algorithm allows the detection of overlapping clusters and optionally considers edge weights. As edge weights are not available for all interaction data sources, this option was not applicable. Furthermore, a density threshold of 0.7 was chosen to retrieve highly connected clusters. Lower density thresholds were tested but resulted in a high number of small clusters. Only clusters containing at least four genes were retained.
Functional Annotation Data and Analysis
GO functional annotation data were retrieved from TAIR (May 31, 2012) (Rhee et al., 2003). Too general categories (GO:0003674, GO:0008150, GO:0005575, GO:0005623, and GO:0044464) were excluded in the statistics and GO analysis. Gene pairs are considered to have common GO annotation when the genes have either at least one, or at least four GO categories in common, resulting in a more restrictive definition in the latter case (Table 5). Functional enrichment is tested using a hypergeometric distribution (van Helden, 2003). P values are corrected for multiple testing using the Bonferroni method (Philip, 2012). For GO enrichment studies, only GO categories with more than 10 and <5000 genes are considered.
MapMan functional annotation data describing biological pathways and processes were retrieved from the MapMan store (Ath_AFFY_ATH1_TAIR9_Jan2010, mapman.gabipd.org). General categories (35, 35.2, 35.1, 35.1.999, 29, 28, 28.99, 27, 26, 26.1, and 26.11) were excluded from the analysis. Analyses using MapMan data were performed in a similar fashion as for the GO analyses.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Precision Rates in the PLEV Data Set, Plotted against the Confidence Thresholds of the Text Mining Data.
Supplemental Table 1. Overview of the Mid- and Large-Scale PPI Mapping Studies.
Supplemental Table 2. GO Enrichment Results for the Network Presented in Figure 3.
Supplemental Table 3. Illustrative Examples of Event Extraction from Text, Randomly Chosen from the Interactions Depicted in Figure 3.
Supplemental Table 4. An in-Depth Analysis of the Low Confidence Text Mining Events Appearing in Figure 6.
Supplemental Table 5. Overview of the 26 Candidate Genes Associated with Core CC Genes, Based Solely on Text Mining.
Supplemental Data Set 1. The PLEV Corpus, Containing All Manual Evaluations Performed in This Study.
Supplementary Material
Acknowledgments
We thank Filip Ginter and Chih-Hsuan Wei for their work on the recent update of the EVEX data source, Marijn Vandevoorde and Michiel Van Bel for technical assistance with the text mining evaluation framework, and Yvan Saeys and Sampo Pyysalo for fruitful discussions. We acknowledge the support of Ghent University (Multidisciplinary Research Partnership “Bioinformatics: from nucleotides to networks” and “Bijzonder Onderzoeksfonds Methusalem project” no. BOF08⁄01M00408) and the Research Foundation Flanders for funding (S.V.L. and S.D.B.).
AUTHOR CONTRIBUTIONS
S.V.L. and S.D.B. conceived the study and wrote the article with substantial input from Z.J.D., D.I., and Y.V.D.P. S.V.L. processed the text mining data, designed the manual evaluation study, and interpreted the curated data. Z.J.D. performed the manual evaluation. S.D.B. processed the CORNET data sets. S.D.B. and S.V.L. performed the data integration and network analyses. All authors read and approved the final article for publication.
Glossary
- PPI
protein–protein interaction
- PMC
PubMed Central
- GO
Gene Ontology
- miRNA
microRNA
- TAIR
The Arabidopsis Information Resource
- CC
cell cycle
References
- Alonso-Peral M.M., Sun C., Millar A.A. (2012). MicroRNA159 can act as a switch or tuning microRNA independently of its abundance in Arabidopsis. PLoS ONE 7: e34751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoutzias G.D., Pichler E.E., Mian N., De Graaf D., Imsiridou A., Robinson-Rechavi M., Bornberg-Bauer E., Robertson D.L., Oliver S.G. (2007). A protein interaction atlas for the nuclear receptors: Properties and quality of a hub-based dimerisation network. BMC Syst. Biol. 1: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki K., Ogata Y., Shibata D. (2007). Approaches for extracting practical information from gene co-expression networks in plant biology. Plant Cell Physiol. 48: 381–390 [DOI] [PubMed] [Google Scholar]
- Arabidopsis Interactome Mapping Consortium (2011). Evidence for network evolution in an Arabidopsis interactome map. Science 333: 601–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arighi C.N., Lu Z., Krallinger M., Cohen K.B., Wilbur W.J., Valencia A., Hirschman L., Wu C.H. (2011). Overview of the BioCreative III Workshop. BMC Bioinformatics 12 (suppl. 8): S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader G.D., Betel D., Hogue C.W.V. (2003). BIND: The Biomolecular Interaction Network Database. Nucleic Acids Res. 31: 248–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassel G.W., Gaudinier A., Brady S.M., Hennig L., Rhee S.Y., De Smet I. (2012). Systems analysis of plant functional, transcriptional, physical interaction, and metabolic networks. Plant Cell 24: 3859–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardini T.Z., et al. (2004). Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiol. 135: 745–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björne J., Ginter F., Salakoski T. (2012). University of Turku in the BioNLP’11 Shared Task. BMC Bioinformatics 13 (suppl. 11): S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björne, J., Heimonen, J., Ginter, F., Airola, A., Pahikkala, T., and Salakoski, T. (2009). Extracting complex biological events with rich graph-based feature sets. In Proceedings of BioNLP 2009, (Boulder, Colorado: Association for Computational Linguistics), pp. 10–18. [Google Scholar]
- Brady S.M., Provart N.J. (2009). Web-queryable large-scale data sets for hypothesis generation in plant biology. Plant Cell 21: 1034–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfun-Junior A., Franken J., Mes J.J., Marsch-Martinez N., Pereira A., Angenent G.C. (2005). ASYMMETRIC LEAVES2-LIKE1 gene, a member of the AS2/LOB family, controls proximal-distal patterning in Arabidopsis petals. Plant Mol. Biol. 57: 559–575 [DOI] [PubMed] [Google Scholar]
- Chasman D.I., et al. CARDIoGRAM Consortium ICBP Consortium CARe Consortium WTCCC2 (2012). Integration of genome-wide association studies with biological knowledge identifies six novel genes related to kidney function. Hum. Mol. Genet. 21: 5329–5343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-aryamontri A., Ceol A., Palazzi L.M., Nardelli G., Schneider M.V., Castagnoli L., Cesareni G. (2007). MINT: The Molecular INTeraction database. Nucleic Acids Res. 35 (Database issue): D572–D574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bodt S., Carvajal D., Hollunder J., Van den Cruyce J., Movahedi S., Inzé D. (2010). CORNET: A user-friendly tool for data mining and integration. Plant Physiol. 152: 1167–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bodt S., Hollunder J., Nelissen H., Meulemeester N., Inzé D. (2012). CORNET 2.0: Integrating plant coexpression, protein-protein interactions, regulatory interactions, gene associations and functional annotations. New Phytol. 195: 707–720 [DOI] [PubMed] [Google Scholar]
- Faro A., Giordano D., Spampinato C. (2012). Combining literature text mining with microarray data: advances for system biology modeling. Brief. Bioinform. 13: 61–82 [DOI] [PubMed] [Google Scholar]
- Hasson A., Plessis A., Blein T., Adroher B., Grigg S., Tsiantis M., Boudaoud A., Damerval C., Laufs P. (2011). Evolution and diverse roles of the CUP-SHAPED COTYLEDON genes in Arabidopsis leaf development. Plant Cell 23: 54–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay A., Barkoulas M., Tsiantis M. (2006). ASYMMETRIC LEAVES1 and auxin activities converge to repress BREVIPEDICELLUS expression and promote leaf development in Arabidopsis. Development 133: 3955–3961 [DOI] [PubMed] [Google Scholar]
- Hermjakob H., et al. (2004). IntAct: An open source molecular interaction database. Nucleic Acids Res. 32 (Database issue): D452–D455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyndrickx K.S., Vandepoele K. (2012). Systematic identification of functional plant modules through the integration of complementary data sources. Plant Physiol. 159: 884–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschman L., et al. (2012). Text mining for the biocuration workflow. Database (Oxford) 2012: bas020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikezaki M., Kojima M., Sakakibara H., Kojima S., Ueno Y., Machida C., Machida Y. (2010). Genetic networks regulated by ASYMMETRIC LEAVES1 (AS1) and AS2 in leaf development in Arabidopsis thaliana: KNOX genes control five morphological events. Plant J. 61: 70–82 [DOI] [PubMed] [Google Scholar]
- Inzé D., De Veylder L. (2006). Cell cycle regulation in plant development. Annu. Rev. Genet. 40: 77–105 [DOI] [PubMed] [Google Scholar]
- Johnston A.J., Gruissem W. (2009). Gametophyte differentiation and imprinting control in plants: Crosstalk between RBR and chromatin. Commun. Integr. Biol. 2: 144–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullien P.E., Mosquna A., Ingouff M., Sakata T., Ohad N., Berger F. (2008). Retinoblastoma and its binding partner MSI1 control imprinting in Arabidopsis. PLoS Biol. 6: e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.D., Ohta T., Pyysalo S., Kano Y., Tsujii J. (2011). Extracting bio-molecular events from literature–The BioNLP'09 shared task. Comput. Intell. 27: 513–540 [Google Scholar]
- Klopffleisch K., et al. (2011). Arabidopsis G-protein interactome reveals connections to cell wall carbohydrates and morphogenesis. Mol. Syst. Biol. 7: 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler J., Baumbach J., Taubert J., Specht M., Skusa A., Rüegg A., Rawlings C., Verrier P., Philippi S. (2006). Graph-based analysis and visualization of experimental results with ONDEX. Bioinformatics 22: 1383–1390 [DOI] [PubMed] [Google Scholar]
- Kojima S., Iwasaki M., Takahashi H., Imai T., Matsumura Y., Fleury D., Van Lijsebettens M., Machida Y., Machida C. (2011). Asymmetric leaves2 and Elongator, a histone acetyltransferase complex, mediate the establishment of polarity in leaves of Arabidopsis thaliana. Plant Cell Physiol. 52: 1259–1273 [DOI] [PubMed] [Google Scholar]
- Kourmpetis Y.A., van Dijk A.D., van Ham R.C., ter Braak C.J. (2011). Genome-wide computational function prediction of Arabidopsis proteins by integration of multiple data sources. Plant Physiol. 155: 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krallinger M., Rodriguez-Penagos C., Tendulkar A., Valencia A. (2009). PLAN2L: A web tool for integrated text mining and literature-based bioentity relation extraction. Nucleic Acids Res. 37 (Web Server issue): W160–W165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde S., et al. (2010). A membrane protein/signaling protein interaction network for Arabidopsis version AMPv2. Front. Physiol. 1: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani E.R., Kilinc A., Smyth D.R. (2012). PETAL LOSS is a boundary gene that inhibits growth between developing sepals in Arabidopsis thaliana. Plant J. 71: 724–735 [DOI] [PubMed] [Google Scholar]
- Larkin J.C., Oppenheimer D.G., Lloyd A.M., Paparozzi E.T., Marks M.D. (1994). Roles of the GLABROUS1 and TRANSPARENT TESTA GLABRA genes in Arabidopsis trichome development. Plant Cell 6: 1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue C.T., Wen J., Walker J.C. (2009). Genetic interactions between the miRNA164-CUC2 regulatory module and BREVIPEDICELLUS in Arabidopsis developmental patterning. Plant Signal. Behav. 4: 666–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaman R., Gonzalez G. (2008). BANNER: An executable survey of advances in biomedical named entity recognition. Pac. Symp. Biocomput. 2008: 652–663 [PubMed] [Google Scholar]
- Lee I., Ambaru B., Thakkar P., Marcotte E.M., Rhee S.Y. (2010). Rational association of genes with traits using a genome-scale gene network for Arabidopsis thaliana. Nat. Biotechnol. 28: 149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D., Oppenheimer D.G. (1999). Genetic control of trichome branch number in Arabidopsis: The roles of the FURCA loci. Development 126: 5547–5557 [DOI] [PubMed] [Google Scholar]
- Michoel T., Joshi A., Nachtergaele B., Van de Peer Y. (2011). Enrichment and aggregation of topological motifs are independent organizational principles of integrated interaction networks. Mol. Biosyst. 7: 2769–2778 [DOI] [PubMed] [Google Scholar]
- Morohashi K., Grotewold E. (2009). A systems approach reveals regulatory circuitry for Arabidopsis trichome initiation by the GL3 and GL1 selectors. PLoS Genet. 5: e1000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepusz T., Yu H., Paccanaro A. (2012). Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 9: 471–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T., Kim, J.-D., Pyysalo, S., Wang, Y., and Tsujii, J.i. (2009). Incorporating GENETAG-style annotation to GENIA corpus. In Proceedings of the Workshop on Current Trends in Biomedical Natural Language Processing, (Boulder, Colorado: Association for Computational Linguistics), pp. 106–107. [Google Scholar]
- Phelps-Durr T.L., Thomas J., Vahab P., Timmermans M.C. (2005). Maize rough sheath2 and its Arabidopsis orthologue ASYMMETRIC LEAVES1 interact with HIRA, a predicted histone chaperone, to maintain knox gene silencing and determinacy during organogenesis. Plant Cell 17: 2886–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip S. (2012). Multiple significance tests: The Bonferroni correction. BMJ 344: e509 [Google Scholar]
- Rhee S.Y., et al. (2003). The Arabidopsis Information Resource (TAIR): A model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 31: 224–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A.M., et al. (2012). Uncovering the molecular machinery of the human spindle—An integration of wet and dry systems biology. PLoS ONE 7: e31813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salwinski L., Miller C.S., Smith A.J., Pettit F.K., Bowie J.U., Eisenberg D. (2004). The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 32 (Database issue): D449–D451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C., Breitkreutz B.-J., Reguly T., Boucher L., Breitkreutz A., Tyers M. (2006). BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 34 (Database issue): D535–D539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Zhou Q., Zhang W., Fu Y., Huang H. (2002). ASYMMETRIC LEAVES1, an Arabidopsis gene that is involved in the control of cell differentiation in leaves. Planta 214: 694–702 [DOI] [PubMed] [Google Scholar]
- Tranchevent L.C., Capdevila F.B., Nitsch D., De Moor B., De Causmaecker P., Moreau Y. (2011). A guide to web tools to prioritize candidate genes. Brief. Bioinform. 12: 22–32 [DOI] [PubMed] [Google Scholar]
- Usadel B., et al. (2005). Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol. 138: 1195–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Auken K., et al. WormBase Consortium (2012). Text mining in the biocuration workflow: Applications for literature curation at WormBase, dictyBase and TAIR. Database (Oxford) 2012: bas040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandepoele K., Raes J., De Veylder L., Rouzé P., Rombauts S., Inzé D. (2002). Genome-wide analysis of core cell cycle genes in Arabidopsis. Plant Cell 14: 903–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helden J. (2003). Regulatory sequence analysis tools. Nucleic Acids Res. 31: 3593–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Landeghem S., Björne J., Wei C.-H., Hakala K., Pyysalo S., Ananiadou S., Kao H.-Y., Lu Z., Salakoski T., Van de Peer Y., Ginter F. (April 13, 2013). Large-scale event extraction from literature with multi-level gene normalization. PLoS ONE doi/10.1371/journalpone.005581.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Landeghem, S., Ginter, F., Van de Peer, Y., and Salakoski, T. (2011). EVEX: A PubMed-scale resource for homology-based generalization of text mining predictions. In Proceedings of BioNLP 2011, (Portland, Oregon: Association for Computational Linguistics), pp. 28–37. [Google Scholar]
- Van Landeghem S., Hakala K., Rönnqvist S., Salakoski T., Van de Peer Y., Ginter F. (2012). Exploring biomolecular literature with EVEX: Connecting genes through events, homology, and indirect associations. Adv. Bioinforma. 2012: 582765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leene J., et al. (2010). Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Mol. Syst. Biol. 6: 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C.-H., Kao H.-Y., Lu Z. (2012). SR4GN: A species recognition software tool for gene normalization. PLoS ONE 7: e38460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., Lin W.C., Huang T., Poethig R.S., Springer P.S., Kerstetter R.A. (2008). KANADI1 regulates adaxial-abaxial polarity in Arabidopsis by directly repressing the transcription of ASYMMETRIC LEAVES2. Proc. Natl. Acad. Sci. USA 105: 16392–16397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz A., Mejia-Guerra M.K., Kurz K., Liang X., Welch L., Grotewold E. (2011). AGRIS: The Arabidopsis Gene Regulatory Information Server, an update. Nucleic Acids Res. 39 (Database issue): D1118–D1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.V., King O.D., Wong S.L., Goldberg D.S., Tong A.H., Lesage G., Andrews B., Bussey H., Boone C., Roth F.P. (2005). Motifs, themes and thematic maps of an integrated Saccharomyces cerevisiae interaction network. J. Biol. 4: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Harashima H., Dissmeyer N., Pusch S., Weimer A.K., Bramsiepe J., Bouyer D., Rademacher S., Nowack M.K., Novak B., Sprunck S., Schnittger A. (2012). A general G1/S-phase cell-cycle control module in the flowering plant Arabidopsis thaliana. PLoS Genet. 8: e1002847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.